

Abstract
Redox balance underlies cellular homeostasis. Cancer initiation and progression has been linked to the disruption of redox balance and oxidative stress. Recent findings exemplify the distinctive roles of intra- and extraceullar redox state in the etiology and maintenance of oxidative stress associated with malignancy and metastasis. Within these compartments, redox sensitive cysteines play a critical role in regulating cell signaling events that act to promote the malignant phenotype via the activation of survival pathways, disruption of cell-death signaling, and increases in cell proliferation. New approaches that aim to accurately evaluate subcellular and microenvironment redox potential may be useful in developing cancer diagnostics and therapeutics.
Introduction
The balance between oxidation and reduction reactions plays an essential role in numerous cell signaling cascades including those associated with proliferation, inflammatory responses, apoptosis, and senescence. Reactive oxygen and nitrogen species (ROS; RNS) are invariable components of aerobic metabolism and are key contributors to cellular redox state. However, due to the fact that oxygen and nitrogen radicals readily interact with nucleic acids, proteins and lipids, the disruption of cellular redox homeostasis has emerged as a critical component in the etiology and prognosis of a variety of disease pathologies. Oxidative stress, defined as the result of the imbalance between the production rate of pro-oxidants and that of their removal by antioxidants, has been shown to play a major role in the origination, progression, and malignancy of a number of cancers [1,2]. Similarly, nitrosative stress occurs when the generation of RNS exceeds the ability to neutralize and eliminate them. Within these definitions, it is critical to incorporate the recently coined “redox hypothesis”, which emphasizes the importance of non-radicals and thiols in oxidative and nitrosative stresses [3]. Despite emphasis on free radicals, disruption of redox signaling producing stress is largely non-radical based.
Elevated oxidative stress is observed in many solid tumors and carcinoma cell lines. Redox imbalance in cancer cells may be tied to a number of pathways including mitochondrial dysfunction, the activation of oncogenes, aberrant oxidative metabolism, and hypoxia/reoxygenation cycles. Low levels of oxidative stress may be advantageous for cancer progression, as they may lead to increased rates of genetic mutation that could contribute to the acquisition of a malignant phenotype [4]. Higher levels of ROS are more likely to lead to cell senescence and/or death. Each may be influenced by subcellular localization. Recent studies have identified certain ROS as second messengers in signaling pathways, thus allowing the potential to regulate cell phenotype directly by acting as effector molecules [5]. Understanding the origin of various ROS/RNS, and their roles in cancer initiation and progression, as well as in the cell signaling pathways involved in these, could facilitate the development of therapies that might act to prevent and/or counteract malignancy.
Extracellular vs. Intracellular redox state
Intracellular sources of ROS/RNS
Endogenous ROS can regulate redox signaling and may have independent roles in malignancy as compared to those produced, for example, at the apical membrane corresponding to extracellular ROS. Intracellularly, ROS may be generated by a variety of sources, including non-mitochondrial electron transport chains and redox systems in the cytosol, nuclear envelope or ER, as well as phase I reactions through p450 metabolism, β-oxidation in peroxisomes and inflammatory cytokines. While each can contribute meaningfully to cellular redox homeostasis, mitochondria are traditionally considered the major endogenous source of ROS in mammalian cells. During aerobic respiration, mitochondrial ROS are formed via the univalent reduction of molecular oxygen mediated through the escape of electrons from complexes I and III in the mitochondrial electron transport chain. Of the oxygen consumed by mitochondria about 2% is reduced by these bifurcated electrons to form superoxide and subsequently hydrogen peroxide [6,7]. As such, mitochondrial DNA is intrinsically vulnerable to ROS-mediated injury, partially mitigated by the separate glutathione (GSH) pools maintained by active transport of the tripeptide across the mitochondrial membrane. Mutations in cancer cell mitochondrial DNA have not only been implicated in tumor pathogenesis and metastasis but have been linked to enhanced mitochondrial generation of superoxide as compared to their normal counterparts [8]. Novel methodologies permitting targeting of mitochondrial ROS have identified them as key regulators in NF-kB-dependent anti-apoptotic signaling [9]. Recent studies have focused on new methodologies to evaluate mitochondrial bioenergetics in attempts to use ‘bioenergetic signatures’ as cancer biomarkers and to further unravel the role of a family of inner mitochondrial membrane proteins, termed uncoupling proteins, in cancer progression [10,11]. These studies further emphasize the heterogeneous nature of the tumor microenvironment and support the underlying principles of individualized approaches to anticancer treatment.
Extracellular sources of ROS/RNS
Extracellular redox states are influenced by factors distinct from intracellular and are frequently a consequence of modifications of plasma membrane proteins and the proximal milieu around the cell. This includes ROS directly resulting from exposure to external environmental agents including irradiation, chlorinated compounds, metal ions, barbiturates, phorbol esters and peroxisome proliferating agents [12] or from membrane associated redox modulating proteins such as the NADPH oxidase family, in particular Nox1 [13]. Based on immunostaining, recent studies of the Nox family have not only identified a role for various Nox members as intracellular and/or extracellular signaling oxidases, but have also determined that based on sub-cellular location of oxidase components different ROS species can be produced [14]. Membrane associated γ-glutamyltransferase (GGT) also plays a critical role in controlling redox conditions by degrading extracellular GSH, thus providing cysteine to cells; or alternatively, by acting as a pro-oxidant. Beyond the plasma membrane, antioxidants such as extracellular superoxide dismutase (EC-SOD), glutathione peroxidase 3 (GPX3), and thioredoxin reductase-1 (TR1) as well as the extracellular supply of plasma thiol/disulfide couples, such as glutathione/glutathione disulfide (GSH/GSSG), play a major part in balancing redox homoestasis (reviewed in [15]). This balance is further impacted by glutathione-S-transferases that are involved in maintaining glutathione homeostasis, as well as protein S-glutathionylation and kinase regulation [16,17].
The balance between extracellular and intracellular redox states in cancer metastasis has recently been discussed. Chaiswing et al. determined that extracellular redox-related proteins, GSH/GSSG levels, and the ROS/RNS levels of the extracellular space are altered in prostate cancer cells [18]. Furthermore, alterations in extracellular thiol/disulfide couples and GSH/GSSG also affect proliferation of colorectal carcinoma and lung fibroblast cells [19,20]. It may prove viable to adapt the expression of certain extracellular redox parameters to use as potential biomarkers. For example, cancer patients with high levels of generalized oxidative stress markers in their sera also exhibit markers of constitutive oxidative stress within tumors. Therefore, assessing serum redox state may be useful in the prediction or prognosis of the response/progression of various human cancers (see Table 1).
Table 1.
| Carcinoma and Evaluated Redox State | Marker of Oxidative Stress | Detection Method | Reference | |
|---|---|---|---|---|
| Extracellular | Prostate | ↑ H2O2; ↑ GSH/↓ GSSG; ↑ EC-SOD | Dihydroxyphenoxazine fluorescence; 5.5′-dithiobis - (2-nitrobenzoic acid)-GSSG recycling; Western blot | [18] |
| Hepatocellular | ↑ Thioredoxin | ELISA | [45] | |
| Pancreatic | ↑ Thioredoxin | ELISA | [46] | |
| Liver Metastasis | ↑ Hydroxyl Radicals, ↑ H2O2, ↑ Superoxide anions | Electron spin resonance spectrometry | [47] | |
| Colon | Thiol/disulfide balance | HPLC detection of N,N bis-dansyl or S- carboxymethyl-N-dansyl derivatives with fluorescence detection | [20] | |
| Lymphoma | ↑ Glutathione S-transeferase P | ELISA | [48] | |
| Breast, Gastric. Colorectal | ↓ Glutathione peroxidase 3 activity | Absorbance assay based on reduction of GSH to GSSG | [49] | |
| Intracellular | Pancreatic | ↑ Thioredoxin, ↑ Glutaredoxin | Immunohistochemistry | [46] |
| Squamous | ↑ γ-glutamyltransferase | Immunohistochemistry | [50] | |
| Ovarian, Lung, Breast, Colon, Pancreas, Lymphoma | ↑ Glutathione S-transeferase P | RT-PCR; Immunohistochemistry Western blot | [51] | |
| Breast | ↑ Thioredoxin, ↑ Peroxiredoxins | RT-PCR; Western blot | [52] | |
| Ovarian, Prostate Melanoma, Colon, Pancreatic, Neuroblastoma, Breast, Ovarian | ↑ H2O2 | CM2-DCFHDA staining | [53] | |
| ↑ H2O2 | Horseradish peroxidase-assay using hydrogen donors | [54] | ||
Oxidative Stress in Cancer Initiation and Progression
Chronic oxidative stress in cancer cells
Low levels of superoxide or hydrogen peroxide can enhance cellular survival and stimulate proliferation. However, when this is concomitant with chronic ROS production, redox homeostasis can become imbalanced and normal cells may become transformed [5]. Current evidence supports the hypothesis that cancer cells are characterized by enhanced ROS generation, increased ROS accumulation, and the deregulation of antioxidant enzymes; thus existing in a state of perpetually elevated stress. Constitutively produced NOS have also been found in several human tumor cell lines, an observation further complicated by the fact that RNS can be chemically heterogeneous in different tumor cell types [21].
Methods currently used to evaluate redox status include measurements of pro-oxidants, quantification of antioxidants, detection of oxidized nucleic acids, or evaluation of redox potential based on thiol/disulfide couples via the Nernst equation. Markers of oxidative stress, such as DNA adducts e.g. 8-oxo-7,8 dihydro-2′-deoxyguanosine, as well as generalized biological anti-oxidant capacity of plasma might be developed as tools in disease prognosis. However, general quantification of redox remains a complicated analysis of a plethora of integrated pathways and cross talk imbued by the oxidation/reduction system. Moreover, the pathways are fluid and often unstable.
Chronic oxidative stress in cancer is influenced by numerous factors. Oncogene expression, including that of Ras2, Bcr-Abl, and c-Myc contribute to persistent ROS production in addition to the disruption of p53 function [22,23]. Recent studies have linked ROS produced as a result of chronic inflammation to the neoplastic process [24]. Furthermore, because oxidative stress plays a major role in the induced adaptive expression of antioxidants, malignant cells are often characterized by differential expression in the levels of a number of these enzymes, including superoxide dismutases (SOD), catalase, glutathione-S-transferases, GGT, and EC-SOD [25,26]. Dysregulation in expression of such enzymes can influence cancer therapy and may contribute to metastasis and drug resistance. These characteristics further perpetuate a state of oxidative stress and result in the need to either adapt, or apoptose. Because oxidative stress may induce apoptosis, cell cycle arrest or cellular senescence, eventual cellular fate may be a delicate balance contingent upon factors such as cell type, tissue microenvironment and levels of free radical production/accumulation.
Results of chronic oxidative stress
Increases in ATP requirements in metabolically active cancer cells result from the need to support activities such as rapid proliferation. Such demands in association with ROS induced mitochondrial damage may result in amplified electron ‘leakage’ and ROS generation. In addition to mitochondrial damage, high levels of ROS can produce DNA damage and genetic instability. Paradoxically, this damage further encourages the development of a malignant phenotype when normal repair mechanisms are overwhelmed and result in the accumulation of mutations. Disruption of redox homeostasis impacts cell signaling pathways that regulate cell proliferation, apoptosis, metastasis and angiogenesis and ultimately result in cancer progression and poor clinical outcome (Figure 1).
Figure 1.
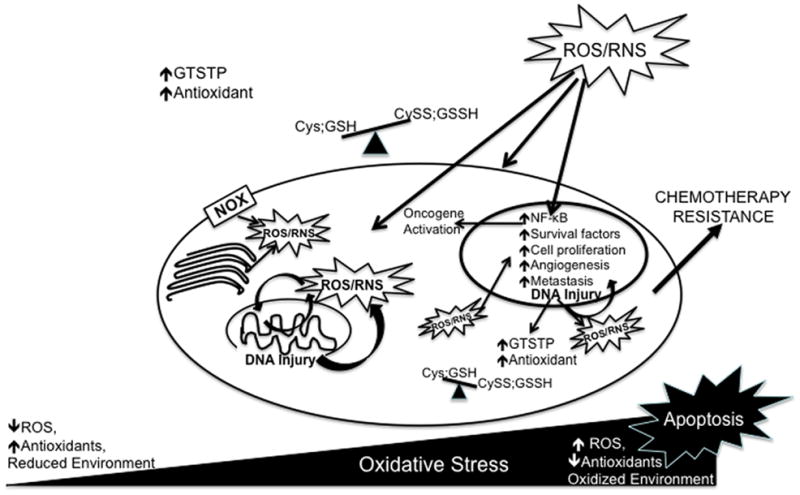
Accumulation of reactive oxygen species (ROS) and/or reactive nitrogen species (RNS), derived either endogenously or exogenously, results in oxidative stress. Disruption of thiol and non-radical circuits may also result in oxidative stress. The extent of this stress will either result in lethal damage and apoptosis or in cell adaptation. In cancer cells chronic oxidative stress activates redox sensitive transcription factors and signaling pathways that act to increase the expression of antioxidants, increase expression of survival factors as well inhibit the expression of pro-apoptotic pathways. ROS/RNS induced DNA injury promotes genomic instability and further provides opportunity to adapt to oxidative stress. Cancer progression occurs via the regulation of redox dependent expression of genes that play roles in proliferation, senescence evasion, metastasis, and angiogenesis. These features in association with the disruption in antioxidant profile may contribute to altered drug sensitivity and chemotherapy resistance. Definition of abbreviations: NOX, NADPH oxidase; nuclear factor-κB; NF-κB; Cys, cysteine; Cyss; cystine; GSH, glutathione; GSSH, glutathione disulfide, GSTP, glutathione-S-transferase P
Redox and thiol dependent cell signaling
Post-translational modification of proteins via redox sensitive cysteine residues can occur as a consequence of alterations in their oxidation state. There are over 200,000 unique cysteines in the human proteome and it is estimated that approximately 10% of these may be oxidizable [3]. Numerous factors determine which properties of these cysteines make them available for this reaction. The range of cellular processes under redox regulation is extensive and includes both the proliferative and apoptotic pathways. However, in cancer cells, excess superoxides and hydrogen peroxide can promote cell growth and proliferation and disrupt thiol redox circuits, thus contributing to oxidative stress.
More than 127 genes and signal transducing proteins have been reported to be directly affected by redox state [27]. Transcription factors, tumor suppressors as well as members of the mitogen activated protein kinase (MAPK) family and anti-apoptotic pathways such as PI 3-K and NF-κB are regulated by ROS and have roles in stimulating cell proliferation, sensitizing cancer cells to electrophilic agents and contributing to treatment resistance [28,29]. These signaling events may be further controlled by protein:protein interactions, such as through GSTP interactions with the MAPK c-jun NH2 terminal kinase (JNK). Oxidative stress can result in the reversal of GSTP regulated intrinsic JNK inhibitory activity via dissociation of the GSTP:JNK complex [30]. Under this system, GSTP serves as a sensor of intracellular changes in redox potential and has the potential to directly regulate kinase pathways, perhaps explaining the drug–resistance phenotypes of many GSTP over-expresssing cancers. Gene expression is modulated by ROS and thiol redox circuits through the interplay of extra-, intra- and even intercellular signaling pathways. In fact, ROS, in association with the mandatory presence of GSH, have been shown to reversibly inhibit gap junction inter-cellular communication (GJIC), which along with the induction of early-response genes is a hallmark of tumor promotion [31]. Furthermore, extracellular thiol/disulfide, GSH/GSSG, and overall redox potential has been linked to cell proliferation pathways mediated by epidermal growth factor (EGF) and MAPK signaling [19], two plausible targets in cancer therapy.
S-glutathionylation, which occurs when a protein cysteine forms a disulfide bond with GS•, serves as a reversible mechanism of post-translational protein regulation that has the potential to selectively regulate the function of enzymes, receptors, structural proteins, transcription factors, transport proteins, and protein-protein interactions (reviewed in [32]). As the most abundant non-protein thiol in cells, GSH plays a key role in oxidative stress. The presence of ROS/RNS directly mediates GSH/GSSG balance as well as interaction with available reactive cysteine residues. During cancer inititation, glutathionylation of the tumor suppressor p53 prevents DNA binding [33]. Furthermore, glutathionylation also has an anti-apoptotic role by preventing caspase cleavage [34]. Alternatively, glutathionylation of NF-κB influences apoptosis of hypoxic tumor cells [35]. Modulation of GSH and/or GST isozymes is an ongoing therapeutic strategy in cancer chemotherapy [30].
Both non-radical based oxidative and nitrosative species are important in regulating signaling pathways in normal and cancer cells (reviewed in [3]. However, where signaling is regulated through thiol:disulfide reactions, it should be noted that their turnover rates and consequent flux rates are quite small when compared to general cellular redox buffering. Ongoing research in redox systems biology will need to focus on the quantification of redox buffering. Stoichiometry of competing reactions and their sub-cellular localization will control how tumor and normal cells may differ in their regulatory pathways. In particular, the mitochondrial compartment seems to be the most reduced in terms of steady state redox potential, while the endoplasmic reticulum has the most oxidized environment, a condition determinant of the function of this organelle in protein folding [36]. Specifically, ER oxidative stress is linked to increases in protein S- glutathionylation as well as to the accumulation of misfolded proteins and a cascade of transcriptional and translational events that attempt to manage this accumulation [32,37]. This phenomenon, termed the unfolded protein response (UPR), is a potential therapeutic target in some cancers [38].
The oncogene, Rac has been shown to induce ROS production and cause loss of cell-cell adhesion and ROS-mediated actin cytoskeleton reorganization eventually impacting metastasis [39]. Furthermore, ROS can induce ICAM-1 and activate matrix metalloproteinases that can act as tumor promoters [40]. Recently, ROS produced via the transcriptional activation of Nox family members have also been linked to the formation of invadapodia and to tumor cell motility/migration [41]. Cancer cells under oxidative stress exhibit decreased attachment to basal lamina as modulation of integrin function, suggesting a propensity to enter blood vessels. Additionally, constitutive ROS production in metastatic cancer cells has been linked to resistance to anoikis, or detachment-induced apoptosis, through persistent EGFR or Src kinase activation [42]. It has been proposed that metastasis is an oxidative stress triggered “escape program” that enables cells to avoid the oxidative stress levels associated with the primary tumor. Additionally, extracellular redox potential has been shown to influence matrix expression, mediated through TGF-β1 fibronectin expression [19]. Specific signaling pathways that are influenced by ROS have been reviewed recently [5,28].
Malignancy and cell environment:hypoxia
The ability of tumors to survive in a hypoxic state is supported by the capacity to up-regulate proteins that favor anaerobic metabolism and improve pH buffered-oxygen sensing. These include HIF-1α, as well as glucose transporters and glycolytic enzymes for anaerobic energy production, erythropoietin and iron metabolism proteins for red blood cell production, and a number of factors that promote angiogenesis. The resulting angiogenesis-induced oxidative stress then acts in a positive feedback loop resulting in increased metastasis and aggressive tumor progression. In a recent study, Cannito et al. showed that during low oxygen tension mitochondrial produced ROS have a direct role in hypoxia-dependent epithelial-mesenchymal transition [43].
Not only does hypoxia result in glucose deprivation followed by the depletion of intracellular pyruvate and the inability to dispose of existing ROS but re-oxygenation of hypoxic tissues, such as during tumor angiogenesis, increases the concentrations of free radicals. This ROS production can further increase the production of the angiogenic factors IL-8 and VEGF as well as factors that promote vessel growth, such as MMP-1, and angiogenesis, such as iNOS. Members of the ROS producing Nox family can also contribute to pathways that lead to tumor angiogenesis and neovascularization [13]. Of recent interest is the role of tumor stem cells and the hypoxic microenvironment in activating quiescent stem cells via HIF/VEGF pathways as well as effectors such as Notch, Wnt and Oct4 and the promotion of tumorigenesis [44].
Conclusion
The role of oxidative stress in metastasis and tumor progression is complex and involves a number of factors including cell type, cellular microenvironment, and free radical type and compartmentalization. Tumor survival depends on a number of processes involving proliferation, motility, apoptosis and senescence, all of which are influenced by changes in redox metabolism. Complexity lies in the fact that individual cancers may be characterized by different redox-based signaling mechanisms. However, as new approaches emerge, e.g. the discrete roles of extracellular vs. intracellular redox state; the importance of non-radicals in redox metabolism; the recognition of the impact of tumor microenvironment on metastasis, the utility of targeted redox-modulating therapeutics may flourish.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
• of special interest
•• of outstanding interest
- 1.Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25(4):695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 2.Cerutti PA. Prooxidant states and tumor promotion. Science. 1985;227(4685):375–381. doi: 10.1126/science.2981433. [DOI] [PubMed] [Google Scholar]
- 3.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295(4):C849–868. doi: 10.1152/ajpcell.00283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; •A comprehensive review of the redox biology of thiol systems that highlights the importance of non-radicals in oxidative stress. This “redox hypothesis” redefines oxidative stress as the result of thiol redox circuit disruption. A focus on GSH- and thioredoxin is used to demonstrate the importance of reversible thiol oxidation/reduction and evidence for the nonequilibrium of thiol-disulfide couples is discussed
- 4.Jackson AL, Loeb LA. The contribution of endogenous sources of DNA damage to the multiple mutations in cancer. Mutat Res. 2001;477(1–2):7–21. doi: 10.1016/s0027-5107(01)00091-4. [DOI] [PubMed] [Google Scholar]
- 5.Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox regulation of cell survival. Antioxid Redox Signal. 2008;10(8):1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]; •A recent review discussing the roles of redox systems in regulating transcription factors, signal-transduction pathways, and cell-death regulators in affecting cell survival. Redox homeostasis in the development of diseases is discussed, as well as how redox regulation of cell survival may be useful in therapeutic strategies
- 6.Staniek K, Gille L, Kozlov AV, Nohl H. Mitochondrial superoxide radical formation is controlled by electron bifurcation to the high and low potential pathways. Free Radic Res. 2002;36(4):381–387. doi: 10.1080/10715760290021225. [DOI] [PubMed] [Google Scholar]
- 7.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134(3):707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konstantinov AA, Peskin AV, Popova E, Khomutov GB, Ruuge EK. Superoxide generation by the respiratory chain of tumor mitochondria. Biochim Biophys Acta. 1987;894(1):1–10. doi: 10.1016/0005-2728(87)90206-4. [DOI] [PubMed] [Google Scholar]
- 9.Hughes G, Murphy MP, Ledgerwood EC. Mitochondrial reactive oxygen species regulate the temporal activation of nuclear factor kappaB to modulate tumour necrosis factor-induced apoptosis: evidence from mitochondria-targeted antioxidants. Biochem J. 2005;389(Pt 1):83–89. doi: 10.1042/BJ20050078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sastre-Serra J, Valle A, Company MM, Garau I, Oliver J, Roca P. Estrogen down-regulates uncoupling proteins and increases oxidative stress in breast cancer. Free Radic Biol Med. 48(4):506–512. doi: 10.1016/j.freeradbiomed.2009.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Henderson JR, Swalwell H, Boulton S, Manning P, McNeil CJ, Birch-Machin MA. Direct, real-time monitoring of superoxide generation in isolated mitochondria. Free Radic Res. 2009;43(9):796–802. doi: 10.1080/10715760903062895. [DOI] [PubMed] [Google Scholar]; •This is a novel study that investigates the application of a cytochrome c functionalized amperometric sensor for monitoring superoxide generation in isolated mitochondrial fractions. A non-invasive sensor system that allows the real-time comparison of superoxide production following specific inhibition of complex I and complex III of the mETC is described
- 12.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 13.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 14.Leto TL, Morand S, Hurt D, Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal. 2009;11(10):2607–2619. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaiswing L, Oberley TD. Extracellular/Microenvironmental Redox State. Antioxid Redox Signal. 2009 [Google Scholar]; •A comprehensive review that focuses on the importance of extracellular redox state in cell-microenvironmental interactions and in cancer. This includes the discussion of redox-modulating proteins that are located on the plasma membrane or outside of cells, extracellular thiol/disulfide couples, and reactive oxygen species (ROS)/reactive nitrogen species (RNS) that are capable of traveling across plasma membranes into the extracellular space
- 16.Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J Biol Chem. 2009;284(1):436–445. doi: 10.1074/jbc.M805586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, et al. Regulation of JNK signaling by GSTp. EMBO J. 1999;18(5):1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaiswing L, Zhong W, Cullen JJ, Oberley LW, Oberley TD. Extracellular redox state regulates features associated with prostate cancer cell invasion. Cancer Res. 2008;68(14):5820–5826. doi: 10.1158/0008-5472.CAN-08-0162. [DOI] [PubMed] [Google Scholar]; •One of the first studies to indicate a role for extracellular redox status in regulation of cell invasion
- 19.Ramirez A, Ramadan B, Ritzenthaler JD, Rivera HN, Jones DP, Roman J. Extracellular cysteine/cystine redox potential controls lung fibroblast proliferation and matrix expression through upregulation of transforming growth factor-beta. Am J Physiol Lung Cell Mol Physiol. 2007;293(4):L972–981. doi: 10.1152/ajplung.00010.2007. [DOI] [PubMed] [Google Scholar]; •The importance of extracellular thiol/disulfide redox environment is demonstrated in lung fibroblast proliferation. An oxidized extracellular Cys/CySS redox potential (E(h) Cys/CySS) was found to induce intracellular signals that stimulate proliferation and fibronectin expression through protein kinase C activation and the upregulation of TGF-beta1
- 20.Jonas CR, Ziegler TR, Gu LH, Jones DP. Extracellular thiol/disulfide redox state affects proliferation rate in a human colon carcinoma (Caco2) cell line. Free Radic Biol Med. 2002;33(11):1499–1506. doi: 10.1016/s0891-5849(02)01081-x. [DOI] [PubMed] [Google Scholar]
- 21.Duenas-Gonzalez A, Isales CM, del Mar Abad-Hernandez M, Gonzalez-Sarmiento R, Sangueza O, Rodriguez-Commes J. Expression of inducible nitric oxide synthase in breast cancer correlates with metastatic disease. Mod Pathol. 1997;10(7):645–649. [PubMed] [Google Scholar]
- 22.Hlavata L, Aguilaniu H, Pichova A, Nystrom T. The oncogenic RAS2(val19) mutation locks respiration, independently of PKA, in a mode prone to generate ROS. EMBO J. 2003;22(13):3337–3345. doi: 10.1093/emboj/cdg314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9(5):1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 24.Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31(1):37–49. doi: 10.1093/carcin/bgp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pompella A, Corti A, Paolicchi A, Giommarelli C, Zunino F. Gamma-glutamyltransferase, redox regulation and cancer drug resistance. Curr Opin Pharmacol. 2007;7(4):360–366. doi: 10.1016/j.coph.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Teoh ML, Fitzgerald MP, Oberley LW, Domann FE. Overexpression of extracellular superoxide dismutase attenuates heparanase expression and inhibits breast carcinoma cell growth and invasion. Cancer Res. 2009;69(15):6355–6363. doi: 10.1158/0008-5472.CAN-09-1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28(3):463–499. doi: 10.1016/s0891-5849(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 28.Giles GI. The redox regulation of thiol dependent signaling pathways in cancer. Curr Pharm Des. 2006;12(34):4427–4443. doi: 10.2174/138161206779010549. [DOI] [PubMed] [Google Scholar]; •Summarizes the biochemistry of redox reactions and incorporates a discussion of subcellular location of these reactions. The differences in redox metabolism between cancer cells and non-malignant cells are also outlined
- 29.Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidative stress and cell signalling. Curr Med Chem. 2004;11(9):1163–1182. doi: 10.2174/0929867043365323. [DOI] [PubMed] [Google Scholar]
- 30.Tew KD. Redox in redux: Emergent roles for glutathione S-transferase P (GSTP) in regulation of cell signaling and S-glutathionylation. Biochem Pharmacol. 2007;73(9):1257–1269. doi: 10.1016/j.bcp.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 31.Azzam EI, de Toledo SM, Little JB. Oxidative metabolism, gap junctions and the ionizing radiation-induced bystander effect. Oncogene. 2003;22(45):7050–7057. doi: 10.1038/sj.onc.1206961. [DOI] [PubMed] [Google Scholar]
- 32.Townsend DM. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol Interv. 2007;7(6):313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Velu CS, Niture SK, Doneanu CE, Pattabiraman N, Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46(26):7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]; •One of the first studies to determine the redox state of p53 tumor suppressor. Cancer cells were shown to have marginal levels of glutathionylated p53 which increased after oxidant and DNA-damaging treatments. Furthermore, shielding of identified reactive cysteines was shown to contribute to p53 negative regulation, thus suggesting an acute defensive response
- 34.Huang Z, Pinto JT, Deng H, Richie JP., Jr Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem Pharmacol. 2008;75(11):2234–2244. doi: 10.1016/j.bcp.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qanungo S, Starke DW, Pai HV, Mieyal JJ, Nieminen AL. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J Biol Chem. 2007;282(25):18427–18436. doi: 10.1074/jbc.M610934200. [DOI] [PubMed] [Google Scholar]; •Identifies a novel role for glutaredoxin (GRx) S-glutathionylation of p65-NFkappaB in NAC-mediated NFkappaB inactivation and enhanced hypoxic apoptosis
- 36.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780(11):1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; •Discusses the importance of thiol/disulfide redox couples, thioredoxin-1, thioredoxin-2, GSH/GSSG and cysteine/cystine (Cys/CySS) non-equilibrium in separate subcelluar compartments including the mitochondria, nuclei, the secretory pathway and the extracellular space. Extracellular redox environment in association with cytoplasmic GSH/GSSG redox potential and cellular proliferation, differentiation and apoptosis is discussed
- 37.Townsend DM, Manevich Y, He L, Xiong Y, Bowers RR, Jr, Hutchens S, Tew KD. Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Res. 2009;69(19):7626–7634. doi: 10.1158/0008-5472.CAN-09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]; •Identifies the S-glutathionylation of protein disulfide isomerase (PDI) as an upstream signaling event in the unfolded protein response (UPR). This study indicates possible therapeutic potential of targeting UPR mediators
- 38.Davenport EL, Morgan GJ, Davies FE. Untangling the unfolded protein response. Cell Cycle. 2008;7(7):865–869. doi: 10.4161/cc.7.7.5615. [DOI] [PubMed] [Google Scholar]; •The need to develop new therapeutics that aim to target families of proteins and multiple signaling pathways on which cancers rely for their ability to survive stress is reviewed. Emphasis is placed on heat shock proteins (HSP), and proteins involved in the unfolded protein response (UPR) in multiple myeloma
- 39.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436(7047):123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savaraj N, Wei Y, Unate H, Liu PM, Wu CJ, Wangpaichitr M, Xia D, Xu HJ, Hu SX, Tien Kuo M. Redox regulation of matrix metalloproteinase gene family in small cell lung cancer cells. Free Radic Res. 2005;39(4):373–381. doi: 10.1080/10715760400029694. [DOI] [PubMed] [Google Scholar]
- 41.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Sci Signal. 2009;2(88):ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]; •Discusses the role of reactive oxygen species (ROS) generated by the NADPH oxidase (Nox) system in the formation and function of cancer cell invadopodia. Specifically, a role for Tks5 in facilitating the production of ROS is determined to be necessary for invadopodia formation, and in turn ROS modulated Tks5 tyrosine phosphorylation in a positive feedback loop is proposed
- 42.Giannoni E, Fiaschi T, Ramponi G, Chiarugi P. Redox regulation of anoikis resistance of metastatic prostate cancer cells: key role for Src and EGFR-mediated pro-survival signals. Oncogene. 2009;28(20):2074–2086. doi: 10.1038/onc.2009.77. [DOI] [PubMed] [Google Scholar]; •Anoikis, as an emerging hallmark of metastatic malignancies is examined. Metastatic prostate carcinoma cells are demonstrated to undergo a constitutive deregulated production of reactive oxygen species (ROS) due to sustained activation of 5-lipoxygenase and thus lack suicidal pathways in response to lack of matrix contact. This was shown to lead to maintenance of Src oxidation and activation in the absence of adhesion and sustain a ligand-independent phosphorylation of epidermal growth factor receptor (EGFR)
- 43.Cannito S, Novo E, Compagnone A, Valfre di Bonzo L, Busletta C, Zamara E, Paternostro C, Povero D, Bandino A, Bozzo F, Cravanzola C, et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis. 2008;29(12):2267–2278. doi: 10.1093/carcin/bgn216. [DOI] [PubMed] [Google Scholar]
- 44.Mazumdar J, Dondeti V, Simon MC. Hypoxia-inducible factors in stem cells and cancer. J Cell Mol Med. 2009;13(11–12):4319–4328. doi: 10.1111/j.1582-4934.2009.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; •A new role for hypoxia inducible factors (HIFs) in stem cell and tumor regulation is discussed. The role of HIFs in the modulation of specific stem cell effectors, such as Notch, Wnt, and Oct4, that control stem cell proliferation, differentiation and pluripotency are reviewed. Furthermore links have also between HIFs and critical cell signaling pathways such as cMyc and p53 are discussed
- 45.Miyazaki K, Noda N, Okada S, Hagiwara Y, Miyata M, Sakurabayashi I, Yamaguchi N, Sugimura T, Terada M, Wakasugi H. Elevated serum level of thioredoxin in patients with hepatocellular carcinoma. Biotherapy. 1998;11(4):277–288. doi: 10.1023/a:1008032703468. [DOI] [PubMed] [Google Scholar]
- 46.Nakamura H, Bai J, Nishinaka Y, Ueda S, Sasada T, Ohshio G, Imamura M, Takabayashi A, Yamaoka Y, Yodoi J. Expression of thioredoxin and glutaredoxin, redox-regulating proteins, in pancreatic cancer. Cancer Detect Prev. 2000;24(1):53–60. [PubMed] [Google Scholar]
- 47.Nonaka Y, Iwagaki H, Kimura T, Fuchimoto S, Orita K. Effect of reactive oxygen intermediates on the in vitro invasive capacity of tumor cells and liver metastasis in mice. Int J Cancer. 1993;54(6):983–986. doi: 10.1002/ijc.2910540620. [DOI] [PubMed] [Google Scholar]
- 48.Katahira T, Takayama T, Miyanishi K, Hayashi T, Ikeda T, Takahashi Y, Takimoto R, Matsunaga T, Kato J, Niitsu Y. Plasma glutathione S-Transferase P1-1 as a prognostic factor in patients with advanced non-Hodgkin’s lymphoma (stages III and IV) Clin Cancer Res. 2004;10 (23):7934–7940. doi: 10.1158/1078-0432.CCR-03-0679. [DOI] [PubMed] [Google Scholar]
- 49.Pawlowicz Z, Zachara BA, Trafikowska U, Maciag A, Marchaluk E, Nowicki A. Blood selenium concentrations and glutathione peroxidase activities in patients with breast cancer and with advanced gastrointestinal cancer. J Trace Elem Electrolytes Health Dis. 1991;5(4):275–277. [PubMed] [Google Scholar]
- 50.Groves RW, Fracchia JN, MacDonald DM. Gamma glutamyl transpeptidase expression in foetal skin, inflammatory dermatoses and cutaneous neoplasia. Br J Dermatol. 1991;125(1):1–5. doi: 10.1111/j.1365-2133.1991.tb06028.x. [DOI] [PubMed] [Google Scholar]
- 51.Tew KD. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994;54(16):4313–4320. [PubMed] [Google Scholar]
- 52.Cha MK, Suh KH, Kim IH. Overexpression of peroxiredoxin I and thioredoxin1 in human breast carcinoma. J Exp Clin Cancer Res. 2009;28:93. doi: 10.1186/1756-9966-28-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67(22):10823–10830. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 54.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–798. [PubMed] [Google Scholar]