

Abstract
Cytochrome P450 (CYP) enzymes of the CYP101 and CYP111 families from the oligotrophic bacterium Novosphingobium aromaticivorans DSM12444 are heme monooxygenases that receive electrons from NADH via Arx, a [2Fe-2S] ferredoxin, and ArR, a ferredoxin reductase. These systems show fast NADH turnovers (kcat = 39–91 s−1) that are efficiently coupled to product formation. The three-dimensional structures of ArR, Arx, and CYP101D1, which form a physiological class I P450 electron transfer chain, have been resolved by x-ray crystallography. The general structural features of these proteins are similar to their counterparts in other class I systems such as putidaredoxin reductase (PdR), putidaredoxin (Pdx), and CYP101A1 of the camphor hydroxylase system from Pseudomonas putida, and adrenodoxin (Adx) of the mitochondrial steroidogenic CYP11 and CYP24A1 systems. However, significant differences in the proposed protein-protein interaction surfaces of the ferredoxin reductase, ferredoxin, and P450 enzyme are found. There are regions of positive charge on the likely interaction face of ArR and CYP101D1 and a corresponding negatively charged area on the surface of Arx. The [2Fe-2S] cluster binding loop in Arx also has a neutral, hydrophobic patch on the surface. These surface characteristics are more in common with those of Adx than Pdx. The observed structural features are consistent with the ionic strength dependence of the activity.
Keywords: Bacteria, Crystal Structure, Cytochrome P450, Electron Transfer, Protein-Protein Interactions, Class I P450 Systems, Novosphingobium aromaticivorans DSM12444
Introduction
Cytochrome P450 (CYP)4 enzymes constitute a superfamily of heme-containing monooxygenases (1, 2) that participate in a variety of biological processes such as carbon source assimilation, biosynthesis and biodegradation, xenobiotic detoxification, and metabolism of medicines (1, 2). The most common activity of P450 enzymes is the insertion of an oxygen atom from dioxygen into chemically inert carbon-hydrogen bonds, but other reaction types including dealkylation, desaturation, heteroatom oxidation, epoxidation, phenol coupling, and reductive dehalogenation are also known (3–6). The various activities of P450 enzymes are of great interest due to their potential applications in, for example, synthesis of fine chemicals and drug metabolites under mild conditions with high specificity.
P450 enzymatic activity requires two electrons that are usually derived from NAD(P)H and delivered to the P450s by electron transfer proteins which are broadly divided into two classes (7, 8). Class I systems are diverse, usually consisting of an oxygenase-coupled NAD(P)H-dependent ferredoxin reductase (ONFR) and an iron-sulfur ferredoxin. Such systems are the predominant forms in prokaryotes but are also found in eukaryotic mitochondrial membranes. ONFRs typically contain an FAD cofactor. Ferredoxin cluster types include [2Fe-2S], [3Fe-4S], [4Fe-4S], and combinations of these (7, 9). Non-ferredoxin FMN proteins have also been identified (10). Class II P450 enzymes are most common in eukaryotes and utilize an NADPH-cytochrome P450 reductase (CPR) containing prosthetic groups FAD and FMN (11). Recently other more diverse electron transfer systems for P450 enzymes have been discovered and these have been defined into several new classes (7, 8).
An important difference between the two main classes is that, whereas a single CPR supports the activity of all 57 human P450s and yeast CPRs support the activity of numerous P450s heterologously expressed in the organism, most class I systems show redox partner specificity. Putidaredoxin (Pdx) is well known to have an effector role in CYP101A1 activity (12, 13). The activity of CYP199A2 from Rhodopseudomonas palustris CGA009 has been reconstituted with palustrisredoxin (Pux), a [2Fe-2S] ferredoxin genomically associated with CYP199A2, and an ONFR, palustrisredoxin reductase (PuR) (14). The high demethylation activity of this system is severely compromised in the hybrid PdR/Pdx/CYP199A2 system (14) due to weak ferredoxin/P450 binding (14, 15). Numerous P450 enzymes with potentially interesting and desirable activities are orphaned in the genome; the electron transfer proteins are not located nearby in the gene sequence. Known reductase/redoxin systems have been recruited to reconstitute the activity in vitro and in vivo, with notable successes (16, 17) but generally such cross-reactions are slow (18, 19).
We have reported the heterologous expression and purification of the majority of the 16 P450 enzymes from Novosphingobium aromaticivorans DSM12444 (20–23). A ferredoxin reductase (ArR) and a [2Fe-2S] ferredoxin (Arx) have been identified that reconstitute monooxygenase activities of five of these enzymes (CYP101B1, CYP101C1, CYP101D1, CYP101D2, and CYP111A2) for the fast and efficient oxidation of terpenoid compounds (20, 21). Whole cell systems capable of product formation on the grams per liter scale have been constructed (20).
Arx, and the five P450 enzymes it supports, offer a unique platform for studying CYP electron transfer partner recognition. Here we report the kinetics and properties of these P450 systems and the crystal structures of the complete ArR/Arx/CYP101D1 class I system. The only class I systems that have been completely structurally characterized are the PdR/Pdx/CYP101A1 and the adrenodoxin reductase (AdR)/Adx/CYP24A1 systems (24–28). The structures of the proteins in a hybrid PuR/PuxB/CYP199A2 system have also been reported (14, 15, 29). The protein recognition interactions in the CYP101D1, CYP101A1, CYP199A2, and the mitochondrial CYP11 and CYP24 systems are compared. A thorough understanding of ferredoxin/P450 recognition and electron transfer may allow the efficient reconstitution of P450 activity by modification of either the P450 enzyme or a stable ferredoxin in instances where the natural ferredoxin is unknown, poorly expressed, or the activity is low (15, 30, 31).
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of ArR, Arx, and CYP101D1
General DNA manipulations and microbiological experiments were carried out by standard methods (32). The cloning and expression of all the genes in this study and protocols for protein production and purification have been reported elsewhere (supplemental data) (20, 21). The concentrations of the P450 enzymes were calculated as follows: CYP101B1 ϵ417 = 113 mm−1 cm−1; CYP101C1 ϵ417 = 121 mm−1 cm−1; CYP101D1 ϵ418 = 107 mm−1 cm−1; CYP101D2 ϵ418 = 109 mm−1 cm−1; and CYP111A2 ϵ418 = 107 mm−1 cm−1 (20). The ArR and Arx concentrations were calculated using ϵ458 = 10.0 mm−1 cm−1 and ϵ414 = 9.3 mm−1 cm−1, respectively (20).
Activity Assays
Assays were carried out using a Cary 1E or Cary 50 spectrophotometer. NADH turnover rate assays were performed with mixtures (1.2 ml) containing 50 mm Tris, pH 7.4 (200 mm KCl where required), 0.5 μm CYP, 5 μm Arx, 0.5 μm ArR, and 100 μg ml−1 of bovine liver catalase. The mixtures were oxygenated and then equilibrated at 30 °C for 2 min. Substrates were added as 100 mm stock solutions in ethanol to a final concentration of 1 mm (0.5 mm for β-ionone). NADH was added to about 320 μm (final A340 = 2.00) and the absorbance at 340 nm was monitored. The rate of NADH turnover was calculated using ϵ340 = 6.22 mm−1 cm−1 (Table 1). The analysis of product formation using gas chromatography and the determination of coupling efficiency were carried out as described previously (supplemental data) (20).
TABLE 1.
Substrate binding, steady state turnover activity with a ArR:Arx:CYP concentration ratio of 1:10:1 (0.5 μm CYP enzyme, 50 mm Tris, pH 7.4), and coupling for the five N. aromaticivorans CYP enzymes with their respective substrates
Coupling is the percentage efficiency of NADH utilization for the formation of products. Data for the assays with 200 mm KCl present are given in parentheses. Rates are reported as mean ± S.D. (n ≥ 3) and given in nmol(nmol CYP)−1 s−1. The NADH consumption rate, product formation rate, and coupling of CYP101D2 with camphor in the presence of 200 mm NH4Cl were 13.5 ± 1.0, 12.6 ± 0.2, and 93%, respectively.
| P450 enzyme/substrate | HS heme | Kd | NADH consumption rate | Product formation rate | Coupling |
|---|---|---|---|---|---|
| % | μm | ||||
| CYP101D2/camphor | 30% (40%) | 5.2 ± 0.7 (3.1 ± 0.5) | 33.7 ± 1.3 (12.4 ± 0.8) | 33.5 ± 1.7 (12.4 ± 0.5) | 99% (99%) |
| CYP101D1/camphor | 30% (40%) | 9.1 ± 1.1 (5.9 ± 0.5) | 30.8 ± 0.6 (7.2 ± 0.03) | 29.3 ± 0.6 (6.8 ± 0.08) | 95% (90%) |
| CYP101D1/2-adamantanone | 50% (60%) | 9.5 ± 1.0 (4.8 ± 0.4) | 27.8 ± 0.6 (7.1 ± 0.3) | 24.0 ± 1.1 (6.4 ± 0.2) | 86% (90%) |
| CYP101B1/β-ionone | ≥95% (≥95%) | 0.23 ± 0.1 (0.09 ± 0.03) | 26.7 ± 1.6 (3.5 ± 0.1) | 16.8 ± 1.0 (2.4 ± 0.03) | 63% (69%) |
| CYP101C1/β-ionone | 20% (20%) | 26.6 ± 4.0 (23.7 ± 1.6) | 44.2 ± 1.3 (7.2 ± 0.2) | 33.7 ± 1.4 (4.7 ± 0.4) | 76% (66%) |
| CYP111A2/linalool | ≥95% (≥95%) | 0.47 ± 0.1 (0.26 ± 0.04) | 48.3 ± 3.3 (11.3 ± 0.6) | 30.7 ± 2.1 (7.7 ± 0.5) | 63% (68%) |
To determine the apparent Km and kcat parameters for electron transfer from a ferredoxin to a P450 enzyme, the NADH turnover assays were carried out as above but with a lower P450 concentration (0.1 μm) and the ferredoxin concentration was varied from 1 to 60 μm. The cytochrome c reduction assay to determine the apparent kinetic parameters for the reduction of ferredoxins by the ferredoxin reductases were performed with mixtures (1.2 ml final assay volume) containing 50 mm Tris, pH 7.4 (200 mm KCl where required), 1 nm of the ferredoxin reductase, and a range of ferredoxin concentrations from 1 to 40 μm. The mixtures were equilibrated at 30 °C for 2 min. Cytochrome c was added from a 20 mg ml−1 stock solution in 50 mm Tris, pH 7.4, to a final concentration of 30 μm. NADH was added to about 320 μm (final A340 = 2.00) and the absorbance at 550 nm was monitored. The initial rate of cytochrome c reduction was calculated using ϵ550 = 22.1 mm−1 cm−1. Apparent Km and kcat values were obtained by fitting the initial catalytic turnover rate of cytochrome c reduction or NADH oxidation against the ferredoxin concentration to a hyperbolic function using the Origin 8 software (Origin Labs). All the data showed good fits to hyperbolic behavior with the exception of CYP101C1 with β-ionone, which showed positive cooperativity and was analyzed using the Hill equation.
Substrate Binding Titrations
The P450 enzymes were diluted to 0.5–2.0 μm using 50 mm Tris, pH 7.4, in 2.5 ml and 0.5–2-μl aliquots of substrate were added using a Hamilton syringe from a 1, 10, or 100 mm stock solution in ethanol. The sample was mixed and the peak to trough difference in absorbance was recorded between 700 and 250 nm. Further aliquots of substrate were added until the peak to trough difference did not shift further. The apparent dissociation constants, Kd, were obtained by fitting the peak to trough difference against substrate concentration to a hyperbolic function,
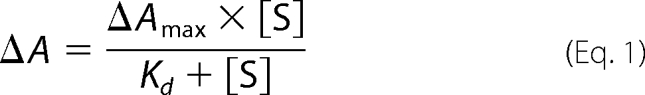 |
where ΔA is the peak to trough absorbance difference, ΔAmax is the maximum absorbance difference, [S] is the substrate concentration. Several substrates exhibited tight binding, with Kd < 1 μm. In these instances the data were fitted to the tight binding quadratic equation (20, 33),
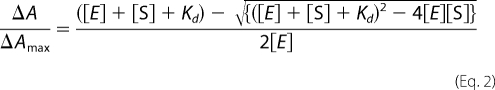 |
where ΔA is the peak to trough absorbance difference, ΔAmax is the maximum absorbance difference, [S] is the substrate concentration, and [E] is the enzyme concentration.
Crystallization and Data Collection
Protein production and purification for crystallographic studies were carried out under modified conditions as described below. The genes encoding full-length ArR and CYP101D1 were subcloned into the vector pET28a(+) (Novagen Inc.) to incorporate an N-terminal His6 tag, and were expressed in Escherichia coli strain BL21(DE3). Cells were grown at 37 °C in 1 liter of Luria-Bertani broth (LB) medium containing 50 μg ml−1 of kanamycin for 5 h to an A600 of 0.6–0.8. Recombinant protein production was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 18 h at 20 °C. Cells were harvested by centrifugation and resuspended in phosphate-buffered saline (1× PBS) and then lysed by sonication at 4 °C. The crude extracts were centrifuged at 27,000 × g for 30 min to remove the cell debris and the supernatant was loaded onto a Ni2+-chelating affinity column (GE Healthcare). The column was washed with 200 ml of wash buffer (1× PBS, pH 7.4, 20 mm imidazole) and the target protein was eluted from the column with elution buffer (1× PBS, pH 7.4, 300 mm imidazole). The pooled fractions were concentrated and then buffer-exchanged into buffer A (20 mm Tris, pH 8.0, 1 mm DTT) and further purified on a Resource Q column (GE Healthcare) using a linear gradient of 0–1 m NaCl in buffer A. Gel filtration chromatography on a Superdex-200 (GE Healthcare) column was used to further purify CYP101D1, eluting with 20 mm Tris, pH 8.0, 150 mm NaCl, 1 mm DTT.
The gene encoding Arx was subcloned into the vector pET-26b(+) (Novagen Inc.) for expression. The growth and expression conditions were the same as for CYP101D1 and ArR. The brown cell pellet from the centrifugation was resuspended in buffer B (20 mm Tris, pH 7.4, 1 mm DTT) and lysed by sonication at 4 °C. The crude extracts were centrifuged at 27,000 × g for 30 min and the supernatant loaded onto a Q Fastflow-Sepharose column (GE Healthcare), and the target protein was eluted using a linear salt gradient of 0–0.5 m NaCl in buffer B. Resource Q and Superdex-75 chromatography (GE Healthcare) were used for further purification with the same buffers and conditions as for CYP101D1 and ArR. The purity of all three proteins was estimated to be >95% by SDS-PAGE analysis (supplemental Fig. S1).
For crystallization the proteins were concentrated to ∼30 mg ml−1 in 20 mm Tris, pH 8.0. All crystals were obtained using the hanging-drop vapor diffusion method at 18 °C using 1 μl of protein solution mixed with 1 μl of reservoir solution and equilibrated with 200 μl of reservoir solution. Crystal screening was carried out with Hampton Research Crystal Screen kits. Good quality crystals of ArR were obtained in 3 days from 0.1 m HEPES, pH 7.6, 17% PEG 8000, and 0.1 m potassium phosphate. Arx and CYP101D1 crystals were obtained in about 1 week from 0.1 m HEPES, pH 7.5, 1.4 m sodium citrate, and 0.1 m Tris, pH 8.2, 12% 1,4-dioxane, 1.6 m ammonium sulfate, respectively. The camphor substrate was soaked into crystals of substrate-free CYP101D1 by adding solid camphor to the drops containing the crystals. Camphor saturates the crystal solution after several days and the soaking time was varied from 1 week to 1 month.
X-ray diffraction data of ArR, Arx, and CYP101D1 in complex with camphor were collected in-house on a Rigaku R-AXIS IV++ image plate using CuKα radiation (λ = 1.5418 Å) from a Rigaku MicroMax-007 rotating anode x-ray generator operating at 40 kV and 20 mA. Diffraction data from native CYP101D1 crystals were collected at −173 °C using an ADSC Quantum 315 detector on beam line BL-5 at the Photon Factory (Tsukuba, Japan). All crystals were cryoprotected by the addition of 20% (v/v) glycerol to the crystallization solutions. All diffraction data were indexed, integrated, and scaled with the HKL2000 package (34). The space groups of ArR, Arx, and CYP101D1 were determined as P43212, C2, and P6422, respectively. Complete data collection statistics for the three proteins are summarized in Table 3.
TABLE 3.
X-ray data collection and structure refinement statistics for ArR, Arx, and substrate-free and camphor-bound CYP101D1 from N. aromaticivorans
| Data collection statistics | ||||
| Data set | ArR | Arx | CYP101D1 | CYP101D1:cam |
| Wavelength (Å) | 1.5418 | 1.5418 | 1.0000 | 1.5418 |
| Space group | P43212 | C2 | P6422 | P6422 |
| Cell dimensions a/b/c (Å) | 140.1/140.1/52.2 | 137.6/78.8/89.2 | 150.9/150.9/195.3 | 151.6/151.6/195.4 |
| α/β/γ (°) | 90.0/90.0/90.0 | 90.0/125.0/90.0 | 90.0/90.0/120.0 | 90.0/90.0/120.0 |
| Resolution (Å) | 50-2.5 (2.59-2.50) | 50-2.3 (2.38-2.30) | 50-2.2 (2.28-2.20) | 50-2.2 (2.28-2.20) |
| Average I/σ(I)a | 21.1 (3.6) | 22.8 (2.5) | 24.5 (2.0) | 35.2 (7.4) |
| Completeness (%)a | 99.8 (99.6) | 99.9 (99.7) | 98.5 (86.9) | 100.0 (100.0) |
| Redundancya | 7.1 (6.6) | 4.1 (3.8) | 9.9 (5.4) | 17.6 (17.6) |
| Rmerge (%)a,b | 9.2 (45.5) | 7.0 (45.1) | 8.0 (44.4) | 8.9 (39.3) |
| Structure refinement statistics | ||||
| Resolution (Å) | 50-2.5 | 50-2.3 | 50-2.2 | 50-2.2 |
| Average B-factor (Å2) | 34.1 | 37.4 | 34.3 | 21.4 |
| Rwork/Rfree (%)c | 18.9/24.4 | 20.0/23.9 | 20.0/24.0 | 18.9/23.6 |
| R.m.s. deviation bond lengths (Å) | 0.007 | 0.016 | 0.016 | 0.013 |
| R.m.s. deviation bond angles (°) | 1.125 | 1.754 | 1.758 | 1.604 |
| Ramachandran plot | ||||
| Most favored (%) | 89.9 | 90.9 | 88.7 | 90.0 |
| Allowed (%) | 9.5 | 9.1 | 10.6 | 9.7 |
| Generously allowed (%) | 0.6 | 0 | 0.7 | 0.3 |
| Disallowed (%) | 0 | 0 | 0 | 0 |
a Values in parentheses correspond to the highest-resolution shell.
b Rmerge = Σi|Ii − 〈I〉|/Σ〈I〉, where Ii is an individual intensity measurement and 〈I〉 is the average intensity for all the reflections.
c Rwork/Rfree = Σ ‖Fo|−|Fc‖/Σ|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
Structure Determination and Refinement
For the structure determination of ArR and native and camphor-bound CYP101D1, the phases were solved using the molecular replacement method with the program Phaser (35) in the CCP4 suite (36). PdR (PDB code 1Q1R) and CYP101A1 (PDB code 2CPP) with camphor removed were used as initial search models (native CYP101D1 was used as a search model for camphor-bound CYP101D1). Electron density maps of ArR and CYP101D1 were obtained after initial model building. These initial models were then rebuilt using COOT (37) and refined by ARP/wARP (38) and Refmac5 (39).
Molecular replacement methods produced poor density maps for Arx. However, Arx contains two intrinsic iron atoms that could be used for initial determination of experimental phases. A single-wavelength anomalous diffraction data set was collected using an in-house Rigaku MM-007 x-ray source. Ten heavy atom positions were identified from the strong anomalous signal with the program Phenix.hyss (40). Resolve (41) was then used for further density modification. The initial Arx model was built using ARP/wARP (38), and COOT (37) was used to make further manual adjustments and model refinement was performed by Refmac5 (39).
The stereochemical qualities of the four refined structures were checked with the program PROCHECK (42). A detailed summary of the refinement statistics are provided in Table 3. The coordinates for the crystal structures of ArR, Arx, substrate-free CYP101D1, and camphor-bound CYP101D1 have been deposited in the Protein Data Bank with the accession codes 3LXD, 3LXF, 3LXH, and 3LXI, respectively.
RESULTS
Electron Transfer Activity of ArR and Arx
Arx supports the monooxygenase activity of five CYP enzymes (CYP101B1, CYP101C1, CYP101D1, CYP101D2, and CYP111A2) from N. aromaticivorans DSM12444 using either PdR as the surrogate ferredoxin reductase (21) or the cognate reductase ArR (20). The ArR-supported hydroxylation activities are 3–5-fold higher than PdR, and the ArR/Arx electron transfer chain is 2 orders of magnitude more active than the PdR/Pdx system with these CYP enzymes (supplemental Table S1).
Potassium binding to CYP101A1 increases the shift of the heme spin state to high spin and cooperatively strengthens camphor binding (43–46). The activity studies on the N. aromaticivorans CYP101 family enzymes in our initial reports were carried out in the presence of 200 mm KCl (20, 21). The effect of KCl concentration has been further studied. Addition of 200 mm KCl results in marginally increased shifts (∼10%) of the heme to the high-spin form and slightly tightens substrate binding (less than 2-fold) for the five P450 enzymes tested here (Table 1). On the other hand, the NADH turnover activities are 3–8-fold higher in the absence of KCl under standard assay conditions (e.g. 33.7 ± 1.3 versus 12.4 ± 0.8 nmol (nmol-CYP)−1 s−1 for CYP101D2, henceforth abbreviated to s−1). Analysis of the organic extracts by gas chromatography showed that substrate oxidation by the Novosphingobium P450 enzymes in the absence of KCl results in the same product distribution as when KCl (up to 500 mm) is present and with similar high product yields based on NADH consumed. With CYP101D2 as a test system, the NADH turnover activity decreases as the salt concentration increases (supplemental Fig. S2a), and the optimal pH of the system is found to be around pH 7.4 (supplemental Fig. S2b).
The apparent kinetic parameters for the electron transfer activity of ArR and Arx have been determined. Using cytochrome c as the terminal electron acceptor (14, 47, 48), the reduction of Arx by ArR shows a kcat of 280 ± 12 s−1 with a Km of 2.9 ± 0.41 μm (Table 2). The kcat is increased slightly to 320 ± 29 s−1 at higher ionic strength (200 mm KCl) but the Km is 5-fold higher (15 ± 2.5 μm). For the hybrid systems, the reduction of Pdx by ArR shows an apparent kcat of 37 ± 5.0 s−1 with a Km of 69 ± 14.6 μm. The reduction of Arx with PdR was not saturated at high concentrations of ferredoxin (40 μm) and the rate of cytochrome c oxidation was also low (<20 s−1). Therefore the greatly reduced activities of the hybrid PdR/Arx and ArR/Pdx electron transfer chains arise directly from the much slower formation of the reduced ferredoxin due to the lower kcat and higher Km compared with the cognate reductase. On the other hand, the reduction of Arx by PuR from R. palustris CGA009 shows a similar kcat to ArR, whereas the Km value of 17 ± 2.2 μm is similar to that for the ArR/Arx reaction in the presence of 200 mm KCl. The reduction of Pux by ArR shows an apparent kcat of 66 ± 1.2 s−1 with a Km of 0.41 ± 0.06 μm.
TABLE 2.
Apparent kinetic parameters (50 mm Tris, pH 7.4, with and without the addition of 200 mm KCl) for reduction of Fdx by FdR using cytochrome c as the terminal electron acceptor, and NADH turnover by the P450 enzymes with different substrates for which the first electron transfer from Arx to the P450 is the slow step
| kcat | Km | ||
|---|---|---|---|
| s−1 | μm | ||
| FdR/Fdx/cyt c | |||
| ArR/Arx | 280 ± 12 | 2.9 ± 0.41 | |
| ArR/Arx, KCl | 320 ± 29 | 15 ± 2.5 | |
| PdR/Arx | –a | –a | |
| PuR/Arx | 250 ± 15 | 17 ± 2.2 | |
| ArR/Pdx | 37 ± 5.0 | 70 ± 14.6 | |
| PuR/Pdx | 600 ± 10 | 12 ± 0.5 | |
| PdR/Pdx | 410 ± 9.0 | 23 ± 1.0 | |
| ArR/Pux | 66 ± 1.2 | 0.4 ± 0.06 | |
| PdR/Pux | 160 ± 10 | 29 ± 3.4 | |
| PuR/Pux | 260 ± 6 | 4 ± 0.3 | |
| ArR/Arx/P450 | |||
| CYP101B1 β-ionone | 68 ± 1.9 | 11 ± 0.74 | |
| CYP101B1 β-ionone. KCl | 83 ± 8.8 | 160 ± 22 | |
| CYP101C1 β-iononeb | 48 ± 0.8 | 0.93 ± 0.04 | n = 1.9 ± 0.1 |
| CYP101C1 β-ionone, KClb | 46 ± 1.8 | 16 ± 1.1 | n = 1.6 ± 0.1 |
| CYP101D1 camphor | 41 ± 0.6 | 2.9 ± 0.14 | |
| CYP101D1 camphor, KCl | 31 ± 0.8 | 30 ± 1.5 | |
| CYP101D2 camphor | 39 ± 0.8 | 1.7 ± 0.12 | |
| CYP101D2 camphor, KCl | 30 ± 0.5 | 9.0 ± 0.38 | |
| CYP111A2 linalool | 91 ± 2.7 | 3.7 ± 0.31 | |
| CYP111A2 linalool, KCl | 63 ± 8.8 | 130 ± 24 | |
a When PdR was used with Arx the rates of reduction of cytochrome c was very slow. When 40 μm Arx was used the turnover rate was less than 20 s−1 and the system did not show saturation suggesting the kcat is low and the Km is high for PdR/Arx, which agree with data observed previously (supplemental Table S1) (20).
b All data showed good fits to hyperbolic behavior with the exception of CYP101C1 with β-ionone, which was fitted to the Hill equation.
For the ArR/Arx/CYP systems, the apparent kcat for the oxidation of reduced Arx by the CYP enzymes varies between 39 ± 0.8 s−1 for CYP101D2 and 91 ± 2.7 s−1 for CYP111A2 under saturating substrate concentrations (Table 2 and Fig. 1). Arx oxidation by CYP101C1 with β-ionone as substrate shows non-hyperbolic behavior, and a fit to the Hill equation gives n = 1.9. For CYP101C1, CYP101D1, and CYP101D2 the turnover rates observed in the steady state assays (ArR:Arx:CYP, 0.5:5:0.5 μm, Table 1) approach the kcat values (e.g. 34 versus 41 s−1 for CYP101D1 and 44 versus 48 s−1 for CYP101C1). However, kcat for CYP111A2 and in particular CYP101B1, exceeds the activities observed under the conditions of the steady state assay (68 versus 27 s−1 for CYP101B1 and 91 versus 48 s−1 for CYP111A2) because these two systems operate further away from saturation kinetics due to the higher apparent Km values (Table 2). The highest activities were observed for CYP111A2 and CYP101B1 whose substrates, linalool and β-ionone, respectively, induce ≥95% spin-state shifts with submicromolar binding constants. In the presence of 200 mm KCl the kcat values vary by 20–30% compared with 5–28-fold increases in Km (e.g. 1.7 ± 0.12 versus 9.0 ± 0.38 μm for CYP101D2 and 3.7 ± 0.3 versus 130 ± 24 μm for CYP111A2). These data show that the dominant factor in the higher activities of the ArR/Arx/CYP systems at lower ionic strength is tighter CYP-Arx binding. A decrease in activity with increasing ionic strength has been observed with other class I CYP systems such as the bovine mitochondrial CYP11 system and the bacterial CYP199A2 and CYP199A4 systems from R. palustris (14, 17, 49).
FIGURE 1.

Kinetic analysis of Arx oxidation with the CYP/substrate combinations: CYP101B1/β-ionone, CYP101C1/β-ionone, CYP101D1/camphor with and without 200 mm KCl, and CYP111A2/linalool. The rate of NADH consumption is given in nmol-nmol P450−1 s−1. Michaelis-Menten kinetics were observed for all the CYP/substrate combinations with the exception of CYP101C1/β-ionone, which showed sigmoidal behavior and a Hill plot analysis gave n = 1.9. The inset shows data for the Arx/CYP101C1/β-ionone system at low ferredoxin concentrations (fitted to the Hill equation).
Crystal Structure of the Ferredoxin Reductase ArR
The crystal structure of ArR was solved at 2.5-Å resolution and refined to a working R-factor of 18.9% and a free R-factor of 24.4% (Table 3), and is composed of an ArR monomer (residues 6–414), 401 water molecules, and one FAD molecule. A disulfide bond exists between Cys123 and Cys218 (Fig. 2a). The overall structure of ArR is similar to those of the glutathione reductase-like ONFR proteins BphA4 (50), PdR (25), and PuR (14). The r.m.s. deviations for Cα atoms are 1.61, 1.49, and 1.59 Å, respectively. As with glutathione reductase-like ONFRs, ArR consists of three distinct domains: an FAD-binding domain (residues 6–115 and 251–328), an NAD-binding domain (residues 116–250), and a C-terminal domain (residues 329–414) (Fig. 2a and supplemental Fig. S3). The FAD-binding domain is structurally the most conserved domain between these ONFR proteins. Minor differences are observed in the NAD-binding domain and the C-terminal domain of ArR (Fig. 2a and supplemental Fig. S4). The presence of a Cys123–Cys218 disulfide bond did not significantly affect the structure.
FIGURE 2.

a, the crystal structure of ArR. The FAD-binding domain, NAD-binding domain, and the C-terminal domain are colored in salmon, green, and blue, respectively. The FAD molecule and the disulfide bond are shown in yellow stick and gray stick representation, respectively. The electron density (2Fo − Fc contoured at 1σ) of the FAD molecule is shown in blue. Regions in which the secondary structure differs from other ONFRs are colored in red. Two random loops (residues 116–118 and 248–250) are found in ArR, whereas the corresponding regions are antiparallel β-sheets in PdR, BphA4, and PuR. The C-terminal helix of ArR is shorter when compared with PdR, BphA4, and PuR. b, the FAD domain of ArR shown from the re side of the FAD molecule. The FAD molecule is shown in yellow and the residues in contact with FAD are shown in gray and the water molecules in red. Residues Leu52, Ser53, Ile162, and Phe330 and some water molecule labels have been omitted for clarity. c, the ArR surface at the top of the adenine ribose moiety. In ArR, residues Arg41 and Asp117 form a salt bridge and hydrogen bond that partially block the cavity found in the more open conformations of PdR and PuR (supplemental Fig. S5).
The orientation of the FAD molecule and its interactions with the polypeptide are similar to those found in the other structurally characterized ONFRs (Fig. 2b). The isoalloxazine ring is buried in the ArR protein and the si side surface is shielded from the solvent by the side chains of Pro50, Ser302, Val303, and Trp331. These residues are conserved in other ONFRs. A large solvent-accessible cavity on the re side surface is filled with water molecules and forms the binding site in ONFR proteins for the nicotinamide ring of the NADH cofactor. The O2 atom of the isoalloxazine ring forms hydrogen bonds with the main chain amide nitrogen of Val303 (3.0 Å) and Wat62 (2.9 Å), whereas the O4 atom is hydrogen-bonded to the Nη atom of Lys54 (2.8 Å) and Wat5 (3.2 Å). Lys54, a key residue in FAD binding and conserved across ONFR proteins, is stabilized by an ionic interaction with Glu165 and a hydrogen bond (2.9 Å) with the carbonyl oxygen of Pro50, and it also contacts the N5 atom of the isoalloxazine ring (3.2 Å). Unique among ONFR proteins with known structure, the ArR adenine ribose moiety interacts with Arg41, whereas the equivalent position in PdR, PuR, and BphA4 is an aspartic acid residue. Arg41 forms a salt bridge and a hydrogen bond with Asp117 (equivalent to Arg in PdR and PuR, and Ala in BphA4) that closes off the cavity above the FAD molecule (Fig. 2c) in ArR. The corresponding residues in PdR (Asp37 and Arg113) and PuR (Asp34 and Arg109) are too distant to interact, and therefore the regions above the FAD in these proteins are more open to bulk solvent (supplemental Fig. S5, a and b).
ArR has a preference for NADH over NADPH (20). There was no evidence of bound NAD+ in the NAD domain of ArR from the experimental electron density map. However, the three loops of ONFR proteins, a Rossmann loop between residues 157 and 163 and the two loops from residues 180 to 188 and 241 to 247, which contact NAD+ (14, 25, 50, 51), are found in similar locations in ArR (supplemental Fig. S6). The residues within these loops are generally well conserved across glutathione reductase-like ONFRs, in particular the three acidic residues Glu165, Glu181, and Glu301 that interact with the NAD cofactor (50). Further details of the structural features of the NAD-binding site in ArR are provided under supplemental data.
The FAD si side surface of ONFRs is the binding site for the ferredoxin electron acceptor. This is supported by a computer model of the PdR-Pdx complex (52), mutagenesis studies of the PuR/Pux interaction (14), the crystal structures of BphA4-BphA3 complexes (51), and the crystal structures of cross-linked PdR-Pdx and AdR-Adx (28, 53). The si side surfaces of ONFRs structurally characterized to date have largely positive electrostatic potential surrounding a central patch of neutral residues located directly above the FAD isoalloxazine ring (Fig. 3a and supplemental Fig. S7, a and b). This patch, consisting of Pro50, Pro51, Val303, Gln304, Asn307, Trp329, and Trp331 in ArR, is highly conserved among ONFRs and is pivotal to ferredoxin binding and dissociation, and intra-complex electron transfer (25, 51–55). Ionic interactions involving charged residues on the periphery of this central patch can also be a determining factor. For example, in the reduction of Pux by PuR, substitution of Lys328 in PuR with Gly increased Km 9-fold, whereas kcat was lowered by just 34% (14).
FIGURE 3.

The electrostatic surface potentials of (a) the FAD si side surface of ArR, (b) the interaction face of Arx surrounding the [2Fe-2S] cluster region, and (c) the proximal faces of CYP101D1. All electrostatic surface potentials were calculated using the program PyMOL. Negatively and positively charged surface areas are colored red and blue, respectively. Compared with their counterparts (supplemental Figs. S7, S11, and S14), residues that contribute to the different surface potential distributions and that may be involved in protein recognition are clearly highlighted.
The electrostatic potentials of the FAD si side surface of ArR, PdR, and PuR (Fig. 3a and supplemental Fig. S7, a and b) have a prominent region of positive charge, around Lys388 and Lys410 in ArR, Lys387 and Lys409 in PdR, and Arg378 and Lys400 in PuR. Among the mainly positive potentials on all the surfaces there are three areas of differences that are potentially significant. First, the potential surrounding Trp331 in ArR is more positive than the corresponding Trp330 in PdR and Trp321 in PuR. This is due to the side chain of Lys338 (Gly337 in PdR, Lys328 in PuR, and notably the equivalent residue is an Arg in both BphA4 and AdR) and Asn333 (Asp332 in PdR and Asp323 in PuR). This latter substitution of an Asp residue for a neutral Asn also leads to a reduced negative potential around the conserved acidic residue Asp336 in ArR (Glu335 in PdR, Asp326 in PuR). Second, the potential around the conserved FAD-binding residue Lys54 in ArR (Lys50 in PdR and Lys47 in PuR) is significantly more negative. This switch is due to the acidic residues Glu48 and Glu60, which correspond to His44 and Lys56 in PdR, and Gln41 and Gly53 in PuR. The third area of difference is around the side chain of Gln304 in ArR (Pro303 in PdR and Gln294 in PuR), which is flanked by Asn307, Thr311, and Tyr324, leading to a slightly negative potential, whereas residues Leu306, Arg310, and Arg322 in PdR, and Thr297, Arg301, and Tyr314 in PuR, impart positive potential to this region in these two proteins.
Crystal Structure of the [2Fe-2S] Ferredoxin Arx
The structure of Arx was refined at 2.3-Å resolution to a working R-factor of 20.3% and a free R-factor of 24.6% (Table 3). Ten iron atom positions were identified within each asymmetric unit, indicating that five Arx molecules were present (Fig. 4a). All five molecules of Arx in the asymmetric unit have very similar folds, with all pairwise r.m.s. deviations for Cα atoms being less than 0.3 Å. Disulfide bonds form between each Cys42 in a pentameric unit and the equivalent residue in an adjacent pentameric unit. Hydrophobic interactions also contribute to this back-to-back packing pattern in the crystals (Fig. 4b and supplemental Fig. S8). However, gel filtration showed that Arx was a monomer in solution, and the pentameric form of Arx is believed to be an artifact of crystal packing (supplemental Fig. S9).
FIGURE 4.

a, the structure of the Arx pentamer in one asymmetric unit. The different Arx molecules are colored in green, yellow, salmon, pink, and cyan; the [2Fe-2S] clusters are shown as brown and yellow spheres. b, the view of the five disulfide bridges (indicated by red circles), which contribute to the back-to-back crystal packing of the Arx pentamers.
Each molecule of Arx was traced from residues 1–104 in the electron density map. Arx has a similar α/β-fold to other vertebrate-type ferredoxins, with five major β-sheets, three α-helices and two 310-helices (Fig. 5a). The r.m.s. deviations for Cα atoms between Arx and the P450-associated ferredoxins Adx and Pdx are 1.37 and 0.97 Å, respectively. The [2Fe-2S] cluster environment, including the conformation of the cluster binding loop between Cys39 and Cys48, is similar to that found in other vertebrate-type ferredoxins (Fig. 5b and supplemental Fig. S10). Cys39, Cys45, Cys48, and Cys85 provide the thiolate ligands for the two irons (supplemental Fig. S10a). Arx also has two cysteines (Cys42 and Cys43) located in the cluster binding loop that do not interact with the [2Fe-2S] cluster (supplemental Fig. S10b). The Cys42 side chain forms disulfide bonds between adjacent pentameric Arx units. The side chain of Cys43 is located on the protein surface and contacts Gln86 (supplemental Fig. S10b). Cysteine residues are also found at the equivalent position to Cys43 of Arx in Fe-S cluster biogenic ferredoxins, e.g. FDVI from Rhodobacter capsulatus (56) and Fdx from E. coli (57), and in the P450-associated ferredoxin terpredoxin (58). The corresponding residue in both Pdx and Pux is alanine while it is a leucine in Adx (24, 29, 59).
FIGURE 5.

a, overlay of the secondary structure of an Arx monomer (green) and Pdx (salmon). The sulfur and iron atoms of the [2Fe-2S] clusters are shown as yellow and brown spheres, respectively. The positional shifts of the C terminus and the two loop regions between residues 6–10 and 73–78 are obvious. b, sequence alignment of Arx with Pdx from P. putida, FdVI from R. capsulatus, Pux and PuxB from R. palustris CGA009, bovine adrenodoxin Adx (truncated at the N and C termini), and terpredoxin from Pseudomonas sp. The alignment was performed using ClustalW and ESPript. Black arrows and cylinders indicate the β-sheets and α-helices, respectively. Conserved residues are highlighted. Two non-ligand cysteines (Cys42 and Cys43) are labeled with red and blue triangles, respectively. c, overlay of the secondary structure of an Arx monomer (green) and Adx (cyan) from the bovine adrenal P450 system.
The most significant secondary structure differences between Arx and other vertebrate-type ferredoxins are observed at the C termini and two loop regions comprising residues 6–10 between β1 and β2, and residues 73–78 between α3 and β4 (Fig. 5, a–c). The C-terminal arms of P450-associated ferredoxins have been shown to be important for recognition and electron transfer with their cognate protein partners. The C terminus of Arx is truncated by one residue compared with other bacterial ferredoxins (except terpredoxin), which all have an additional C-terminal hydrophobic residue. The two acidic residues Glu103 and Asp104 found at the C terminus of Arx are significantly different than those in the other ferredoxins; they align with basic Arg or Lys residues and neutral Asn residues, respectively (Fig. 5b). The side chains of Glu103 and Asp104 point toward the solvent and contribute to the negative electrostatic potential of the Arx surface (Fig. 3b). The C-terminal arm is linked to the cluster binding loop via a series of water molecules, and to loop regions comprising residues 6–10 and 73–78 via hydrogen bonds (supplemental data and Fig. S10c), resulting in these loop regions occupying slightly different positions compared with those in other ferredoxins (Fig. 5, a and c).
The surface potential of Arx has more negative and neutral regions when compared with Pdx, and is more similar in some respects to Adx (Fig. 3b and supplemental Fig. S11, a and b). The surface of Arx around the α1 helix and the cluster binding loop is more neutral, whereas the α3 helix and the C-terminal region are more negative, when compared with Pdx (Fig. 3b and supplemental Fig. S11a). Arg66 in Pdx corresponds to Asp66 in Arx and Glu73 in Adx (Fig. 5b and supplemental Fig. S11b). Within the cluster binding loop the region around Leu38 is predominantly hydrophobic, in common with Adx, which has an Ala residue at the equivalent position, whereas Asp38 gives rise to a prominent area of negative potential in Pdx. Acidic residues dominate the surface potential in the α3 helix and the α3/β4 loop in both Arx and Adx. On the other hand, whereas the Adx surface shows a neutral region extending from the cluster binding loop to the C-terminal region (supplemental Fig. S11b), the C-terminal Asp104 of Arx gives rise to a larger area of negative potential extending from the α3 helix. These differing surface charge distributions will likely play significant roles in binding and recognition of these P450-associated ferredoxins.
Crystal Structures of Native and Camphor-bound CYP101D1
The crystal structures of native and camphor-bound CYP101D1 were both solved at 2.2-Å resolution. The final models contained two molecules per asymmetric unit and were refined to working R-factors of 20.1 and 19.2% and free R-factors of 23.9 and 24.4% for the native and camphor-bound forms, respectively. All molecules were traced from residues 9–416 (see further details under supplemental data). CYP101D1 shows an apparent molecular mass of 45 kDa in gel filtration experiments (supplemental Fig. S12), indicating that the enzyme is a monomer in solution.
CYP101D1 shares 44% sequence identity with CYP101A1 (supplemental Fig. S15), and the structure of the two enzymes are very similar (Fig. 6a and supplemental Fig. S13, a and b), with an r.m.s. deviation of 1.2 Å for Cα atoms. CYP101D1 has a more closed conformation and its G helix is longer at the N-terminal, resulting in a different position of the F-G loop (supplemental Fig. S13, a and b). Other minor differences include two short 310-helices in the N-terminal of CYP101D1, which are absent in CYP101A1 and the β2 and C-terminal β-sheets of CYP101A1 are random loops in CYP101D1 (supplemental Fig. S13, a and b).
FIGURE 6.

a, overall structure of CYP101D1. The more extended N terminus of the G helix and the different position of the F-G loop in CYP101D1 are shown in red. Other differences observed in the N-terminal helical region, the β2 strand, and the C-terminal β sheet are highlighted in supplemental Fig. S13. b, the active site of CYP101D1. The heme (yellow) and the active site residues (green) are shown. The water molecule (red) bound to the heme iron and a single 1,4-dioxane molecule (green), which hydrogen bonds with this water, are also shown. The electron density (2Fo − Fc contoured at 1σ) of the 1,4-dioxane molecule is shown in marine. c, the active site of camphor-bound CYP101D1. The residues that interact with camphor are shown in green stick representation and the electron density (2Fo − Fc contoured at 1σ) of the camphor molecule is shown in marine. d, overlay of the residues in I helix between CYP101D1 (green) and CYP101A1 (salmon). The conformation of the carbonyl oxygen of Asp258 is flipped by ∼90° toward Asn262 due to the formation of a hydrogen bond to the side chain nitrogen of Asn262. The two water molecules that hydrogen bond to Asp258 are also shown as red spheres.
The iron is coordinated on the proximal side of the heme to the sulfur of Cys364 (Fe-S 2.3 Å). To the distal side is a hydrophobic substrate pocket that is defined by the residues Ile87, Trp88, Tyr97, Met99, Thr102, Thr186, Leu251, Leu254, Gly255, Thr259, Val302, Asp304, Ile402, and Val403 (Fig. 6b). These residues are well conserved between CYP101D1 and CYP101A1 (supplemental Table S2). Phe87 in CYP101A1 aligns with Trp88 in CYP101D1; the larger Trp side chain sits high up in the active site and the 6-membered rings of the two residues overlay well, whereas the 5-membered ring of Trp88 points away from the heme and the active site. Phe98 in CYP101A1 aligns with Met99 in CYP101D1. The Met99 side chain S-methyl points away from the heme and the active site. Leu254 in CYP101D1 aligns with Val247 in CYP101A1 and its side chain extends slightly further into the active site (Fig. 6b). The active site of molecule A (of the native CYP101D1 structure) has additional electron density that can be readily modeled as 1,4-dioxane (present in the crystallization reservoir solution) (Fig. 6b). The two oxygen atoms of 1,4-dioxane form hydrogen bonds with Wat463 (2.9 and 3.4 Å), the sixth ligand of the heme iron (Fe-O, 3.0 Å). In molecule B, Wat462 is ligated to the heme iron (2.4 Å) and another region of unidentified density, which is presumed to be 1,4-dioxane, occupies the active site. Unfortunately, partial occupancy did not allow refinement of 1,4-dioxane bound to this chain.
The structure of camphor-bound CYP101D1 is virtually identical to the native structure (r.m.s. deviation for Cα atoms, 0.203 Å). The binding position and orientation of d-(+)-camphor are very similar to the CYP101A1-camphor complex (Fig. 6c) (26). Tyr97 in CYP101D1 provides the hydrogen bond (2.6 Å) to the camphor carbonyl oxygen atom and other residues form van der Waals interactions with the carbon atoms of camphor (supplemental Table S3). The camphor C5 carbon is closest to the heme iron, at 4.3 Å distant, whereas the next nearest camphor carbon atoms are further away (C9, 4.8 Å and C4, 4.8 Å), in agreement with the regioselectivity observed in camphor oxidation (≥98% 5-exo-hydroxycamphor).
CYP101D1 has a closed conformation in both the native and camphor-bound forms with no obvious path for substrate access into the active site. There is a cavity on the F helix side of the F-G loop on the surface of both CYP101D1 and CYP101A1 that may be the opening for substrate access (26). The salt bridge between Arg186 and Asp251 in CYP101A1 (60) that blocks substrate access into the active site via this route is conserved in CYP101D1 (Arg187 and Asp258). However, the Lys178 residue, which forms a salt bridge with Asp251 in CYP101A1, is replaced with a glycine residue (Gly179) in CYP101D1. The residues in the I helix are well conserved between CYP101D1 and CYP101A1, especially between Gly255 and Thr259 of CYP101D1 where the oxygen binding groove is located (26, 61). However, the conformation of the carbonyl oxygen of Asp258 in both native and camphor-bound CYP101D1 is flipped by ∼90° toward Asn262 due to the formation of a hydrogen bond to the side chain nitrogen of Asn262. Moreover, in both CYP101D1 structures, the δ2 side chain oxygen of Asp258 forms hydrogen bonds with the two ordered water molecules nearest to the oxygen binding groove (Fig. 6d). A similar rotation of this carbonyl backbone is found in the ferrous deoxy form of CYP101A1 (61).
The characteristic potassium binding site in CYP101A1, comprising the backbone carbonyl oxygens of Glu84, Gly93, Glu94, and Tyr96, and two solvent water molecules in an octahedral arrangement (26, 45), is absent from CYP101D1. The carboxylate side chain of Glu84 in CYP101A1, which forms a hydrogen bond to one of the water ligands, is replaced with Arg85 in CYP101D1. Substitution of Glu84 with a Lys in the E84K mutant of CYP101A1 has been shown to abolish the effect of potassium ions (45).
The electrostatic potentials of the heme proximal surfaces of CYP101D1 and CYP101A1 show significant differences (Fig. 3c and supplemental Fig. S14a) that may contribute to the specificity for their redox partners. The surface of CYP101D1 is more positive due to the basic residues Arg77, Arg113, Arg126, Arg290, Arg350, Arg363, and Arg371; the corresponding residues in CYP101A1 are Glu76, Arg112, Asp125, Ala283, Gln343, Leu356, and Arg364. The CYP199A2 proximal surface, with equivalent residues Gln71, Arg111, Lys124, Arg285, Lys347, Met360, and Arg368, has an electrostatic potential that is similarly positive (supplemental Fig. S14b) (29). Recently, the crystal structure of CYP24A1 from rat mitochondria was determined (27). Four basic residues (Lys378, Lys382, Arg465, and Arg466), which are conserved among mitochondrial P450 enzymes, are located on the proximal surface of CYP24A1 close to the heme (supplemental Fig. S14c). The mitochondrial CYP11A1, which also receives electrons from Adx, has not been structurally characterized but basic residues, including Lys267, Lys339, Lys343, Lys403, Lys405, and Arg426, have been shown to be important for the Adx/CYP11A1 interaction (49, 62, 63).
DISCUSSION
The full structural characterization of ArR, Arx, and CYP101D1, the three proteins in a physiological class I P450 system from N. aromaticivorans, is reported. Arx is genomically associated with CYP101D2 but not ArR. Nevertheless, the ArR reduction of Arx shows apparent kcat (280 s−1) and Km (2.9 μm) values that are comparable with those of other Class I FdR/Fdx systems. The ArR/Arx electron transport chain supports monooxygenase activity of five different Novosphingobium CYP enzymes for the fast and efficient oxidation of a range of terpenoid compounds in vitro and in vivo.
The oxidation of Arx by the CYP enzymes shows high apparent kcat (39–91 s−1) and low Km (μm range) values. The highest Km (11 μm) is observed with CYP101B1, but the kcat value of 68 s−1 is also among the highest, leading to a high turnover rate under the standard assay conditions with an Arx:CYP101B1 ratio of 10:1. The kcat values for the different CYP enzymes show a reasonable correlation with high-spin heme content. Further analysis on the basis of the rate constant of the first electron transfer and the heme reduction potential will be of interest. CYP101C1 oxidation of Arx unexpectedly shows sigmatropic behavior. The low value of KH (0.93 μm) and a Hill coefficient n of ∼2 indicate positive cooperativity in CYP101C1/Arx binding, leading to a fast NADH turnover rate of 44.2 s−1 under the standard assay conditions. The detailed interactions involved and whether two Arx molecules might bind cooperatively to CYP101C1 remain to be elucidated.
The overall structures of the ArR, Arx, and CYP101D1 proteins are similar to their counterparts in the structurally characterized CYP101A1 and CYP199A2 systems (14, 15, 24–26, 29). ArR belongs to the glutathione reductase-like family of ONFRs and the structure is consistent with its specificity for NADH. Of interest is the more closed conformation of the FAD domain in ArR compared with the other ONFRs due to the salt bridge between Arg41 and Asp117, which could result in more stable FAD binding. Arx adopts the typical structure of vertebrate-type ferredoxins that encompass P450-associated and Fe-S cluster biogenesis functions. The structures of native and camphor-bound CYP101D1 resemble their respective CYP101A1 counterparts. The similar orientation of the I helix residues Asp258 and Asn262 in both forms of CYP101D1 to that of the equivalent residues in the ferrous deoxy form of CYP101A1 (61) is interesting and indicates that protein conformations found in catalytic intermediates are accessible even in the resting state of the enzyme. The highly hydrophobic nature of the CYP101D1 active site is consistent with the entry and binding of 1,4-dioxane from the buffer to the native form. The camphor/enzyme interactions are virtually identical to those found in CYP101A1, with the camphor carbonyl oxygen in CYP101D1 forming a hydrogen bond to Tyr97, the equivalent residue to Tyr96 in CYP101A1 (26). The octahedral cation binding site in CYP101A1 is absent in the CYP101D1 structures, which accounts for the minor effects of KCl addition on the heme spin state and camphor binding by CYP101D1. The data for the other four enzymes (Tables 1 and 2) suggest that the cation binding site is also not properly constituted in these enzymes.
The Ferredoxin Reductase/Ferredoxin Interaction
The activities of the ArR/Arx/CYP systems are 50–400% higher than a hybrid system using PdR and Arx (20). This increase is greater than that observed when the ferredoxin reductases (PdR and PuR) are exchanged in the CYP101A1 and CYP199A2 systems (14). Large differences are observed between the kcat and Km values observed when Arx or ArR is substituted for PdR or Pdx compared with exchange between the PuR/Pux and PdR/Pux or the ArR/Arx and PuR/Pux hybrid systems. This suggests that the features involved in ArR/Arx and PdR/Pdx protein-protein recognition may be more divergent than those of PuR/Pux. Ionic strength has a much greater effect on the Km values of the ArR/Arx and PuR/Pux systems compared with the PdR/Pux system. The Lys328 residue in PuR, which is important in Pux recognition, is conserved in ArR but not PdR (14). The FAD si side surfaces of ArR and PuR in the vicinity of this Lys residue are also more positively charged compared with PdR (Fig. 3a and supplemental Fig. S7, a and b).
The general features of class I electron transfer protein recognition have been established with studies on the CYP101A1 (52, 64–67) and CYP11 systems (49, 68). Structural models of PdR-Pdx and Pdx-CYP101A1 complexes have been proposed from the structures of the individual components and spectroscopic data (52, 65, 69, 70). The structures of cross-linked AdR and Adx, and a related ONFR-ferredoxin (BphA4-BphA3) complex from the biphenyl dioxygenase system of Acidocorax sp. KKS102, have been reported (28, 51). Recently, the structure of a covalently cross-linked PdR-Pdx complex (via Lys409 and Glu72) has been solved, and the resultant investigations indicated that there is a high degree of fine tuning in the protein/protein interactions to allow efficient electron transfer (53, 71). The crystal structures of PuR and PuxB have also provided insight into the recognition interactions (14, 15, 29). Although BphA3 is a Rieske ferredoxin with a different fold from Arx, and AdR has a significantly different structure to ArR, PuR, and PdR, all models and structures indicate extensive non-polar contacts between residues in the [2Fe-2S] cluster binding loop and those closest to the FAD isoalloxazine ring on the si side surface, together with ionic contacts between acidic residues on the α3 helix of the ferredoxin and basic residues on the ferredoxin reductase.
The cluster binding loop of P450-associated ferredoxins is composed of neutral or polar residues (Fig. 3b, 5b and supplemental Fig. S11, a and b) that complement the non-polar region on the FAD si side surfaces of ferredoxin reductases. Of the potential ionic contacts, a PuR-PuxB complex structure determined from double electron-electron resonance spectroscopy suggested that the Lys328 residue of PuR forms an ionic contact with Glu38 in Pux (72). The cross-linked PdR-Pdx complex is linked through Glu72 and Lys409, and a second salt bridge, formed by Asp38 and Arg310, is located at the interface. Besides the salt bridge interaction, both hydrophobic and polar interactions are important in PdR/Pdx binding (53, 71). Glu72 in Pdx and Lys409 in PdR align with Asp72 in Arx and Lys410 in ArR, but Asp38 in Pdx aligns with Leu38 in Arx, and Arg310 in PdR aligns with Thr311 in ArR (the corresponding residues in Pux and PuR are Glu38 and Arg301, respectively). The inability of the Leu38 side chain of Arx to interact in this way with Lys328 may be the cause for the increased Km (17 versus 2.9 μm) for Arx reduction by PuR compared with ArR. Comparison of the FAD si side surfaces (Fig. 3a and supplemental Fig. S7, a and b) also suggests that Asn333 in ArR may have a significant effect. ArR has two regions of negative potential, in the vicinity of Lys54 and Gln304. PuR and PdR both have positive potentials in these areas and yet PuR shows much higher Arx reduction activity than PdR, which points to the region surrounding Trp331 of ArR as being important. The main difference is the substitution of Asp323 in PuR (Asp332 in PdR) for Asn333 in ArR, rendering the region more neutral and accommodating of the Leu38 side chain of Arx (Fig. 3, a and b, and supplemental Figs. S7, a and b, and S11, a and b).
The Ferredoxin/CYP Interaction
The ability of Arx to support electron transfer to five different P450 enzymes indicates that these enzymes should have certain common features for protein recognition and electron transfer. However, the different values of kcat and Km, and the varied sensitivity of the activity of the systems to ionic strength (Table 2), point to effects of specific residues on the recognition surfaces. The decrease in the steady state activities of the five P450 enzymes with increasing ionic strength is mainly due to increased Km values (Table 2) and is indicative of significant electrostatic interactions in the P450/Arx recognition (49, 73, 74). The Arx surface potential in the vicinity of the [2Fe-2S] cluster is mainly negative, with a neutral area immediately surrounding the cluster binding loop (Fig. 3b and supplemental Fig. S11, a and b). The potential of the heme proximal surface of CYP101D1 is mainly positive with some neutral regions, suggesting complementary interactions between the two enzymes (Fig. 3c and supplemental Fig. S14, a–c).
Extensive studies suggested that the interaction between CYP101A1 and Pdx is dominated by contacts between Trp106 (C-terminal residue) and Asp38 (before the cluster binding loop) on Pdx with residues on CYP101A1 (65, 69, 70). Other Pdx residues, on helices α1 (Val28, Tyr33) and α3 (Glu65, Arg66), and the cluster binding loop (Ser42, Ser44) are also proposed to be involved (52, 64). Trp106 has been proposed to interact with Leu356, the residue before the proximal Cys357 in CYP101A1, and this specific contact is suggested to alter the proximal loop structure that is related to the effector requirement of CYP101A1 for Pdx (75). The equivalent residue in CYP101D1 is Arg363, which would match the C-terminal Asp104 residue of Arx. Arg363 is conserved in CYP101D2 and CYP101C1 but it aligns with Met345 in CYP101B1 and Val351 in CYP111A2. Residues Arg112 and Arg364 in CYP101A1, which are predicted to interact with Pdx, are conserved in CYP101D1 (Arg113 and Arg371) as well as the other Novosphingobium enzymes supported by Arx with the exception of CYP111A2 where the equivalent to Arg364 is Glu359 (Table 4 and supplemental Fig. S15).
TABLE 4.
Comparison of the positively charged proximal surface residues of CYP101D1 to those in P450cam (CYP101A1), CYP101D2, CYP101B1, CYP101C1, and CYP111A2
Similar residues across all of the N. aromaticivorans CYP enzymes are highlighted in bold.
| CYP101D1 | CYP101A1 | CYP101D2 | CYP101C1 | CYP101B1 | CYP111A2 |
|---|---|---|---|---|---|
| Arg77 | Glu76 | Ser76 | Gly61 | Ser80 | –a |
| Arg113 | Arg112 | Arg112 | Arg97 | Arg116 | Arg100 |
| Arg126 | Asp125 | Arg125 | Val110 | Arg129 | Gln113 |
| Arg290 | Ala283 | Arg296 | Ala268 | Pro288 | Ala277 |
| Arg349 | Arg342 | Arg348 | Arg327 | Arg347 | Arg337 |
| Arg350 | Gln343 | Arg349 | Ala330b | Lys348 | Pro338 |
| His368 | His361 | His367 | Gly348 | Glu366 | Arg356 |
| Arg371 | Arg364 | Arg370 | Arg351 | Arg369 | Glu359 |
a The equivalent region to the B helix in which Arg77 is situated is absent.
b In CYP101C1 there is a 2-residue insert (glycine and leucine) before the Ala330 residue.
In contrast, the CYP11A1/Adx interaction involves acidic residues on the α3 helix in Adx (Asp72, Glu73, Asp76, and Asp79) and basic residues on CYP11A1 (49, 62, 63). Tyr82 on Adx has also been implicated in CYP11A1 recognition (49). Asp38 of Pdx aligns with Ala45 in Adx (supplemental Table S4) and mutagenesis studies have shown that this residue does not play a significant role in Adx/CYP11A1 binding (49). The structure of CYP24A1 from rat mitochondria revealed several positively charge residues on the proximal face including Arg120, Arg159, Lys164, Lys168, Lys378, Lys382, Lys443, Arg465, and Arg466 (supplemental Fig. S14c) (27). Many of these residues are conserved across mitochondrial P450 enzymes. It has been suggested that ferredoxin/P450 binding in the CYP101A1 and CYP11A1 systems utilize different regions on the P450 protein surfaces (49). Given that the hydrophobic Ala45 of Adx is Leu38 in Arx, that the surfaces around the cluster binding loop in these two ferredoxins are non-polar, and the presence of negatively charged residues in the α3 helix, it appears that the Arx/CYP101D1 interactions resemble those in the Adx-CYP11A1 complex rather than the Pdx/CYP101A1.
In addition to Arg113 and Arg371, there are other positively charged residues on the proximal face of CYP101D1 (Arg77, Arg126, Arg290, and Arg350, Fig. 3c and supplemental Fig. S14) and these are partly conserved across the five P450 enzymes supported by Arx (supplemental Fig. S15). The closest is CYP101D2 (Ser76, Arg112, Arg125, Arg289, Arg349, and Arg370, Table 4) with which Arx is genomically associated. The Arx/CYP101D2 interaction is slightly stronger than that with CYP101D1 (Km 1.7 versus 2.9 μm in 50 mm Tris, pH 7.4) and shows less of an ionic strength dependence (Km 9.0 versus 30 μm in 50 mm Tris, pH 7.4, 200 mm KCl). This suggests that Arg77 may have a functional role. Among the other three P450 enzymes, the variations commonly involve substituting the basic Arg residue for a non-polar, but not an acidic residue. CYP111A2 is more unusual in that sequence alignment suggests that the equivalent region to the B helix in which Arg77 is situated has been deleted, and Arg371 in CYP101D1 aligns with Glu359 in CYP111A2. However, the CYP101D1 surface residue His368 is close to Arg371 (Fig. 3c and supplemental Fig. S15). His368 aligns with Arg356 in CYP111A2, and the side chain of this basic residue may reduce the repulsion between Glu359 and surface acidic residues on Arx. His368 of CYP101D1 aligns with Glu366 in CYP101B1. A negatively charged residue at this position, directly above the heme, and the replacement of Arg363 with Met345 may weaken binding to Arx, which is consistent with the higher Km observed for CYP101B1 turnover.
The structures of Arx and CYP101D1 have enabled structural, sequence, and surface potential comparisons with related enzymes. However, as the analysis of the heme proximal residues shows, although residues likely to play significant role have been identified, differences at one or more residues might be overcome by a compensatory substitution nearby, which makes the identification of the detailed ferredoxin binding interactions by sequence alignment alone, difficult. Moreover, these proteins may also undergo redox-induced conformational changes that can differentially affect the sequential electron transfer from NADH to the CYP enzyme (12, 52). The crystal structures of the other P450 enzymes supported by Arx will be informative for a fuller analysis. The different strengths of Arx/P450 binding and the differential effects of ionic strength on the P450 systems offers numerous opportunities for targeted mutagenesis studies to clarify the role of P450 proximal face regions and residues within them, and to provide a detailed picture of the ferredoxin-P450 electron transfer complex.
Conclusion
The ArR/Arx system can support the activity of at least five of the P450 enzymes from N. aromaticivorans DSM12444 resulting in fast product formation rates. A complete physiological class I electron transfer system (ArR/Arx/CYP101D1) has been structurally characterized. The ArR/Arx interaction is similar to those previously observed with ONFR proteins and P450-associated ferredoxins but the Arx/CYP101D1 interactions appear to have more in common with the bovine Adx/CYP11A1 and rat Adx/CYP24A1 mitochondrial systems than the bacterial Pdx/CYP101A1 system. The ability of a single ferredoxin to support activity of multiple CYP enzymes offers a platform to study ferredoxin-P450 recognition and electron transfer in detail.
Supplementary Material
Acknowledgment
We are grateful to Dr. Zhiyong Lou for help with data collection and processing.
The work was supported by Grant 2007CB914301 from the Ministry of Science and Technology of China Project 973 (to M. B.), Tianjin Municipal Science and Technology Commission Grant 08SYSYTC00200, and a grant from the Higher Education Funding Council for England, UK (to L.-L. W.).
The atomic coordinates and structure factors (codes 3LXD, 3LXF, 3LXH, and 3LXI) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S15 and Tables S1–S4.
- CYP
- cytochrome P450
- P450
- cytochrome P450
- ArR
- ferredoxin reductase
- Arx
- [2Fe-2S] ferredoxin
- PdR
- putidaredoxin reductase
- Pdx
- putidaredoxin
- AdR
- adrenodoxin reductase
- Adx
- adrenodoxin
- BphA4
- NADH dependent ferredoxin reductase
- BphA3
- Rieske-type [2Fe-2S] ferredoxin
- ONFR
- coupled NADH-ferredoxin reductase
- PuR
- palustrisredoxin reductase
- Pux
- palustrisredoxin
- FdVI
- ferredoxin from R. capsulatus
- r.m.s.
- root mean square.
REFERENCES
- 1.Ortiz de Montellano P. R. (ed) (2005) Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd Ed., Kluwer Academic/Plenum Press, New York [Google Scholar]
- 2.Sigel A., Sigel H., Sigel R. (2007) The Ubiquitous Roles of Cytochrome P450 Proteins, 1st Ed., John Wiley & Sons, New York [Google Scholar]
- 3.Cryle M. J., Stok J. E., De Voss J. J. (2003) Aust. J. Chem. 56, 749–762 [Google Scholar]
- 4.Guengerich F. P. (2001) Chem. Res. Toxicol. 14, 611–650 [DOI] [PubMed] [Google Scholar]
- 5.Guengerich F. P. (2001) Curr. Drug Metab. 2, 93–115 [DOI] [PubMed] [Google Scholar]
- 6.Isin E. M., Guengerich F. P. (2007) Biochim. Biophys. Acta 1770, 314–329 [DOI] [PubMed] [Google Scholar]
- 7.Hannemann F., Bichet A., Ewen K. M., Bernhardt R. (2007) Biochim. Biophys. Acta 1770, 330–344 [DOI] [PubMed] [Google Scholar]
- 8.Munro A. W., Girvan H. M., McLean K. J. (2007) Biochim. Biophys. Acta 1770, 345–359 [DOI] [PubMed] [Google Scholar]
- 9.Mandai T., Fujiwara S., Imaoka S. (2009) FEBS J. 276, 2416–2429 [DOI] [PubMed] [Google Scholar]
- 10.Kimmich N., Das A., Sevrioukova I., Meharenna Y., Sligar S. G., Poulos T. L. (2007) J. Biol. Chem. 282, 27006–27011 [DOI] [PubMed] [Google Scholar]
- 11.Hanukoglu I. (1996) Adv. Mol. Cell. Biol. 14, 29–56 [Google Scholar]
- 12.Pochapsky S. S., Dang M., OuYang B., Simorellis A. K., Pochapsky T. C. (2009) Biochemistry 48, 4254–4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brazeau B. J., Wallar B. J., Lipscomb J. D. (2003) Biochem. Biophys. Res. Commun. 312, 143–148 [DOI] [PubMed] [Google Scholar]
- 14.Xu F., Bell S. G., Peng Y., Johnson E. O., Bartlam M., Rao Z., Wong L. L. (2009) Proteins 77, 867–880 [DOI] [PubMed] [Google Scholar]
- 15.Bell S. G., Xu F., Johnson E. O., Forward I. M., Bartlam M., Rao Z., Wong L. L. (2010) J. Biol. Inorg. Chem. 15, 315–328 [DOI] [PubMed] [Google Scholar]
- 16.Nodate M., Kubota M., Misawa N. (2006) Appl. Microbiol. Biotechnol. 71, 455–462 [DOI] [PubMed] [Google Scholar]
- 17.Bell S. G., Tan A. B., Johnson E. O., Wong L. L. (2010) Mol. BioSyst. 6, 206–214 [DOI] [PubMed] [Google Scholar]
- 18.Agematu H., Matsumoto N., Fujii Y., Kabumoto H., Doi S., Machida K., Ishikawa J., Arisawa A. (2006) Biosci. Biotechnol. Biochem. 70, 307–311 [DOI] [PubMed] [Google Scholar]
- 19.Momoi K., Hofmann U., Schmid R. D., Urlacher V. B. (2006) Biochem. Biophys. Res. Commun. 339, 331–336 [DOI] [PubMed] [Google Scholar]
- 20.Bell S. G., Dale A., Rees N. H., Wong L. L. (2010) Appl. Microbiol. Biotechnol. 86, 163–175 [DOI] [PubMed] [Google Scholar]
- 21.Bell S. G., Wong L. L. (2007) Biochem. Biophys. Res. Commun. 360, 666–672 [DOI] [PubMed] [Google Scholar]
- 22.Romine M. F., Fredrickson J. K., Li S. M. (1999) J. Ind. Microbiol. Biotechnol. 23, 303–313 [DOI] [PubMed] [Google Scholar]
- 23.Romine M. F., Stillwell L. C., Wong K. K., Thurston S. J., Sisk E. C., Sensen C., Gaasterland T., Fredrickson J. K., Saffer J. D. (1999) J. Bacteriol. 181, 1585–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sevrioukova I. F., Garcia C., Li H., Bhaskar B., Poulos T. L. (2003) J. Mol. Biol. 333, 377–392 [DOI] [PubMed] [Google Scholar]
- 25.Sevrioukova I. F., Li H., Poulos T. L. (2004) J. Mol. Biol. 336, 889–902 [DOI] [PubMed] [Google Scholar]
- 26.Poulos T. L., Finzel B. C., Howard A. J. (1987) J. Mol. Biol. 195, 687–700 [DOI] [PubMed] [Google Scholar]
- 27.Annalora A. J., Goodin D. B., Hong W. X., Zhang Q., Johnson E. F., Stout C. D. (2010) J. Mol. Biol. 396, 441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Müller J. J., Lapko A., Bourenkov G., Ruckpaul K., Heinemann U. (2001) J. Biol. Chem. 276, 2786–2789 [DOI] [PubMed] [Google Scholar]
- 29.Bell S. G., Xu F., Forward I., Bartlam M., Rao Z., Wong L. L. (2008) J. Mol. Biol. 383, 561–574 [DOI] [PubMed] [Google Scholar]
- 30.Koo L. S., Immoos C. E., Cohen M. S., Farmer P. J., Ortiz de Montellano P. R. (2002) J. Am. Chem. Soc. 124, 5684–5691 [DOI] [PubMed] [Google Scholar]
- 31.Schiffler B., Kiefer M., Wilken A., Hannemann F., Adolph H. W., Bernhardt R. (2001) J. Biol. Chem. 276, 36225–36232 [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, New York [Google Scholar]
- 33.Williams J. W., Morrison J. F. (1979) Methods Enzymol. 63, 437–467 [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 35.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Project T. C. (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 37.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38.Perrakis A., Morris R., Lamzin V. S. (1999) Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 39.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 40.Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 41.Terwilliger T. C. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2144–2149 [DOI] [PubMed] [Google Scholar]
- 42.Laskowski R. A., Macarthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 43.Deprez E., Di Primo C., Hoa G. H., Douzou P. (1994) FEBS Lett. 347, 207–210 [DOI] [PubMed] [Google Scholar]
- 44.Deprez E., Gill E., Helms V., Wade R. C., Hui Bon Hoa G. (2002) J. Inorg. Biochem. 91, 597–606 [DOI] [PubMed] [Google Scholar]
- 45.Westlake A. C., Harford-Cross C. F., Donovan J., Wong L. L. (1999) Eur. J. Biochem. 265, 929–935 [DOI] [PubMed] [Google Scholar]
- 46.Denisov I. G., Frank D. J., Sligar S. G. (2009) Pharmacol. Ther. 124, 151–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lambeth J. D., Seybert D. W., Kamin H. (1979) J. Biol. Chem. 254, 7255–7264 [PubMed] [Google Scholar]
- 48.Roome P. W., Peterson J. A. (1988) Arch. Biochem. Biophys. 266, 32–40 [DOI] [PubMed] [Google Scholar]
- 49.Grinberg A. V., Hannemann F., Schiffler B., Müller J., Heinemann U., Bernhardt R. (2000) Proteins 40, 590–612 [DOI] [PubMed] [Google Scholar]
- 50.Senda T., Yamada T., Sakurai N., Kubota M., Nishizaki T., Masai E., Fukuda M., Mitsuidagger Y. (2000) J. Mol. Biol. 304, 397–410 [DOI] [PubMed] [Google Scholar]
- 51.Senda M., Kishigami S., Kimura S., Fukuda M., Ishida T., Senda T. (2007) J. Mol. Biol. 373, 382–400 [DOI] [PubMed] [Google Scholar]
- 52.Kuznetsov V. Y., Blair E., Farmer P. J., Poulos T. L., Pifferitti A., Sevrioukova I. F. (2005) J. Biol. Chem. 280, 16135–16142 [DOI] [PubMed] [Google Scholar]
- 53.Sevrioukova I. F., Poulos T. L., Churbanova I. Y. (2010) J. Biol. Chem. 285, 13616–13620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crowley P. B., Carrondo M. A. (2004) Proteins 55, 603–612 [DOI] [PubMed] [Google Scholar]
- 55.Prudêncio M., Ubbink M. (2004) J. Mol. Recognit. 17, 524–539 [DOI] [PubMed] [Google Scholar]
- 56.Sainz G., Jakoncic J., Sieker L. C., Stojanoff V., Sanishvili N., Asso M., Bertrand P., Armengaud J., Jouanneau Y. (2006) J. Biol. Inorg. Chem. 11, 235–246 [DOI] [PubMed] [Google Scholar]
- 57.Kakuta Y., Horio T., Takahashi Y., Fukuyama K. (2001) Biochemistry 40, 11007–11012 [DOI] [PubMed] [Google Scholar]
- 58.Mo H., Pochapsky S. S., Pochapsky T. C. (1999) Biochemistry 38, 5666–5675 [DOI] [PubMed] [Google Scholar]
- 59.Müller A., Müller J. J., Muller Y. A., Uhlmann H., Bernhardt R., Heinemann U. (1998) Structure 6, 269–280 [DOI] [PubMed] [Google Scholar]
- 60.Deprez E., Gerber N. C., Di Primo C., Douzou P., Sligar S. G., Hui Bon Hoa G. (1994) Biochemistry 33, 14464–14468 [DOI] [PubMed] [Google Scholar]
- 61.Schlichting I., Berendzen J., Chu K., Stock A. M., Maves S. A., Benson D. E., Sweet R. M., Ringe D., Petsko G. A., Sligar S. G. (2000) Science 287, 1615–1622 [DOI] [PubMed] [Google Scholar]
- 62.Usanov S. A., Graham S. E., Lepesheva G. I., Azeva T. N., Strushkevich N. V., Gilep A. A., Estabrook R. W., Peterson J. A. (2002) Biochemistry 41, 8310–8320 [DOI] [PubMed] [Google Scholar]
- 63.Wada A., Waterman M. R. (1992) J. Biol. Chem. 267, 22877–22882 [PubMed] [Google Scholar]
- 64.Holden M., Mayhew M., Bunk D., Roitberg A., Vilker V. (1997) J. Biol. Chem. 272, 21720–21725 [DOI] [PubMed] [Google Scholar]
- 65.Kuznetsov V. Y., Poulos T. L., Sevrioukova I. F. (2006) Biochemistry 45, 11934–11944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Roitberg A. E., Holden M. J., Mayhew M. P., Kurnikov I. V., Beratan D. N., Vilker V. L. (1998) J. Am. Chem. Soc. 120, 8927–8932 [Google Scholar]
- 67.Sevrioukova I. F., Hazzard J. T., Tollin G., Poulos T. L. (2001) Biochemistry 40, 10592–10600 [DOI] [PubMed] [Google Scholar]
- 68.Schiffler B., Bernhardt R. (2003) Biochem. Biophys. Res. Commun. 312, 223–228 [DOI] [PubMed] [Google Scholar]
- 69.Zhang W., Pochapsky S. S., Pochapsky T. C., Jain N. U. (2008) J. Mol. Biol. 384, 349–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karyakin A., Motiejunas D., Wade R. C., Jung C. (2007) Biochim. Biophys. Acta 1770, 420–431 [DOI] [PubMed] [Google Scholar]
- 71.Churbanova I. Y., Poulos T. L., Sevrioukova I. F. (2010) Biochemistry 49, 58–67 [DOI] [PubMed] [Google Scholar]
- 72.Lovett J. E., Bowen A. M., Timmel C. R., Jones M. W., Dilworth J. R., Caprotti D., Bell S. G., Wong L. L., Harmer J. (2009) Phys. Chem. Chem. Phys. 11, 6840–6848 [DOI] [PubMed] [Google Scholar]
- 73.Hamamoto I., Kurokohchi K., Tanaka S., Ichikawa Y. (1993) J. Steroid Biochem. Mol. Biol. 46, 33–37 [DOI] [PubMed] [Google Scholar]
- 74.Lambeth J. D., Kriengsiri S. (1985) J. Biol. Chem. 260, 8810–8816 [PubMed] [Google Scholar]
- 75.Pochapsky S. S., Pochapsky T. C., Wei J. W. (2003) Biochemistry 42, 5649–5656 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.