

Abstract
The S100B-p53 protein complex was discovered in C8146A malignant melanoma, but the consequences of this interaction required further study. When S100B expression was inhibited in C8146As by siRNA (siRNAS100B), wt p53 mRNA levels were unchanged, but p53 protein, phosphorylated p53, and p53 gene products (i.e. p21 and PIDD) were increased. siRNAS100B transfections also restored p53-dependent apoptosis in C8146As as judged by poly(ADP-ribose) polymerase cleavage, DNA ladder formation, caspase 3 and 8 activation, and aggregation of the Fas death receptor (+UV); whereas, siRNAS100B had no effect in SK-MEL-28 cells containing elevated S100B and inactive p53 (p53R145L mutant). siRNAS100B-mediated apoptosis was independent of the mitochondria, because no changes were observed in mitochondrial membrane potential, cytochrome c release, caspase 9 activation, or ratios of pro- and anti-apoptotic proteins (BAX, Bcl-2, and Bcl-XL). As expected, cells lacking S100B (LOX-IM VI) were not affected by siRNAS100B, and introduction of S100B reduced their UV-induced apoptosis activity by 7-fold, further demonstrating that S100B inhibits apoptosis activities in p53-containing cells. In other wild-type p53 cells (i.e. C8146A, UACC-2571, and UACC-62), S100B was found to contribute to cell survival after UV treatment, and for C8146As, the decrease in survival after siRNAS100B transfection (+UV) could be reversed by the p53 inhibitor, pifithrin-α. In summary, reducing S100B expression with siRNA was sufficient to activate p53, its transcriptional activation activities, and p53-dependent apoptosis pathway(s) in melanoma involving the Fas death receptor and perhaps PIDD. Thus, a well known marker for malignant melanoma, S100B, likely contributes to cancer progression by down-regulating the tumor suppressor protein, p53.
Keywords: Apoptosis, Calcium-binding Proteins, p53, Protein-Protein Interactions, Tumor Suppressor, S100 Proteins, S100B
Introduction
In addition to regulating numerous genes and pathways involved in cell cycle control (1), the tumor suppressor protein, p53, is an important component for inducing apoptosis (2–4). The p53 protein activates the transcription of pro-apoptotic factors (BAX, Bak, Fas/APO-1, PIDD, etc.) as well as suppresses the transcription of anti-apoptotic genes (Bcl-2, Bcl-XL, etc.) (5, 6). In addition, p53 itself up-regulates apoptosis, without transcription activation, by directly localizing to mitochondria following DNA damage and interacting with anti-apoptotic proteins such as Bcl-XL to free pro-apoptotic proteins like BAX (2, 3, 5, 7). Under stress, the p53 protein can also contribute to apoptosis by facilitating the transport of death receptors such as Fas/APO-1 and/or Killer/DR5 from cytoplasmic stores to the cell surface as required for programmed cell death (5, 8). Although, it is now clear that p53-dependent pathways of apoptosis are numerous and are regulated in a cell-type and signal-specific manner (5).
Apoptosis is initiated by a variety of stimuli, including withdrawal of growth factors, activation of specific receptors, such as Fas antigen and TNF receptor, and/or by exposure to UV radiation, γ irradiation, DNA-damaging reagents, and chemotherapeutic drugs. In skin, UV radiation is a major source of DNA damage, and sunlight exposure is now recognized as a primary trigger for the occurrence of malignant melanoma (MM),4 the most aggressive type of skin cancer. In most cells, apoptosis is induced following exposure to UV radiation as part of a protective mechanism, which eliminates severely damaged cells and subsequent malignant transformations; however, apoptosis pathways are suppressed in malignant melanoma despite the fact that this cancer usually retains the gene for wild-type p53 (9–11). Although it is not completely understood why malignant melanoma is resistant to p53 apoptotic pathways, a possibility explored in this study involves the protein S100B, a tumor marker that is highly elevated in malignant melanoma.
S100B is a member of the S100 protein family of Ca2+-binding proteins initially named because of their solubility in 100% saturated ammonium sulfate (12). There are now over twenty S100 proteins widely distributed in human tissue (13–15), including a large numbers of cancers (16–18), making it the largest family of EF-hand calcium-binding proteins (19). S100B, a 21.5-kDa symmetric homodimer, is overexpressed in malignant melanoma (18), anaplastic astrocytomas (20), and glioblastomas (21), and in the case of malignant melanoma, high concentrations of S100B correlate directly with poor prognosis in patients (22). More recently, S100B has gained attention, because it binds directly to the p53 tumor suppressor protein in melanoma (23–26), reduces p53 protein levels (26, 27), and inhibits wild-type p53 functions in malignant melanoma (26–28). Such S100B-dependent effects on p53 are cell-specific, because there is also evidence that S100B rescued a temperature-sensitive p53val135 mutant in fibroblast cell lines (25, 29, 30). As pointed out by Fersht and colleagues (31–33), several S100 proteins bind both the transactivation domain of p53 as well as the C terminus of p53 with varying affinities, and these S100-p53 protein-protein interactions are regulated by post-translational modifications (i.e. phosphorylation and acetylation) (28, 33). Such data provide additional explanations for why the effects of S100 proteins on p53 may be different in various cell types particularly, because S100 proteins themselves are cell specifically distributed (13). Therefore, it is important that specific S100-p53 complexes are considered, in defined cell types, to clearly understand their affect on specific cellular function(s). In this study, we focused our work on how the melanoma marker, S100B, affected p53 activity in several wild-type p53 containing malignant melanoma cells. Our data show that down-regulation of S100B via siRNA (siRNAS100B) decreased the survival of three human malignant melanoma cell lines (C8146A, UACC-2571, and UACC-62) harboring a p53 wild-type genotype (34), but not of a p53 mutant cell line (SK-MEL-28) (34) when exposed to UV radiation. In addition, the induction of apoptosis through an extrinsic pathway was observed in cells with wild-type p53 (C8146A) after UV treatment when S100B expression was inhibited via siRNA. On the other hand, apoptosis activity was significantly lower, under the identical conditions, in scrambled siRNA controls or in cells with mutant p53 (SK-MEL-28). Taken together, these data showed that high levels of S100B found in malignant melanoma contribute to cancer progression by down-regulating wild-type p53 activity and conversely that extrinsic p53-dependent apoptosis pathways could be rescued in malignant melanoma when S100B production is specifically inhibited by siRNA.
EXPERIMENTAL PROCEDURES
Cells and Cell Treatments
The human malignant melanoma C8146A cell line was obtained from Dr. Frank L. Meyskens (University of California, Irvine, CA) (35–38), and the LOX-IM VI (LOX-MM), UACC-62, and UACC-2571 cells were purchased from the Division of Cancer Treatment and Diagnosis, National Cancer Institute at Frederick, MD). With the LOX-MM as an exception, which has low levels of S100B and wild-type p53, the other melanoma cells all have elevated levels of S100B and wild type of p53 (26, 34). Malignant melanoma cells (SK-MEL-28), which have similarly elevated S100B levels and mutated p53 (p53R145L), were purchased from the American Type Culture Collection (Fig. 1). The p53 status in both the C8146A and SK-MEL-28 cell lines was confirmed (26, 39), and both the SK-MEL-28 and the C8146A cells were found to encode p53 with an arginine residue at position 72, which is the more abundant residue at this polymorphic site. Melanoma cells were grown in RPMI 1640 media (Invitrogen) containing 10% fetal bovine serum in the absence of antibiotics.
FIGURE 1.
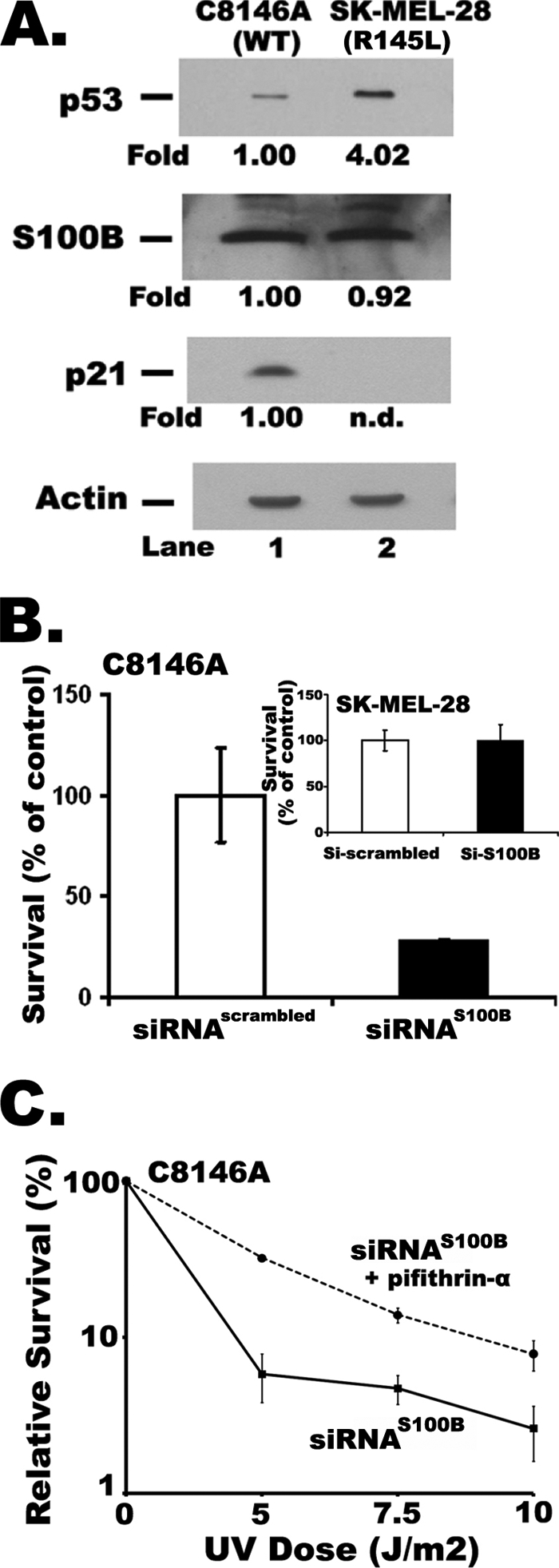
Inhibiting S100B expression via siRNAS100B decreased the survival of malignant melanoma cells after UV treatment in a p53-dependent manner. A, Western blots of endogenous p53, S100B, and p21 levels in cells with wild-type p53 (C8146A, lane 1) and the R145L p53 mutant (SK-MEL-28, lane 2). B, after treatment with UV radiation (10 J/m2), cell survival is lowered with siRNAS100B treatment for cells with wild-type p53 (C8146A), but the cells with the R145L mutant p53 (SK-MEL-28; inset of B) were not affected by siRNAS100B when compared with siRNAscrambled. Survival is expressed as the percentage of colonies obtained with the untreated cells transfected with the scrambled siRNA control (siRNAscrambled). Similar survival results for C8146A and SK-MEL-28 cells were observed at 14 J/m2 UV radiation (see supplemental Fig. 1S), and the % survival ratio of siRNAS100B/siRNAscrambled at 5 J/m2 was 45 ± 5% (not shown); C, the relative survival of C8146A cells was monitored as a function of UV dose (0–10 J/m2) for cells transfected with siRNA directed against S100B (filled squares; siRNAS100B) and siRNAS100B plus 20 μm pifithrin-α (filled circles). These values were recorded as % of survival versus controls without UV treatment (i.e. survival with UV/survival without UV treatment). The error for the 5 J/m2 point with si-S100B plus 20 μm pifithrin-α treatment (filled circle) was smaller in value than the size of the symbol.
siRNA
As previously described (26), three siRNA constructs (termed siRNAS100B) reduced S100B mRNA by 5- to 7-fold as determined by qPCR. However, the highly stable S100B protein was only reduced by ∼2-fold when dually transfected at 24 and 48 h, as previously reported (26). When the siRNAS100B was only transfected once up to 48 h, the reduction of S100B was minimal, if observed at all, due to the high stability of the S100B protein. Of the three siRNA constructs that reduced S100B levels, the 21-nucleotide double-stranded RNA sequence (5′-GGAAUUCAUGGCCUUUGUU-3′) was used extensively for studies here, because it corresponds to the S100B C-terminal end (nucleotides 216–234) plus a 2-dT 3′-overhang, which does not have homology with any other S100 protein, and was shown to be specific for S100B (i.e. versus S100A1 and S100A4) (26). Scrambled siRNA was purchased from Ambion (Silencer negative control number 1 siRNA, catalogue number 4611; antisense 5′-CCGUAUCGUAAGCAGUACUTT-3′). The siRNA directed against S100B (siRNAS100B) or scrambled siRNA was transfected as described above with the Ambion siPORT lipid transfection agent according to the manufacturer's instructions (Ambion, Austin, TX).
Cell Survival Assay
C8146A, UACC-62, UACC-2571, and SK-MEL-28 melanoma cells were grown to confluence and then plated at 1000 cells/100-mm dish. 24 h later, the cells were transfected with 20 nm scrambled siRNA or siRNAS100B, and the next day the transfections were repeated with the same amount of siRNA. On the third day, the cells were rinsed with phosphate buffer, exposed to UV radiation or sham irradiated, and replenished with growth media. Cells were then allowed to grow for one (SK-MEL-28) or 2 weeks (C8146A, UACC-2571, LOX-MM, and UACC-62). For treatments of C8146A cells with the p53 reversible inhibitor pifithrin-α (Calbiochem), cells were treated with 20 μm pifithrin-α 4 h prior to UV treatment, as previously described (40). Pifithrin-α is a small water-soluble p53 inhibitor that was isolated in 1999 (41). It prevents transactivation of p53-responsive genes and suppresses p53-dependent apoptosis (41). It is commonly used to distinguish p53-dependent and independent apoptosis (42–48). Although other proteins are also targeted by pifithrin-α, they required much higher doses than what is necessary to inhibit wild-type p53 (49). Therefore pifithrin-α is considered a specific p53 inhibitor when used at these low doses (i.e. 20 μm). Colonies were then fixed, stained with crystal violet (0.5% in 20% methanol) for 1 h, rinsed in water, and counted. The survival fraction was determined by dividing the number of colonies formed in cells transfected with siRNAS100B by those transfected with scrambled siRNA (±UV). The UV source was a Philips 30-watt germicidal lamp emitting at 253.7 nm and calibrated with a UVX Radiometer (UVP Inc., Upland, CA).
Internucleosomal Fragmentation Assay
C8146A and SK-MEL-28 cells were grown to confluence, plated at 1.8 × 105 cells/100-mm dish, and transfected with siRNA as described above. 4 h after UV exposure (14 J/m2), siRNA-transfected cells were lysed in buffer containing 500 mm Tris-HCl (pH 9.0), 20 mm EDTA (pH 8.0), 10 mm NaCl, 1% SDS, 1 mg/ml Proteinase K, and incubated at 50 °C for 24 h. The 4-h time point after UV treatment was chosen to minimize p53-independent apoptotic effects that arise at later time points as was observed for fibroblasts (50, 51), A549 cells (52), and U2OS cells (40). DNA was purified from the cell extract via phenol extraction/isopropanol precipitation, re-suspended in H2O, digested with RNase A, loaded onto a 1.5% agarose gel, and stained with ethidium bromide.
Caspase 3 Assays
C8146A and SK-MEL-28 cells were grown to confluency, plated at 4.2 × 105 cells/100-mm dish, and transfected with siRNA as described above. Where indicated, cells were incubated at 4 °C for 30 min to prevent Fas aggregation (40) before exposure to UV radiation treatment (14 J/m2). For caspase 3 assays of LOX-MM cells, the siRNAscrambled and siRNAS100B transfections and UV treatment were as described above, and the pCMV5 S100B mammalian expression vector and vector alone were transfected into the cells (5 × 105 cells/100-mm dish) prior to the assay, as described previously (27). In all cases, fresh media were added to the cells 5 h after transfection, and the cells were harvested 48 h later. For measurement of cellular apoptosis, caspase 3 activity was assayed 4 h after UV exposure in 96-well plates according to the manufacturer's recommendation (Promega, Madison, WI) by monitoring the fluorescence of the cleaved substrate with a multi-mode microplate reader (BioTek, Synergy HT). Briefly, the harvested cell pellets were re-suspended in hypotonic cell lysis buffer (25 mm HEPES, pH 7.5, 5 mm MgCl2, 5 mm EDTA, 5 mm DTT, 2 mm PMSF, 10 μg/ml pepstatin A, and 10 μg/ml leupeptin) and lysed using four freeze/thaw cycles. Enzymatic activity from 25 μg of cellular extract was normalized by subtracting background fluorescence values obtained in the presence of a specific caspase 3 inhibitor, Ac-DEVD-CHO (aldehyde) (acetyl-Asp-Glu-Val-Asp-CHO).
UV Radiation or Bleomycin Treatment for Western Blots
C8146A and SK-MEL-28 cells were grown to confluency, plated at 4.2 × 105 cells/10-mm dish, and transfected with siRNA as described above. Cells were exposed to UV radiation (0–20 J/m2) or treated with 10 μg of bleomycin (Enzo Life Sciences) for 4 h. Protein samples were separated on NuPAGE 4–12%, Bis-Tris Gel (Invitrogen) and transferred to 0.2-μm PVDF (Millipore), and Western blot analyses were performed, as described previously (27) using the following antibodies: p53 mouse monoclonal antibody (DO-1, Calbiochem), phospho-p53(Ser-15) polyclonal antibody (Cell Signaling), S100B rabbit monoclonal antibody (Abcam), p21 rabbit monoclonal antibody (Calbiochem), cytochrome c mouse monoclonal antibody (BD Biosciences), caspase 8 and 9 mouse monoclonal antibody (Cell Signaling Technology), caspase 3 rabbit polyclonal antibody (Cell Signaling Technology), BID rabbit polyclonal antibody (Cell Signaling Technology), PIDD rabbit polyclonal antibody (also termed LRDD, Abcam), VDAC (also called Porin) mouse monoclonal antibody (Calbiochem), and actin mouse monoclonal antibody (Calbiochem).
Total RNA Isolation
Total RNA was isolated from cells by the TRIzol plus the PureLink Micro-to-midi total RNA purification system according to the manufacturer (Invitrogen Inc.).
RT-PCR and qPCR
Reverse transcription-PCR was performed to assess the expression levels of multiple transcripts using the SuperScript One-Step RT-PCR kit with Platinum Taq polymerase (Invitrogen, Inc.) and the following DNA primers: p53-forward, 5′-CCCCTCCTGGCCCCTGTCATCTTC-3′; p53-reverse, 5′-GCAGCGCCTCACAACCTCCGTCAT-3′; p21-forward, 5′-GAGGCCGGGATGAGTTGGGAGGAG-3′; p21-reverse, 5′-CAGCCGGCGTTTGGAGTGGTAGAA-3′; HDM2-forward, 5′-TAGTATTTCCCTTTCCTTTGATGA-3′; HDM2-reverse, 5′-CACTCTCCCCTGCCTGATAC-3′; S100B-forward, 5′-AGCTGGAGAAGGCCATGGTG-3′; S100B-reverse, 5′-GAACTCGTGGCAGGCAGTAG-3′; GAPDH-forward, 5′-ACCACAGTCCATGCCATCAC-3′; and GADPH-reverse, 5′-TCCACCACCCTGTTGCTGTA-3′.
Briefly, 100 ng of total RNA and 0.2 μm of each primer (final concentration) were used for each reaction. The cDNA syntheses were performed at 50 °C for 30 min and pre-denaturation at 94 °C for 2 min. PCR amplification was performed with 24–32 cycles (depending on the gene) using a Robocycler Gradient 96 PCR instrument (Stratagene Inc.) with the following protocol: first, the sample was denatured at 94 °C for 30 s, then annealed at 55 °C for 30 s, and lastly extended at 72 °C for 1 min with the final extension cycle performed at 72 °C for 10 min rather than for 1 min.
Quantitative PCR (qPCR) was completed with total RNA isolated using the RNeasy system (Qiagen, Valencia, CA). Single-stranded cDNA was generated using an iScript cDNA Synthesis Kit (Bio-Rad) following the manufacturer's directions. A 1:50,000 dilution of the intercalating dye SYBR Green I (Molecular Probes) was added to the PCR reactions, and amplification products were quantified in real-time using an iCycler (Bio-Rad). For qPCR, the GAPDH primers used were 5′-CGGAGTCAACGGATTTGGTCGTAT-3′ and 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′, and the S100B primers were 5′-ATGTCTGAGCTGGAGAAGGC-3′ and 5′-TTCAAAGAACTCGTGGCAGG-3′ rather than those listed above.
Mitochondrial Membrane Potential Measurements
Changes in the mitochondrial membrane potential were detected using the dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide (JC-1, Sigma). Transfection and UV treatment were as described above. Four hours after UV treatment (14 J/m2), siRNA transfected cells were treated with trypsin, spun down, mixed with the staining solution, incubated for 20 min at 37 °C, centrifuged, and re-suspended in 1 × JC-1 staining buffer on ice. Changes in fluorescence, as a result of varying mitochondria membrane potential in the cells, were then detected by flow cytometry within 30 min.
Sub-cellular Fractionation
Transfection and UV treatments were as described above. Specifically, 4 h after UV treatment (14 J/m2), adherent cells (∼2 × 107) were harvested, and the mitochondrial and cytosolic fractions were isolated with a Mitochondria Isolation Kit for Cultured Cells according to the manufacturer (Pierce).
Confocal Laser Scanning Microscopy
Cells were first seeded on coverslips and transfected with siRNA as described above. Four hours after UV treatment (14 J/m2), siRNA-treated cells were fixed in 4% paraformaldehyde and washed three times with PBS. Cells were washed once with 0.1 m glycine for 10 min and then three times with PBS. Cells were blocked in PBS with 2% BSA for 1 h at room temperature and incubated with Fas antibody (Calbiochem) diluted in PBS with 1% BSA at 4 °C overnight. The samples were further processed by an indirect immunofluorescence technique using FITC-conjugated goat anti-mouse antibodies (KPL, Gaithersburg, MD). Nuclear DNA was counterstained with 4′,6′-diamidineno-a-phenylindole (DAPI, Vector Laboratories, Burlingame, CA). The specimens were analyzed with an LSM 410 Zeiss confocal laser scanning microscope with an ×63 objective lens.
RESULTS
Inhibiting S100B Expression (via siRNAS100B) Sensitizes Human Melanoma Cells with Wild-type p53 to UV Radiation but Has No Effect on Cells with Mutant p53 (SK-MEL-28 and p53R145L)
Malignant melanoma cells, which have similarly elevated S100B levels (Fig. 1A), were selected for comparison, because the C8146A cells have wild-type p53 and the SK-MEL-28 cells have an inactive p53 mutant (p53R145L) (34). As a result of the R145L mutation, the SK-MEL-28 cells cannot induce expression of the cell cycle inhibitor p21 (Fig. 1A, lane 2) or any other p53 downstream target as reported previously (26, 35, 36, 39). Although these two cell lines are not isogenic, they provided useful correlations with regard to p53 function when S100B protein production was inhibited by siRNA (siRNAS100B).
The survival of the C8146A and SK-MEL-28 cells was compared first (±UV treatment) after transfection with either siRNAscrambled or siRNAS100B (Fig. 1 and supplemental Fig. 1S). In these assays, C8146A cells transfected with siRNAS100B were 3- to 4-fold more sensitive to UV radiation (0–20 J/m2, p value < 0.03) and thus were less capable of surviving than cells transfected with siRNAscrambled (Fig. 1B). On the other hand, survival of the SK-MEL-28 cells, with the inactive p53 mutant, was not affected by siRNAS100B as compared with siRNAscrambled at any dose of UV tested (Fig. 1B). Thus, inhibiting S100B significantly reduced the survival of wild-type p53-containing cells (C8146A) after UV treatment; whereas, inhibiting S100B production had no impact on the survival of mutant p53 containing cells (SK-MEL-28, Fig. 1B). To obtain more direct evidence that wild-type p53 that was responsible for sensitizing siRNAS100B-treated C8146A cells to UV treatment (Fig. 1B), we next completed these same survival assays in the presence or absence of the specific p53 inhibitor, pifithrin-α (Fig. 1C). In the absence of pifithrin-α, the data confirmed that down-regulating S100B via siRNA (closed squares) decreased the survival of C8146A cells when compared with transfections with siRNAscrambled. However, when p53 was inhibited by pifithrin-α, cells expressing reduced levels of S100B (si-S100B plus pifithrin-α, closed circles) were significantly more resistant to cell death at every UV dose tested versus in the absence of the p53-targeting drug (si-S100B, closed squares). Specifically, an increase of >6-fold in the capacity of the cells to survive 5 J/m2 of UV radiation was observed when the p53 inhibitor, pifithrin-α, was added together with siRNAS100B (i.e. compare closed circle and closed square, p value < 0.006, Fig. 1C). It was therefore concluded that the ability of siRNAS100B to reduce cell survival in C8146A cells was dependent on the presence of the wild-type tumor suppressor protein, p53 (Fig. 1, B and C).
As with C8146A cells, UV-dependent survival assays were repeated with two additional human melanoma cell lines harboring a wild-type p53 genotype (UACC-2571 and UACC-62) (34). The data shown in Fig. 2 indicated that down-regulation of S100B via siRNA also sensitized UACC-2571 (p value < 0.004 at all doses) and UACC-62 melanoma cells to UV radiation; however, the effect was most pronounced at lower UV doses for the UACC-62 cells (5, 7.5 J/m2, p value < 0.03). The lesser sensitivity to higher UV doses for these two cell lines was not completely understood (Fig. 2), but it was likely related to the different levels of S100B, other S100 proteins, and wild-type p53 protein levels in these two cell lines, as was observed previously (26). In summary, the cell survival data for all three melanoma cell lines containing wild-type p53 (Figs. 1 and 2) demonstrated that elevated S100B levels, as is found in most human malignant melanoma, contributed to UV resistance to cell death in a p53-dependent manner (i.e. for C8146A cells), and thus it was not surprising that lowering S100B did not affect survival of SK-MEL-28 cells, because these cells harbor the inactive p53R145L mutant.
FIGURE 2.

Transfections of siRNA directed against S100B reduce the survival of two additional wild-type containing p53 melanoma cell lines (UACC-2571 and UACC-62 cells). The relative survival data are illustrated as a function of UV dose (0–10 J/m2) in the presence of either scrambled siRNA (filled diamonds; siRNAscrambled) or siRNA directed against S100B (filled squares; siRNAS100B). These data with wild-type p53-containing cells (UACC-2571, UACC-62, and C8146A data from Fig. 1) are in contrast to cell survival data for cells containing mutant p53 (SK-MEL28), which show no difference in cell survival at any dose of UV tested when transfections of si-S100B (siRNAS100B: 0 J/m2, 100%; 5 J/m2, 63 ± 11%; 7.5 J/m2, 45 ± 14%; 10 J/m2, 32 ± 12%) or scrambled siRNA are compared (siRNAscrambled: 0 J/m2, 100%; 5 J/m2, 61 ± 12%; 7.5 J/m2, 46 ± 15%; and 10 J/m2, 28 ± 14%). These values were recorded as % of survival versus controls without UV treatment (i.e. percentage of survival with UV/survival without UV treatment).
Inhibiting S100B via siRNA Restored Wild-type p53 Protein Levels and Transcription Activation in Wild-type C8146A Human Malignant Melanoma Cells
In the absence of UV radiation, previous studies have indicated that a direct interaction between S100B and p53 contributed to the down-regulation of the tumor suppressor at the protein level, and correspondingly, p53 transcription activation function was significantly impaired (26, 27). In an effort to delineate how down-regulation of S100B affects cell survival with UV (Fig. 1), we next monitored the p53 protein, phosphorylated p53 protein, and one of its downstream targets, p21, in C8146A and SK-MEL-28 cells (±UV) when S100B was inhibited by siRNA for (Fig. 3). The mRNA levels for p53 were also monitored under identical conditions, but no change in the levels of p53 mRNA was observed as a function of siRNAS100B under any of the conditions tested (data not shown).
FIGURE 3.

Western blots of C8146A (wild-type p53, lanes 1–4) and SK-MEL-28 (L145Rp53, lanes 5–8) melanoma cells transfected with either siRNAS100B or siRNAscrambled (±UV) using antibodies specific for S100B, p53, phosphorylated p53 (at Ser-15 or Ser-392), and two downstream p53 targets (i.e. p21 and hdm2). UV radiation was administered to cells in lanes (+UV, lanes 3, 4, 7, and 8) labeled as described under “Experimental Procedures.” To calculate the relative protein levels, blots were analyzed using densitometry and normalized to cells transfected with scrambled siRNA and not UV treated (i.e. fold = 1.0), with actin blots used as loading controls (C8146A, lane 1; SK-MEL-28, lane 5). Lanes in which no protein could be detected are labeled nd (i.e. not detected). For the C8146A and SK-MEL-28 cells, the blot of phospho-p53 (at Ser-15) for the scrambled siRNA controls (lanes 1 and 5) were weak at the exposure time illustrated, but readily detected with longer exposure times.
In the absence of UV treatment, lowering the S100B protein via siRNA in C8146A cells corresponded to elevated levels of wild-type p53 protein and its downstream targets, p21 and hdm2 (Fig. 3, compare lanes 1 and 2), as previously reported (26). For comparison in the absence of UV, a parallel set of siRNAS100B experiments was completed with SK-MEL-28 cells containing the p53R145L mutant. As found with C8146A cells, transfection of siRNAS100B into SK-MEL-28 cells resulted in higher basal levels of p53R145L mutant protein (Fig. 3, compare lanes 5 and 6). That the siRNAS100B did not restore expression of the p53 targets, p21 or hdm2, in SK-MEL-28 cells (Fig. 3, lanes 5 and 6) was consistent with the p53 mutant genotype of this cell line (39).
Protein expression levels in the p53 wild-type (C8146A) and p53 mutant (SK-MEL-28) containing cells were examined next after being exposed to UV radiation. These experiments were completed to determine whether lowering S100B protein levels could also affect p53 induction in response to UV radiation. As previously established by others (50, 51), we also observed increases in total and phosphorylated p53 proteins in response to UV radiation in C8146A cells and increases in total p53 in SK-MEL-28 melanoma cells (Fig. 3). Thus, 4 h after UV radiation treatment of C8146A cells, p53 and phosphorylated p53 protein levels (i.e. at Ser-15 and Ser-392) were increased when compared with untreated cells (Fig. 3, compare lanes 1 and 3). In SK-MEL-28 cells, p53 levels were also elevated upon UV treatment, although to a lesser degree (Fig. 3, compare lanes 5 and 7). Importantly, additional increases in p53 protein levels and phosphorylated p53 protein were observed in response to UV radiation when C1846A were transfected with siRNAS100B (Fig. 3, compare lanes 3 and 4). Also, the combination of siRNAS100B together with UV treatment gave the largest increases in p53 and phosphorylated p53 protein when compared with sham irradiated cells transfected with scrambled siRNA (Fig. 3, compare lanes 1 and 4). Likewise, transfections with siRNAS100B minimized UV-induced down-regulation of the p21 protein (Fig. 3, compare lanes 1, 3, and 4), which was observed here and by others in UV treatments alone (53). UV-treated SK-MEL-28 cells containing p53R145L also showed increased mutant p53, when transfections with siRNAscrambled and siRNAS100B were compared (Fig. 3, compare lanes 7 and 8). As expected, such increases in the p53R145L mutant protein did not correspond to any detectable p21 (Fig. 3A, lanes 7 and 8), because this p53 mutant in SK-MEL-28 cells has no functional activity. Thus, activation of p53 and phosphorylated p53 protein resulted from inhibiting S100B via siRNA (±UV); however, only in the C8146A cells did the activation of p53 levels result in transcription activation leading to the accumulation of the p53 target protein, p21.
We have previously shown by structural analysis (NMR) that S100B binding to p53 blocks sites of p53 phosphorylation and acetylation that are important for transcription activation (54). It is thus possible that the inhibitory effect of S100B on p53 activation is specific for UV-induced signal transduction mechanisms. To test this possibility, we next analyzed the effect of down-regulating endogenous S100B protein on p53 activation in response to the DNA-damaging reagent, bleomycin, in C8146A cells (supplemental Fig. 2S). As with UV treatment, data with bleomycin implicated S100B in the down-regulation of phosphorylated p53 (supplemental Fig. 2S, compare lanes 3 and 4 for p53P-S15). However, unlike what is observed with UV, bleomycin treatment alone did not result in lower p21 levels (supplemental Fig. 2S, compare lanes 1 and 3). Nonetheless, as expected, the downstream gene product, p21, was activated with siRNAS100B and corresponded to increases observed in p53 levels (supplemental Fig. 2S, compare lanes 3 and 4 for p21). Thus, regardless of the triggering mechanism to activate p53 (i.e. UV versus bleomycin treatment), lowering S100B levels contributed to the restoration of p53 protein, phospho-p53, and the p53 target, p21, even when different types of cellular stress were administered.
Although, a protein-protein interaction was previously detected between S100B and p53 in melanoma, it remained important to show whether or not down-regulation of S100B affected levels of p53 message. Thus, the effect of siRNAS100B on p53 and p53 target gene mRNA expression levels was monitored semi-quantitatively via a reverse-transcriptase PCR and more quantitatively via real-time PCR methods (qPCR). Together, these data indicated that siRNAS100B treatment lowered S100B mRNA levels by 2- to 8-fold in both C8146A and SK-MEL-28 cells resulting in only a 2-fold loss of S100B protein, at best (Fig. 3). That mRNA levels were largely affected with lesser affects on S100B protein levels were attributed to the high stability of S100 proteins, such as S100B (13). However, when mRNA levels were monitored, no changes in p53 mRNA levels were detected in either cell line under any conditions after the siRNAS100B transfections (siRNAS100B versus siRNAscrambled; ±UV). For the downstream p53 gene target, p21, the mRNA was found to be up-regulated (±UV) in C8146A cells by 2- to 3-fold when treated with siRNAS100B. This result with p21 mRNA was consistent with the increasing levels of wild-type p53 and p21 proteins observed in these cells when S100B was inhibited by siRNAS100B (Fig. 3). When cells were treated with scrambled siRNA, down-regulation of p21 protein levels (Fig. 3) but not mRNA levels were observed following UV exposure. This apparent disconnect between p21 mRNA and p21 protein levels following UV radiation has been reported before and is attributed to a p53-independent translational/post-translational events involving the p21 protein (48). In SK-MEL-28 cells, the mRNA for p53R145L gene was also constant under all conditions of siRNA treatment (siRNAS100B versus siRNAscrambled; ±UV). Furthermore, the elevated levels of the p53R145L mutant protein from siRNAS100B treatment (Fig. 3) did not affect p21 mRNA levels because of the deleterious mutation of p53 in these cells, as expected. Lastly, the siRNA treatments did not trigger RNA degradation under any of the conditions tested, because ribosomal RNA was well preserved in all the samples (data not shown).
In summary, it was concluded that lowering S100B protein levels via siRNA did not affect p53 mRNA levels under any conditions (siRNAS100B versus siRNAscrambled; ±UV), so the S100B-dependent down-regulation of p53 protein levels and function observed here (Figs. 1–3, supplemental Figs. 2S and 3S), and previously (26, 27), was likely the result of a direct S100B-p53 protein-protein interaction detected in vitro (23, 24, 26, 28, 31–33) and via coimmunoprecipitation from C8146A cells (26).
S100B Inhibits UV-induced Apoptosis in Cells with Wild-type p53 (C8146A) but Not in Cells with Mutant p53 (SK-MEL-28 and p53R145L)
We next explored whether the up-regulation of wild-type p53 and phosphorylated p53 proteins via siRNAS100B (Fig. 3) was sufficient to restore apoptosis in cells containing wild-type p53 (i.e. C8146A cells, Fig. 4). As a control, apoptosis was also assayed by monitoring internucleosomal DNA cleavage under identical conditions for melanoma cells with similarly elevated levels of S100B, but mutant p53 (i.e. in SK-MEL-28 cells, Fig. 4). These data indicated that down-regulation of S100B protein via siRNA (siRNAS100B) significantly increased DNA fragmentation in C8146A when exposed to UV radiation (Fig. 4, lane 7). This effect was specific, because no comparable increase was observed in transfections with scrambled siRNA (Fig. 4, lane 6). Furthermore, the apoptotic effect was likely p53-dependent, because transfections of siRNAS100B into SK-MEL-28 cells, harboring a deleterious p53 mutant, did not induce genomic DNA fragmentation whether or not UV radiation was applied (Fig. 4, lanes 10–14).
FIGURE 4.

Assays measuring internucleosomal DNA cleavage in C8146A and SK-MEL-28 cells transfected with either siRNAS100B or siRNAscrambled (±UV). Ethidium bromide staining of agarose gels with genomic DNA extracted from C8146A cells (lanes 3–7) and SK-MEL-28 cells (lanes 10–14) 4 h after UV exposure to the cells transfected with either siRNAS100B or siRNAscrambled. Molecular weight markers (lanes 1 and 8) and media controls were also included on each gel (lanes 3 and 10).
Additional methods to measure apoptosis were also performed on both C8146A and SK-MEL-28 cells when S100B expression was inhibited by siRNAS100B (±UV). This included comparing the activation of the DNA repair associated enzyme poly(ADP-ribose) polymerase (PARP) and by examining caspase 3, 8, and 9 activities (Fig. 5). Densitometry analysis indicated that ∼8 times more PARP cleavage occurred in C8146A cells (wild-type p53) pretreated with siRNAS100B and then exposed to UV radiation (Fig. 5A, lanes 4) than was observed in cells pretreated with scrambled siRNA under similar conditions (Fig. 5A, lane 3). In the absence of UV, PARP cleavage was not readily observed in C8146A cells whether or not S100B expression was inhibited (Fig. 5, lanes 1 and 2). As was found with PARP cleavage and DNA ladder formation, the effect of S100B on UV-induced caspase 3 and 8 activation was also clear when treatments with siRNAS100B and siRNAscrambled were compared (Fig. 5, A and B, lanes 3 and 4). Furthermore, evidence that apoptosis was induced by siRNAS100B via an extrinsic pathway was provided when caspase 8 activation, but not caspase 9 activation, was observed in C8146A cells (Fig. 5A, lane 4, +UV). In SK-MEL-28 cells (p53R145L mutant), neither caspase 3, 8, or 9 activity nor PARP cleavage was observed in cells transfected with siRNAS100B (Fig. 5, A and B, lanes 5–8), a result that was consistent with the absence of DNA fragmentation under the same conditions (Fig. 4, lanes 10–14). As a secondary control, we also demonstrated that LOX-MM melanoma cells, which have little or no detectable S100B protein (Fig. 6A, inset), were not affected by siRNAS100B as monitored by caspase 3 activity (±UV, Fig. 6A). Likewise, transfection of S100B into these cells reduced caspase 3 activity (>7-fold), consistent with S100B-dependent down-regulation of wild-type p53 and its associated apoptosis activities (Fig. 6B).
FIGURE 5.

Assays for PARP cleavage and caspase 3, 8, and 9 activation in C8146A and SK-MEL-28 cells transfected with either siRNAS100B or siRNAscrambled (±UV). A, Western blots of C8146A (lanes 1–4) and SK-MEL-28 (lanes 5–8) cells transfected with either siRNAS100B (lanes 2, 4, 6, and 8) or siRNAscrambled (lanes 1, 3, 5, and 7) using antibodies specific for PARP, caspase 8, and caspase 9. Cells were either mock irradiated (lanes 1, 2, 5, and 6) or exposed to UV radiation (+UV, lanes 3, 4, 7, and 8). Labeled with asterisks are regions on the gels where cleavage products of PARP, caspase 8, and caspase 9 arise in apoptotic cells. To calculate the relative protein levels, blots were analyzed using densitometry and normalized to cells transfected with scrambled siRNA and not UV-treated (i.e. fold = 1.0), with actin blots used as loading controls (C8146A (lane 1); SK-MEL-28 (lane 5)). B, bar graphs illustrating caspase 3 enzymatic activity (±UV) in C81461A cells and SK-MEL-28 cells transfected with scrambled siRNA (open bars) or siRNAS100B (solid bars). In each assay, the relative caspase 3 activities (units × 10−4) were reported after subtracting basal fluorescence intensity in the presence of a specific caspase 3 inhibitor (Ac-DEDV-CHO).
FIGURE 6.

siRNAS100B does not affect apoptosis activity in LOX-IM VI cells (LOX-MM) with little or no exogenous S100B and normal levels of wild-type p53 (±UV); however, transfection of S100B into LOX-MM cells reduced UV-induced apoptosis. A, apoptosis was monitored by the measurement of caspase 3 activity in LOX-MM cells in the presence of scrambled and siRNAS100B (±UV). The inset shows the relative amount of p53 and S100B proteins in LOX-MM, and C8146A cells for comparison, as determined by densitometry measurements of Western blots of p53 and S100B. B, the ratio of caspase 3 activity in the presence or absence of UV treatment is plotted for LOX-MM cells after transfection of either an empty pCMV or a pCMV vector containing the S100B gene (+S100B). These data illustrate that the introduction of the S100B protein into LOX-MM significantly reduces UV-dependent apoptosis (>10-fold).
The results from these five apoptosis assays (DNA ladder, PARP, and caspase 3, 8, and 9; Figs. 4 and 5) indicated that lowering S100B expression with siRNAS100B activated UV-induced apoptosis via a caspase 8 pathway (versus caspase 9 pathway). That the extrinsic pathway was induced was confirmed when it was observed that siRNAS100B induced expression and aggregation of Fas in C8146A cells (Fig. 7A); whereas, Fas aggregation was not observed in p53 mutant cell lines despite the fact that Fas was elevated under all conditions tested (Fig. 7A). This was consistent with a previous report indicating that p53 can regulate sensitivity to apoptosis by allowing cytoplasmic death receptors redistribution to the cell surface (8). To evaluate further the role of Fas aggregation in the siRNAS100B sensitization to UV-induced apoptosis, we incubated the cells at 4 °C prior to UV exposure to prevent Fas aggregation (55). The data shown in Fig. 7B (lane 4) indicated that preventing Fas aggregation in C8146A cells by lowering the temperature (4 °C, dotted bar, lane 4) was sufficient to abrogate the activation of caspase 3 observed when S100B is down-regulated via siRNA and treated with UV radiation (black bar, lane 4). The effect was specific, because cold exposure had no significant effect on apoptosis in cells transfected with the control siRNA (Fig. 7B, lane 3). That p53 was involved in the UV-induced apoptosis recorded when S100B was inhibited by siRNA was also supported by the inability to generate any apoptosis activity in SK-MEL-28 cells containing mutant p53 (Figs. 4–6). Other possible extrinsic pathways activated by p53 were also examined, and it was found that the p53-induced protein with a death domain (PIDD (56–58)) was up-regulated with siRNAS100B transfections (supplemental Fig. 3S) in response to UV radiation indicating that more than one stress-induced extrinsic apoptosis pathway is likely activated when S100B levels are reduced.
FIGURE 7.

Expression and activation of Fas in C8146A via siRNAS100B but not in SK-MEL-28 cells. A, C8146A cells were monitored by indirect immunofluorescence for levels of Fas and Fas clustering in the presence of either siRNAS100B or siRNAscrambled (±UV). In addition, SK-MEL-28 cells were similarly monitored for levels of Fas and Fas clustering in the presence of either siRNAS100B or siRNAscrambled (±UV). DNA content is visualized by DAPI. B, C8146A cells were transfected with either siRNAscrambled or siRNAS100B and incubated at 37 °C (filled bars; w/Fas clustering) or 4 °C (dotted bars; Fas clustering inhibited) for 30 min prior to UV exposure or sham irradiation. Four hours after UV or sham irradiation, the cells were harvested, and caspase 3 activity was measured as described under “Experimental Procedures.”
Next it was found that down-regulation of S100B restored UV-induced apoptosis solely via extrinsic versus intrinsic pathways. This question was explored first by examining whether changes in the expression of the pro- and anti-apoptotic proteins (BAX, Bcl-XL, and Bcl-2) could be detected when siRNAS100B and scrambled siRNA were transfected into C8146A and SK-MEL-28 cells, respectively (Fig. 8). Likewise, in parallel experiments, cytochrome c release and mitochondrial membrane potential were examined in both C8146A and SK-MEL-28 cells to determine whether or not inhibiting S100B production affected p53-dependent apoptosis pathways involving the mitochondria (59, 60). These experiments also established that S100B did not affect mitochondrial events, which can occur via UV-dependent H2O2 production under some conditions (61). Specifically, under the conditions tested here, it was found that no significant changes in the levels of anti- or pro-apoptotic proteins (BAX, Bcl-XL, and Bcl-2) were detected when transfections of siRNAscrambled and siRNAS100B were compared in C8146A and SK-MEL-28 malignant melanoma cells (Fig. 8). Likewise, no changes in the mitochondrial transmembrane electrical potential (Δψ) were observed in siRNAS100B experiments performed with these cells (Fig. 8), a result that further demonstrated the lack of involvement of the mitochondria in S100B apoptotic events. A change in the mitochondrial membrane potential did however occur for the positive controls using valinomycin, which efficiently permeabilizes the mitochondrial membrane, and dissipated the mitochondrial electrochemical potential in both C8146A and SK-MEL-28 cells (Fig. 8). In a third experiment, no changes in cytochrome c release were detected when C8146A and SK-MEL-28 cells transfected with siRNAS100B and scrambled siRNA were compared (data not shown), which is yet another indication that the apoptosis activity observed for C8146A cells treated with siRNAS100B (Figs. 4–6) did not involve any components of an intrinsic apoptotic pathway involving the mitochondria. Thus, the data here demonstrated that inhibiting S100B via siRNA restored the extrinsic apoptosis pathway(s) via the Fas death receptor and perhaps via PIDD but had little or no affect on the mitochondrial-dependent intrinsic pathways.
FIGURE 8.

The expression of pro- or anti-apoptotic proteins (i.e. BAX, Bcl-2, and Bcl-XL) or the mitochondrial membrane potential were not affected by transfections with siRNAS100B or siRNAscrambled (±UV) in either C8146A or SK-MEL-28 cells. A, Western blots of C8146A (wild-type p53, lanes 1–4) and SK-MEL-28 (L145Rp53, lanes 5–8) melanoma cells transfected with either siRNAS100B or siRNAscrambled (±UV) using antibodies specific for BAX, Bcl-2, and Bcl-XL proteins. To calculate the relative protein levels (i.e. -fold changes), blots were analyzed using densitometry and normalized to cells transfected with scrambled siRNA and not UV treated (i.e. -fold = 1.0), with actin blots on the same blot used as loading controls. B, line graphs illustrating no change in the ratio of BAX: Bcl-2 proteins (◇, open diamonds) or the ratio of BAX:Bcl-XL proteins (▵, open triangles) as analyzed from Western blots of C81461A cells and SK-MEL-28 cells transfected with siRNAscrambled or siRNAS100B (±UV; 14 J/m2). C, no changes in the mitochondrial membrane potential (i.e. little if any monomeric JC-1) were observed in C81461A or SK-MEL-28 cells transfected with siRNAscrambled or siRNAS100B (±UV). Cells that were not transfected with siRNA (±UV) were included as additional negative controls and C8146A and SK-MEL-28 cells treated with valinomycin alone were included as positive controls.
DISCUSSION
Most malignant melanoma strains have a wild-type p53 genotype (62), and yet they fail to properly induce cell cycle arrest or have an adequate apoptosis response even when treated with DNA-damaging agents or UV radiation (9–11). Here, we examined whether high levels of S100B expression, as found in malignant melanoma, was sufficient to prevent such p53-dependent functions (16–18). This investigation was initiated based on previous studies, which showed that elevated levels of S100B correlated with reduced levels of p53 protein (26, 27). Additionally, it was found that S100B interacts directly with p53 resulting in lower p53 protein levels and a corresponding loss of its transcriptional activity in cells (26, 27).
In cell survival assays performed here, melanoma cells containing wild-type p53 were found to be more sensitive to UV damage than SK-MEL-28 melanoma cells with mutant p53, which is in good agreement with previous comparisons of SK-MEL-28 cells to lymphoblastoid and other wild-type p53-containing melanoma cells (63, 64). That p53 was important in such longer term survival assays was supported here, because a significant increase in the sensitivity of C8146A, UACC-2571, and UACC-62 melanoma cells to UV-induced cell death was observed when these malignant melanoma cells were treated with siRNAS100B; whereas, no effect was observed for SK-MEL-28 cells treated similarly (Figs. 1 and 2). Further evidence was also provided when the p53-specific inhibitor, pifithrin-α, which abolished this affect, and the C8146A cells once again became resistant to UV radiation even when wild-type p53 levels were elevated as a result of siRNAS100B treatments. The cell survival assays (Figs. 1 and 2 and supplemental Fig. 1S) taken together with the apoptosis data discussed below (Figs. 4–7) indicated that elevated levels of S100B contribute to cancer progression by inhibiting p53-dependent apoptosis in melanoma.
To examine this idea more directly, it was next discovered that decreasing the S100B calcium-binding protein via siRNA was sufficient to restore p53, phosphorylated p53, and its associated activities, including apoptosis in C8146A cells with wild-type p53 but not in SK-MEL-28 cells, with an inactive mutant of p53 (Figs. 4, 5, and 7). The impact of S100B on p53-dependent apoptosis was verified using apoptosis assays such as DNA ladder formation, caspase 3 and 8 activation, PARP cleavage, and the activation of Fas (Figs. 4–7). Further, the restored apoptotic activity was found to arise at a relatively short time point (4 h) after treatment with UV radiation. That apoptosis measured here was via an extrinsic pathway was concluded, because transfections with siRNAS100B induced activation of Fas and caspase 8, but had no affect on caspase 9 activation. Additional experiments at this time point, including measurements of the anti- and pro-apoptotic protein levels (BAX, Bcl2, and Bcl-XL), lack of Bid cleavage (data not shown), changes in mitochondrial membrane potential, and cytochrome c release, all of which were not affected by transfections with siRNAS100B, confirmed that the mitochondria was not involved in the apoptotic response observed in C8146A cells transfected with siRNAS100B (Fig. 8). That the extrinsic apoptotic pathway(s) (Fas and PIDD) were activated in C8146A cells when S100B levels were reduced was particularly interesting in light of the fact that a peptide from the C terminus of p53, which binds to S100B and inhibits the S100B-p53 interaction (28), also restored Fas-mediated apoptosis in glioma, another cancer that has highly elevated levels of S100B and wild-type p53 (65). Therefore, in light of these studies, the activation of Fas and perhaps other p53-regulated death receptors (i.e. PIDD, supplemental Fig. 3S) should be characterized further in C8146A and other melanoma and glioma cell lines containing wild-type p53.
Closer examination of the relative protein and mRNA levels of p53 and p21 in C8146A cells, as a function of UV radiation alone, and together with siRNAS100B, provided some additional insights regarding the resistance of melanoma cells to apoptosis. First, UV radiation treatments alone affected C8146A cells in a manner similar to other wild-type p53 cells (40, 51, 52, 63, 66–72). As was reported for A549 cells (52) and several other wild-type p53 containing melanomas (40, 53, 73–75), the C8146A cells monitored shortly after UV treatment (<6 h) were also found to have lower p21 protein levels (Fig. 3A, compare lanes 1 and 3). Interestingly, in UV-treated C8146A cells, transfection of siRNAS100B restored p21 levels by 3-fold (Fig. 3A, compare lanes 3 and 4), whereas no changes in BAX, Bcl-2, or Bcl-XL expression were observed (Fig. 8). Thus, at early time points after UV treatment, p21 levels, a relatively strong p53 target gene product, was significantly restored, whereas other more weakly targeted p53 genes (i.e. BAX, Bcl-2, and others) were not appreciably affected. Nonetheless, at the 4-h time point, siRNAS100B up-regulated total p53 and phosphorylated p53 (Figs. 3A), which was sufficient for the restoration of apoptosis activity via extrinsic apoptotic pathway(s) (Figs. 4–7 and supplemental Fig. 3S). Previous reports have indicated that p53 can facilitate the redistribution of the death receptors from the Golgi to the cell surface (8). Our data are consistent with these findings and indicate that S100B is preventing p53 from inducing apoptosis through the extrinsic pathways.
There are some conflicting reports on the functional significance of the S100B-p53 interaction (29, 30). However, studies from the Fersht group have begun to identify the source of these discrepancies, because they have discovered that several S100 proteins can interact with p53 and that these S100 proteins interact with the transactivation and/or the C terminus of p53 with affinities that vary by as much as an order of magnitude for two specific sites on p53 (31–33, 76). With this information in mind, it now becomes important to know the relative amount of each member of the S100 protein family member within a particular cell type, because their abundance could result in different functional outcomes with regard to p53 activity.
In this study, we concentrated on melanoma cell lines expressing very high levels of endogenous S100B and low levels of wild-type p53 as a paradigm for the development of future therapies for malignant melanomas (26, 27). However, such an approach of inhibiting S100B also warrants further investigation for other cancers with elevated S100B and wild-type p53 such as several very aggressive brain cancers. In particular, many astrocytomas and gliomas have elevated S100B and harbor wild-type p53, and previous studies have demonstrated that reducing S100B levels in C6 glioma cells is sufficient to elevate p53 at the protein level (77), as found here for malignant melanoma.
With respect to p21, our results in C8146A cells are also reminiscent of data from other laboratories in which sufficient levels of p21, in addition to having sufficient levels of p53, were required for apoptosis via both p53-dependent (78, 79) and p53-independent mechanisms (80–82); so these data with p21 should also be considered further in future works.
In summary, the data presented here are consistent with a mechanism in which elevated S100B levels found in malignant melanoma contribute to the down-regulation of the p53 tumor suppressor via a direct S100B-p53 protein-protein interaction observed previously (28). Such an interaction may occur by S100B destabilizing p53 and/or preventing the formation of p53 oligomers, as observed previously (23, 31–33) and by NMR from our group (data not shown). Additionally, it is possible that elevated S100B blocks covalent modification of p53 (i.e. phosphorylation and acetylation) in cells as was observed previously in vitro, which could also contribute to p53 down-regulation (28). Thus, in either case, melanoma cells overexpressing S100B are unable to efficiently undergo apoptosis even when treated with UV radiation, because they have lower than normal levels of p53 and/or phosphorylated p53 protein. However, when wild-type p53 protein, phosphorylated p53 protein, and protein levels of early gene targets such as p21 are increased beyond a minimal threshold, by lowering S100B levels via siRNA, then the p53 protein and phospho-p53 levels are sufficiently elevated as is necessary to activate extrinsic apoptosis pathways (Figs. 4, 5, and 7). Furthermore, that diminishing S100B levels, by as little as 2-fold (Fig. 3 and supplemental Fig. 2S), was sufficient to activate wild-type p53 and reduce the ability of malignant melanoma to survive (Figs. 1, 2) was very encouraging and may represent a new therapeutic approach for activating apoptosis in cancers with elevated S100B and wild-type p53, such as for malignant melanoma and glioma. Fersht and colleagues (31, 32) demonstrated several calcium-dependent S100-p53 interactions, necessitating the importance of examining the levels and role(s) of other S100 proteins in regulating p53 function in (i) malignant melanoma, (ii) other cancers, and (iii) in healthy cells where as many as 20 or so S100 family members can be variably expressed in a cell-specific manner (13).
Supplementary Material
Acknowledgments
We thank Jin Hyen Baek for technical advice on DNA ladder formation and Michele Vitolo for checking the p53 status of the cell lines.
This work was supported, in whole or in part, by National Institutes of Health Grants GM58888 and CA107331 (to D. J. W.). This work was also supported by American Cancer Society Grant CDD107745 (to D. J. W.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1S–3S.
- MM
- malignant melanoma
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PARP
- poly(ADP-ribose) polymerase
- PIDD
- p53-induced protein with a death domain
- JC-1
- 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide.
REFERENCES
- 1.Levine A. J., Hu W., Feng Z. (2006) Cell Death Differ. 13, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 2.Marchenko N. D., Zaika A., Moll U. M. (2000) J. Biol. Chem. 275, 16202–16212 [DOI] [PubMed] [Google Scholar]
- 3.Mihara M., Erster S., Zaika A., Petrenko O., Chittenden T., Pancoska P., Moll U. M. (2003) Mol. Cell. 11, 577–590 [DOI] [PubMed] [Google Scholar]
- 4.Bates S., Vousden K. H. (1996) Curr. Opin. Genet. Dev. 6, 12–18 [DOI] [PubMed] [Google Scholar]
- 5.Benchimol S. (2001) Cell Death Differ. 8, 1049–1051 [DOI] [PubMed] [Google Scholar]
- 6.Hoffman W. H., Biade S., Zilfou J. T., Chen J., Murphy M. (2002) J. Biol. Chem. 277, 3247–3257 [DOI] [PubMed] [Google Scholar]
- 7.Chipuk J. E., Kuwana T., Bouchier-Hayes L., Droin N. M., Newmeyer D. D., Schuler M., Green D. R. (2004) Science 303, 1010–1014 [DOI] [PubMed] [Google Scholar]
- 8.Bennett M., Macdonald K., Chan S. W., Luzio J. P., Simari R., Weissberg P. (1998) Science 282, 290–293 [DOI] [PubMed] [Google Scholar]
- 9.Geara F. B., Ang K. K. (1996) Surg. Clinics North Am. 76, 1383–1398 [DOI] [PubMed] [Google Scholar]
- 10.Satyamoorthy K., Chehab N. H., Waterman M. J., Lien M. C., El-Deiry W. S., Herlyn M., Halazonetis T. D. (2000) Cell Growth Differ. 11, 467–474 [PubMed] [Google Scholar]
- 11.Sawa H., Murakami H., Ohshima Y., Sugino T., Nakajyo T., Kisanuki T., Tamura Y., Satone A., Ide W., Hashimoto I., Kamada H. (2001) Brain Tumor Pathol. 18, 109–114 [DOI] [PubMed] [Google Scholar]
- 12.Moore B. W. (1965) Biochem. Biophys. Res. Commun. 19, 739–744 [DOI] [PubMed] [Google Scholar]
- 13.Zimmer D. B., Cornwall E. H., Landar A., Song W. (1995) Brain Res. Bull. 37, 417–429 [DOI] [PubMed] [Google Scholar]
- 14.Donato R. (2001) Int. J. Biochem. Cell Biol. 33, 637–668 [DOI] [PubMed] [Google Scholar]
- 15.Heizmann C. W. (2002) Methods Mol. Biol. 172, 69–80 [DOI] [PubMed] [Google Scholar]
- 16.Hansson L. O., von Schoultz E., Djureen E., Hansson J., Nilsson B., Ringborg U. (1997) Anticancer Res. 17, 3071–3073 [PubMed] [Google Scholar]
- 17.Maelandsmo G. M., Flørenes V. A., Mellingsaeter T., Hovig E., Kerbel R. S., Fodstad O. (1997) Int. J. Cancer 74, 464–469 [DOI] [PubMed] [Google Scholar]
- 18.Böni R., Burg G., Doguoglu A., Ilg E. C., Schäfer B. W., Müller B., Heizmann C. W. (1997) Br. J. Dermatol. 137, 39–43 [PubMed] [Google Scholar]
- 19.Ravasi T., Hsu K., Goyette J., Schroder K., Yang Z., Rahimi F., Miranda L. P., Alewood P. F., Hume D. A., Geczy C. (2004) Genomics 84, 10–22 [DOI] [PubMed] [Google Scholar]
- 20.Camby I., Nagy N., Lopes M. B., Schäfer B. W., Maurage C. A., Ruchoux M. M., Murmann P., Pochet R., Heizmann C. W., Brotchi J., Salmon I., Kiss R., Decaestecker C. (1999) Brain Pathol. 9, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davey G. E., Murmann P., Heizmann C. W. (2001) J. Biol. Chem. 276, 30819–30826 [DOI] [PubMed] [Google Scholar]
- 22.Hauschild A., Michaelsen J., Brenner W., Rudolph P., Gläser R., Henze E., Christophers E. (1999) Melanoma Res. 9, 155–161 [DOI] [PubMed] [Google Scholar]
- 23.Baudier J., Delphin C., Grunwald D., Khochbin S., Lawrence J. J. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 11627–11631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rustandi R. R., Drohat A. C., Baldisseri D. M., Wilder P. T., Weber D. J. (1998) Biochemistry 37, 1951–1960 [DOI] [PubMed] [Google Scholar]
- 25.Delphin C., Ronjat M., Deloulme J. C., Garin G., Debussche L., Higashimoto Y., Sakaguchi K., Baudier J. (1999) J. Biol. Chem. 274, 10539–10544 [DOI] [PubMed] [Google Scholar]
- 26.Lin J., Yang Q., Yan Z., Markowitz J., Wilder P. T., Carrier F., Weber D. J. (2004) J. Biol. Chem. 279, 34071–34077 [DOI] [PubMed] [Google Scholar]
- 27.Lin J., Blake M., Tang C., Zimmer D., Rustandi R. R., Weber D. J., Carrier F. (2001) J. Biol. Chem. 276, 35037–35041 [DOI] [PubMed] [Google Scholar]
- 28.Wilder P. T., Lin J., Bair C. L., Charpentier T. H., Yang D., Liriano M., Varney K. M., Lee A., Oppenheim A. B., Adhya S., Carrier F., Weber D. J. (2006) Biochim. Biophys. Acta 1763, 1284–1297 [DOI] [PubMed] [Google Scholar]
- 29.Scotto C., Deloulme J. C., Rousseau D., Chambaz E., Baudier J. (1998) Mol. Cell. Biol. 18, 4272–4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scotto C., Delphin C., Deloulme J. C., Baudier J. (1999) Mol. Cell. Biol. 19, 7168–7180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez-Fernandez M. R., Veprintsev D. B., Fersht A. R. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 4735–4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Dieck J., Fernandez-Fernandez M. R., Veprintsev D. B., Fersht A. R. (2009) J. Biol. Chem. 284, 13804–13811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Dieck J., Teufel D. P., Jaulent A. M., Fernandez-Fernandez M. R., Rutherford T. J., Wyslouch-Cieszynska A., Fersht A. R. (2009) J. Mol. Biol. 394, 922–930 [DOI] [PubMed] [Google Scholar]
- 34.O'Connor P. M., Jackman J., Bae I., Myers T. G., Fan S., Mutoh M., Scudiero D. A., Monks A., Sausville E. A., Weinstein J. N., Friend S., Fornace A. J., Jr., Kohn K. W. (1997) Cancer Res. 57, 4285–4300 [PubMed] [Google Scholar]
- 35.Meyskens F. L., Jr. (1981) Front. Radiat. Ther. Oncol. 16, 55–61 [DOI] [PubMed] [Google Scholar]
- 36.Meyskens F. L., Jr., Soehnlen B. J., Saxe D. F., Casey W. J., Salmon S. E. (1981) Stem Cells 1, 61–72 [PubMed] [Google Scholar]
- 37.Thomson S. P., Meyskens F. L., Jr. (1982) Cancer Res. 42, 4606–4613 [PubMed] [Google Scholar]
- 38.Bregman M. D., Funk C., Fukushima M. (1986) Cancer Res. 46, 2740–2744 [PubMed] [Google Scholar]
- 39.Kramer D. L., Vujcic S., Diegelman P., Alderfer J., Miller J. T., Black J. D., Bergeron R. J., Porter C. W. (1999) Cancer Res. 59, 1278–1286 [PubMed] [Google Scholar]
- 40.Allan L. A., Fried M. (1999) Oncogene 18, 5403–5412 [DOI] [PubMed] [Google Scholar]
- 41.Komarov P. G., Komarova E. A., Kondratov R. V., Christov-Tselkov K., Coon J. S., Chernov M. V., Gudkov A. V. (1999) Science 285, 1733–1737 [DOI] [PubMed] [Google Scholar]
- 42.Camphausen K., Moses M. A., Ménard C., Sproull M., Beecken W. D., Folkman J., O'Reilly M. S. (2003) Cancer Res. 63, 1990–1993 [PubMed] [Google Scholar]
- 43.Culmsee C., Zhu X., Yu Q. S., Chan S. L., Camandola S., Guo Z., Greig N. H., Mattson M. P. (2001) J. Neurochem. 77, 220–228 [DOI] [PubMed] [Google Scholar]
- 44.Schäfer T., Scheuer C., Roemer K., Menger M. D., Vollmar B. (2003) FASEB J. 17, 660–667 [DOI] [PubMed] [Google Scholar]
- 45.Leker R. R., Aharonowiz M., Greig N. H., Ovadia H. (2004) Exp. Neurol. 187, 478–486 [DOI] [PubMed] [Google Scholar]
- 46.Kuo P. C., Liu H. F., Chao J. I. (2004) J. Biol. Chem. 279, 55875–55885 [DOI] [PubMed] [Google Scholar]
- 47.Walton M. I., Wilson S. C., Hardcastle I. R., Mirza A. R., Workman P. (2005) Mol. Cancer Ther. 4, 1369–1377 [DOI] [PubMed] [Google Scholar]
- 48.Fraser M., Chan S. L., Chan S. S., Fiscus R. R., Tsang B. K. (2006) Oncogene 25, 2203–2212 [DOI] [PubMed] [Google Scholar]
- 49.Murphy P. J., Galigniana M. D., Morishima Y., Harrell J. M., Kwok R. P., Ljungman M., Pratt W. B. (2004) J. Biol. Chem. 279, 30195–30201 [DOI] [PubMed] [Google Scholar]
- 50.Latonen L., Kurki S., Pitkänen K., Laiho M. (2003) Cell. Signal. 15, 95–102 [DOI] [PubMed] [Google Scholar]
- 51.Latonen L., Taya Y., Laiho M. (2001) Oncogene. 20, 6784–6793 [DOI] [PubMed] [Google Scholar]
- 52.Magrini R., Russo D., Fronza G., Inga A., Menichini P. (2007) J. Cell. Biochem. 100, 1276–1287 [DOI] [PubMed] [Google Scholar]
- 53.Rieber M., Strasberg Rieber M. (2000) Int. J. Cancer 86, 462–467 [DOI] [PubMed] [Google Scholar]
- 54.Rustandi R. R., Baldisseri D. M., Weber D. J. (2000) Nat. Struct. Biol. 7, 570–574 [DOI] [PubMed] [Google Scholar]
- 55.Kulms D., Pöppelmann B., Yarosh D., Luger T. A., Krutmann J., Schwarz T. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 7974–7979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baptiste-Okoh N., Barsotti A. M., Prives C. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 1937–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin Y., Ma W., Benchimol S. (2000) Nat. Genet. 26, 122–127 [DOI] [PubMed] [Google Scholar]
- 58.Wu Z. H., Mabb A., Miyamoto S. (2005) Cell 123, 980–982 [DOI] [PubMed] [Google Scholar]
- 59.Chen Y., Kramer D. L., Diegelman P., Vujcic S., Porter C. W. (2001) Cancer Res. 61, 6437–6444 [PubMed] [Google Scholar]
- 60.Cheng L. H. (2001) Head Neck 23, 972–978 [DOI] [PubMed] [Google Scholar]
- 61.Takai D., Park S. H., Takada Y., Ichinose S., Kitagawa M., Akashi M. (2006) Free Radic. Res. 40, 1138–1148 [DOI] [PubMed] [Google Scholar]
- 62.Montano X., Shamsher M., Whitehead P., Dawson K., Newton J. (1994) Oncogene 9, 1455–1459 [PubMed] [Google Scholar]
- 63.Zhang H. (2006) Cancer Lett. 244, 229–238 [DOI] [PubMed] [Google Scholar]
- 64.Kaina B. (2003) Biochem. Pharmacol. 66, 1547–1554 [DOI] [PubMed] [Google Scholar]
- 65.Senatus P. B., Li Y., Mandigo C., Nichols G., Moise G., Mao Y., Brown M. D., Anderson R. C., Parsa A. T., Brandt-Rauf P. W., Bruce J. N., Fine R. L. (2006) Mol. Cancer Ther. 5, 20–28 [DOI] [PubMed] [Google Scholar]
- 66.Wu L., Levine A. J. (1997) Mol. Med. 3, 441–451 [PMC free article] [PubMed] [Google Scholar]
- 67.Bulavin D. V., Saito S., Hollander M. C., Sakaguchi K., Anderson C. W., Appella E., Fornace A. J., Jr. (1999) EMBO J. 18, 6845–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maiguel D. A., Jones L., Chakravarty D., Yang C., Carrier F. (2004) Mol. Cell. Biol. 24, 3703–3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saucedo L. J., Carstens B. P., Seavey S. E., Albee L. D., 2nd, Perry M. E. (1998) Cell Growth Differ. 9, 119–130 [PubMed] [Google Scholar]
- 70.Zeng X., Keller D., Wu L., Lu H. (2000) Cancer Res. 60, 6184–6188 [PubMed] [Google Scholar]
- 71.Kurki S., Peltonen K., Laiho M. (2004) Cell Cycle 3, 976–979 [PubMed] [Google Scholar]
- 72.Haapajärvi T., Kivinen L., Heiskanen A., des Bordes C., Datto M. B., Wang X. F., Laiho M. (1999) Exp. Cell Res. 248, 272–279 [DOI] [PubMed] [Google Scholar]
- 73.Butz K., Geisen C., Ullmann A., Zentgraf H., Hoppe-Seyler F. (1998) Oncogene 17, 781–787 [DOI] [PubMed] [Google Scholar]
- 74.Izumaru S., Arima N., Toyozumi Y., Kato S., Morimatsu M., Nakashima T. (2004) Int. J. Oncol. 24, 1245–1255 [PubMed] [Google Scholar]
- 75.Wang J. A., Fan S., Yuan R. Q., Ma Y. X., Meng Q., Goldberg I. D., Rosen E. M. (1999) Int. J. Radiat. Biol. 75, 301–316 [DOI] [PubMed] [Google Scholar]
- 76.van Dieck J., Brandt T., Teufel D. P., Veprintsev D. B., Joerger A. C., Fersht A. R. (2010) Oncogene 29, 2024–2035 [DOI] [PubMed] [Google Scholar]
- 77.Lin J., Weber D. J., Carrier F. (2006) AACR Meeting Abstracts 2006, 1168-a- [Google Scholar]
- 78.Hastak K., Agarwal M. K., Mukhtar H., Agarwal M. L. (2005) FASEB J. 19, 789–791 [DOI] [PubMed] [Google Scholar]
- 79.Fan X., Liu Y., Chen J. J. (2005) Apoptosis 10, 63–73 [DOI] [PubMed] [Google Scholar]
- 80.Huo J. X., Metz S. A., Li G. D. (2004) Cell Death Differ. 11, 99–109 [DOI] [PubMed] [Google Scholar]
- 81.Levkau B., Koyama H., Raines E. W., Clurman B. E., Herren B., Orth K., Roberts J. M., Ross R. (1998) Mol. Cell. 1, 553–563 [DOI] [PubMed] [Google Scholar]
- 82.Chopin V., Toillon R. A., Jouy N., Le Bourhis X. (2004) Oncogene 23, 21–29 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.