

Abstract
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels mediate hyperpolarization-activated currents (Ih). In hippocampus, these currents contribute greatly to intrinsic cellular properties and synchronized neuronal activity. The kinetic and gating properties of HCN-mediated currents are largely determined by the type of subunits–for example, HCN1 and HCN2–that assemble to form homomeric channels. Recently, functional heteromeric HCN channels have been described in vitro, further enlarging the potential Ih repertoire of individual neurons. Because these heteromeric HCN channels may promote hippocampal hyperexcitability and the development of epilepsy, understanding the mechanisms governing their formation is of major clinical relevance. Here, we find that developmental seizures promote co-assembly of hippocampal HCN1/HCN2 heteromeric channels, in a duration-dependent manner. Long-lasting heteromerization was found selectively after seizures that provoked persistent hippocampal hyperexcitability. The mechanism for this enhanced heteromerization may involve increased relative abundance of HCN2-type subunits relative to the HCN1 isoform at both mRNA and protein levels. These data suggest that heteromeric HCN channels may provide molecular targets for intervention in the epileptogenic process.
Keywords: Ion channel, Epilepsy, HCN, Hyperpolarization, Ih, Febrile seizures, Animal model, Epileptogenesis, Heteromeric, Co-immunoprecipitation, Rat, Kainic acid
Introduction
HCN channels mediate hyperpolarization-activated (Ih) currents (DiFrancesco, 1993; Pape, 1996). In hippocampus, these currents contribute to maintenance of resting membrane potential (Lupica et al., 2001), shape rhythmic and synchronized neuronal activity (Maccaferri and McBain, 1996; Magee, 1999) and regulate the temporal summation of dendritic depolarization (Magee, 1998; Poolos et al., 2002). Short-term modulation of HCN channel function is mediated by cAMP, influencing channel kinetics and voltage dependent activation curves (DiFrancesco, 1993; Waigner et al., 2001). Recently, long-term modulation of the properties of Ih currents has been suggested to result from regulated changes in channel subunit expression (Bräuer et al., 2001; Brewster et al., 2002; Santoro and Baram, 2003).
Of the four characterized genes encoding HCN channels, two (HCN1 and 2) are substantially expressed in rodent hippocampus (Bender et al., 2001; Moosmang et al., 1999; Santoro et al., 2000) and both isoforms can reside in a single neuron (Brewster et al., 2002; Franz et al., 2000). HCN4 is expressed almost exclusively in subcortical regions, whereas levels of HCN3 expression within neurons are generally low (Bender et al., 2001; Moosmang et al., 1999; Santoro et al., 2000). As described for other members of the voltage-gated cation channel family, four HCN subunits assemble to form a channel (for recent reviews, see Robinson and Siegelbaum, 2003; Santoro and Baram, 2003). Homomeric HCN1 channels conduct fast-kinetic currents with modest cAMP gating, consistent with currents recorded in hippocampal pyramidal cells and CA1 interneurons where HCN1 expression is high (Bender et al., 2001; Brewster et al., 2002; Santoro et al., 2000). By contrast, homomeric HCN2 channels conduct Ih currents with slower kinetics and robust cAMP-evoked shifts in voltage dependence (Ludwig et al., 1998; Robinson and Siegelbaum, 2003). More recently, the formation of heteromeric channels in vitro (Chen et al., 2001b; Er et al., 2003; Much et al., 2003; Proenza et al., 2002; Ulens and Tytgat, 2001; Xue et al., 2002) as well as in vivo (Er et al., 2003; Much et al., 2003) has been described. However, the mechanisms promoting heteromerization have remained unclear.
Because the isoform composition of both homomeric and heteromeric HCN channels determines their physiological characteristics (Robinson and Siegelbaum, 2003; Santoro and Baram, 2003), the relative abundance of the HCN isoforms, as well as the degree to which heteromerization occurs, will contribute to the properties of the Ih of an individual neuron (Franz et al., 2000; Santoro et al., 2000; Vasilyev and Barish, 2002), and thus to its intrinsic firing patterns and network responses. Differential regulation of HCN1 and HCN2 mRNA expression has been found in pathological states (Bender et al., 2003; Bräuer et al., 2001; Brewster et al., 2002), including experimental febrile and kainate-induced seizures, and has been associated with altered properties of the Ih (Chen et al., 2001a). Interestingly, the properties of Ih after experimental febrile seizures resembled neither those of heterologously expressed homomeric HCN1 or HCN2 channels, nor their arithmetic intermediates (Chen et al., 2001a,b; Ulens and Tytgat, 2001). Therefore, we considered the possibility that these currents may be a result of heteromeric HCN1/HCN2 channels, and tested the hypothesis that pathological neuronal activity such as developmental seizures promotes formation of heteromeric HCN1/HCN2 channels, at least in part by altering the relative abundance of cellular HCN1 and HCN2 subunits at the mRNA and protein level. This constitutes a novel mechanism for activity-induced plasticity of the ion channel complement of a neuron. In addition, it highlights an as yet undescribed molecular foundation for the evolution of neurological disease, such as seizure-evoked epilepsy.
Materials and methods
Seizure induction
Experimental procedures were approved by the UCI Animal Care Committee in accordance to National Institutes of Health (NIH) guidelines. Immature (postnatal day 10) Sprague–Dawley rats were subjected to experimental prolonged febrile seizures as previously described (Brewster et al., 2002; Dubé et al., 2000; Toth et al., 1998). Briefly, hyperthermia was induced using a stream of warm air directed ∼30 cm above the animals. Core temperatures of pups were measured prior to hyperthermia induction (33.9 ± 0.3°C), at 2-min intervals, and at the onset of hyperthermia-provoked seizures (40.8 ± 0.9°C). Hyperthermia was maintained for 30 min resulting in seizure duration of approximately 24 min. Following hyperthermia, animals were moved to a cool surface for 10–15 min, then returned to their home cages. Hyperthermic controls were generated by subjecting littermates to the same hyperthermia procedure, but blocking the resulting seizures using diazepam (10 mg/kg). Longer and more intense seizures were evoked in rats of the same age by intraperitoneal injection of the glutamate receptor agonist kainic acid (KA, 1.2 mg/kg) as described previously (Brewster et al., 2002). Experimental groups (Feb, n = 37; KA, n = 15; diazepam + Feb, n = 4; diazepam alone, n = 4) were compared to littermate controls (n = 40).
Tissue harvesting
For immunoprecipitation and Western blot analyses (1, 2, 4 and 8 weeks after seizures) rats were rapidly decapitated, the hippocampus and thalamus were quickly dissected and homogenized in a glass/Teflon homogenizer in ice cold 0.32 M Sucrose, 0.1 M Tris–HCl (pH 7.4) containing Protease Inhibitor Cocktail (PIC Complete™; Roche). Following homogenization, samples were centrifuged at 1000 × g for 10 min at 4°C. The supernatant was centrifuged at 16,000 × g for 20 min at 4°C and the pellet was resuspended in radioimmunoprecipitation (RIPA) buffer (50 mM Tris–HCl, pH 7.4, 1% NP-40, 1% Triton X-100, 1 mM EDTA, 150 mM NaCl, 1 × PIC) for immunoprecipitation procedures or in artificial cerebrospinal fluid (aCSF; 124 mM NaCl, 3 mM KCl, 1.25 mM KH2PO4, 2.5 mM MgSO4, 3.4 mM CaCl2, 26 mM NaHCO3, 10 mM glucose) for direct Western blot analyses. Protein concentration was determined using Bio-Rad Protein assay (Bio-Rad Laboratories, Inc., Hercules, CA). For in situ hybridization procedures, rats were quickly decapitated and brains dissected and placed on powdered dry ice as described (Brewster et al., 2002; Eghbal-Ahmadi et al., 1999).
Immunoprecipitation
An equal amount of protein for each hippocampal and thalamic sample (1 mg/ml), diluted in 750 μl of RIPA buffer, was incubated with rabbit anti-HCN1 or anti-HCN2 antisera (AB5884 HCN1, AB5378 HCN2; 5 Al of 15 μg/μl, Chemicon, Temecula, CA) on a rotator overnight at 4°C, following previously described procedures (Kramar et al., 2002). After incubation with primary antibodies, samples were incubated with 80 μl of Protein A Agarose (Upstate Biotech, Lake Placid, NY) on a rotator for 2 h at 4°C and collected in spin-filter columns (CytoSignal Research Products, Irvine, CA). Samples were then washed twice with RIPA+ 1 × PIC, followed by three washes in phosphate buffered saline (PBS)+ 1 × PIC. Following the washes, samples were eluted from the columns with 50 μl of 2× Laemmli buffer and processed for Western blot analyses (Kramar et al., 2002).
Western blotting
Protein samples were denatured at 100°C for 5 min in Laemmli buffer and separated on a 4–12% gradient SDS-PAGE gel. After SDS-PAGE, the proteins were transferred to Hybond-P Polyvinyl Difluoride (PVDF) membranes (Amersham Pharmacia Biotech) at 100 V for 1 h at 4°C. Membranes were probed with rabbit anti-HCN1 or anti-HCN2 serum (1:5000 and 1:3000, respectively) in PBS with 5% non-fat milk overnight at 4°C followed by washes in PBS-1% Tween (PBS-T) (3 × 5 min). Membranes were then incubated with anti-rabbit immunoglobulin-horseradish peroxidase (1:10,000 Amersham Pharmacia Biotech) in PBS for 1 h at room temperature, followed by incubation with the enhanced chemiluminescence ECL-Plus kit (Amersham Pharmacia Biotech) for 5 min. Immunoreactive bands were visualized by apposing membranes to Hyperfilm™ ECL (Amersham Pharmacia Biotech). To control for equal sample loading, membranes were probed with an antibody to beta actin.
In situ hybridization
Semi-quantitative analysis of HCN1 and HCN2 mRNA levels was accomplished using in situ hybridization (ISH) of 35S-cRNA probes and slide-mounted frozen sections (20 μm) as previously described (Brewster et al., 2002; Eghbal-Ahmadi et al., 1999), with the most stringent wash at 0.03 × SSC, at 62°C for 60 min. Sections were then dehydrated and apposed to Kodak Biomax film. Exposure time was monitored using 14C standards to maintain the signal in the linear range.
Analysis
Data acquisition and analysis of in situ hybridizations were carried out as described elsewhere (Bender et al., 2003; Brewster et al., 2002) on sections run concurrently, and always without knowledge of treatment. Semi-quantitative mRNA analysis was done by measuring optical density of the incorporated radioactivity using the image analysis program ImageTool. Western blot data acquisition and analysis were accomplished by measuring optical density of immunoreactive bands of experimental and control samples from the ECL films using the AIS imaging system (Imaging Res., St. Catherine, Ontario, Canada). Control and experimental samples for each time point were run concurrently on the same blot. When multiple blots from immunoprecipitation experiments were compared, the mean value of the control samples of each blot was considered 100%, and experimental sample values expressed as ‘percent control samples’ within each blot. This permitted analysis across blots. In addition, absolute amounts of HCN1 and HCN2 channel proteins were quantified using normal rabbit IgG standards (0.1 ng–4 μg; XP-5001, ProSci Incorporated, Poway, CA) that were run with experimental and control samples in the same gel. Significance level for t tests and ANOVA was set at 0.05, and data are presented as means with standard errors.
Results
Developmental ‘febrile’ seizures promote co-immunoprecipitation of HCN1 and HCN2 channel isoforms
To determine if co-assembly of HCN1 and HCN2 isoforms was induced by experimental ‘febrile’ seizures to generate modified h channels, immunoprecipitation (IP) experiments were conducted at several time points after the seizures (Kramar et al., 2002). Experimental febrile seizures were induced in 10-day-old rat pups (Brewster et al., 2002; Dubé et al., 2000). One and 7 days post-seizures, precipitation of hippocampal homogenates with an antiserum to HCN2 followed by Western blots probed with anti-HCN1 was utilized to demonstrate HCN1 molecules associated with the HCN2 isoforms. Experimental febrile seizures led to a significant (74%) increase in the amount of HCN1 that co-precipitated with HCN2, as compared with levels in paired controls (Fig. 1A; n = 7 animals per group) at the 7-day time-point, but not earlier. This finding was corroborated by IP using anti-HCN1 followed by Western blot detection of HCN2 (Fig. 1B). The enhanced appearance of HCN1/HCN2 co-association after seizures was not attributable to cross-reactivity of their respective antisera for several reasons: first, IP using anti-HCN2 followed by probing for the same isoform resulted in an HCN2-immunoreactive band with the expected molecular weight of 96 kDa. As described for HCN2, this protein was more abundant in thalamic homogenates compared to those from equal-weight hippocampal tissue (Santoro and Baram, 2003; Santoro et al., 2000) (Fig. 1C, top). In contrast, IP with anti-HCN2 followed by probing for HCN1 yielded a protein band with the expected molecular weight of ∼112 kDa (Much et al., 2003) and higher expression levels in hippocampus (Fig. 1C, bottom). In addition, when hippocampal and thalamic homogenates were subjected to direct Western blotting and probed with either HCN1 or HCN2 antisera, each antiserum recognized a single protein entity (Fig. 1D) of the appropriate molecular weight.
Fig. 1.
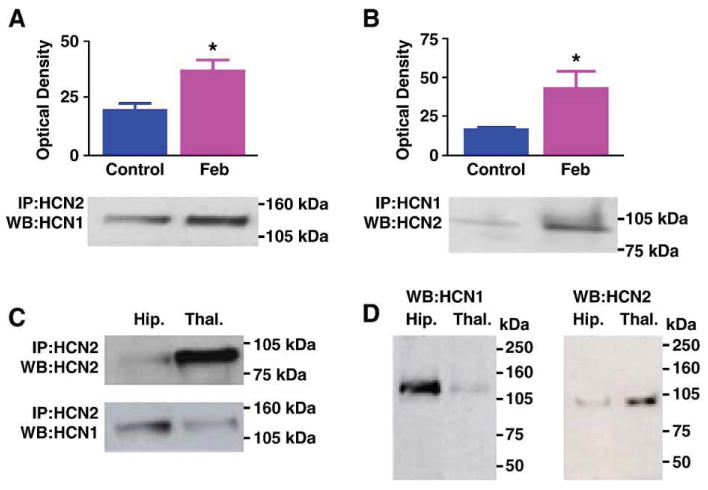
(A) Immunoprecipitation (IP) with antiserum to HCN2 followed by Western blot (WB) analysis for HCN1 shows enhanced co-association of HCN1/HCN2 in hippocampi from animals subjected to experimental prolonged febrile seizures on postnatal day 10 (Feb). The immunoreactive HCN1 band has an apparent molecular weight of approximately 110 kDa. Quantitative analysis of optical density of HCN1 immunoreactive bands reveals a significant increase in HCN1/HCN2 heteromerization 1 week after the induction of the seizures. (B) IP with antiserum to HCN1 followed by Western blot analysis for HCN2 corroborates the greater co-association of the two isoforms in the Feb group. (C) IP with antiserum to HCN2 followed by Western blot analysis for the same isoform shows greater HCN2 precipitation and immunoreactivity in thalamus compared with hippocampus (top blots). By contrast, hippocampal homogenates contain higher levels of HCN1 that is co-immunoprecipitated with the HCN2 antiserum (bottom blots). Note also different molecular weights of the HCN1 and HCN2 bands. (D) Western blots (without prior IP) for HCN1 (left) and HCN2 (right) demonstrate that the antisera used here detect HCN1 (abundant in hippocampus) and HCN2 (abundant in thalamus), and bind to antigens of the expected molecular weights, further supporting the specificity of these antisera.
HCN1/HCN2 co-association requires seizure activity and is not an effect of elevated brain temperatures
The Febrile seizure group was essentially subjected to (a) elevated brain temperature that (b) resulted in seizures. To determine which of these two factors led to increased HCN1/HCN2 co-immunoprecipitation, we evaluated also groups where these parameters were dissociated. As shown in Fig. 2, hippocampi from rats sustaining hyperthermia but in whom seizures were prevented (using diazepam) were not enriched in co-immunoprecipitated HCN1/HCN2 isoforms, suggesting that seizure activity rather than elevated temperature was required for heteromerization. Heteromerization was not increased in the group treated with diazepam alone (without temperature elevation).
Fig. 2.
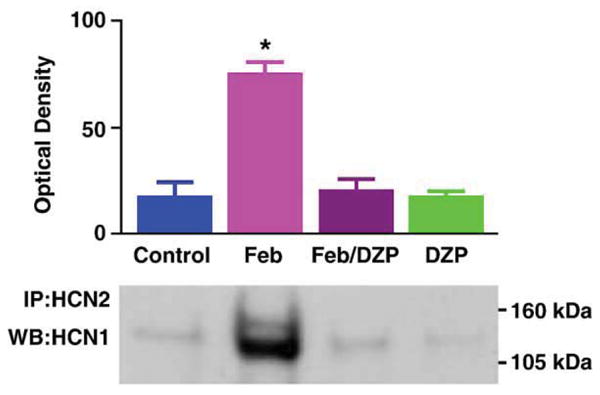
Hyperthermic (febrile) seizures rather than the elevation of brain temperature per se are required for increased HCN1/HCN2 co-association. To dissociate ‘fever’ from ‘febrile seizures’, rats sustaining hyperthermia were pre-treated with the benzodiazepine diazepam (DZP; 10 mg/kg) to block the seizures. As shown, HCN1/HCN2 co-immunoprecipitation (co-IP) was increased in hippocampi of rats sustaining seizures (Feb), but not in those from hyperthermic rats that did not have seizures (Feb/DZP) or in the normothermic, diazepam-treated group (DZP). * Significantly different from controls (P = 0.01).
HCN1/HCN2 co-assembly is associated with reduced HCN1 channel protein and mRNA expression and increased HCN2/HCN1 ratio
Augmented HCN1/HCN2 heteromerization could be a result of an increased HCN2/HCN1 ratio (Hadley et al., 2003; Levitan and Takimoto, 1998; Liao et al., 1996), leading to increased combinatorial interaction of the normally more abundant HCN1-molecules with HCN2 isoforms. Alternatively, enhanced heteromerization could be an independent effect of the seizures. To help distinguish between these possibilities, we determined the effects of the seizures on the expression of HCN1 and HCN2 proteins. HCN1 levels were significantly reduced in ‘febrile’ seizure pups as compared with littermate controls (49.4 ± 6.6 ng/mg vs. 78 ± 11.5 ng/mg; Fig. 3A; P < 0.05). Reduced HCN1 protein levels in the seizure group were apparent also when determined in comparison to β-actin content of the same samples (not shown). In accord with the reduction of HCN1 protein, the quantity of HCN1 channels immunoprecipitated with anti-HCN1 followed by probing for the same isoform was reduced in the seizure animals (Fig. 3B). The reduced HCN1 protein levels after ‘febrile’ seizures were consistent with our previous findings of decreased HCN1 mRNA levels, data that were reaffirmed in the current series of experiments (Fig. 4A).
Fig. 3.
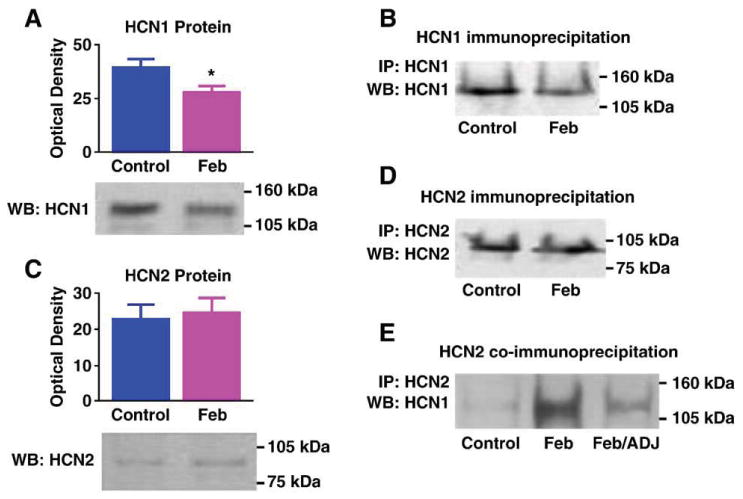
Differential regulation of the HCN1 and HCN2 protein levels promotes heteromerization: (A) Western blot (WB) showing a significant decrease on HCN1 protein levels 7 days after experimental ‘febrile’ seizures (Feb, P = 0.03). (B) Reduced HCN1 protein after seizures is apparent also when IP is carried out with anti-HCN1 followed by WB probing with the same antiserum: a fainter immunoreactive band is seen in a Feb hippocampal homogenate, compared with a hippocampus from a control animal. Note that the samples shown here are from the same hippocampi used in Fig. 1B. Thus, increased HCN1/HCN2 (Fig. 1B) is found despite reduced total HCN1 immunoreactivity. (C) HCN2 protein levels are not changed after experimental febrile seizures, as shown in the WB using anti-HCN2. (D) In accord with the WB, IP for HCN2 followed by probing for the same isoform does not lead to increased HCN2-immunoreactive band intensity. For each gel, every lane was loaded with equal amounts (25 μg) of extract from a single hippocampus. (E) When equal amounts of HCN2 protein derived from a Feb or a control hippocampus are immunoprecipitated with anti-HCN2 and then probed for HCN1, more HCN1 is co-associated with HCN2 in the Feb group. A Feb homogenate in which a mild increase of HCN2 protein occurred was subjected to WB for HCN2, compared with a control. Amount of this Feb homogenate, adjusted to include the same HCN2 quantity as in the control (Feb/ADJ) was immunoprecipitated with anti-HCN2, together with the control and the non-adjusted Feb homogenate (Feb). As shown, in Feb hippocampus, higher levels of HCN1 still co-precipitated with HCN2 even when HCN2 amounts were carefully adjusted to equal those in the control, suggesting that reduced HCN1 in the homogenate or other seizure-evoked factors govern enhanced HCN1/HCN2 co-association in the Feb group.
Fig. 4.
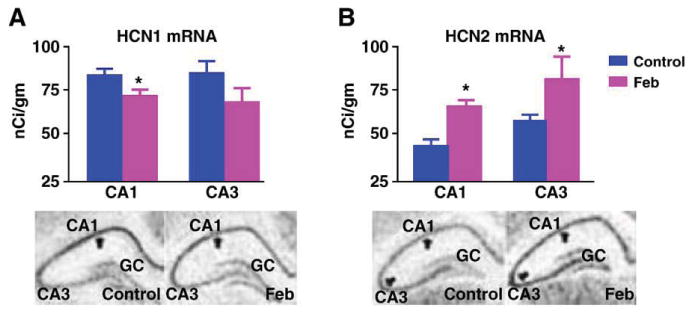
Reduced HCN1 protein levels after ‘febrile’ seizures are attributable to lower mRNA levels. (A) Representative photomicrographs and quantitative analysis of HCN1 mRNA in hippocampal CA1 and CA3 a week after the seizures reaffirm diminished expression of this channel isoform (Brewster et al., 2002). (B) HCN2 mRNA levels are greater in the Feb group, as seen in the representative hippocampal photomicrographs and the analysis. However, increased mRNA expression is not reflected in increased protein levels (Fig. 3C), potentially because of increased channel turnover. * Denotes significant difference from the control group.
In contrast to HCN1, HCN2 protein levels were not consistently influenced by the seizures, as shown in the Western blots from control and seizure-experiencing animals (9.86 ± 2.34 ng/mg vs. 9.4 ± 1.55 ng/mg in experimental and controls, respectively; Fig. 3C). Thus, in the case of HCN2, the increased mRNA levels (Fig. 4B) were not consistently translated into increased protein, potentially because of increased protein turnover. In support of this finding, precipitation with anti-HCN2 followed by probing for the same isoform on Western blot failed to show increases in total HCN2-immunoreactivity (Fig. 3D). Importantly, in homogenates from control and febrile seizures (Feb) hippocampi that were titrated to contain the same amounts of HCN2, increased HCN1 co-immunoprecipitation was still observed (see example in Fig. 3E). These findings exclude the possibility that increased HCN2 protein levels in some hippocampal precipitates from seizure-experiencing rats account for the increase of co-precipitated HCN1, and suggest that the enhanced association of HCN1 and HCN2 after seizures is not driven by altered HCN2 levels, but by either reduced HCN1 abundance or independent factors. Indeed, although HCN2 protein levels were not reduced, the ratio of HCN2/HCN1 tended to be higher in the seizure-experiencing rats (0.124 ± 0.02 in controls vs. 0.245 ± 0.05 in the Feb group; P = 0.09).
Seizure-evoked co-association of HCN1/HCN2 is not model-specific, but endures only in seizures promoting long-lasting hyper-excitability
We next asked whether enhanced HCN1/HCN2 co-assembly was unique to developmental febrile seizures, or resulted from pathological network activity in the developing hippocampus per se. In the latter case, the degree of heteromerization should correlate to the duration and severity of the seizures. Longer and more intense hippocampal seizures were elicited using the glutamatergic agonist kainic acid (Holmes et al., 1988; Tremblay et al., 1984) and HCN co-assembly was quantified. Kainic acid provoked seizures lasting for ∼180 min (Brewster et al., 2002; Holmes et al., 1988; Stafstrom et al., 1992; Tremblay et al., 1984), compared with ∼20 min for ‘febrile’ seizures (Brewster et al., 2002; Dubé et al., 2000), and led to greater co-precipitation of HCN1 and HCN2 7 days (but not 24 h) later (Fig. 5A). In addition, the extent of HCN1/HCN2 co-precipitation was higher after kainic acid-evoked seizures compared with the shorter ‘febrile’ seizures. Together with the effects of blocking the seizures (Fig. 2), this suggests that seizures per se, rather than the manner by which they are elicited, govern HCN1/HCN2 interaction in a duration-dependent manner.
Fig. 5.
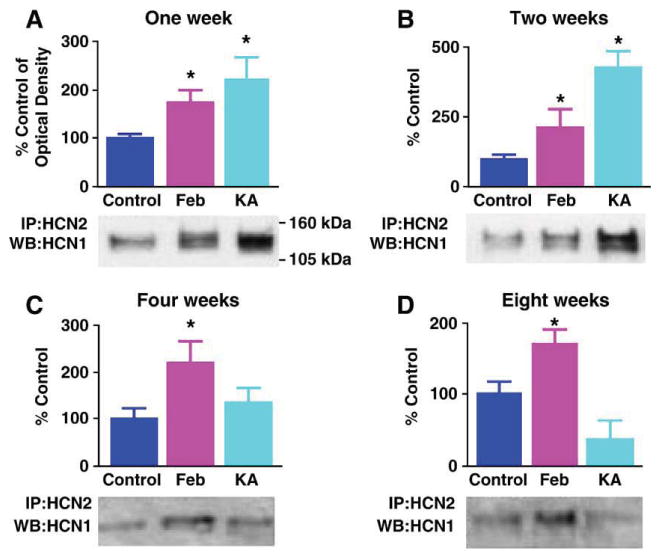
HCN1/HCN2 co-precipitation is greater in prolonged kainic acid (KA)-evoked seizures as compared to the shorter experimental febrile seizures. However, the increased HCN1/HCN2 heteromerization endures long term only after the latter. Quantitative analysis of Western blots (WB) probed for HCN1 of hippocampal extracts immunoprecipitated (IP) with anti-HCN2 at several time-points after ‘febrile’ and KA seizures. (A and B) One and 2 weeks after seizures, the Western blot (bottom) and quantitative analysis (top) show significant co-association of HCN1/HCN2 in both seizure groups, that is significantly more robust after ∼180 min of KA seizures (n = 5 per group). (C) By 4 weeks after the seizures, enhanced heteromerization of HCN1/HCN2 is significant only in the febrile seizure group (n = 4 per group). (D) Eight weeks after these developmental seizures, co-association of HCN1/HCN2 persists in the group subjected to experimental prolonged febrile seizure (P = 0.04), whereas the KA group is not significantly different from the controls (n = 4 per group). *Denotes significant difference from the control group. The KA group is significantly different also from the Feb group at this time point (P = 0.01).
The striking HCN1/HCN2 co-immunoprecipitation after kainic acid seizures was surprising because, unlike developmental febrile seizures (Dubé et al., 2000) kainic acid-evoked seizures at the same age do not lead to long-lasting hippocampal hyperexcitability (Stafstrom et al., 1992). Therefore, we considered the possibility that if heteromeric h channels contribute to the enduring aberrant hippocampal excitability, then such channels should be persistently augmented after ‘febrile’ but not kainic acid-evoked seizures. For both seizure types, HCN1/HCN2 co-IP was enhanced 1 (Fig. 5A) and 2 (Fig. 5B) weeks after the seizures, (and tended to be more so after the kainic acid-evoked seizures compared to the ‘febrile’ ones at the two week time-point; P = 0.13). This enhanced co-association endured at 4 and 8 weeks after experimental febrile seizures. In contrast, co-IP of HCN1 with HCN2 waned in hippocampi from kainic acid-treated animals and was indistinguishable from that in the control group at the later time-points (Figs. 5C and D; febrile- vs. kainate-treated groups at 8 weeks P = 0.01). These findings are in line with the duration of transcriptional dysregulation of the HCN channel isoform expression: Reduced HCN1 mRNA levels endured for at least 3 months after ‘febrile’ seizures, but not in rats subjected to kainic acid seizures early in life (Brewster et al., 2002). This enduring increase of HCN channel heteromerization after ‘febrile’ seizures was associated with alterations of Ih that promote hippocampal hyperexcitability (Chen et al., 2001a; Santoro and Baram, 2003). While it is tempting to speculate that heteromeric channels underlie the modified Ih, other potential mechanisms might be involved.
Discussion
The major findings of this study are: (1) Seizures increase the co-immunoprecipitation of HCN1 and HCN2 channel isoforms in developing rat hippocampus. (2) This effect requires synchronized paroxysms of neuronal activity (seizures), and is not model-specific. (3) The increased HCN1/HCN2 co-association may be driven by the doubling of the HCN2/HCN1 ratio (from ∼0.12 to ∼0.24), increasing the probability that the more abundant HCN1 subunit will interact with HCN2. (4) Both the quantitative changes in HCN expression and the enhanced Co-IP endure after ‘febrile’—but not after KA induced seizures. Taken together with the fact that experimental febrile seizures persistently modulate the Ih current and enhance seizure susceptibility, these findings implicate the HCN channels in the underlying mechanisms.
Potential mechanisms for the formation of heteromeric HCN channels
In naive rat hippocampus, amounts of HCN1 protein are over 8-fold higher than those of HCN2 (∼78 versus 9.4 ng/mg), so that most HCN1 channel molecules are likely to interact with other HCN1 subunits and form homomeric channels. Developmental seizures virtually doubled the hippocampal HCN2/HCN1 ratio (from ∼0.12 to 0.24). Other factors being equal, this should increase the stochastic probability of the HCN1 isoform's interaction with HCN2. This finding is reflected in Fig. 3E, where increased in vivo co-association of HCN1 and HCN2 was evident in Feb homogenates controlled for levels of HCN2. The increased HCN1/HCN2 co-assembly will result in a proportionately higher contribution of heteromeric HCN channels to the macroscopic Ih of CA1 pyramidal cells. Because the properties of heteromeric channels are distinct from those of homomeric ones, their contribution should significantly modify the properties of the neuronal Ih. In this scenario, the larger effects of the (longer and more severe) KA seizures on HCN isoform expression would lead to increased heteromerization compared with the milder ‘febrile’ seizures, as was found here (Fig. 5A). In contrast, the persistent reduction of HCN1 protein levels after the ‘febrile’, but not the kainic acid seizures, would favor long-lasting co-assembly of heteromeric channels after the former type of seizures (Fig. 5D).
Whereas in vivo evidence for this abundance-dependent dynamic interaction of HCN channels isoforms is not yet available, in vitro studies, transfecting different ratios of HCN1/HCN2 constructs into heterologous expression systems, support this notion (S. Chen et al., 2001b). In addition, several types of voltage- and G-protein-gated cationic channels can exist as homomeric or heteromeric structures (Hadley et al., 2003; Levitan and Takimoto, 1998; Liao et al., 1996). The extent of their heteromerization depends on the presence of the interacting isoforms in the same neuron (Liao et al., 1996) and may be a function of the relative abundance of the involved subunits.
Alternative mechanisms for seizure-driven enhanced HCN1/HCN2 heteromerization may involve activity-evoked changes in the intracellular trafficking or membrane insertion of channels. Whereas the post-translational regulation of the HCN channel molecules is not fully understood, recent evidence supports activity-dependent subcellular distribution of the HCN1 subunit (Bender et al., 2004) and interaction of the channels with scaffolding proteins (Gravante et al., 2004). Potentially, seizure activity may alter glycosylation or other post-translational editing of the HCN isoform molecules that will influence their cellular fate.
Why do changes of HCN isoform interaction endure after ‘febrile’ seizures?
The basis for the unique persistence of reduced HCN1 protein expression and ensuing HCN1/HCN2 heteromerization after the relatively short developmental ‘febrile’ seizures remains speculative. The majority of molecular changes after developmental seizures are transient (Holmes and Ben-Ari, 1998), so that the mechanism for these unique persistent effects of febrile seizures may be attributable to the recruitment of additional molecular messengers that are not typically activated by developmental seizures. Among these, the pro-inflammatory cytokines such as interleukin-1β are attractive—they are recruited by hippocampal seizures in adult but generally not in developing animals (Rizzi et al., 2003); remarkably, these cytokines are involved in the mechanisms of febrile seizure generation in immature hippocampus (Dubé et al., 2005). Alternatively, the hyperthermia that is integral to febrile seizures, though not sufficient to evoke heteromerization in itself, may modulate the duration of HCN channel expression or trafficking. For example, hyperthermia may influence the release or actions of thyroid hormone, which regulates the expression of cardiac isoforms of the HCN channels (Gloss et al., 2001).
Significance of activity-evoked heteromerization of the HCN channel isoforms
The experiments described above demonstrate that pathological network activity (seizure) promotes co-immunoprecipitation of HCN1 and HCN2, very likely reflecting the formation of heteromeric channels. While the ability of HCN1 and HCN2 to co-assemble into functional channels has been demonstrated (Chen et al., 2001b; Er et al., 2003; Much et al., 2003; Proenza et al., 2002; Ulens and Tytgat, 2001; Xue et al., 2002), we find only modest co-association of HCN1/HCN2 in the ‘normal’ hippocampal formation of both developing and adult rats (control groups, Figs. 1 and 5), consistent with the single report on whole mouse brain (Much et al., 2003). However and importantly, we demonstrate that this apparent heteromerization is a dynamic, malleable process that is governed by network activity. Thus, pathological neuronal firing elicited by either KA or hyperthermia provoked HCN1/HCN2 assembly, and the effects of both types of seizures endured for weeks, representing a novel mechanism by which activity may alter neuronal Ih. The resulting changes in the Ih properties of individual neurons, a form of intrinsic neuronal plasticity, may contribute to long-term pathological alteration of the hippocampal network.
Acknowledgments
The authors thank Drs. B. Santoro, A. Ludwig, F. Hofmann and M. Biel for the HCN isoform probes, and appreciate critical comments by Dr. R.A. Bender. Supported by NlH grants NS 35439, NS 28912 (TZB), NS047993 (AB) and AG00358 and NS37799 (CMG).
References
- Bender RA, Brewster AL, Santoro B, Ludwig A, Hofmann F, Biel M, Baram TZ. Differential and age-dependent expression of hyperpolarization-activated, cyclic nucleotide-gated cation channel isoforms 1–4 suggests evolving roles in the developing rat hippocampus. Neuroscience. 2001;106:689–698. doi: 10.1016/s0306-4522(01)00314-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Soleymani SV, Brewster AL, Nguyen ST, Beck H, Mathern GW, Baram TZ. Enhanced expression of a specific hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J Neurosci. 2003;23:6826–6836. doi: 10.1523/JNEUROSCI.23-17-06826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Kirschstein T, González-Vega R, Beck H, Baram TZ. Age- and pathway-specific axonal expression of hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in the rat perforant path. Soc Neurosci Abst. 2004;52:15. [Google Scholar]
- Bräuer AU, Savaskan NE, Kole MH, Plaschke M, Monteggia LM, Nestler EJ, Simburger E, Deisz RA, Ninnemann O, Nitsch R. Molecular and functional analysis of hyperpolarization-activated pacemaker channels in the hippocampus after entorhinal cortex lesion. FASEB J. 2001;15:2689–2701. doi: 10.1096/fj.01-0235com. [DOI] [PubMed] [Google Scholar]
- Brewster AL, Bender RA, Chen Y, Dubé C, Eghbal-Ahmadi M, Baram TZ. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform and cell-specific manner. J Neurosci. 2002;22:4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med. 2001a;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001b;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D. Pacemaker mechanisms in cardiac tissue. Annu Rev Physiol. 1993;55:455–472. doi: 10.1146/annurev.ph.55.030193.002323. [DOI] [PubMed] [Google Scholar]
- Dubé C, Chen K, Eghbal-Ahmadi M, Brunson K, Soltesz I, Baram TZ. Prolonged febrile seizures in the immature rat model enhance hippocampal excitability long term. Ann Neurol. 2000;47:336–344. [PMC free article] [PubMed] [Google Scholar]
- Dubé C, Vezzani A, Behrens M, Bartfai T, Baram TZ. Interleukin 1β contributes to the generation of experimental febrile seizures. Ann Neurol. 2005;57:152–155. doi: 10.1002/ana.20358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghbal-Ahmadi M, Avishai-Eliner S, Hatalski CG, Baram TZ. Differential regulation of the expression of corticotropin-releasing factor receptor type 2 (CRF2) in hypothalamus and amygdala of the immature rat by sensory input and food intake. J Neurosci. 1999;19:3982–3991. doi: 10.1523/JNEUROSCI.19-10-03982.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Er F, Larbig R, Ludwig A, Biel M, Hofmann F, Beuckelmann DJ, Hoppe UC. Dominant-negative suppression of HCN channels markedly reduces the native pacemaker current I(f) and undermines spontaneous beating of neonatal cardiomyocytes. Circulation. 2003;107:485–489. doi: 10.1161/01.cir.0000045672.32920.cb. [DOI] [PubMed] [Google Scholar]
- Franz O, Liss B, Neu A, Roeper J. Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization-activated cyclic nucleotide-gated ion channels (Ih) in central neurons. Eur J Neurosci. 2000;12:2685–2693. doi: 10.1046/j.1460-9568.2000.00151.x. [DOI] [PubMed] [Google Scholar]
- Gloss B, Trost S, Bluhm W, Swanson E, Clark R, Winkfein R, Janzen K, Giles W, Chassande O, Samarut J, Dillmann W. Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor alpha or beta. Endocrinology. 2001;142:544–550. doi: 10.1210/endo.142.2.7935. [DOI] [PubMed] [Google Scholar]
- Gravante B, Barbuti A, Milanesi R, Zappi I, Viscomi C, DiFrancesco D. Interaction of the pacemaker channel HCN1 with filamin A. J Biol Chem. 2004;279:43847–43853. doi: 10.1074/jbc.M401598200. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci. 2003;23:5012–5019. doi: 10.1523/JNEUROSCI.23-12-05012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GL, Ben-Ari Y. Seizures in the developing brain: perhaps not so benign after all. Neuron. 1998;21:1231–1234. doi: 10.1016/s0896-6273(00)80642-x. [DOI] [PubMed] [Google Scholar]
- Holmes GL, Thompson JL, Marchi T, Feldman DS. Behavioral effects of kainic acid administration on the immature brain. Epilepsia. 1988;29:721–730. doi: 10.1111/j.1528-1157.1988.tb04226.x. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Bernard JA, Gall CM, Lynch G. Alpha3 integrin receptors contribute to the consolidation of long-term potentiation. Neuroscience. 2002;110:29–39. doi: 10.1016/s0306-4522(01)00540-1. [DOI] [PubMed] [Google Scholar]
- Levitan ES, Takimoto K. Dynamic regulation of K+ channel gene expression in differentiated cells. J Neurobiol. 1998;37:60–68. doi: 10.1002/(sici)1097-4695(199810)37:1<60::aid-neu5>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Liao YJ, Jan YN, Jan LY. Heteromultimerization of G-protein-gated inwardly rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in weaver brain. J Neurosci. 1996;16:7137–7150. doi: 10.1523/JNEUROSCI.16-22-07137.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- Lupica CR, Bell JA, Hoffman AF, Watson PL. Contribution of the hyperpolarization-activated current (Ih) to membrane potential and GABA release in hippocampal interneurons. J Neurophysiol. 2001;86:261–268. doi: 10.1152/jn.2001.86.1.261. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ. The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens-alveus interneurons. J Physiol. 1996;497:119–130. doi: 10.1113/jphysiol.1996.sp021754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic lh normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:508–514. doi: 10.1038/9158. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Biel M, Hofmann F, Ludwig A. Differential distribution of four hyperpolarization-activated cation channels in mouse brains. Biol Chem. 1999;380:975–980. doi: 10.1515/BC.1999.121. [DOI] [PubMed] [Google Scholar]
- Much B, Wahl-Schott C, Zong X, Schneider A, Baumann L, Moosmang S, Ludwig A, Biel M. Role of subunit heteromerization and N-linked glycosylation in the formation of functional hyperpolarization-activated cyclic nucleotide-gated channels. J Biol Chem. 2003;278:43781–43786. doi: 10.1074/jbc.M306958200. [DOI] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- Proenza C, Tran N, Angoli D, Zahynacz K, Balcar P, Accili EA. Different roles for the cyclic nucleotide binding domain and amino terminus in assembly and expression of hyperpolarization-activated, cyclic nucleotide-gated channels. J Biol Chem. 2002;277:29634–29642. doi: 10.1074/jbc.M200504200. [DOI] [PubMed] [Google Scholar]
- Rizzi M, Perego C, Aliprandi M, Richichi C, Ravizza T, Colella D, Veliskova J, Moshe SL, De Simoni MG, Vezzani A. Glia activation and cytokine increase in rat hippocampus by kainic acid-induced status epilepticus during postnatal development. Neurobiol Dis. 2003;14:494–503. doi: 10.1016/j.nbd.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Santoro B, Baram TZ. The multiple personalities of h-channels. Trends Neurosci. 2003;26:550–554. doi: 10.1016/j.tins.2003.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, Thompson JL, Holmes GL. Kainic acid seizures in the developing brain: status epilepticus and spontaneous recurrent seizures. Dev Brain Res. 1992;65:227–236. doi: 10.1016/0165-3806(92)90184-x. [DOI] [PubMed] [Google Scholar]
- Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ. Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay E, Nitecka L, Berger ML, Ben-Ari Y. Maturation of kainic acid seizure-brain damage syndrome in the rat: I. Clinical, electrographic and metabolic observations. Neuroscience. 1984;13:1051–1072. doi: 10.1016/0306-4522(84)90288-4. [DOI] [PubMed] [Google Scholar]
- Ulens C, Tytgat J. Functional heteromerization of HCN1 and HCN2 pacemaker channels. J Biol Chem. 2001;276:6069–6072. doi: 10.1074/jbc.C000738200. [DOI] [PubMed] [Google Scholar]
- Vasilyev DV, Barish ME. Postnatal development of the hyperpolarization-activated excitatory current Ih in mouse hippocampal pyramidal neurons. J Neurosci. 2002;22:8992–9004. doi: 10.1523/JNEUROSCI.22-20-08992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waigner BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–809. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- Xue T, Marban E, Li RA. Dominant-negative suppression of HCN1- and HCN2-encoded pacemaker currents by an engineered HCN1 construct: insights into structure–function relationships and multimerization. Circ Res. 2002;90:1267–1273. doi: 10.1161/01.res.0000024390.97889.c6. [DOI] [PubMed] [Google Scholar]