

Abstract
Excitotoxic cell death is the fundamental process responsible for many human neurodegenerative disorders, yet the basic mechanisms involved are not fully understood. Here, we exploited the fact that the immature brain is remarkably resistant to seizure-induced excitotoxic cell death and examined the underlying protective mechanisms. We found that, unlike in the adult, seizures do not increase the formation of reactive oxygen species or result in mitochondrial dysfunction in neonatal brain, because of high levels of the mitochondrial uncoupling protein (UCP2). UCP2 expression and function were basally increased in neonatal brain by the fat-rich diet of maternal milk, and substituting a low-fat diet reduced UCP2, restored mitochondrial coupling, and permitted seizure-induced neuronal injury. Thus, modulation of UCP2 expression and function by dietary fat protects neonatal neurons from excitotoxicity by preventing mitochondrial dysfunction. This mechanism offers novel neuroprotective strategies for individuals, greater than 1% of the world’s population, who are affected by seizures.
Prolonged seizures kill neurons in the adult limbic circuit including perirhinal cortex and hippocampus,1,2 and this neuronal death may contribute to seizure-induced cognitive deficits and the pathogenesis of epilepsy.3,4 In contrast, although prolonged limbic status epilepticus is readily provoked in immature rat during the first 2 postnatal weeks,5–8 neuronal death is limited.5–7,9 The mechanisms for this remarkable resistance of the immature brain to seizure-induced neuronal death have not been delineated. However, fundamental differences in neuronal metabolism under conditions of high demand, that is, the repetitive neuronal firing during a prolonged seizure, have been postulated.10,11
Recently, the key role of mitochondrial function and dysfunction in the mechanisms of excitotoxic cell death has been unfolding.12–14 Specifically, these organelles have been shown to contribute crucially to calcium homeostasis of the cell, and to the handling of calcium influx during intense neuronal excitation.15,16 Conditions of intense energy demand and increased calcium load (i.e., those promoting excitotoxicity) lead to a marked increase in the formation of reactive oxygen species (ROS) in mitochondria.17–20 Accumulation of these compounds, coupled with progressive mitochondrial dysfunction and disruption of ATP production, play a critical role in excitotoxic neuronal injury and death.16,20–22
The magnitude of ROS production is largely dependent on, and correlates with, the mitochondrial membrane potential.23,24 This is because increased negative potential inside the mitochondria hinders further extrusion of positively charged protons, slowing electron transport. The resulting increased half-life of electron-rich compounds promotes electron shunting into ROS.23,24 Therefore, reduction of this potential via increased proton conductance across the mitochondrial inner membrane (uncoupling) reduces ROS formation. Uncoupling is mediated by members of the UCP family, which function to dissociate ATP production from oxygen consumption in mitochondria of muscle and fat tissues,13,25 leading to heat generation. Among characterized uncoupling proteins, UCP2 is significantly expressed in brain, including seizure-sensitive regions.25,26 The physiological roles of UCP2 expression in hippocampus, amygdala, and limbic (perirhinal and piriform) cortex are unclear.27,28 However, UCP2-mediated mitochondrial uncoupling and reduced ROS formation during seizures could be neuroprotective.25,27,29 In addition, during the developmental period when seizure-induced cell death is limited,6,8,9,30 the key brain energy substrate and a principal component of the suckling rat’s diet is fat,10 which induces and activates UCPs.29,31 Therefore, we tested the hypothesis that the resistance of limbic neurons of immature rat to seizure-induced cell death stems from the partial uncoupling of mitochondria in these neurons as a result of the high levels of UCP2 expression, which are enhanced by the fat-rich maternal milk. To further test this hypothesis, we then artificially reduced UCP activity and expression by providing immature rats with a low-fat synthetic diet; this manipulation provoked seizure-induced death of select limbic neurons.
Materials and Methods
Experimental Protocols
All animal experiments conformed to National Institute of Health guidelines and were performed with permission of the institutional animal care committee. To generate seizures of similar severity,32,33 we gave adult Sprague-Dawley rats (n = 56) 10 to 15mg/kg kainic acid (Opika; Ocean Produce International, Shelbourne, Nova Scotia) in 5mg/kg increments and immature rats (10–11 days; total n = 426) rats 1.5mg/kg,34 both intraperitoneally. At both ages, severe seizures ensued, progressing from automatisms to clonus and loss of balance.2,32–34 The kainic acid doses used here led to approximately 20% mortality in adults and minimal (<1%) in the immature rats.34 In the latter group, neither the duration nor the intensity of the seizures (scored behaviorally according to scales previously correlated with electroencephalogram seizures34) were influenced by the experimental and dietary manipulations (see below).
Dietary Manipulations
Adult rats were maintained on ad libitum laboratory chow. Control immature rats were kept with the dams, nursing normally. The isocaloric low-fat diet was administered to P10 rats for 24 hours as described in detail previously.35 The diet consisted of 10ml fat-free milk (Nestle Carnation) supplemented with nonfat powdered milk (Carnation; 14mg/100ml), supplying 16Kcal/day. Both experimental animals and nursing littermate controls were weighed before and after the experiment, and only those gaining or maintaining their weight were used, to avoid fasting-induced breakdown of body fat.36 When kainic acid was administered, experimental animals (n = 90) were maintained on the low-fat diet for the duration of the experiment (30 hours total).
Mitochondria Isolation and Measurements of Respiration and Reactive Oxygen Species Production
Total mitochondria (hippocampal) and purified somatic mitochondria (perirhinal and piriform cortex) were isolated using differential centrifugation or a discontinuous ficoll gradient.22 Synaptic mitochondria were released using a nitrogen cell disrupter (1,000psi for 5 minutes). Mitochondrial respiration was assessed using standard polarography methods at 37°C with pyruvate and malate (5 and 2.5mM) as oxidative substrates.22,29 UCP-mediated proton conductance was measured as increased fatty acid–induced respiration,29 which then was compared with maximum respiration induced by the chemical uncoupler FCCP.29 Mitochondrial ROS production was measured using the H2DCFDA (DCF) dye22 and quantified by comparing the relative amounts of ROS produced in the presence of oligomycin (to maximize membrane potential) to basal ROS production (indicated as 100%). For all experiments, tissue from two to three adults and four to six immature brains was combined per sample. There were four to nine samples per assay.
Immunocytochemistry
UCP2 immunoreactivity was determined using a rabbit anti–UCP2 polyclonal antibody (Chemicon, Temecula, CA) diluted 1:10,000, and routine biotin/avidin complex methods (Elite kit; Vector Laboratories, Burlingame, CA), as described in detail previously.33,37 Immunoreactive neurons in cornu ammonis 3 region of the hippocampus (CA3) were counted, without knowledge of group, in six sections from each of a minimum of four animals per group. These groups included adult and infant rat controls and adult and infant seizure-experiencing animals.
Determination of Neuronal Injury
Animals were perfused transcardially under deep barbiturate anesthesia as described elsewhere.6,37 Brains were sectioned and processed for silver staining and Fluoro-Jade using established methods.6,9,38,39 Neuronal injury was evaluated using argyrophilia6,38 or uptake of Fluoro-Jade.9,39
Statistical Considerations
Cell counts were performed without knowledge of treatment. For all analyses, the significance of differences among groups was set at p value of 0.05. Data were evaluated using Student’s t test, with Welch’s correction when required.
Results
Seizures Injure Limbic Neurons in Adult, but Not in Immature Rat Brain
Prolonged limbic seizures induced by systemic administration of kainic acid to adult rats32 resulted in the typical pattern of limbic cell death.1,2 Significant, widespread injury occurred in piriform and perirhinal cortex (Fig 1D, E, G, H), and a less pronounced injury involved hippocampal CA3 pyramidal cells (see Fig 1A, B) and hilar neurons (not shown). Kainate led to seizures that were as severe and prolonged (approximately 3 hours) in immature (10-day-old) rats,30,33,34 but to no observable neuronal death (see Fig 1C, F, I).
Fig 1.
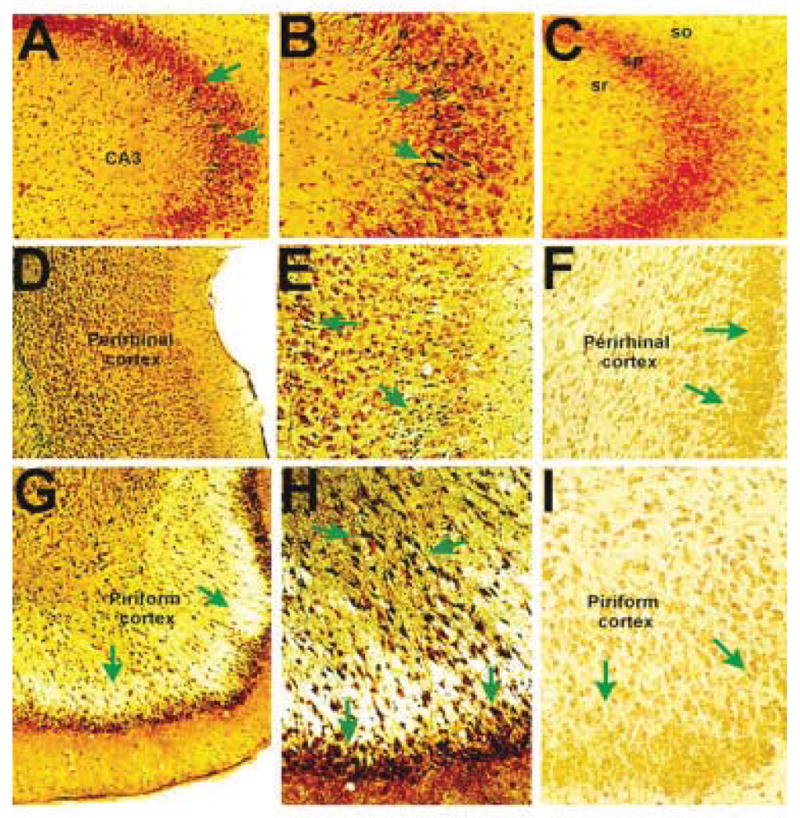
Neuronal injury in seizure-sensitive limbic regions of mature and postnatal day 10 (P10) rats after systemic administration of kainic acid.2,33 (A, B) Hippocampal CA3 from adult rats demonstrates neurons with silver affinity within the pyramidal cell layer (green arrows), indicating their injury.1,2,6,7,9 Such cells are not found in the P10 rat CA3 (C) including the pyramidal (sp), radiatum (sr), or oriens (so) cell layers. (D, E) Low and higher magnification views of the perirhinal cortex of mature rat show excitotoxicity in both deep and superficial layers of this seizure-vulnerable limbic region, whereas the corresponding region from a P10 rat (F) is free of silver-stained cells. (G–I) The piriform cortex, a highly seizure-vulnerable region1,2 demonstrates seizure-injured neurons in adult (G, H) but not immature (I) brain.
Neuronal Mitochondria of Immature Hippocampus Are Uncoupled and Are Enriched in UCP2
The potential contribution of mitochondrial uncoupling to this resistance of the immature rat to seizure-induced limbic cell death was examined by conducting functional analyses of free fatty acid (FFA)–induced increases in proton conductance, a measure of the activation of mitochondrial UCPs.29,40 UCP-mediated proton conductance was measured as increased FFA-induced respiration,22 which then was compared with maximum respiration induced by the chemical uncoupler FCCP (Fig 2A, inset). Hippocampal mitochondria from adult rats showed a modest response to FFA; this indicates their limited uncoupling capacity, that is, a limited availability of UCPs that can be activated by FFAs.29 In contrast, mitochondria from immature animals exhibited drastic increase in proton conductance in the presence of FFA, indicating UCP activation (see Fig 2A, top graph). Somatic mitochondrial fractions from perirhinal and piriform cortex demonstrated a similar pattern (see Fig 2A, bottom graph). These findings indicate that the UCP function and uncoupling capacity of neuronal mitochondria from immature rat brain are significantly greater than in mitochondria from limbic neurons of the adult.
Fig 2.

Uncoupling protein (UCP) function and UCP2 expression in seizure-vulnerable limbic brain regions of immature (P10) compared with adult rat. (A) Mitochondrial fractions from hippocampus (top) and perirhinal/piriform cortex (bottom) of the P10 rat are highly uncoupled on exposure to the UCP-activating free fatty acid (FFA) palmitate, when respiration is compared with the maximum respiration induced by the chemical uncoupler FCCP (bottom inset). Adult hippocampal and perirhinal/piriform cortex mitochondria respond modestly to FFA (indicating little UCP available for activation), whereas mitochondria from immature rats are completely uncoupled in the presence of palmitic acid (bars represent group means ± SEM; asterisk indicates p < 0.02). (B) When brain sections from immature and adult rats are processed for UCP2 immunocytochemistry,37 fewer UCP2-immunoreactive neurons (arrows) are evident in adult CA3 (8.7 ± 0.9) compared with the corresponding region of the P10 rat hippocampus (17.6 ± 0.6; C, D). (E, F) Compared with the rare UCP2-immunoreactive neurons in adult perirhinal cortex (E, arrows), dense staining is found in the limbic cortex of immature rats (F, arrows).
Among characterized members of the UCP family of proteins, UCP2 is significantly expressed in brain, including seizure-sensitive regions.25,26 To determine whether the mitochondrial uncoupling activity in immature rat limbic neurons might derive from high levels of UCP2 expression, we compared UCP2 immunoreactivity in neonatal and adult brain.37 UCP2 protein was concentrated in cell bodies in hippocampal CA3 (see Fig 2B, C, D). A more quantitative analysis indicated that the numbers of neurons expressing this protein were significantly higher in immature CA3 compared with the corresponding adult region (17.6 ± 0.6 vs 8.7 ± 0.9). In perirhinal and piriform cortices, where neuronal cell bodies are smaller, UCP2 immunoreactivity was more notable in neuronal processes, likely representing mitochondria-rich terminals and postsynaptic elements25,26 (see Fig 2E and F).
Uncoupled Mitochondria from Immature Brain Produce Little Reactive Oxygen Species during Severe Limbic Seizures
Enhanced UCP2 levels and mitochondrial uncoupling in immature neurons would predict reduced ROS production during seizures, compared with adult rat neurons. Indeed, consistent with previous reports22,41 seizures led to striking increases in ROS production in adult (Fig 3A), but not immature rat mitochondria (see Fig 3B). Increased ROS production in adult brain mitochondria was evident both 6 and 24 hours after the seizures (see Fig 3A). Interestingly, seizures increased both the expression (see Fig 3C) and the function of UCP2 (see Fig 3D) in mature but not neonatal rat hippocampus. This ROS-induced upregulation of UCP2 might be a compensatory, neuroprotective phenomenon,25,26 limiting further free radical formation.29
Fig 3.

Differential effects of severe seizures on reactive oxygen species (ROS) production in mitochondria from immature and adult rat limbic neurons. (A) Evaluation of adult rat ROS production showed a significant increase at 6 and 24 hours after kainic acid administration, whereas (B) infant rat ROS production was not enhanced by these severe seizures.33 (C) Prolonged seizures significantly increased the number of UCP2-expressing neurons in the CA3 hippocampal region of mature rats. (D) Concomitantly, adult rat mitochondria demonstrated a significant increase in fatty acid–induced respiration (a measure of UCP function) 24 hours after the seizures. For all graphs, bars represent group means ± SEM; asterisks indicate p < 0.05).
UCP expression and function are enhanced by fatty acids,29,40 which are maintained at high levels in neonatal animals by the high fat content of maternal milk.10 Furthermore, fat breakdown products are taken up into the neonatal brain at rates far higher than in older immature and adult rats.10 These facts raised the possibility that the high expression and function of UCP2 in neonatal brain might be caused by the neonatal diet. This hypothesis would predict that UCP2 levels and activity, and the UCP2-mediated neuroprotection, would diminish in neonatal rats deprived of their normally fat-rich diet.
Reducing Uncoupling Protein Expression and Function in Immature Brain Provokes Seizure-induced Production of Reactive Oxygen Species, Leading to Neuronal Injury
To test this hypothesis, we fed neonatal rats an isocaloric low-fat diet for 24 hours.35 In mitochondria isolated after this acute dietary fat restriction, FFA-induced proton conductance was significantly decreased, indicating reduced mitochondrial uncoupling capacity (Fig 4A). As expected from this diet-induced decrease of mitochondrial UCP activity, basal ROS production was significantly enhanced, approaching adult levels (see Fig 4A, inset).
Fig 4.

Reduction of dietary fat by substitution of an isocaloric, low-fat diet to immature rats reduces UCP function and promotes ROS production as well as seizure-induced excitotoxicity. (A) UCP function, measured as fatty acid–induced respiration, is significantly reduced in neonatal rats fed low-fat diet compared with maternal milk–fed littermates and resembles those in adult mitochondria. (inset) Basal ROS production in the presence of oligomycin (to maximize membrane potential) in isolated mitochondria from the group fed a low-fat diet are significantly increased compared with milk-fed littermates, approaching the basal levels found in adult mitochondria. (B) Energetic demand induced by severe seizures provokes striking increases in ROS production in UCP-suppressed (low-fat diet fed) neonatal rats, but not in those maintained on maternal milk. Bars represent means ± SEM; asterisks indicate p < 0.05). (C–H) Seizures provoke neuronal injury (visualized using Fluro-Jade)9 in several highly seizure-vulnerable regions of infant rats with suppressed UCP function. In adults, both perirhinal (C) and piriform (D) cortex demonstrated excitotoxic injury (arrows), whereas none was evident in the corresponding limbic regions of P10 rats on a “normal” high-fat diet (E, F). In striking contrast, excitotoxic injury occurred in perirhinal (G) and piriform (H) cortex of the low-fat diet fed infant rats (arrows).
The enhancement of ROS production was even more remarkable when kainic acid was administered to immature rats; seizure duration and intensity were not influenced by the immature rats’ diet. However, whereas seizures elicited little ROS production in neonatal rats maintained on fat-rich maternal milk, these seizures provoked drastic increases in ROS production in mitochondria of the low-fat diet fed group (see Fig 4B), resembling the findings in adult mitochondria (see Fig 3A).
If UCP-mediated mitochondrial uncoupling underlies the resistance of the infant brain to seizure-induced excitotoxic cell death, then animals fed a low-fat diet should no longer be “protected.” In line with this prediction, brains of animals that had been fed a low-fat diet and then treated with kainic acid demonstrated striking seizure-induced neuronal injury, visualized by Fluoro-Jade staining. Fluoro-Jade–positive neurons were numerous in perirhinal and piriform cortex in most (7 of 13) immature rats in the low-fat diet group (see Fig 4G and H) and were seen in only one of the 9 animals in the high-fat diet control group (see Fig 4E and F). Thus, reduction of UCP2 function via dietary manipulation rendered immature rat limbic neurons vulnerable to seizure-induced neuronal injury.
Discussion
The major findings of these studies are as follows. (1) Neuronal injury in mature hippocampus and cortex is associated with ROS production, whereas little ROS production is induced in resistant neurons in immature brain. (2) Mitochondrial uncoupling is a key determinant of the absence of seizure-induced ROS production in the immature limbic neurons and is associated with high basal expression of the uncoupling protein UCP2. (3) Reduction of UCP2 function and the resulting mitochondrial “coupling” endows the immature brain with vulnerability to seizure-induced neuronal injury.
Seizure-induced cell death has been a major topic of research and debate for several decades.1–11,42 Neuronal death resulting from status epilepticus,43,44 and perhaps even from numerous recurrent seizures45–47 (but see Pitkanen and colleagues48), have been implicated in the process of epileptogenesis and in the declining cognitive function in populations of individuals with persistent epilepsy.49,50 Therefore, prevention of seizure-induced “damage” is a primary goal for neurologists and neuroscientists, a goal that requires understanding of the mechanisms involved.3–5,44,49,51 Remarkably, early in postnatal life, whereas limbic neurons are vulnerable to direct application of excitotoxins,52 seizure-induced cell death, secondary to extreme activation of a neuronal circuit1,2,53 is uncommon in both humans and animal models.3,5–9,11,30,51,54 The mechanisms for this resistance, which disappears during maturation,54 have not been well resolved; clearly, knowledge of these mechanisms should be highly applicable to amelioration or prevention of seizure-induced cell death in the adult, and perhaps for other forms of excitotoxicity.
Whereas the role of ROS and of mitochondrial dysfunction in excitotoxic cell death has been rapidly unfolding, the relationship of mitochondrial function to specific vulnerability or resistance of neurons to seizure-induced cell death are not fully understood. Here, we highlight the role of mitochondrial uncoupling, driven by the UCPs, in the striking resistance of limbic regions of immature brain to prolonged seizures, including status epilepticus. Indeed, elicitation of seizure-provoked injury of limbic neurons by reducing mitochondrial uncoupling provides direct evidence for the critical role of this UCP-governed process in the mechanisms that protect the immature central nervous system from excitotoxic injury.55
How do UCPs protect neurons from excitotoxicity? UCPs dissipate mitochondrial membrane potential that governs ROS production (see introduction). Reduced membrane potential also decreases mitochondrial uptake of [Ca2+].15 Because mitochondrial Ca2+ overload is considered a major trigger of mitochondrial dysfunction leading to ROS production, UCPs may reduce ROS formation also by limiting mitochondrial [Ca2+]. UCP’s expression is increased by fatty acids, independent of caloric overload, and the mechanism might involve fatty acid oxidation.56 Thus, the high fatty acid content of the neonatal rat diet and the excellent penetration of FFAs into the immature brain might account for the increased UCP levels shown here. In addition, FFAs increase UCP function, by “chaperoning” proton transport through the UCP “pore.”
In this study, neuronal injury occurred in limbic (perirhinal and piriform) cortex but not in hippocampus of immature rats with lowered UCP2 function. This might be a result of the increased vulnerability of perirhinal and piriform neurons to kainic acid–induced seizures compared with hippocampal pyramidal cells1,2 or of subtle differences in the pattern of seizure activity induced in the neonatal brain. Alternatively, this observation suggests additional determinants of the vulnerability of these neurons.
In summary, the experiments described here suggest straightforward strategies for manipulating mitochondrial uncoupling in seizure-vulnerable brain regions, which is of major importance to human health. Seizures affect 1 in 25 infants and children and more than 1% of adults,57,58 where they may be associated with progressive regional loss of brain volume.47 Therefore, understanding the mechanisms utilized by the immature brain to prevent seizure-induced cell death should facilitate interventional strategies aimed at preventing excitotoxic injury in the mature brain.
Acknowledgments
This work was supported by grants from the NIH (NS35439; NS28912, T.Z.B.) and NS32280 (O.S.), a NeoTherapeutics Fellowship (P.G.S.), and a predoctoral training grant (NS07444, K.D.).
We thank Drs. T. Bartfai, T. Horvath, and J. M. Rho for support and helpful discussions and M. Eghbal-Ahmadi for excellent technical advice and help with the dietary manipulations.
References
- 1.Schwob JE, Fuller T, Price TL, Olney JW. Widespread patterns of neuronal damage following system or intracerebral injections of kainic acid: a histological study. Neuroscience. 1980;5:991–1014. doi: 10.1016/0306-4522(80)90181-5. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Ari Y, Tremblay E, Riche D, et al. Electrographic, clinical and pathological alterations following systemic administration of kainic acid, bicuculline or pentetrazole: metabolic mapping using the deoxyglucose method with special reference to the pathology of epilepsy. Neuroscience. 1981;6:1361–1391. doi: 10.1016/0306-4522(81)90193-7. [DOI] [PubMed] [Google Scholar]
- 3.Holmes GL, Ben-Ari Y. Seizures in the developing brain: perhaps not so benign after all. Neuron. 1998;21:1231–1234. doi: 10.1016/s0896-6273(00)80642-x. [DOI] [PubMed] [Google Scholar]
- 4.Sutula TP, Pitkanen A. More evidence for seizure-induced neuron loss: is hippocampal sclerosis both cause and effect of epilepsy? Neurology. 2001;57:169–170. doi: 10.1212/wnl.57.2.169. [DOI] [PubMed] [Google Scholar]
- 5.Baram TZ, Hatalski CG. Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 1998;21:471–476. doi: 10.1016/s0166-2236(98)01275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toth Z, Yan XX, Haftoglou S, et al. Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haas KZ, Sperber EF, Opanashuk LA, et al. Resistance of immature hippocampus to morphologic and physiologic alterations following status epilepticus or kindling. Hippocampus. 2001;11:615–625. doi: 10.1002/hipo.1076. [DOI] [PubMed] [Google Scholar]
- 8.Holmes GL, McCabe B. Brain development and generation of brain pathologies. Int Rev Neurobiol. 2001;45:17–41. doi: 10.1016/s0074-7742(01)45005-7. [DOI] [PubMed] [Google Scholar]
- 9.Kubová H, Druga R, Lukasiuk K, et al. Status epilepticus causes necrotic damage in the mediodorsal nucleus of the thalamus in immature rats. J Neurosci. 2001;21:3593–3599. doi: 10.1523/JNEUROSCI.21-10-03593.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nehlig A, Boyet S, Pereira de Vasconcelos A. Autographic measurement of local cerebral beta-hydroxybutyrate uptake in the rat during postnatal development. Neuroscience. 1991;40:871–878. doi: 10.1016/0306-4522(91)90018-j. [DOI] [PubMed] [Google Scholar]
- 11.Fernandes MJ, Dubé C, Boyet S, et al. Correlation between hypermetabolism and neuronal damage during status epilepticus induced by lithium and pilocarpine in immature and adult rats. J Cereb Blood Flow Metab. 1999;19:195–209. doi: 10.1097/00004647-199902000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Patel M, Day BJ, Crapo JD, et al. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- 13.Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- 14.Cock HR, Tong X, Hargreaves LP, et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res. 2002;48:157–168. doi: 10.1016/s0920-1211(01)00334-5. [DOI] [PubMed] [Google Scholar]
- 15.Stout AK, Raphael HM, Kanterewicz BI, et al. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- 16.Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. Calcium-induced cytochrome C release from CNS mitochondria is associated with the permeability transition and rapture of the outer membrane. J Neurochem. 2002;80:207–218. doi: 10.1046/j.0022-3042.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- 17.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schulz JB, Henshaw DR, Siwak D, et al. Involvement of free radicals in excitotoxicity in vivo. J Neurochem. 1995;64:2239–2247. doi: 10.1046/j.1471-4159.1995.64052239.x. [DOI] [PubMed] [Google Scholar]
- 19.Rong Y, Doctrow SR, Tocco G, Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci USA. 1999;96:9897–9902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–305. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 21.Melov S, Coskun P, Patel M, Tuinstra R. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sullivan PG, Geiger JD, Mattson MP, Scheff S. Dietary supplement creatine protects against traumatic brain injury. Ann Neurol. 2000;48:723–729. [PubMed] [Google Scholar]
- 23.Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q Rev Biophys. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
- 24.Votyakova TV, Reynolds IJ. DeltaPsi(m)-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem. 2001;79:266–277. doi: 10.1046/j.1471-4159.2001.00548.x. [DOI] [PubMed] [Google Scholar]
- 25.Richard D, Clavel S, Huang Q, et al. Uncoupling protein 2 in the brain: distribution and function. Biochem Soc Trans. 2001;29:812–817. doi: 10.1042/0300-5127:0290812. [DOI] [PubMed] [Google Scholar]
- 26.Horvath TL, Craig H, Warden CH, et al. Brain uncoupling protein-2: uncoupled neuronal mitochondria predict thermal synapses in homeostatic centers. J Neurosci. 1999;19:10417–10427. doi: 10.1523/JNEUROSCI.19-23-10417.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arsenijevic D, Onuma H, Pecqueur C, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 28.Vidal-Puig AJ, Grujic D, Zhang CY, et al. Energy metabolism in uncoupling protein 3 gene knockout mice. J Biol Chem. 2000;275:16258–16266. doi: 10.1074/jbc.M910179199. [DOI] [PubMed] [Google Scholar]
- 29.Echtay KS, Roussel D, St-Pierre J, et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 30.Nitecka L, Tremblay E, Charton G, et al. Maturation of kainic acid seizure-brain damage syndrome in the rat. II. Histopathological sequelae. Neuroscience. 1984;13:1073–1094. doi: 10.1016/0306-4522(84)90289-6. [DOI] [PubMed] [Google Scholar]
- 31.Richard D, Rivest R, Huang Q, et al. Distribution of uncoupling protein 2 mRNA in the mouse brain. J Comp Neurol. 1998;397:549–560. [PubMed] [Google Scholar]
- 32.Dubé C, Chen K, Eghbal-Ahmadi M, et al. Prolonged febrile seizures in the immature rat model enhance hippocampal excitability long-term. Ann Neurol. 2000;47:336–344. [PMC free article] [PubMed] [Google Scholar]
- 33.Brewster A, Bender RA, Chen Y, et al. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform and cell-specific manner. J Neurosci. 2002;22:4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunson K, Schultz L, Baram TZ. The in vivo proconvulsant effects of CRH in developing rat are independent of ionotropic glutamate receptor activation. Brain Res. 1998;111:119–128. doi: 10.1016/s0165-3806(98)00130-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eghbal-Ahmadi M, Avishai-Eliner S, Hatalski CG, et al. Regulation of the expression of corticotropin releasing factor receptor type 2 (CRF2) in the hypothalamus and amygdala of the immature rat. J Neurosci. 1999;19:3982–3991. doi: 10.1523/JNEUROSCI.19-10-03982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stafstrom CE. Animal models of the ketogenic diet: what have we learned, what can we learn? Epilepsy Res. 1999;37:241–259. doi: 10.1016/s0920-1211(99)00067-4. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Bender R, Frotscher M, et al. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci. 2001;21:7171–7181. doi: 10.1523/JNEUROSCI.21-18-07171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmued LCY, Albertson C, Slikker W., Jr Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- 40.Garlid KD, Jaburek M, Jezek P. Mechanism of uncoupling protein action. Biochem Soc Trans. 2001;29:803–806. doi: 10.1042/0300-5127:0290803. [DOI] [PubMed] [Google Scholar]
- 41.Liang LP, Ho YS, Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience. 2000;101:563–570. doi: 10.1016/s0306-4522(00)00397-3. [DOI] [PubMed] [Google Scholar]
- 42.Sagar HJ, Oxbury JM. Hippocampal neuron loss in temporal lobe epilepsy: correlation with early childhood convulsions. Ann Neurol. 1987;22:334–340. doi: 10.1002/ana.410220309. [DOI] [PubMed] [Google Scholar]
- 43.Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med. 1998;338:970–976. doi: 10.1056/NEJM199804023381407. [DOI] [PubMed] [Google Scholar]
- 44.Coulter DA, DeLorenzo RJ. Basic mechanisms of status epilepticus. Adv Neurol. 1999;79:725–733. [PubMed] [Google Scholar]
- 45.Mathern GW, Adelson PD, Cahan LD, Leite JP. Hippocampal neuron damage in human epilepsy: Meyer’s hypothesis revisited. Prog Brain Res. 2002;135:237–251. doi: 10.1016/s0079-6123(02)35023-4. [DOI] [PubMed] [Google Scholar]
- 46.Kotloski R, Lynch M, Lauersdorf S, Sutula T. Repeated brief seizures induce progressive hippocampal neuron loss and memory deficits. Prog Brain Res. 2002;135:95–110. doi: 10.1016/S0079-6123(02)35010-6. [DOI] [PubMed] [Google Scholar]
- 47.Kalviainen R, Salmenpera T, Partanen K, et al. Recurrent seizures may cause hippocampal damage in temporal lobe epilepsy. Neurology. 1998;50:1377–1382. doi: 10.1212/wnl.50.5.1377. [DOI] [PubMed] [Google Scholar]
- 48.Pitkanen A, Nissinen J, Nairismagi J, et al. Progression of neuronal damage after status epilepticus and during spontaneous seizures in a rat model of temporal lobe epilepsy. Prog Brain Res. 2002;135:67–83. doi: 10.1016/S0079-6123(02)35008-8. [DOI] [PubMed] [Google Scholar]
- 49.Sutula TP, Hermann B. Progression in mesial temporal lobe epilepsy. Ann Neurol. 1999;45:553–556. [PubMed] [Google Scholar]
- 50.Meador KJ. Cognitive outcomes and predictive factors in epilepsy. Neurology. 2002;58(suppl 5):S21–S26. doi: 10.1212/wnl.58.8_suppl_5.s21. [DOI] [PubMed] [Google Scholar]
- 51.Baram TZ, Eghbal-Ahmadi M, Bender RA. Is neuronal death required for seizure-induced epileptogenesis in the immature brain? Prog Brain Res. 2002;135:365–375. doi: 10.1016/S0079-6123(02)35033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trescher WH, McDonald JW, Johnston MV. Quinolinate-induced injury is enhanced in developing rat brain. Dev Brain Res. 1994;83:224–232. doi: 10.1016/0165-3806(94)00141-3. [DOI] [PubMed] [Google Scholar]
- 53.Olney JW, Collins RC, Sloviter RS. Excitotoxic mechanisms of epileptic brain damage. Adv Neurol. 1986;44:857–877. [PubMed] [Google Scholar]
- 54.Sankar R, Shin DH, Liu H, et al. Patterns of status epilepticus-induced neuronal injury during development and long-term consequences. J Neurosci. 1998;18:8382–8393. doi: 10.1523/JNEUROSCI.18-20-08382.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bechmann I, Diano S, Warden CH, et al. Brain mitochondrial uncoupling protein 2 (UCP2) a protective stress signal in neuronal injury. Biochem Pharmacol. 2002;64:363–367. doi: 10.1016/s0006-2952(02)01166-8. [DOI] [PubMed] [Google Scholar]
- 56.Li LX, Skorpen F, Egeberg K, et al. Induction of uncoupling protein 2 mRNA in β-cells is stimulated by oxidation of fatty acids but not by nutrient oversupply. Endocrinology. 2002;143:1371–1377. doi: 10.1210/endo.143.4.8717. [DOI] [PubMed] [Google Scholar]
- 57.Hauser WA. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994;35(suppl 2):S1–S6. doi: 10.1111/j.1528-1157.1994.tb05932.x. [DOI] [PubMed] [Google Scholar]
- 58.Nevo Y, Shinnar S, Samuel E, et al. Unprovoked seizures and development disabilities: clinical characteristics of children referred to a child development center. Pediatric Neurol. 1995;13:235–241. doi: 10.1016/0887-8994(95)00185-i. [DOI] [PubMed] [Google Scholar]