

Abstract
In addition to regulating the neuroendocrine stress response, corticotropin-releasing hormone (CRH) has been implicated in both normal and pathological behavioral and cognitive responses to stress. CRH-expressing cells and their target neurons possessing CRH receptors (CRF1 and CRF2) are distributed throughout the limbic system, but little is known about the regulation of limbic CRH receptor function and expression, including regulation by the peptide itself. Because CRH is released from limbic neuronal terminals during stress, this regulation might play a crucial role in the mechanisms by which stress contributes to human neuropsychiatric conditions such as depression or posttraumatic stress disorder. Therefore, these studies tested the hypothesis that CRH binding to CRF1 influenced the levels and mRNA expression of this receptor in stress-associated limbic regions of immature rat. Binding capacities and mRNA levels of both CRF1 and CRF2 were determined at several time points after central CRH administration. CRH downregulated CRF1 binding in frontal cortex significantly by 4 h. This transient reduction (no longer evident at 8 h) was associated with rapid increase of CRF1 mRNA expression, persisting for >8 h. Enhanced CRF1 expression—with a different time course—occurred also in hippocampal CA3, but not in CA1 or amygdala, CRF2 binding and mRNA levels were not altered by CRH administration. To address the mechanisms by which CRH regulated CRF1, the specific contributions of ligand-receptor interactions and of the CRH-induced neuronal stimulation were examined. Neuronal excitation without occupation of CRF1 induced by kainic acid, resulted in no change of CRF1 binding capacity, and in modest induction of CRF1 mRNA expression. Furthermore, blocking the neuroexcitant effects of CRH (using pentobarbital) abolished the alterations in CRF1 binding and expression. These results indicate that CRF1 regulation involves both occupancy of this receptor by its ligand, as well as “downstream” cellular activation and suggest that stress-induced perturbation of CRH–CRF1 signaling may contribute to abnormal neuronal communication after some stressful situations.
Keywords: stress, mechanisms of depression, PTSD, corticotropin-releasing hormone, CRF, receptor regulation
INTRODUCTION
The neuropeptide corticotropin-releasing hormone (CRH) functions as a key mediator of the responses to stress (57). It has been increasingly recognized, however, that, in addition to its neuroendocrine effects, CRH acts as a neuromodulator in stress-associated limbic regions to propagate and integrate stress-induced behaviors. Specifically, at the cellular level, CRH has been shown to enhance synaptic efficacy [e.g., LTP (35, 38)]. In addition, abnormally high CRH levels at the synapse may lead to excessive excitability and contribute to some developmental seizures (7). CRH functions via activation of specific membrane-spanning G-protein coupled receptors, and both CRH-expressing cells and target neurons possessing these specific receptors (CRF1 and CRF2) are abundant throughout the limbic system (3, 16–18, 22, 53).
Whereas information about the regulation of the expression of CRH itself, both in the hypothalamus and in stress-related limbic regions, has been forthcoming (26, 27, 40, 50), the regulation of CRF1 expression in these regions has remained relatively unexplored. The majority of studies investigating regulation of CRH receptors have centered on the hypothalamic paraventricular nucleus (PVN), where, in response to stress, clusters of CRH-producing cells release the peptide into hypothalamic-pituitary portal vessels. In PVN, stress has been shown to increase gene expression of CRF1 (33, 41, 48). Other studies have addressed this issue in pituitary corticotrophs bearing CRH receptors, which are stimulated by CRH to secrete ACTH (1, 28, 59): CRF1 expression in pituitary corticotrophs was modulated by stress in a biphasic manner—a reduction at 2 h was followed by an increase at 4 h (47)—raising the possibility of altered receptor turnover. The only published investigation of the regulation of brain CRH receptor mRNA after acute stress found changes in CRH receptor expression that were confined to the hypothalamus (48).
Thus, limited information is available on the regulation of CRH receptors in limbic structures such as hippocampus, amygdala, and cortex. In essence, a single study has suggested that chronic stress in adult rats resulted in a decrease in CRF1 mRNA in frontal cortex while increasing CRF1 expression in hippocampus (34).
An important aspect of the regulation of CRH receptors involves the consequences of their occupation and activation by the ligand, CRH. Indeed, modulation of receptor expression by the cognate neurotransmitter has been well established. A number of studies have shown that endogenous CRH is probably the ligand for CRH receptors in the limbic system: CRH release by physiological stimuli (e.g., during stress) has been shown in amygdala (43, 54), hippocampus (7, 27, 50), and other regions containing CRF1. Therefore, the changes in CRF1 expression that have been reported after stress may be mediated directly by stress-induced CRH release and binding to these receptors, or alternatively, by other stress-related molecules, e.g., glucocorticoids.
To begin addressing these alternative regulatory mechanisms, the current experiments were designed to (i) determine if CRH directly regulated limbic CRF1 and CRF2 mRNA levels using in situ hybridization, (ii) determine if CRH directly regulated its receptors at the protein level by analyzing receptor binding capacity in tissue homogenates, and (iii) dissect out the specific factors (i.e., ligand–receptor interaction, stress, or CRH-induced activation of signal transduction cascades) involved in the mechanisms of CRH effects on CRF1 and CRF2 expression and abundance.
EXPERIMENTAL PROCEDURES
Animals and Tissue Preparation
Immature rats were used in these studies for two reasons: (i) Previous work from the authors’ laboratories had characterized hippocampal, amygdala, and hypothalamic CRH expression during this age (26, 27, 60). (ii) Limbic CRH and CRH receptor expression may be particularly robust during the second and third postnatal week in the rat (3, 17, 22, 46). Ten-day old rats of both sexes used in this study were offspring of timed-pregnancy Sprague–Dawley-derived dams (Zivic-Miller, Zelienople, PA). Animals were maintained in NIH-approved facilities on a 12-h light/dark cycle with access to unlimited lab chow and water. Cages were monitored every 12 h for the presence of pups and the date of birth was considered day 0. Litters were culled to 12 pups and mixed among experimental groups (n = 5–10 per group). Potential diurnal variability was addressed by initiating all experiments between 0800 h and 1000 h, and cages were undisturbed for 24 h prior to initiation of experiments. For obvious technical reasons, experiments were performed in “batches.” However, each included both experimental animals and littermate controls, and tissue processing and data analyses were performed to include all experiments. All experimental procedures were approved by the Institutional Animal Care Committee and conformed to National Institutes of Health guidelines.
Surgical Procedures and CRH Administration
For CRH infusion, pups were implanted with stainless steel cannulae into the left lateral cerebral ventricle, using halothane anesthesia, on the day preceding the experiments, as described in detail elsewhere (6, 13). Stereotaxic coordinates using Bregma as landmark were: AP −0.7, L 2.0, V 3.3. Peptide infusion or the infusion of an equal volume of vehicle, was carried out on postnatal days 10–12 to freely moving pups, using a Kd-Scientific multi-microinfusion pump (Boston, MA) via a Hamilton syringe attached to flexible tubing (6, 13).
The dose of CRH (0.75 nmol in 1.3 μ,L) was selected based on previous dose–response studies (6). The dose used here has been shown to lead to activation of limbic neuronal populations, i.e., behavioral, as well as by electrographic seizures (Fig. 1). The convulsant effects of the dose of CRH used here lasted ~3 h (see Ref. 11 for EEG procedures). The specificity of the effects of this dose of CRH was evident from their prevention by preadministration of nonselective antagonists of CRH that block both CRF1 and CRF2 receptors. In addition, the activatory effects of this CRH dose in limbic regions have been blocked by nonpeptide, selective antagonists of the CRF1 receptor subtype (6).
FIG. 1.

Effect of pentobarbital on electrophysiological activity induced by CRH in the dorsal hippocampus. The tracings depict bipolar EEG recordings from 10-day-old rats. Baseline recordings were taken 5 min prior to CRH administration. EEG recordings were obtained 30–40 min following CRH or KA administration, and were correlated with limbic seizures consisting of oral automatisms and forepaw clonus. Intraperitoneal injection of pentobabital (P) abolished all CRH-induced electrophysiological activity in dorsal hippocampus within 5 min of drug administration. Arrowheads indicate spike-wave discharges and the arrow indicates motion artifact. Horizontal bar, 1 s; vertical bar, 50 μV.
An additional group of animals received an anesthetic dose (40 mg/kg) of the short-acting barbiturate, pentobarbital, via intraperitoneal (ip) injection 5 min prior to infusion of CRH or vehicle. This dose was calibrated to block both the behavioral and the electrographic convulsant action of CRH (Fig. 1). Another group of cannula-carrying animals received an ip injection of the limbic excitant kainic acid (KA) at a dose of 1.2 mg/kg. This dose was chosen to approximate the magnitude and duration of CRH-induced excitation (seizures) of immature rat neurons [Fig. 1 (11)].
Animals were decapitated at 2, 3, 4, 6, or 8 h after drug infusion and brains were rapidly dissected onto powdered dry ice and stored at −80°C. For in situ hybridization, sections were cut coronally at 20 μm using a cryostat, mounted on gelatin-coated slides and stored at −80°C. For homogenate-binding studies, frontal cortices were isolated and frozen on powdered dry ice. Trunk blood was collected for measurement of plasma corticosterone levels using a commercial radioimmunoassay (RIA) kit (ICN, Irvine, CA).
Radioligand Binding Studies
Membrane preparation
On the day of the assay, frontal cortex tissues were thawed on ice, weighed and placed in 5 to 7-mL ice-cold PBS containing 10 mM MgCl2, 2 mM EGTA, pH 7.0 (assay buffer), at 22°C and homogenized at maximum setting on a tissue tearer (Model 985–370; Biospec Products, Inc.) for 20 s on ice. Membrane homogenates were then washed twice by centrifugation (30,000 g for 10 min at 4°C) in the same buffer (5–7 mL). These extensive membrane washes virtually exclude the possibility of residual peptide after CRH administration in vivo. The final pellets were resuspended in buffer to a working concentration of approximately 25 mg/mL protein original wet weight. Final protein concentrations were determined using the BCA protein determination assay from Pierce Biochemical Co. (Rockford, IL) and quantified using bovine serum albumin (BSA) as a standard. Typically the final protein concentrations in the assay ranged from 200–300 μg/tube.
Tyr°-sauvagine binding studies
Radioligand binding of [125I]Tyr°-sauvagine ([125I]sauvagine) in rat brain membranes was performed essentially as previously described (25). Briefly, 1.5-mL Eppendorf tubes received, in order, 50 μL buffer, 50 μL competing drug, 50 μL of [125I]sauvagine, and 150 μL membrane suspension as described above for a total assay volume of 300 μL. (All drugs and reagents were made up in assay buffer). The assay was incubated at equilibrium routinely for 2 h at 22°C as determined by direct kinetic experiments for the association of [125I] sauvagine binding (25). Nonspecific binding was defined in the presence of 1 μM unlabeled peptide antagonist D-Phe12, Nle21,38, Ala32 human CRF-[12–41] (D-Phe-CRF). Reactions were terminated by centrifugation in Beckman microfuge for 10 min at 12,000 g. The resulting pellets were washed gently with 1 mL of ice-cold PBS containing 0.01% Triton X-100 and centrifuged again for 10 min at 12,000 g. The supernatants were aspirated and the tubes cut just above the pellet and placed into 12 × 75-mm polystyrene tubes and monitored for radioactivity in a Packard Cobra II γ-counter at approximately 80% efficiency.
Quantification of CRF1 and CRF2 receptor concentration
The determination of [125I]sauvagine binding to the CRF1 receptor was specifically defined in the presence of 3 μM NBI 27914, a compound that has been shown to have no binding activity to the CRF2 receptor subtype (15). In tissues such as the olfactory bulb, where both CRF1 and CRF2 receptors exist (14), 3 μM NBI 27914 completely blocked [125I] sauvagine binding to the CRF1 receptor subtype, leaving a substantial amount of [125I] sauvagine bound. In the same tissues, 1 μM D-Phe-CRF was able to completely block the binding to both receptor subtypes. The difference between the binding in the presence of NBI 27914 and the binding in the presence of D-Phe-CRF was thus defined as specific binding of [125I] sauvagine to the CRF2 receptor subtype. The concentration of [125I]sauvagine used in these studies was 600–800 pM, achieving >85% fractional receptor occupancy of both receptor subtypes based on their KD values (~ 150 pM). Values therefore are listed as receptor concentration approximating Bmax levels. In this way, direct comparisons of the effects of treatment on the relative receptor levels could be obtained.
In Situ Hybridization Histochemistry (ISH) Procedures for CRF1 and CRF2 Messenger RNAs
cRNA probe synthesis and preparation have been described previously (14, 23). Briefly, a plasmid containing a 600-bp fragment of CRF1 cDNA or 461-bp fragment of CRF2 cDNA was linearized with HindIII. Radioactive antisense cRNA was synthesized using a T3 transcription system in a standard labeling reaction mixture consisting of 1 μg linearized plasmid, 5× transcription buffer, 250 μCi of [35S]CTP, 30 nmol NTPs, 50 nmol dithiothreitol, 40 U of Rnasin, and 34 U of RNA polymerase. The reaction was incubated at 37°C for 1 h. The probes were subjected to alkaline hydrolysis and purified by column chromatography [Select-D(RF), 5 Prime-3 Prime, Boulder, CO]. Probe specificities for both CRF1 and CRF2 mRNA have been established (14).
ISH was performed as described previously for cRNA probes (23). Briefly, 20-μm coronal sections were collected on gelatin-coated slides and stored at −80°C. Sections were thawed, air-dried, fixed in paraformaldehyde, dehydrated and rehydrated through graded ethanols, exposed to 0.25% acetic anhydride in 0.1 M triethanolamine (pH 8), and dehydrated. Prehybridization (1 h) and hybridization steps were performed in a humidified chamber in a solution of 50% formamide, 5× SET (0.75 M NaCl, 0.15 M Tris, 0.01 M EDTA), 0.2% SDS, 5× Denhardt’s, 0.5 mg/mL salmon sperm DNA, 0.25 mg/mL yeast tRNA, 100 mM dithiothreitol, and 10% dextran sulfate. Following prehybridization, sections were hybridized overnight. For detection of CRF1 and CRF2 mRNA, sections were hybridized at 55°C with 1 × 106 cpm of 35S-labeled ribonucleotide probe complementary to CRF1 or CRF2 mRNA (37). After hybridization, sections were digested with 200 μg/mL RNase A (Calbiochem, La Jolla, CA) for 30 min at 37°C. Sections underwent serial washes of increasing stringency at 55°C, the most stringent at 0.03 × SSC for 1 h (23). Hybridized sections were apposed to film (Hyperfilm βMax, Amersham, IL) for 7–14 days.
Semiquantitative Analysis of CRF1 and CRF2 mRNA and Statistical Considerations
Analysis of CRH mRNA ISH signal was performed on digitized films using the Image Tool software program (University of Texas Health Science Center, San Antonio, TX) as described elsewhere (23). Five anatomically matched sections per region per brain were analyzed, without knowledge of treatment. Unbiased methods for section sampling have been described in detail elsewhere (23). Cingulate cortex was sampled at the coronal level 2.9–3.2 mm anterior to Bregma in the 10-day-old rat (49); frontal cortex was sampled at 2.9–3.2 mm anterior and 4–5 mm lateral with reference to bregma. Densities were calibrated using 14C standards and are expressed in nCi/g after correcting for background by subtracting the density of the hybridization signal over the corpus callosum. Differences among groups were determined using a one-way analysis of variance (ANOVA) or Student t test, as appropriate (Prism GraphPad, San Diego, CA) and significance levels were set at P < 0.05. Further analysis used Tukey’s multiple comparison post hoc tests to determine the source of the detected significance in the ANOVAs.
RESULTS
CRH Administration Reduces CRF1 Binding in Frontal Cortex
CRF1-binding capacity in homogenates of frontal cortex was significantly reduced by 4 h after administration of CRH (CRH, 22.3 ± 1.4; controls, 28.4 ± 2 fmol/mg protein; Fig. 2A), whereas no significant changes were found in CRF2 binding (CRH; 5.7 ± 0.4; controls, 5.3 ± 0.6 fmol/mg protein; Fig. 2B). The effect on CRF1 binding in frontal cortex was transient and no longer evident at 8 h after CRH administration (CRH, 28.9 ± 1.2; controls, 30.8 ± 1.5 fmol/mg protein; t test, P = 0.36).
FIG. 2.

CRH administration reduces CRF1 binding in frontal cortex of immature rats (5–6/group). (A) CRF1 binding capacity in homogenates of frontal cortex was selectively downregulated, 4 h after CRH administration (*P = 0.02, t test). (B) In contrast, no significant changes were found in CRF2 binding capacity (P = 0.58, t test). Values are means ± SEM. (Note: the CRF1 binding values also contribute to Fig. 7.)
CRF1 mRNA Expression Is Enhanced in Select Regions of Frontal Cortex Following Activation of the Receptor by CRH
CRF1 mRNA expression was robust in neocortical and hippocampal regions of the developing rat brain (Fig. 3A). As a result of CRH administration, CRF1 mRNA levels were significantly upregulated in specific neocortical regions. Figures 4A and 4B show enhanced levels in cingulate cortex from CRH-infused animals (120 ± 7 nCi/g) as compared to vehicle-treated controls (50 ± 4 nCi/g) at the 4-h time point. Interestingly, the onset of upregulation of CRF1 mRNA levels was evident by 2 h and this CRH-induced increase persisted for at least 8 h posttreatment (36 ± 7.1% over controls, Fig. 4C).
FIG. 3.

(A) CRF1 and (B) CRF2 mRNA expression patterns in immature (10-day-old) rat forebrain. Representative computer-generated pseudo-color images of coronal hemisections following in situ hybridization for CRF1 or CRF2 mRNA (red, high; yellow-green, medium; dark blue, low mRNA levels) are shown. Cing, cingulate; Cx, cortex; HIPP, hippocampus; BLA, basolateral amygdala; MEA, medial amygdala; BMA, basomedial amygdala; VMH, ventromedial hypothalamus.
FIG. 4.

CRF1 mRNA expression is selectively upregulated in cortical regions following CRH administration. Semiquantitative analysis was performed after in situ hybridization for CRF1 on coronal sections from immature animals given CRH or vehicle (n = 5–10/group/time point for A and C). The graph in (A) represents maximal CRH-induced increases of CRF1 mRNA levels in cingulate cortex and cortical layer II/III (peak at 4 and 2 h, respectively). No significant changes in CRF1 mRNA expression were detected in the deeper cortical layers V/VI at any time point analyzed (4 h shown in graph). (B) Representative coronal sections from animals administered CRH or vehicle, showing CRF1 mRNA expression. (C) The inverse temporal relationship between binding capacity (protein levels) and mRNA levels of CRF1 receptors after CRH administration; values given as percentage difference from those of vehicle-treated rats. Values are means ± SEM. (Note: values for frontal cortex layer II/III are also incorporated in Fig. 8) *Significant difference from vehicle treated (P < 0.05, one-way ANOVA with Tukey’s multiple comparison analysis). HIPP, hippocampus; BLA, basolateral amygdala; Cing, cingulate; Cx, cortex.
A significant, rapid (by 2 h) upregulation (150 ± 25.2%) of CRF1 mRNA expression occurred also in layers II/III of frontal cortex (Figs. 4A and 4B). This enhancement was short-lived and returned to control values by 4 h after CRH administration, but a second wave of significant elevation in CRF1 mRNA levels in these neuronal populations was observed at the 8 h time point (44 ± 7.5%; Fig. 4C). In contrast to the superficial layers, no significant difference in receptor expression between experimental and control groups was observed in layers V/VI of neocortex, where CRF1 mRNA expression levels were much lower (Figs. 3A, 4A, and 4B).
In addition to frontal neocortex, CRF1 mRNA levels were analyzed in the CA1 and CA3 pyramidal layers of hippocampus proper and in the granule cell layer of dentate gyrus (Fig. 4B) at 2, 4, 6, and 8 h after CRH administration. In hippocampus, significant upregulation of CRF1 mRNA expression was observed in the CA3 pyramidal cell layer, peaking at the 4-h time point (Fig. 5A). Time-course experiments demonstrated that the onset of CRH-induced enhancement of CRF1 mRNA expression in the hippocampal CA3 region began later than in neocortical regions, peaked at 4 h and returned to basal levels by 8 h following CRH administration (not shown). No significant changes in CRH receptor expression were observed for other analyzed hippocampal regions at any time point. In the amygdaloid complex, robust CRF1 expression was noted in the basolateral nucleus (BLA, Figs. 3A and 4B), but this expression was not altered by CRH administration at any time point analyzed (Fig. 5B).
FIG. 5.

CRF1 mRNA expression is selectively upregulated in select hippocampal regions (A), but not in amygdala (B) after CRH administration. Semiquantitative analysis of CRF1 mRNA signal in coronal sections of immature rats (5–10/group) 4 h after infusion with CRH or vehicle. DG, dentate gyrus. Values are means ± SEM. (Note: values for CA3 are also incorporated in Fig. 8) *Significant difference from vehicle treated (P < 0.05, one-way ANOVA with Tukey’s multiple comparison analysis).
CRF2 mRNA Expression Is Not Modulated by CRH Administration
CRF2 mRNA was confined primarily to subcortical regions of the immature rat brain (Fig. 3B), consistent with previous reports (14, 22, 23). No changes were observed in CRF2 mRNA expression in hypothalamic and amygdalar regions with significant mRNA expression of this receptor (22, 23). Although only the 4-h time point is shown in Fig. 6, CRH administration did not result in significant alteration of CRF2 mRNA levels in the ventromedial hypothalamic nucleus or in the medial and basomedial amygdala nuclei at any of the analyzed time points.
FIG. 6.

CRH administration does not result in significant alterations of CRF2 mRNA levels in hypothalamic or amygdala regions with high expression levels of this receptor. Semiquantitative analysis of CRF2 mRNA signal in coronal sections of immature rats (5–10/group) 4 h postinfusion with CRH or vehicle. VMH, ventromedial hypothalamus; MEA, medial amygdala; BMA, basomedial amygdala. Values are means ± SEM.
Changes in CRF1 Expression and Binding Require Occupancy and Activation of This Receptor by Its Ligand (CRH)
Central administration of CRH to immature rats results in several effects. These include: (i) occupancy of CRH receptors, leading to activation of molecular signaling cascades in several neuronal populations (21, 31); (ii) behavioral and electrographic evidence of intense neuronal stimulation [i.e., seizures (4)]; (iii) potentially, activation of the neuroendocrine responses to stress, including elevation of plasma glucocorticoid levels, although the doses administered have previously been shown not to enhance plasma glucocorticoids by themselves (4). To determine which of these factors contributed to the mechanisms by which CRH modulated CRF1 expression and binding capacity, several experiments were carried out.
The first experiment demonstrated that CRH binding to the receptor, by itself, was not sufficient to modulate binding capacity (Fig. 7) or CRF1 mRNA expression (Fig. 8): to prevent molecular and electrophysiological measures of neuronal activation, a group of animals were given the short-acting barbiturate, pentobarbital, prior to CRH administration (11, 56). As shown in Fig. 7, CRH-induced changes in CRF1 binding capacity in frontal cortex were eliminated by pentobarbital administration, as was the enhancement of CRF1 mRNA levels in frontal cortex layers II/III and hippocampal CA3 pyramidal cell layer (Figs. 8A and 8B).
FIG. 7.

Regulation of CRF1 binding capacity by CRH requires both neuronal activation and receptor occupancy by the ligand. CRH-induced changes in CRF1 binding capacity in homogenates of frontal cortex are blocked in immature rats given pentobarbital (P) prior to CRH to prevent neuronal activation. In addition, administration of the limbic convulsant kainic acid (KA) to immature rats does not significantly alter CRF1 binding capacity in homogenates from frontal cortex. *Significant difference from vehicle (n = 5 pups/group; P < 0.05, one-way ANOVA with Tukey’s multiple comparison analysis). Values are means ± SEM. (Note: vehicle and CRH values are also shown in Fig. 2.)
FIG. 8.

Modulation of CRF1 mRNA levels in frontal cortex (A) and hippocampus (B) requires neuronal activation. (A) Administration of CRH or kainic acid (KA) induced significant upregulation of CRF1 mRNA expression in cortical layer II/III. (B) CRH, but not KA, enhances CRF1 mRNA expression in hippocampal CA3. In both frontal cortex and hippocampus, blocking the excitatory effects of CRH with preadministration of pentobarbital (P) abolished the effects of CRH on CRF1 mRNA levels. *Significant difference from vehicle and “significant difference from vehicle and CRH + P (n = 5/group; P < 0.05, one-way ANOVA with Tukey’s multiple comparison analysis). Values are means ± SEM. (Note: values for vehicle and CRH in (A) and (B) are also shown in Figs. 4 and 5, respectively.)
Second, to determine whether the intense activation of limbic neurons (also induced by CRH) and the resultant induction of signal-transduction cascades (27) were sufficient to alter CRF1 expression without the requirement for occupancy of this CRH receptor, a similar pattern and degree of limbic neuronal activation were generated by administration of the limbic stimulant, kainic acid (5, 11). The compound induced prolonged limbic seizures which were of similar phenotype and duration to those induced by CRH (5, 11). In addition, kainic-acid-induced seizures provoked glucocorticoid secretion, resulting in plasma levels similar to those arising from CRH-induced seizures (Fig. 9). As noted above, previous studies have shown that the dose of CRH utilized here is not sufficient to stimulate the pituitary gland to evoke release of glucocorticoids from the adrenals by itself. Once CRH-induced seizures commence, glucocorticoid levels are increased, suggesting that it is endogenous peptide released due to stress from the seizures that triggers steroid secretion (4). Despite the similarities between kainic acid and CRH as limbic stimulatory agents, kainic acid resulted in no significant change of CRF1 binding capacity (Fig. 7). Kainic acid administration did enhance1 CRF mRNA levels significantly in neocortical layers II/II1 (Fig. 8A) and moderately in the hippocampal CA3 pyramidal cell layer (Fig. 8B) as compared to controls.
FIG. 9.
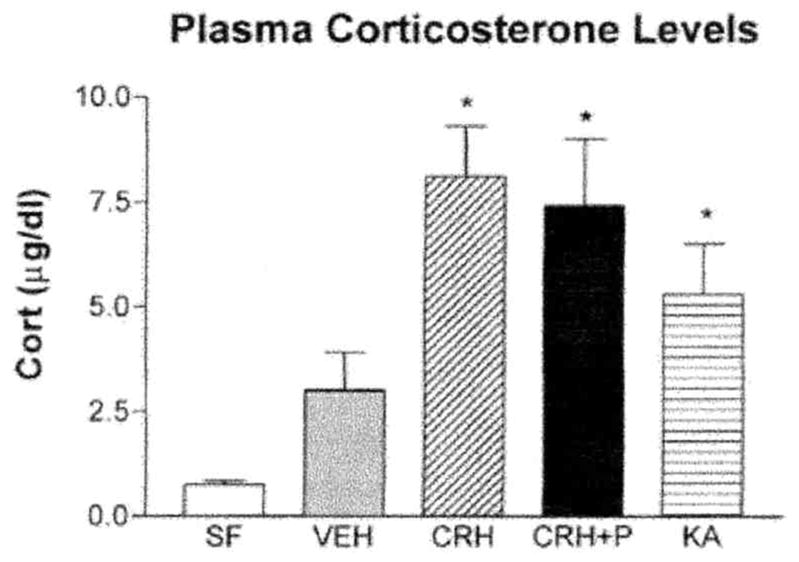
Plasma corticosterone (cort) levels are significantly elevated 30 min after treatment with kainic acid (KA), CRH, or pentobarbital (P) together with CRH (CRH + P). Both CRH- and KA-treated animals demonstrated overt behavioral neuronal activation (seizures) at the time of sacrifice, whereas pentobarbital blocked this activation in CRH + P-treated animals. Thus, peripheral corticosterone levels did not correlate with limbic neuronal activation. *Significant difference from vehicle and stress-free controls (SF; unmanipulated and sacrificed within 20 s of disturbance; n = 5–10/group; P < 0.05, one-way ANOVA with Tukey’s multiple comparison analysis). Values are means ± SEM.
DISCUSSION
The major findings of the present study were (i) CRH administration to immature rats decreased CRF1 binding capacity in frontal cortex, (ii) CRF1 mRNA expression increased in frontal cortex and select hippocampal regions following CRH administration, and (iii) both occupancy of this receptor by its ligand (CRH), and activation of postreceptor, “downstream” signal-transduction cascades were required for this modulation of CRF1 binding levels and mRNA expression.
Two specific, high-affinity receptors mediate the actions of CRH. These have been characterized and localized to discrete brain regions throughout the central nervous system (CNS; 3, 14, 18, 22). The CRF1 receptor, constituting the focus of this study, is distributed primarily in cortical areas, as well as in anterior pituitary, whereas CRF2 is mainly found in subcortical regions, including lateral septum and hypothalamus, as well as in choroid plexus and cerebral vasculature. Consistent with similar studies in the adult (42), this study yielded no evidence for regulation of CRF2 binding capacity or expression by CRH in any of the examined brain regions of the immature rat. Thus, these two members of the CRH receptor family demonstrate differing regulatory mechanisms, consistent with their subserving distinct and separate functions.
CRH administration in the present study downregulated CRF1 binding in the frontal cortex. This is particularly interesting, because alteration of both CRH and its receptor has been demonstrated in a number of human disorders (19). For example, increased CRF1 binding capacity has been shown in humans with Alzheimer’s dementia, where cortical levels of CRH are low (9, 37), suggesting that the increased receptor binding may be a compensatory mechanism attempting to increase CRH-mediated neuronal communication. Furthermore, reduced CRH receptor binding capacity in frontal cortex has been reported in depressed suicide victims (44 but see 32), in whom cerebrospinal fluid CRH levels are increased (44). These reciprocal alterations of the levels of CRH and CRF1 are consistent with regulation of the receptor by its ligand in the human CNS. The relationship between altered binding capacity and mRNA levels of CRF1, however, has not been previously examined (see below).
Whereas decreased binding capacity of CRF1 was shown in homogenates derived from frontal cortex and thus consisting of all cortical layers (I–VI), in situ hybridization permitted a higher resolution, demonstrating specific effects of CRH administration on CRF1 mRNA expression in superficial (II/III) but not in deeper cortical layers (V/VI). Based on previous studies by our laboratory and others (10, 21), it is unlikely that the differential effects on CRF1 mRNA expression were due to the inability of centrally administered CRH to diffuse equally to all populations of CRH receptors. High levels of CRF1 mRNA and protein have been reported in all of these cortical layers (14, 16). Our findings raise the possibility of potential specific roles of CRF1 located in discrete neocortical layers.
CRH also enhanced CRF1 mRNA expression throughout the cingulate cortex (medial prefrontal cortex). Enhanced levels of cingulate-CRF1 receptors could contribute to the proposed role of this region in modulation of HPA axis function in response to stress (20). Specifically, stress levels of CRH may interact with CRF1-bearing neurons in the cingulate cortex to promote negative feedback onto the hypothalamic–pituitary adrenal axis via indirect pathways from this region that project to hypothalamic CRH-expressing neurons (e.g., via septum or nucleus of the solitary tract). This should result in reduction of CRH secretion from these PVN neurons.
It should be noted that no alteration of CRF1, mRNA levels by CRH was observed in the amygdala. CRF1 has been considered a major mediator for many of the central neuromodulatory effects of CRH (6, 54). For example, mice deficient in CRF1 demonstrate reduced anxiety and an impaired stress response (50, 55), attributed to amygdala functions. Thus, using selective antagonists, CRF1 receptors in the amygdala were shown to mediate anxiogenesis (54). Within the amygdala complex, the central nucleus (ACe) has been suggested to be the primary site of these central CRH effects, but although substantial CRF1-like immunoreactivity is found in ACe (16), neurons in this nucleus express low levels of CRF1 mRNA (14). This may suggest that the origin of the immunoreactive CRF1 may lie outside the ACe. In the context of the current findings, the lack of alteration of CRF1 mRNA levels in basolateral amygdala by any of the stressful manipulations performed in this study, as well as by the administration of CRH itself, indicates that in this region, CRF1 mRNA expression is probably not significantly regulated by stress or by CRH binding.
Unlike in the amygdala, CRF1 mRNA levels in the hippocampal CA3 pyramidal layer, a region where high levels of CRF1 mRNA (3, 14) and of CRF1-like immunoreacvity (16) have been demonstrated, were highly enhanced by CRH administration. This upregulation may have important physiological consequences: in hippocampal slice preparations, CRH application leads to excitatory discharges from CA1 and CA3 pyramidal neurons (2, 30). In addition, high doses of CRH infused icv to developing rats lead to hippocampal pyramidal cell injury (13) and it is interesting to note that this injury occurs selectively in CA3 pyramidal cells (5, 13). While the mechanisms for this selective vulnerability of CA3 pyramidal neurons to CRH have not been established, this subfield is particularly rich in CRF1 receptors (16), and as shown here. CRH induces upregulation of CRF1 levels only in CA3 cells (not in CA1; Fig. 5A). The potential importance of this effect of early life administration of CRH for hippocampal pathophysiology during adulthood has recently been emerging: studies from this laboratory (13) demonstrate that CRH binding to its receptor early in life may lead to prolonged upregulation of CRF1 mRNA expression which persists up to 1 year. This receptor upregulation is associated with progressive death of CA3 hippocampal neurons, suggesting that early life modulation of CRH receptor expression and function may be a persistent and important factor in neuronal dysfunction throughout life.
Thus, the findings of these experiments suggest that in CA3 pyramidal cells, CRH may induce enhanced CRF1 expression long-term promoting exaggerated downstream effects of CRF1 activation, such as calcium entry (36, 58), which may result in cellular injury (13) and contribute to stress-related neurological disorders.
The current study demonstrated regulation of CRF1 levels and expression in cortical and limbic regions of the immature rat. It should be noted that in previous studies performed in the adult rat, administration of a dose of CRH 10 times larger than those employed here resulted in upregulation of CRF1 mRNA solely in PVN (42). A possible explanation for these differences may derive from the enhanced potency of CRH in the activation of limbic neurons during development, compared with the adult (4, 7, 12, 21, 24). This activation is mediated specifically by CRF1 (6). Therefore, the current findings are consistent with the notion that in order to regulate CRF1 levels and expression, not only occupancy of the receptor is required, but also activation of downstream signal-transduction cascades, measurable by immediate early genes (12), and electrophysiological or metabolic markers of neuronal activation (21). This notion is further supported by the fact that administration of CRH in the presence of pentobarbital did not lead to alteration of CRF1 binding or mRNA levels (Figs. 7 and 8). Pentobarbital eliminated the central behavioral and electrophysiological measures of hippocampal neuronal activation by CRH (4, 12, 21), while not altering peripheral stress effects, i.e., glucocorticoid secretion (most likely induced by the stress of the injection: Fig. 9). These findings suggest that in addition to receptor occupancy by the ligand (CRH), downstream activation of target neurons is required for modulation of CRF1. Furthermore, the lack of correlation with plasma glucocorticoid levels suggests that these mechanisms may be independent of peripheral steroid actions.
However, neuronal activation per se, in the absence of receptor occupancy by the ligand, does not fully reproduce the effect of CRH administration on CRF1 levels: Provocation of intense activation of limbic neurons using the excitatory agent kainic acid led to no changes in cortical CRF1 binding (Fig. 7) and to an intermediate level of enhancement of mRNA levels (between CRH and vehicle; Fig. 8). While neuronal activation is clearly required for this regulation (since CRH-induced changes in CRF1 expression were suppressed by blocking neuronal activation using pentobarbital) the data suggest that CRH itself also contributes to this regulatory effect. In this context it may be noted that previous work from several laboratories has shown that kainic acid and other agents promoting neuronal activation increase levels of CRH in hippocampus and related limbic regions (27, 45, 50). Therefore, it is likely that, in the current study, kainic acid administration increased CRF1 receptor levels in part via enhancement of CRH levels in hippocampus and frontal cortex. Thus, this effect of kainic acid supports the notion that the mechanism by which CRH regulates levels of CRF1 requires both the ligand, CRH, and activation of postsynaptic signal transduction pathways.
The precise molecular events leading from receptor binding to modulation of receptor levels and expression are not fully understood. The present study demonstrated a decline in CRF1 binding capacity in neocortex, associated with increased CRF1 mRNA expression. These findings are consistent with a mechanism of transient receptor elimination due to internalization, which leads to enhanced receptor mRNA transcription as has been shown in several other G-protein-coupled receptor systems (8, 29, 52). In other systems, this ligand-induced receptor internalization has been proposed to contribute to receptor desensitization via cell surface receptor downregulation (8, 29), as well as by regulating transcription of mRNA (52).
The specificity of the changes in binding capacity and synthesis to CRF1 should be noted. The fact that none of the manipulations in our studies led to changes in the CRF2 receptor reinforces the notion that this receptor must be regulated in a different manner by its cognate ligand. Because it has been shown that CRH has low affinity for this receptor subtype, the effects of urocortin, a native endogenous neuropeptide with high affinity for CRF2, on expression and binding capacity of this receptor will require evaluation.
In conclusion, the current studies strongly support both the presence as well as the clinical importance of the complex regulatory effects of CRH on CRF 1 expression in cortical and limbic regions. Because such reciprocal alterations in CRH-peptide and CRF1-receptor levels have been observed in a number of human neuropathological states, understanding the mechanisms by which CRH modulates its receptors is highly relevant to human neuropsychiatric conditions.
Acknowledgments
We thank Mariam Eghbal-Ahmadi for excellent technical assistance with the radioimmunoassays. Supported by NIH Grants RO1 NS 28912 and NS 39307 (T.Z.B) and R41 HD34975 (T.Z.B. and D.E.G.).
References
- 1.Aguilera G, Millan MA, Hauger RL, Catt KJ. Corticotropin-releasing factor receptors: Distribution and regulation in brain, pituitary, and peripheral tissues. Ann N Y Acad Sci. 1987;512:48–66. doi: 10.1111/j.1749-6632.1987.tb24950.x. [DOI] [PubMed] [Google Scholar]
- 2.Aldenhoff JB, Gruol DL, Rivier J, Vale W, Siggins GR. Corticotropin releasing factor decreases post-burst hyperpolarizations and excites hippocampal neurons. Science. 1983;221:875–877. doi: 10.1126/science.6603658. [DOI] [PubMed] [Google Scholar]
- 3.Avishai-Eliner S, Yi SJ, Baram TZ. Developmental profile of messenger RNA for the corticotropin-releasing hormone receptor in the rat limbic system. Dev Brain Res. 1996;91:159–163. doi: 10.1016/0165-3806(95)00158-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baram TZ, Schultz L. Corticotropin releasing hormone is a rapid and potent convulsant in the infant rat. Dev Brain Res. 1991;61:97–101. doi: 10.1016/0165-3806(91)90118-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baram TZ, Ribak CE. Peptide-induced infant status epilepticus causes neuronal death and synaptic reorganization. Neuroreport. 1995;6:277–280. doi: 10.1097/00001756-199501000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baram TZ, Chalmers DT, Chen C, Koutsoukos Y, De Souza EB. The CRF1 receptor mediates the excitatory actions of corticotropin releasing factor (CRF) in the developing rat brain: In vivo evidence using a novel, selective, non-peptide CRF receptor antagonist. Brain Res. 1997;770:89–95. doi: 10.1016/s0006-8993(97)00759-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baram TZ, Hatalski CG. Neuropeptide-mediated excitability: A key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21:471–476. doi: 10.1016/s0166-2236(98)01275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beaumont V, Hepworth MB, Luty JS, Kelly E, Henderson G. Somatostatin receptor desensitization in NG108–15 cells. A consequence of receptor sequestration. J Biol Chem. 1998;273:33,174–11,183. doi: 10.1074/jbc.273.50.33174. [DOI] [PubMed] [Google Scholar]
- 9.Bohan DP, Hoinrichs SC, Troncoso JC, Liu XJ, Kawas CH, Ling N, De Souza EB. Displacement of CRF from its binding protein as a possible treatment for Alzheimer’s disease. Nature. 1995;378:284–287. doi: 10.1038/378284a0. [DOI] [PubMed] [Google Scholar]
- 10.Bittencourt JC, Sawchenko PE. Do centrally administered neuropeptides access cognate receptors?: An analysis in the central corticotropin-releasing factor system. J Neurosci. 2000;20:1142–1156. doi: 10.1523/JNEUROSCI.20-03-01142.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunson KL, Schultz L, Baram TZ. The in vivo proconvulsant effects of corticotropin releasing hormone in the developing rat are independent of ionotropic glutamate receptor activation. Dev Brain Res. 1998a;111:119–128. doi: 10.1016/s0165-3806(98)00130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brunson KL, Schultz L, Baram TZ. Induction of c-fos expression in limbic neuronal populations of the immature rat by CRH administration. Soc Neurosci Abs. 1998b;24:1846. [Google Scholar]
- 13.Brunson KL, Eghbal-Ahmadi M, Bender RA, Chen Y, Baram TZ. Progressive hippocampal cell loss and long-term cognitive dysfunction induced by early-life administration of corticotropin releasing hormone reproduce the effects of early-life stress. Proc Natl Acad Sci. 2001;98:8856–8861. doi: 10.1073/pnas.151224898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin- releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: Comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;15:6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C, Dagnino R, Jr, De Souza EB, Grigoriadis DE, Huang CQ, Kim KI, Lui Z, Moran T, Webb TR, Whitten JP, Xie YF, McCarthy JR. Design and synthesis of a series of non-peptide high-affinity human corticotropin-releasing factor1 (CRF1) receptor antagonists. J Med Chem. 1996;39:4358–4360. doi: 10.1021/jm960149e. [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Brunson K, Cariaga W, Baram TZ. Immunocytochemical distribution of CRH receptor type-I (CRF1) in mouse brain: Light microscopy analysis. J Comp Neurol. 2000;420:305–323. doi: 10.1002/(sici)1096-9861(20000508)420:3<305::aid-cne3>3.0.co;2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Bender RA, Frotscher M, Baram TZ. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci. 2001;21:7171–7181. doi: 10.1523/JNEUROSCI.21-18-07171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Souza EB, Insel TR, Perrin MH, Rivier J, Vale WW, Kuhar MJ. Corticotropin-releasing factor receptors are widely distributed within the rat CNS: An autoradiographic study. J Neurosci. 1985;5:3189–3203. doi: 10.1523/JNEUROSCI.05-12-03189.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Souza EB, Whitehouse PJ, Price DL, Vale WW. Abnormalities in Corticotropin-releasing hormone (CRH) in Alzheimer’s disease and other human disorders. Ann N Y Acad Sci. 1987;512:237–247. doi: 10.1111/j.1749-6632.1987.tb24964.x. [DOI] [PubMed] [Google Scholar]
- 20.Diorio D, Viau V, Meaney MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci. 1993;13:3839–3847. doi: 10.1523/JNEUROSCI.13-09-03839.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubé C, Brunson KL, Nehligand A, Baram TZ. Corticotropin releasing hormone activates specific neuronal circuits, as indicated by c-fos expression and glucose metabolism. J Cerebral Blood Flow Metab. 2000;20:1414–1424. doi: 10.1097/00004647-200010000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eghbal-Ahmadi M, Hatalski C, Lovenberg TW, Avishai-Eliner S, Chalmers DT, Baram TZ. The developmental profile of the corticotropin releasing hormone receptor (CRF2) in rat brain predicts distinct age-specific functions. Dev Brain Res. 1998;107:81–90. doi: 10.1016/s0165-3806(98)00002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eghbal-Ahmadi M, Avishai-Eliner S, Hatalski CG, Baram TZ. Regulation of the expression of corticotropin releasing factor receptor type 2 (CRF2) in the hypothalamus and amygdala of the immature rat. J Neurosci. 1999;19:3982–3991. doi: 10.1523/JNEUROSCI.19-10-03982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehlers CL, Henriksen SL, Wang M, Rivier J, Vale W, Bloom FE. Corticotropin releasing factor produces increases in brain excitability and convulsive seizures in rats. Brain Res. 1983;278:332–336. doi: 10.1016/0006-8993(83)90266-4. [DOI] [PubMed] [Google Scholar]
- 25.Grigoriadis DE, Liu XJ, Vaughn J, Palmer SF, True CD, Vale WW, Ling N, De Souza EB. 125I-Tyr°-sauvagine: A novel high affinity radioligand for the pharmacological and biochemical study of human corticotropin-releasing factor2α receptors. Mol Pharmacol. 1996;50:679–686. [PubMed] [Google Scholar]
- 26.Hatalski CG, Guirguis C, Baram TZ. Corticotropin releasing factor mRNA expression in the hypothalamic paraventricular nucleus and the central nucleus of the amygdala is modulated by repeated stress in the immature rat. J Neuroendocrinol. 1998;10:663–669. doi: 10.1046/j.1365-2826.1998.00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatalski CG, Brunson KL, Tantayanubutr B, Chen Y, Baram TZ. Neuronal activity and stress differentially regulate hippocampal and hypothalamic corticotropin releasing hormone expression in the immature rat. Neuroscience. 2000;101:571–580. doi: 10.1016/s0306-4522(00)00386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hauger RL, Millan MA, Catt KJ, Aguilera G. Differential regulation of brain and pituitary corticotropin-releasing factor receptors by corticosterone. Endocrinology. 1987;120:1527–1533. doi: 10.1210/endo-120-4-1527. [DOI] [PubMed] [Google Scholar]
- 29.Hipkin RW, Friedman J, Clark RB, Eppler CM, Schonbrunn A. Agonist-induced desensitization, internalization, and phosphorylation of the sst2A somatostatin receptor. J Biol Chem. 1997;272:13,869–13,876. doi: 10.1074/jbc.272.21.13869. [DOI] [PubMed] [Google Scholar]
- 30.Hollrigel GS, Chen K, Baram TZ, Soltesz I. The pro-convulsant actions of corticotropin-releasing hormone in the hippocampus of infant rats. Neuroscience. 1998;84:71–79. doi: 10.1016/s0306-4522(97)00499-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honkaniemi J, Kononen J, Kainu T, Pyykönen I, Pelto-Huikko M. Induction of multiple immediate early genes in rat hypothalamic paraventricular nucleus after stress. Mol Brain Res. 1994;25:234–241. doi: 10.1016/0169-328x(94)90158-9. [DOI] [PubMed] [Google Scholar]
- 32.Hucks D, Lowther S, Crompton MR, Katona CL, Horton RW. Corticotropin releasing factor binding sites in cortex of depressed suicides. Psychopharmacology. 1997;134:174–178. doi: 10.1007/s002130050439. [DOI] [PubMed] [Google Scholar]
- 33.Imaki T, Nahan JL, Rivier C, Sawchenko PE, Vale W. Differential regulation of corticotropin-releasing factor mRNA in rat brain regions by glucocorticoids and stress. J Neurosci. 1991;11:585–599. doi: 10.1523/JNEUROSCI.11-03-00585.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iredale PA, Terwilliger R, Widnell KL, Nestler EJ, Duman RS. Differential regulation of corticotropin-releasing factor1 receptor expression by stress and agonist treatments in brain and cultured cells. Mol Pharmacol. 1996;50:1103–1110. [PubMed] [Google Scholar]
- 35.Koob GF, Heinrichs SC, Pich E, Menzaghi F, Baldwin H, Miczek K, Britton KT. The role of corticotropin-releasing factor in behavioral responses to stress. Ciba Found Symp. 1993;172:277–289. doi: 10.1002/9780470514368.ch14. [DOI] [PubMed] [Google Scholar]
- 36.Kuryshev YA, Childs GV, Ritchie AK. Corticotropin-releasing hormone stimulates Ca2+ entry through L- and P-type Ca2+ channels in rat corticotropes. Endocrinology. 1996;137:2269–2277. doi: 10.1210/endo.137.6.8641175. [DOI] [PubMed] [Google Scholar]
- 37.Leake A, Perry EK, Perry RH, Fairbairn AF, Ferrier IN. Cortical concentrations of corticotropin-releasing hormone and its receptor in Alzheimer type dementia and major depression. Biol Psychiatry. 1990;28:603–608. doi: 10.1016/0006-3223(90)90398-l. [DOI] [PubMed] [Google Scholar]
- 38.Lee EH, Hung HC, Lu KT, Chen WH, Chen HY. Protein synthesis in the hippocampus associated with memory facilitation by corticotropin-releasing factor in rats. Peptides. 1992;13:927–937. doi: 10.1016/0196-9781(92)90051-4. [DOI] [PubMed] [Google Scholar]
- 39.Lovenberg TW, Liaw CW, Grigoriadis DE, Clevenger W, Chalmers DT, De Souza EB, Oltersdorf T. Cloning and characterization of a functionally distinct corticotropin-releasing factor receptor subtype from rat brain. Proc Nat Acad Sci USA. 1995;92:836–840. doi: 10.1073/pnas.92.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makino S, Gold PW, Schulkin J. Corticosterone effects on corticotropin-releasing hormone mRNA in the central nucleus of the amygdala and the parvocellular region of the paraventricular nucleus of the hypothalamus. Brain Res. 1994;640:105–112. doi: 10.1016/0006-8993(94)91862-7. [DOI] [PubMed] [Google Scholar]
- 41.Makino S, Schulkin J, Smith MA, Pacak K, Palkovits M, Gold PW. Regulation of corticotropin-releasing hormone receptor messenger ribonucleic acid in the rat brain and pituitary by glucocorticoids and stress. Endocrinology. 1995;136:4517–4525. doi: 10.1210/endo.136.10.7664672. [DOI] [PubMed] [Google Scholar]
- 42.Mansi JA, Rivest S, Drolet G. Regulation of corticotropin-releasing factor type 1 (CRF1) receptor messenger ribonucleic acid in the paraventricular nucleus of rat hypothalamus by exogenous CRF. Endocrinology. 1996;137:4619–4628. doi: 10.1210/endo.137.11.8895325. [DOI] [PubMed] [Google Scholar]
- 43.Merali Z, Mcintosh J, Kent P, Michaud D, Anisman H. Aversive and appetitive events evoke the release of corticotropin-releasing hormone a bombesin-like peptides at the central nucleus of the amygdala. J Neurosci. 1998;18:4758–4766. doi: 10.1523/JNEUROSCI.18-12-04758.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry. 1988;45:577–579. doi: 10.1001/archpsyc.1988.01800300075009. [DOI] [PubMed] [Google Scholar]
- 45.Piekut DT, Phipps B. Increased corticotropin-releasing factor immunoreactivity in select brain sites following kainate elicited seizures. Brain Res. 1998;781:99–111. doi: 10.1016/s0006-8993(97)01219-5. [DOI] [PubMed] [Google Scholar]
- 46.Pihoker C, Cain ST, Nemeroff CB. Postnatal development of regional binding of corticotropin-releasing factor and adenylate cyclase activity in the rat brain. Prog Neuro Psychopharmacol Biol Psychiatry. 1992;16:581–586. doi: 10.1016/0278-5846(92)90063-k. [DOI] [PubMed] [Google Scholar]
- 47.Rabadan-Diehl C, Kiss A, Camacho C, Aguilera G. Regulation of messenger ribonucleic acid for corticotropin releasing hormone receptor in the pituitary during stress. Endocrinology. 1996;137:3808–381. doi: 10.1210/endo.137.9.8756551. [DOI] [PubMed] [Google Scholar]
- 48.Rivest S, Laflamme N, Nappi RE. Immune challenge and immobilization stress transcription of the gene encoding the CRF receptor in selective nuclei of the rat hypothalamus. J Neurosci. 1995;15:2680–2695. doi: 10.1523/JNEUROSCI.15-04-02680.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherwood NM, Timiras PS. A Sterotaxic Atlas of the Developing Rat Brain. University of California Press; Berkeley: 1970. [Google Scholar]
- 50.Smith MA, Weiss SR, Berry RL, Zhang LX, Clark M, Massenburg G, Post RM. Amygdala-kindled seizures increase the expression of corticotropin-releasing factor and CRF-binding protein in GABAergic interneurons of the dentate hilus. Brain Res. 1997;745:248–256. doi: 10.1016/s0006-8993(96)01157-2. [DOI] [PubMed] [Google Scholar]
- 51.Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- 52.Souazé F, Rostene W, Forgez P. Neurotensin agonist induces differential regulation of neurotensin receptor mRNA. Identification of distinct transcriptional and post-tran-scriptional mechanisms. J Biol Chem. 1997;272:10,087–10,094. doi: 10.1074/jbc.272.15.10087. [DOI] [PubMed] [Google Scholar]
- 53.Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin releasing factor immunoreactive cells and fibers in the rat brain: An immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 54.Swiergel AR, Takahashi LK, Kalin NH. Attenuation of stress-induced behavior by antagonism of corticotropin-releasing factor receptors in the central amygdala in the rat. Brain Res. 1993;623:229–234. doi: 10.1016/0006-8993(93)91432-r. [DOI] [PubMed] [Google Scholar]
- 55.Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JMHM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- 56.Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ. Seizure-induced neuronal injury: Vulnerability to febrile seizures in an immature rat model. J Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- 58.Weiss JH, Yin HZ, Baram TZ. Intracellular calcium is acutely increased in subsets of hippocampal cells exposed to corticotropin releasing hormone. Epilepsia. 1996;37(Suppl 5):27. [Google Scholar]
- 59.Wynn PC, Harwood JP, Catt KJ, Aguilera G. Regulation of corticotropin-releasing factor (CRF) receptors in the rat pituitary gland: Effects of adrenalectomy on CRF receptors and corticotroph responses. Endocrinology. 1985;116:1653–1659. doi: 10.1210/endo-116-4-1653. [DOI] [PubMed] [Google Scholar]
- 60.Yi SJ, Baram TZ. Corticotropin-releasing hormone mediates the response to cold stress in the neonatal rat without compensatory enhancement of the peptide’s gene expression. Endocrinology. 1994;135:2364–2368. doi: 10.1210/endo.135.6.7988418. [DOI] [PMC free article] [PubMed] [Google Scholar]