

Abstract

Toll-like receptor 4 (TLR4), a membrane-spanning receptor protein that functions in complex with its accessory protein MD-2, is an intriguing target for therapeutic development. Herein, we report the identification of a series of novel TLR4 inhibitors and the development of a robust, enantioselective synthesis using an unprecedented Mannich type reaction to functionalize a pyrazole ring. In silico and cellular assay results demonstrated that compound 1 and its analogues selectively block TLR4 activation in live cells. Animal model tests showed that 1 and its derivatives could potentiate morphine-induced analgesia in vivo, presumably by attenuating the opioid-induced TLR4 activation.
Keywords: Toll-like receptor, glial cells, signal transduction, protein−protein interactions, synthesis
The regulation of protein−protein interactions using small-molecule agents is a fast-evolving field in medicinal chemistry and chemical biology.1,2 One of the mechanisms by which the innate immune system senses the invasion of pathogenic microorganisms is through the toll-like receptors (TLRs). TLRs are type I integral membrane glycoproteins3 that recognize specific molecular patterns present in microbial components.4 Stimulation of different TLRs induces distinct patterns of gene expression, which not only leads to the activation of innate immunity but also instructs the development of antigen-specific acquired immunity. TLR4 detects lipopolysaccharide (LPS, a component of Gram-negative bacterial cell walls).5 Most recently, Watkins et al. found that TLR4 plays an essential role in microglial activation that contributes to the development of morphine tolerance and thereby compromises the analgesic effects of morphine.6 This exciting discovery provides a novel avenue for therapeutic development to attenuate morphine tolerance by blocking TLR4 signal transduction in glial cells.7
The structure of TLR4 has recently been solved,8,9 rendering opportunities for the use of structure-based drug design technology to develop potential inhibitors. TLR4 forms a heterodimer with MD-2, regulating a number of critical cell-signaling pathways.10 The protein−protein interactions between TLR4 and MD-2 are essential for TLR4 signaling and are, therefore, intriguing targets for therapeutic development specifically for TLR4 over other TLRs since MD-2 interacts primarily with TLR4 among TLR family proteins.11
To identify novel, druglike small molecule inhibitors for TLR4, we employed a recently developed high-throughput in silico screening method.12 A low molecular weight, druglike lead structure, T5342126 (1), has been idenfitied from the in silico screening that targets the MD-2 binding region on the TLR4 surface. The next step was to develop a synthesis that would be conducive to structure−activity relationship (SAR) studies.
When considering a retrosynthesis for 1 (Figure 1), the most logical disconnection was that of the β-amino alcohol, giving epoxide 2 and tetra-substituted pyrazole 3. Epoxide 2 could be traced back to epichlorohydrin (4) and phenol 5. The pyrazole portion 3 can be dismantled via a Mannich type reaction and SN2 displacement of benzylchloride 7 by pyrazole 6. This retrosynthesis gave us the ability to easily alter four parts of the molecule independently to aid in SAR studies.
Figure 1.

Retrosynthesis of 1.
In the forward direction (Scheme 1), the construction of the top piece commenced with the treatment of 3,5-dimethylpyrazole (6) with KOH and the 2-chloro benzyl chloride (7) to give N-aryl pyrazole 8 in a quantitative yield. The next reaction to append the methylene methylamine was unprecedented in the literature to the best of our knowledge. With guidence from a number of sources,13−16 we were able to develop a robust and high-yielding Mannich type reaction for the synthesis. By treating pyrazole 8 with a 1:2 mixture of methyl amine and paraformaldehyde in refluxing ethanol,17 we were able to obtain the desired amine 3 in a 93% yield.
Scheme 1. Preparation of 1.

The synthesis of the bottom half of the molecule involved stirring 4-ethoxy phenol (5) with epichlorohydrine (4) and potassium carbonate in refluxing acetone18,19 to yield 83% of desired product 2. Coupling of the two pieces was best accomplished by stirring amine 3 and epoxide 2 in a 1.2:1 ratio in refluxing 2 M ethanol, to give the target compound 1 in an 89% yield, with an 82% overall yield from commercially available 3,5-dimethylpyrazole.
To test the stereospecificity of the binding of 1 to TLR4, we made the enantiopure forms of both enantiomers at the single alcohol stereocenter of the molecule. To accomplish this, racemic epoxide 2 was treated with Jacobsen's hydrolytic kinetic conditions20 (Scheme 2) to access either the pure (+)- or the (−)-enantiomers. Because both the (R,R)- and the (S,S)-Jacobsen precatalysts are commercially availible, either enantiomer of 2 could be readily produced. This material could then be used in the final coupling reaction with 3 to give access to both enantiomers of 1. Using this method, both enantiopure isomers of 1 were synthesized with 99% enantiomeric excess (ee).
Scheme 2. Synthesis of Enantiopure (+)-1.

Using this robust, modular synthesis, we also prepared a focused library of analogues of lead compound 1 (Table 1). All of these analogues were made racemically using the same sequence as the parent compound and in comparable yields. This first round of analogues was meant to apply changes only to the periphery of the parent compound.
Table 1. Results of the SEAP Reporter Gene Activation Assaya.
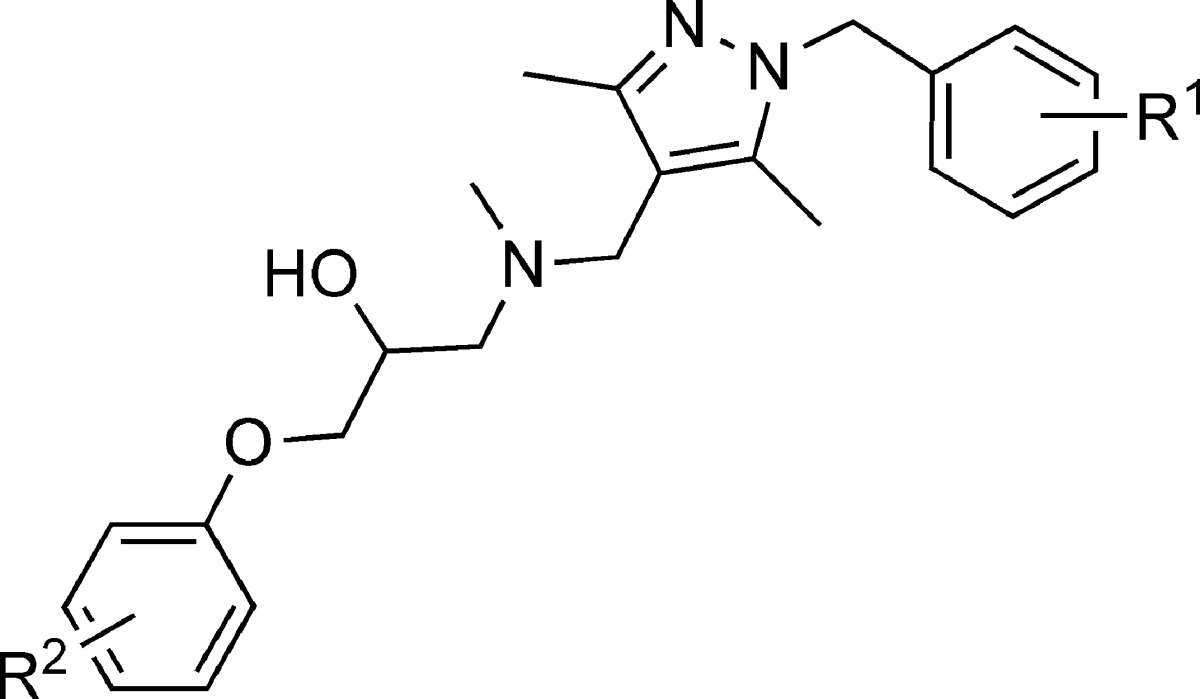
| compound | R1 | R2 | inhibition (%) |
|---|---|---|---|
| 1 | 2-Cl | 4-OEt | 52 |
| (−)-1 | 2-Cl | 4-OEt | 61 |
| (+)-1 | 2-Cl | 4-OEt | 60 |
| 11 | 4-OMe | 4-OEt | 4 |
| 12 | H | H | 1 |
| 13 | 2-Cl | 4-(C2H4)OMe | 32 |
| 14 | 2-Cl | 2-OMe | 36 |
| 15 | 2-Cl | 4-Cl | 99 |
| 16 | 2-F | 4-OEt | 40 |
| 17 | 2-OMe | 4-OEt | 35 |
| 18 | 2-Me | 4-OEt | 31 |
The percent inhibition was determined by measurement of LPS-induced TLR4 activation in HEK293 cells in the presence of 50 μM drug as compared to blank control.
With these analogues in hand, we tested the in vitro inhibitory effects for TLR4 signaling in the HEK 293 cells using a previously described SEAP assay.21 Compound 15 with 2-Cl and 4-Cl substitutes on the top and bottom aromatic rings has been identified with the highest potency to block TLR4 signaling in live cells. At a concentration of 50 μM, racemic 15 showed a complete inhibition of LPS-induced TLR4 activation in HEK 293 cells. By contrast, analogue 12 with no substitution on the aromatic rings showed negligible inhibition, indicating the critical importance of the functional groups. Both enantiomers of 1 showed comparable inhibitory potency, suggesting that its stereocenter does not involve in the recognition of TLR4. These SAR results also demonstrated that the derivatives of 1 inhibit TLR4 activation in a specific manner, ruling out the possibility of nonspecific artifacts by the backbone scaffold. Furthermore, none of these compounds showed any detectable cytotoxicity at concentrations up to 300 μM in various cell lines (e.g., RAW264.7, HEK 293, and HeLa).
Next, we carried out dose dependence studies for the representative inhibitors. Using the SEAP assay, the EC50 values for racemic compounds 15 and 1 were determined to be 18.7 ± 3.2 and 48.1 ± 2.1 μM, respectively (figure in the Supporting Information). Unfortunately, complete inhibition plots for other less potent analogues were not achieved due to the limitation of the cellular assay. TLR4 is an innate immune receptor and can recognize an array of exogenous ligands.22 The addition of a higher dose of inhibitors activates TLR4's signal transduction, which in turn compromises the inhibitory effect analyses. Nonetheless, these results have demonstrated that the inhibitory effects of these analogues are dose-dependent and that the functional groups on the benzene rings are crucial.
In an effort to understand the specificity of our small-molecule inhibitors between different TLRs, we investigated the selectivity of 15 by measuring nitric oxide (NO) production in RAW cells. RAW cells express all TLRs, and each specific TLR can be individually activated by treatment with a receptor-specific ligand.23 The activation of TLRs results in downstream signaling and production of pro-inflammatory mediators such as NO. Compound 15 (27 μM) inhibited TLR4-mediated NO production but showed negligible effects on the signaling of TLR3, TLR 2/6, TLR 2/1, and TLR7 (Figure 2). These results demonstrated that 15 selectively inhibits LPS-induced TLR4 activation without affecting other homologous toll-like receptors.
Figure 2.
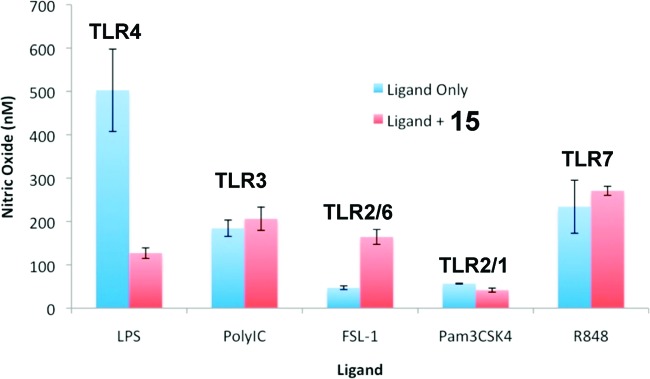
Effect of 15 on TLR-ligand-induced NO production in RAW264.7 cells. LPS, poly(I:C) (polyinosinic-polycytidylic acid), FSL-1 [(S,R)-(2,3-bispalmitoyloxypropyl)-Cys-Gly-Asp-Pro-Lys-His-Pro-Lys-Ser-Phe], Pam3CSK4 {N-palmitoyl-S-[2,3-bis(palmitoyloxy)-(2R,S)-propyl]-[R]-cysteinyl-[S]-seryl-[S]-lysyl-[S]-lysyl-[S]-lysyl-[S]-lysine·3HCl}, and R848 {4-amino-2-(ethoxymethyl)-α,α-dimethyl-1H-imidazo[4,5-c]quinoline-1-ethanol} were used to selectively activate TLR4, TLR3, TLR2/6, TLR2/1, and TLR7, respectively. Compound 15 (25.0 μM) selectively inhibited the NO production induced by LPS but not ligands of other TLRs. Data are means from three independent experiments.
To obtain insights into the possible binding modes of the lead inhibitors, we conducted in silico docking simulations for compounds 1 and 15. Figure 3 shows the top-ranked binding mode of 15 predicted by the AutoDock 4.0 Simulation Suite, implying that 15 recognizes the same cleft on the surface of TLR4 to which a protruding loop region of MD-2 binds. These results suggest that 15 may inhibit the TLR4−MD-2 complex formation by competing with MD-2's binding to TLR4. Docking results for compound 1 suggested a similar binding mode to 15 (data not shown), lending further support that the in silico prediction is consistent and valid.
Figure 3.
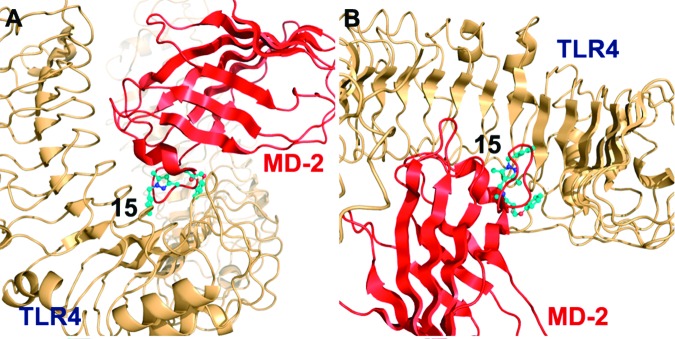
Results of the molecular-docking simulation predict that 15 disrupts the TLR4−MD-2 interaction by competing with MD-2 binding to TLR4. (A) Side and (B) top views of the top-ranked binding modes of 15 (shown in ball-and-stick representation) are shown in complex with full-length TLR4 (orange), competing with MD-2 (red) for the same binding side by mimicking a protruding loop region of MD-2. The structure of TLR4 was taken from the complex with the lymphocyte antigen 96 (PDB ID: 2Z65; resolution, 2.70 Å).
An established rat model was employed to test whether the newly discovered TLR4 signaling antagonist was able to potentiate the analgesic effect of morphine in vivo.23 The Hargreaves test was employed to measure the time taken to observe radiant heat-induced withdrawal responses by the tails of unrestrained rats. Prior to drug administration, two readings were recorded. Following these baseline measurements, drugs were injected intrathecally, and the rats' responses to radiant heat were reassessed across a 3 h time course (Figure 4). While 15 had no effect on pain responsivity in the absence of coadministered morphine, it robustly potentiated the acute analgesic effects of morphine such that the rats exhibited the maximum analgesia recordable on the test. Using the same assay, compound 1 at an elevated dose (injection of 1 μL of 30 mM solution) was shown to have similar effects to potentiate and prolong morphine-induced analgesia, suggesting that the analogues of 1 consistently inhibit morphine-induced TLR4 activation in vivo. It is noticeable that the moderate in vitro inhibitory potency of 1 (EC50 = 48.1 ± 2.1 μM) and 15 (EC50 = 18.7 ± 3.2 μM) is relevant under the physiological conditions in vivo. The volume of cerebrospinal fluid (CSF) in the rat model that we employed has been determined to be around 100 μL,24 and intrathecally administrated morphine, 15, and 1 (injection volume = 1 μL) are estimated to be present at concentrations of 530, 10, and 300 μM, respectively, in the rat CSF, which is consistent with the previously established assay conditions.24
Figure 4.
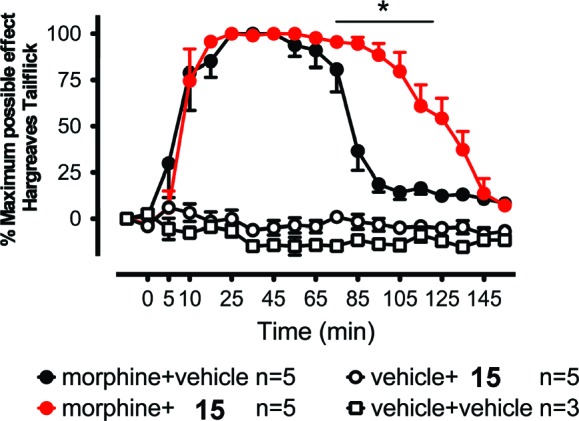
Potentiating intrathecal morphine analgesia by intrathecal 15. Following predrug (baseline) assessment of responsivity to radiant heat (Hargreaves test), rats received intrathecal morphine (1 μL, 15 mg/mL), 15 (1 mM, 1 μL), or the combination of these same morphine and small-molecule doses. While 15 or vehicle alone (5× PBS) had no effect on pain sensitivity, 15 enhanced morphine analgesia. The asterisk denotes the significant difference between morphine + vehicle and morphine + 15 (P < 0.001) from 85 to 125 min. Error bars show the SEM.
In conclusion, we have accomplished our goal of developing and executing an easily modified and scalable synthesis (5 g scale to date) of derivatives of the lead compound 1. In the process, we developed a reliable and previously unreported Mannich type reaction. We have shown that the substituents on the benzene rings of 1 affect the in vitro activity of the molecule and that one of the enantiomers at the alcohol position is more active then the other. Finally, we have shown 1 and its analogues to be effective at blocking TLR4 activation in live cells and at potentiating morphine analgesia in live animal studies.
Supporting Information Available
Experimental procedures and characterization of novel compounds, biological testing procedure, and results. This material is available free of charge via the Internet at http://pubs.acs.org.
We thank the National Institutes of Health (DA026950, DA025740, NS067425, RR025780, and DA029119 to H.Y. and DA023132, DA024044, NS067807, and DE017782 to L.R.W.) for financial support of this work.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yin H.; Hamilton A. D. Angew. Chem., Int. Ed. 2005, 44, 4130. [DOI] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. Nature 2007, 450, 1001. [DOI] [PubMed] [Google Scholar]
- Gangloff M.; Gay N. J. Annu. Rev. Biochem. 2007, 76, 141–165. [DOI] [PubMed] [Google Scholar]
- Kawai T.; Akira S. Trends Mol. Med. 2007, 13, 460–469. [DOI] [PubMed] [Google Scholar]
- Hoshino K.; Takeuche O.; Kawai T.; Sanjo H.; Ogawa T.; Takeda Y.; Takeda K.; Akira S. J. Immunol. 1999, 162, 3749. [PubMed] [Google Scholar]
- Milligan E. D.; Watkins L. R. Nat. Rev. Neurosci. 2009, 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan C. Nat. Biotechnol. 2009, 27, 114. [DOI] [PubMed] [Google Scholar]
- Park B. S.; Song D. H.; Kim H. M.; Choi B. S.; Lee H.; Lee J. O. Nature 2009, 458, 1191. [DOI] [PubMed] [Google Scholar]
- Kim H. M.; Park B. S.; Kim J. I.; Kim S. E.; Lee J.; Oh S. C.; Enkhbayar P.; Matsushima N.; Lee H.; Yoo O. J.; Lee J. O. Cell 2007, 130, 906. [DOI] [PubMed] [Google Scholar]
- Nagai Y.; Akashi S.; Nagafuku M.; Miyake K; Ogata M.; Iwakura Y.; Akira S.; Kitamura T.; Kosugi A.; Kimoto M. Nat. Immunol. 2002, 3, 667. [DOI] [PubMed] [Google Scholar]
- Bsibsi M.; Ravid R.; Gveric D.; van Noort J. M. J. Neuropathol. Exp. Neurol. 2002, 61, 1013. [DOI] [PubMed] [Google Scholar]
- Fedichev P. O. Quantum Pharmaceuticals, Ltd., www.q-pharm.com.
- Kürti L.; Czakó B.. Strategic Applications in Named Reactions in Organic Synthesis; Elsevier Academic Press: Burlington, MA, 2005; p 274. [Google Scholar]
- Tramontini M.; Angiolini L. Tetrahedron 1990, 46, 1791. [Google Scholar]
- Tramontini M. Synthesis 1973, 703. [Google Scholar]
- Ragab F. A. Eur. J. Med. Chem. 2007, 42, 1117. [DOI] [PubMed] [Google Scholar]
- General procedure: A solution of paraformaldehyde (0.820 g, 27.2 mmol) and methylamine.HCl (0.920 g, 13.6 mmol) was dissolved in ethanol (9.06 mL) and stirred for 1 h, then trisubstituted pyrazole (4.53 mmol) was added, and the reaction mixture was stirred at reflux for 12 h. The mixture was then cooled to room temperature and quenched with aqueous NaHCO3 (15 mL). The aqueous layer was extracted three times with chloroform (15 mL), and the combined organic layers were washed with brine (30 mL). The organic layer was dried with MgSO4 and concentrated under reduced pressure. The resulting yellow oil was purified by flash column chromatography using 3% methanol in dichloromethane as the eluting solvent. The product was isolated as a solid with a melting point near room temperature in 93% yield.
- Bevinakatti H. S.; Banerji A. A. J. Org. Chem. 1992, 57, 6003. [Google Scholar]
- Erhardt P. W.; Woo C. M.; Gorczynski R. J.; Anderson W. G. J. Med. Chem. 1982, 25, 1402. [DOI] [PubMed] [Google Scholar]
- Schaus S. E.; Brandes B. D.; Larrow J. F.; Tokunaga M.; Hansen K. B.; Gould A. E.; Furrow M. E.; Jacobsen E. N. J. Am. Chem. Soc. 2002, 124, 1307. [DOI] [PubMed] [Google Scholar]
- Slivka P. F.; Shridhar M.; Lee G. I.; Sammond D. W.; Hutchinson M. R.; Martinko A. J.; Buchanan M. M.; Sholar P. W.; Kearney J. J.; Harrison J. A.; Watkins L. R.; Yin H. ChemBioChem 2009, 10, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan M. M.; Hutchinson M. R.; Watkins L. R.; Yin H.. J. Neurochem. 2010, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson M. R.; Zhang Y.; Shridhar M.; Evans J. H.; Buchanan M. M.; Zhao T. X.; Slivka P. F.; Coats B. D.; Rezvani N.; Wieseler J.; Hughes T. S.; Landgraf K. E.; Chan S.; Fong S.; Phipps S.; Falke J. J.; Leinwand L. A.; Maier S. F.; Yin H.; Rice K. C.; Watkins L. R. Brain, Behav., Immun. 2010, 24, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane E.; Langer S.; Jekich J, B.; Mahoney J.; Huges T.; Frank M.; Selbert W.; Huberty G.; Coats B.; Harrison J.; Klinman D.; Poole S.; Maier S.; Johnson K.; Chavez R.; Watkins L. R.; Leinwand L.; Milligan E. Gene Ther. 2009, 16, 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.