

Abstract
Oxidants generated by eosinophils during chronic inflammation may lead to mutagenesis in adjacent epithelial cells. Eosinophil peroxidase, a heme enzyme released by eosinophils, generates hypobromous acid that damages tissue in inflammatory conditions. We show that human eosinophils use eosinophil peroxidase to produce 5-bromodeoxycytidine. Flow cytometric, immunohistochemical, and mass spectrometric analyses all demonstrated that 5-bromodeoxycytidine generated by eosinophil peroxidase was taken up by cultured cells and incorporated into genomic DNA as 5-bromodeoxyuridine. Although previous studies have focused on oxidation of chromosomal DNA, our observations suggest another mechanism for oxidative damage of DNA. In this scenario, peroxidase-catalyzed halogenation of nucleotide precursors yields products that subsequently can be incorporated into DNA. Because the thymine analog 5-BrUra mispairs with guanine in DNA, generation of brominated pyrimidines by eosinophils might constitute a mechanism for cytotoxicity and mutagenesis at sites of inflammation.
Under normal conditions, eosinophils are a minor component of circulating phagocytic white blood cells. However, they become much more abundant in blood and tissues in a variety of inflammatory disorders, including chronic allergic conditions, helminthic infections, and cancer (1). Moreover, eosinophils are implicated in tissue damage associated with asthma, dermatitis, vasculitis, and the hypereosinophilia syndromes. These granulocytes also may play important roles in host defenses against microbial parasites and tumor cells. One potential mechanism of eosinophil-mediated damage and defense involves their production of oxidizing intermediates.
Chronic inflammation is associated with an increased risk of cancer, raising the possibility that reactive intermediates generated by eosinophils, neutrophils, monocytes, and macrophages might damage nucleic acids and compromise the integrity of the genome (2–6). Activated eosinophils generate such intermediates by first producing superoxide using a membrane-associated NADPH oxidase (7, 8). Superoxide then dismutates to hydrogen peroxide (H2O2), which is the oxidizing substrate for eosinophil peroxidase, a granule enzyme. Reactive intermediates generated by eosinophil peroxidase may represent one pathway for tissue damage by eosinophils (9–14). At plasma concentrations of halide ion (Cl− ≈ 100 mM, Br− 20–100 μM, I− <1 μM; refs. 15 and 16), the major product of eosinophil peroxidase is thought to be hypobromous acid (HOBr; refs. 9, 11, and 13).
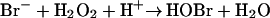 |
1 |
The pseudohalide thiocyanate also has been proposed to be a major substrate (17). Recent studies provide compelling evidence that HOBr generated by activated eosinophils is one pathway for tissue damage in asthma (18).
A striking example in which inflammation-mediated DNA damage may lead to a carcinogenic insult is schistosomiasis (19, 20). This disease is caused by the blood fluke Schistosoma, the eggs of which trigger an intense eosinophilic granulomatous reaction. Many lines of evidence implicate Schistosoma, which is distributed widely in Africa, the Middle East, and Asia in the genesis of bladder cancer, liver cancer, and colon cancer (21). Thus, oxidants generated by eosinophils during a chronic inflammatory response may lead to mutagenesis in adjacent epithelial cells.
Oxidative damage of DNA has been implicated in carcinogenesis (2, 3, 6, 22, 23). In vitro studies have identified numerous modified nucleic acids as products of phagocytic oxidative reactions. For example, activated phagocytes generate the mutagenic nucleobase 8-oxoguanine by pathways that may involve hydroxyl radical or singlet oxygen (24). Nitric oxide, or its degradation products, deaminate nucleobases in vitro and enhance mutagenesis in cultured cells (25, 26). Human neutrophils produce 8-nitrodeoxyguanosine by a pathway involving myeloperoxidase, nitrite, and H2O2 (27). Moreover, we showed recently that myeloperoxidase will also convert deoxycytidine (dC) to 5-chlorodeoxycytidine (28).
Most studies have focused on oxidative damage of chromosomal DNA, but we reasoned that potentially mutagenic adducts might be generated in the abundant cellular pools of ribo- and deoxyribonucleoside precursors. In the current studies, we used deoxynucleosides to determine whether the brominating intermediates generated by eosinophil peroxidase can modify nucleic acid bases. Through MS and NMR spectroscopy, we identified 5-bromodeoxycytidine (BrdC) as a product at physiologically plausible Br− and Cl− concentrations. Activated eosinophils generated BrdC in a reaction that was inhibited by catalase, implicating a peroxidase in the cellular pathway. Cultured cells incorporated the BrdC into genomic DNA as BrdUrd, a well established mutagen. Our observations indicate that brominating intermediates generated by eosinophil peroxidase represent one potential pathway for modifying nucleic acids at sites of inflammation in vivo. They also suggest a mechanism for the oxidative damage of DNA by phagocytes. In this scenario, reactive species halogenate nucleotides and nucleotide precursors that subsequently are incorporated into DNA, in which they can exert cytotoxic and mutagenic effects.
Experimental Procedures
Peroxidase Activity Assay.
Porcine eosinophil peroxidase (ExOxEmis, Little Rock, AR) yielded a single band of active material as assessed by nondenaturing PAGE (29, 30).
Hypobromous Acid.
Bromide-free HOBr was prepared by addition of silver nitrate to ≈80 mM bromine water (1.5:1, mol/mol) and vacuum distillation (31). Reagent taurine monobromamine was prepared by adding HOBr to a 100-fold mole excess of taurine.
Oxidation of dC.
Reactions were performed in gas-tight vials and initiated by adding oxidant from a gas-tight syringe through the septum while vortexing the sample. Reactions were terminated by adding L-methionine to a final concentration of 6 mM. HOBr and taurine bromamine (ɛ240 = 43.6 M−1⋅cm−1), and H2O2 (ɛ288 = 430 M−1⋅cm−1) concentrations were determined spectrophotometrically (13, 32). The pH dependence of product formation was determined as described (28).
Human Eosinophils.
Human polymorphonuclear cells were prepared from blood by density-gradient centrifugation (33). Neutrophils were removed by passage over beads coupled to anti-CD16 antibody (R & D Systems). Cells (>95% eosinophils and <5% lymphocytes) were suspended in PBS (100 mM NaCl/10 mM sodium phosphate supplemented with 2 mM dextrose/1.4 mM CaCl2/1.4 mM MgCl2/1 mM dC/100 μM NaBr/100 μM diethylenetriaminepentaacetic acid), incubated at 37°C for 60 min, and maintained in suspension with intermittent inversion. The reaction was terminated by addition of 6 mM methionine and removal of cells by centrifugation.
Reverse-Phase HPLC.
Reaction mixtures were analyzed by reverse-phase HPLC with a C18 column (Porasil, 5-μm resin, 4.6 × 250 mm; Beckman–Altex) at a flow of 1 ml/min with UV detection at 295 nm (28). For NMR analysis, concentrated reaction mixtures were fractionated isocratically on a semipreparative C18 column (Porasil; 5 μm resin, 10 × 250 mm; Beckman–Altex) (28).
Synthesis of [13C4,15N2]BrUra.
[13C4,15N2]Uracil (1 mM; Cambridge Isotope Laboratories, Cambridge, MA) in 50 mM phosphoric acid and 0.5 M NaBr was exposed to 1 mM HOCl for 15 min at room temperature. The reaction mixture was quenched by addition of 6 mM methionine. [13C4,15N2]BrUra (isotope enrichment >98%) was isolated by HPLC. The synthetic compound was identical to authentic 5-BrUra by HPLC and GC retention times and by MS analysis of its trimethylsilyl derivative.
GC/MS.
DNA bases were converted to trimethylsilyl or dimethyl-tert-butylsilyl derivatives and analyzed in the positive-electron ionization mode by using a Varian Star 3400 CX gas chromatograph equipped with a 12-m DB-1 capillary column (0.2-mm id, 0.33-μm film thickness; J & W Scientific, Folsom, CA) and interfaced with a Finnigan-MAT SSQ 7000 mass spectrometer (San Jose, CA; ref. 28). 5-Fluorodeoxycytidine was used as the internal standard for experiments with eosinophils. For quantification of 5-BrUra and thymine in DNA, [13C415N2]BrUra and 5-fluorouracil were added before hydrolysis.
Electrospray Ionization/MS.
Aliquots from HPLC fractions were analyzed on a Waters Alliance 2670 HPLC equipped with a C18 column (Porasil, 5-μm resin, 2.1 × 150 mm; Beckman–Altex) interfaced with a Finnigan-MAT LCQ as described (28).
Cell Culture, Fluorescence-Activated Cell-Sorter (FACS) Analysis, and Immunohistochemistry.
Asynchronous Chinese hamster fibroblasts (HA1) were seeded at 1 × 105 cells per 60-mm tissue-culture dish; nucleoside incorporation experiments were carried out 48 h later when ≈1 × 106 log phase cells were present. Cells were cultured in Eagle's minimal essential medium supplemented with 10% (vol/vol) FBS and 0.1% penicillin–streptomycin and were maintained at 37°C in a humidified atmosphere of 5% CO2 (34). FACS analysis of nuclei isolated from the cells was performed as described (34) by using a mouse antibromodeoxyuridine monoclonal antibody (Becton-Dickinson Immunocytometry Systems) and FITC-conjugated goat anti-mouse IgG (Sigma). Flow-cytometric analysis was carried out on a Becton-Dickinson FACS 440 equipped with an I-90–5 UV laser (Coherent Radiation, Palo Alto, CA). Data from at least 20,000 nuclei were collected. For immunohistochemistry, HA1 cells were fixed in PBS-buffered formalin, dehydrated through graded ethanol, and processed for immunohistochemistry by using a BrdUrd staining kit (Zymed) with 3,3′-diaminobenzidine tetrahydrochloride as chromogen. Immunostaining was performed as recommended by the manufacturer except that the stock antibody solution was diluted 1:3 (vol/vol). For negative controls, parallel slides were processed with preimmune serum.
Results
The Eosinophil Peroxidase-H2O2-Br− System Brominates dC at Plasma Concentrations of Halide Ions.
We exposed the deoxyribonucleosides of adenine, cytosine, guanine, and thymine to the eosinophil peroxidase-H2O2-Br− system in buffer A (50 mM sodium phosphate/100 mM sodium chloride/100 μM diethylenetriaminepentaacetic acid, pH 4.5). After terminating the reaction with methionine (which scavenges hypohalous acids, halogenated amines, and H2O2), the reaction mixture was analyzed by HPLC using absorbance detection (280 nm for pyrimidines and 254 nm for purines). The dC reaction yielded a major product with a longer retention time than the substrate (Fig. 1), whereas no major new peaks were observed for the other nucleosides. We observed the same product when we replaced eosinophil peroxidase with lactoperoxidase. The HPLC retention time and UV-absorption spectrum of the product were indistinguishable from those of authentic BrdC (Fig. 1).
Figure 1.

Reverse-phase HPLC analyses of dC oxidized by the eosinophil peroxidase-H2O2-Br− system. dC (1 mM) was incubated with 3 nM eosinophil peroxidase (EPO), 100 μM H2O2, and 100 μM NaBr in buffer A (50 mM sodium phosphate/100 mM sodium chloride/100 μM diethylenetriaminepentaacetic acid, pH 4.5) for 60 min at 37°C (EPO/H2O2/Br−). Where indicated, eosinophil peroxidase was omitted from the reaction mixture (H2O2/Br−). Also shown is a chromatogram of 20 μM authentic BrdC. Reactions were initiated by adding H2O2 and terminated with 6 mM l-methionine.
We used electron-ionization GC/MS to characterize further the structure of the modified nucleoside that eosinophil peroxidase generated from dC. This procedure yields information only about the nucleobase because the N-glycoside bond of the nucleoside is hydrolyzed during derivatization. The GC retention times and electron-ionization mass spectra of the trimethylsilyl (TMS) derivative of the oxidation product were identical essentially to those obtained by using authentic BrdC (data not shown). The compound exhibited a molecular ion at m/z 333 and prominent fragment ions at m/z 318, consistent with the ⋅CH3 loss typical of TMS derivatives. Ions with prominent [molecular ion (M) + 2] isotope peaks were present as expected from the natural isotopic abundance of 79Br and 81Br, strongly suggesting monobromination of the products. Ions consistent with loss of the bromine radical were observed at m/z 254 [M− Br]+ and m/z 238 ([M− CH3− HBr]+ or [M− CH4− Br]+). As anticipated for compounds that lack bromine, these fragment ions no longer exhibited the prominent (M + 2) isotope pattern.
To further investigate the structure of the oxidized dC, we examined the electrospray-ionization mass spectrum of the eosinophil peroxidase product. The latter yielded the same major [M + H]+ ions at m/z 306 and 308 as authentic BrdC, strongly suggesting that it was monobrominated dC. The collisionally activated dissociation tandem mass spectrum of the m/z 306 ion from the dC reaction generated a product ion at m/z 190, which suggests that the N-glycoside bond of BrdC had rearranged to yield 79Br-substituted cytosine. The collisionally activated dissociation tandem mass spectrum of the m/z 308 ion likewise generated a product ion at m/z 192, which again suggests fragmentation of the brominated nucleoside to yield 81Br-substituted cytosine. The electrospray-ionization MS/MS spectrum indicated that the cytosine base of the dC was monobrominated.
Eosinophil Peroxidase-Catalyzed Bromination Occurs at C-5 of the Cytosine Ring.
To determine the position of the bromine on the pyrimidine ring, we isolated the product from the eosinophil peroxidase reaction by using HPLC. We then subjected the purified materials to 1H NMR analysis. The reaction-product spectrum was identical essentially to that of commercially available BrdC with a single pyrimidine resonance at 8.23 ppm (singlet, C-6 proton). Significant features when compared with dC included loss of the C-5 proton resonance, a downfield shift in the C-6 proton, and conversion of the C-6 proton resonance from a doublet to a singlet. These findings are consistent with substitution of a bromine atom at the C-5 position. The deoxyribose resonances were similar in the nucleoside substrate, the eosinophil peroxidase-oxidation product, and authentic BrdC.
Reaction Requirements for BrdC Production by Eosinophil Peroxidase.
We used reverse-phase HPLC to characterize the bromination of dC by eosinophil peroxidase. The reaction required enzyme and H2O2; it was blocked by catalase, a scavenger of H2O2, and omission of NaBr from the reaction mixture. Two heme enzyme inhibitors, cyanide and aminotriazole, inhibited product formation. These results demonstrate that bromination of dC by eosinophil peroxidase requires active enzyme, Br−, and H2O2.
Fig. 2 shows the effects of varying the reaction conditions. Enzymatic bromination was proportional to H2O2 concentration up to 100 μM. Thereafter, product yield declined, perhaps because of autoinactivation, substrate inhibition, or consumption of HOBr by H2O2 to generate singlet oxygen (35). When the concentration of Br− was increased, the product yield reached a plateau at 50 μM Br−, the concentration of H2O2 included in the reaction mixture. Bromination was complete by 40 min, and product yields approached 100% (relative to H2O2) at pH 4–5.
Figure 2.

Reaction conditions for oxidation of dC by the eosinophil peroxidase-H2O2-Br− system. The reaction was initiated by adding H2O2 (50 nmol) to 1 ml of buffer A containing 1 mM dC, 3 nM eosinophil peroxidase, and 100 μM NaBr. After a 60-min incubation at 37°C, the reaction was terminated by adding 6 μmoles of l-methionine. Reaction products were analyzed by reverse-phase HPLC. Conditions were varied by performing the reaction with the indicated final concentration of H2O2, Br− ions, hydrogen ions (pH), or for the indicated reaction time (Minutes).
HOBr, Primary Bromamines, and Secondary Bromamines Generate BrdC.
When we replaced the enzymatic system with HOBr, the yields (relative to H2O2) of BrdC were comparable to those obtained when eosinophil peroxidase was included in the reaction mixture. Because HOBr can react readily with amines to form bromamines, we compared the abilities of reagent HOBr, taurine monobromamine, and N-bromosuccinimide to brominate dC in the presence of 100 mM NaCl and 100 μM NaBr. As shown in Fig. 3 for HOBr, N-bromotaurine, and N-bromosuccinimide, the yield and pH dependence of dC bromination were similar for each of the oxidants. We also investigated the ability of HOBr and bromamines to generate N-bromodeoxycytidine. We were unable to observe this postulated species in reaction mixtures lacking methionine when we looked for new peaks of material during HPLC analysis.
Figure 3.

The pH dependence for generation of BrdC by HOBr (●), the primary bromamine N-bromotaurine (○), and the secondary bromamine N-bromosuccinimide (▾). The reaction was initiated by adding oxidant (50 nmol) to 1 ml of buffer A containing 1 mM dC. After a 60-min incubation at 37°C, the reactions were terminated by adding 6 μmol of l-methionine. Reaction products were analyzed by reverse-phase HPLC. The pH of the reaction mixture was determined at the end of the reaction but before adding methionine.
Our results indicate that primary and secondary bromamines will generate BrdC; the yield and pH dependence of the reactions were indistinguishable from those of reagent HOBr. These observations suggest that bromination by eosinophil peroxidase may be facilitated by pyrimidines and free and protein-bound amines, which could catalyze BrdC formation.
Activated Human Eosinophils Generate BrdC.
To determine whether oxidants generated by human eosinophils might brominate dC, we activated human eosinophils (106 per ml) with phorbol myristate acetate in PBS supplemented with 100 μM NaBr and 1 mM dC. HPLC analysis revealed ≈0.2 μM BrdC in the medium of stimulated cells under mildly acidic conditions (pH 6.5). The identity of the oxidation product as BrdC was confirmed by GC/MS and electrospray-ionization/MS analysis (Fig. 4). Cellular BrdC generation was optimal at pH 6.5, but there was substantial production of the brominated product even at neutral pH (Fig. 4). Generation of BrdC required cellular activation, was enhanced by superoxide dismutase (perhaps because of elevated levels of peroxide or protection of the peroxidase from inactivation), and was inhibited by the peroxide scavenger catalase and the heme poison sodium azide (36).
Figure 4.

BrdC generation by human eosinophils. (A) Positive-ion electrospray ionization/MS of the dC oxidation product generated by activated eosinophils. Note the ≈1:1 isotope ratio in the [M + H]+ ions at m/z 306 and 308 characteristic of 79Br and 81Br. (B) Tandem mass analysis of the m/z-306 ion. The product ion at m/z 190 is consistent with loss of deoxyribose to give brominated cytosine. (C) HPLC analysis of media from eosinophils (1 × 106 per ml) stimulated with 200 nM phorbol ester (PMA) at pH 6.5. The retention time of authentic BrdC was 12.9 min. (D) Effect of medium pH on BrdC generation by activated eosinophils. Values represent the mean and ranges of two independent experiments. Superoxide dismutase (SOD), 10 μg/ml; azide, 10 mM; catalase, 10 μg/ml.
Purified BrdC Generated by Eosinophil Peroxidase Is Incorporated as BrdUrd into the DNA of Dividing Mammalian Cells.
To determine whether cells would incorporate the dC oxidation product into DNA, we exposed log-phase hamster HA1 fibroblasts to purified BrdC generated by the eosinophil peroxidase-H2O2-Br− system and analyzed them by flow cytometry (Fig. 5) and histochemistry (Fig. 6). The nuclei of cells incubated with authentic BrdC and the eosinophil peroxidase product were immunoreactive intensely with a monoclonal antibody to BrdUrd (Fig. 6), and the prominent nuclear-staining pattern strongly suggested that the immunoreactive material was incorporated into DNA (Fig. 6). Immunoreactivity was not seen in cells when the antibody was preincubated with BrdUrd but was little affected by preincubation with BrdC. These observations indicate that the antibody reacted selectively with BrdUrd. Flow-cytometric analysis of cells exposed to authentic BrdC for 30 min revealed that this antibody selectively labeled cells that were synthesizing DNA (data not shown). Together, these results suggest that BrdUrd derived from deaminated BrdC is incorporated into the DNA of dividing cells.
Figure 5.

Flow-cytometric analysis of DNA from log phase HA1 cells exposed to the purified eosinophil peroxidase product. Nuclei from cells cultured for 24 h in medium containing 10% FBS were isolated, immunostained with a monoclonal antibody to BrdUrd and an FITC-conjugated secondary antibody, then subjected to flow cytometry. (Top) Control cells. (Middle) Cells exposed for 24 h to 10 μM of purified eosinophil peroxidase product. (Bottom) Cells exposed for 30 min to 10 μM of BrdUrd.
Figure 6.

Immunohistochemical staining of HA1 cells exposed to purified eosinophil peroxidase product. Cells were cultured as described in Fig. 5 and then immunostained with a monoclonal antibody to BrdUrd. (a) Control cells. (b) Cells exposed for 30 min to 3 μM of BrdC. Note that many cells had no detectable staining (arrows mark examples of negative cells). (c) Cells exposed for 24 h to 10 μM of BrdC. Essentially all cells show intense, prominent staining in their nuclei for BrdUrd. (Bar = 48 μm.)
To confirm this idea, we used MS to examine HA1 cells exposed to the purified eosinophil peroxidase-H2O2-Br− system product. GC/MS analysis revealed a substantial quantity of 5-BrUra but not 5-bromocytosine in the DNA of these cells (Fig. 7). In HA1 cells exposed for 24 h to 1 μM and 10 μM of BrdC, respectively, isotope-dilution GC/MS analysis demonstrated that 5-BrUra replaced 0.5 and 4.0% of the thymine residues in the DNA. In contrast, 5-bromocytosine was undetectable (<0.01% substitution) in cellular DNA under any of these conditions. The incorporation of authentic BrdC into cellular DNA was not affected by the presence or absence of 1 mM dC in the medium. These observations indicate that the incorporation of 5-BrUra into DNA was proportional to the concentration of BrdC in the medium.
Figure 7.

Full-scan positive-ion mass spectrum of 5-BrUra in DNA isolated from HA1 cells exposed to purified eosinophil peroxidase product. Cells were cultured as described in Fig. 5 in medium containing 10 μM of BrdC. At the end of the incubation, DNA was isolated from the cells, hydrolyzed to nucleobases with formic acid, and dimethyl-tert-butylsilyl derivatives of the nucleobases were subjected to GC/MS analysis.
Discussion
Our observations indicate that the eosinophil peroxidase-H2O2 system of activated human eosinophils can generate BrdC at plasma concentrations of Cl− and Br− by a reaction pathway that involves HOBr or bromamines. MS, flow-cytometric analysis, and immunohistochemical studies revealed that 5-BrUra is incorporated into the DNA of dividing mammalian cells exposed to the purified eosinophil peroxidase product. These results indicate that eosinophil peroxidase generates brominating intermediates that convert dC to BrdC. The halogenated nucleoside then can be taken up by cultured cells and incorporated into genomic DNA as the mutagenic thymidine analog BrdUrd. The conversion of BrdC to BrdUrd may involve spontaneous or enzymatically catalyzed deamination of the exocyclic amine.
BrdC production by activated human eosinophils was optimal under mildly acidic conditions (pH 6.5). Such conditions are likely to exist in vivo, given that inflammation causes tissue acidosis (37, 38). We also observed a significant level of BrdC production at neutral pH. Thus, although the eosinophil peroxidase-H2O2 system has optimal activity at acidic pH in vitro, the cellular system still possesses activity at physiological pH. Inflamed tissue also may contain high concentrations of nucleosides, because T cells and B cells release dC extracellularly (39), and DNA degradation liberates nucleotides from injured and dead cells. Inflammation also prompts cells to proliferate and repair tissue, creating the opportunity for abnormal bases to be incorporated into new DNA (3). In our study, dividing cells exposed to 1 μM of 5-BrdC for 24 h replaced 0.5% of thymine residues in DNA with 5-BrUra, a 5,000-fold higher substitution level than that observed for guanine oxidation products in normal tissue (40, 41). Optimally stimulated eosinophils (106 per ml, 1 mM dC, pH 6.5) produced ≈0.2 μM of BrdC, whose incorporation into cellular DNA was proportional directly to its concentration in the medium. Moreover, no known repair system excises 5-BrUra from DNA (42), suggesting that chronic exposure to eosinophils in vivo might cause cells to accumulate substantial levels of 5-BrUra. Thus, inflammation promotes a microenvironment—acidosis and the release of nucleosides—that may enable eosinophils to brominate dC and facilitate the incorporation of BrdUrd into the nuclear DNA of proliferating cells.
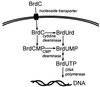
Our observations suggest that halogenation of nucleobases within
a precursor pool provides a mechanism for DNA damage by reactive
intermediates generated by activated phagocytes (Scheme
1). In this pathway, activated phagocytes generate
halogenating intermediates that react with nucleotides or nucleotide
precursors in the extracellular and intracellular milieu. Products of
these reactions include abnormal halogenated or oxidized
deoxynucleotides, which are ferried into the nucleus as substrates for
DNA polymerase. Consequently, potentially cytotoxic and mutagenic
deoxynucleotide derivatives become incorporated into the genomes of
daughter cells. Indeed, the thymidine analogue BrdUrd is a well
established mutagen that acts by mispairing opposite guanine in DNA
(43). If halogenated nucleobases find their way into tumor-suppressor
genes, genes for DNA repair, or potential oncogenes, they might
increase the risk for cancer. A similar incorporational mechanism of
mutagenesis has been suggested by studies of the MutT system in
bacteria, which cleanses the deoxynucleotide pool of 8-oxodGTP. When
MutT is inactivated genetically, the spontaneous-mutation rate
increases by 100- to 10,000-fold (44, 45).
Our demonstration that 5-BrUra is incorporated into the DNA of cultured cells exposed to BrdC is consistent with the activity of cellular cytidine deaminases, which convert dC nucleosides and nucleotides into the corresponding deoxyuridines. Indeed, previous studies have shown that brominated, chlorinated, or fluorinated dC is deaminated to the corresponding deoxyuridine after injection into humans or animals (46, 47). BrdUrd is a thymidine analog with clastogenic and mutagenic activity, causing GC-to-AT and AT-to-GC transition mutations (43). These transitions have been attributed to the ability of the halogen substituent to stabilize the enolic, ionized, or wobble mispairing of the molecule with guanine (48, 49). BrdUrd, BrdC, and oxidants generated by white blood cells may contribute also to mutagenesis by creating an imbalance in the nucleotide pool (50, 51). Moreover, bromination of cytosine has long been recognized to alter the secondary structure of DNA, favoring the formation of Z-DNA (52). Brominated pyrimidine compounds exhibit antiviral activity and alter normal nucleotide metabolism (53, 54). Brominated pyrimidine ribonucleotides can be incorporated also into mammalian RNA, in which they might exert cytotoxic effects by interfering with RNA metabolism (55, 56).
A scenario in which halogenating toxins derived from phagocytic peroxidases promote DNA damage by oxidizing nucleic acids is consistent with the strong association of chronic inflammation and malignancy. Persistent inflammation results in acidosis and tissue injury, which in turn mediates a proliferative, reparative response in resident cells. Thus, not only can phagocytic cells mediate production of modified nucleoside precursors, but they also can create an environment that promotes the incorporation of these mutagenic compounds into parenchymal DNA. Our demonstration that eosinophil peroxidase can brominate dC suggests that pyrimidines indeed may be targets for such damage. Detection of 5-BrUra, 5-bromocytosine, or their nucleosides in tissue would support strongly the hypothesis that activated eosinophils generate potent brominating intermediates in vivo, with important implications for the genesis of tissue injury and perhaps cancer at sites of inflammation.
Acknowledgments
We thank Dr. A. d'Avignon (Department of Chemistry, Washington University) and Dr. R. Higashikubo (Radiation Biology, Washington University) for assistance with NMR and flow cytometry, respectively. MS experiments were performed at the Washington University School of Medicine Mass Spectrometry Resource. This work was supported by grants from the National Institutes of Health (AG19309, AG12293, ES05781, and RR00954), the Veterans Administration Merit Review Program and the Monsanto–Searle/Washington University Biomedical Program. J.P.H. was supported by a Biophysics Training Grant from the National Institutes of Health and a Glenn/American Federation for Aging Research Scholarship for Research in the Biology of Aging.
Abbreviations
- dC
deoxycytidine
- BrdC
5-bromodeoxycytidine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.041146998.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.041146998
References
- 1.Epstein F H. N Engl J Med. 1998;338:1592–1600. [Google Scholar]
- 2.Weitzman S A, Gordon L I. Blood. 1990;76:655–663. [PubMed] [Google Scholar]
- 3.Ames B N, Gold L S, Willett W C. Proc Natl Acad Sci USA. 1995;92:5258–5265. doi: 10.1073/pnas.92.12.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klebanoff S J, Clark R A. The Neutrophil: Function and Clinical Disorders. Amsterdam: North–Holland; 1978. [Google Scholar]
- 5.Marnett L J. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 6.Zhuang J C, Lin C, Lin D, Wogan G N. Proc Natl Acad Sci USA. 1998;95:8286–8291. doi: 10.1073/pnas.95.14.8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tauber A I, Goetzl E J, Babior B M. Inflammation. 1979;3:261–272. doi: 10.1007/BF00914183. [DOI] [PubMed] [Google Scholar]
- 8.Babior B M. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 9.Weiss S J, Test S T, Eckmann C M, Ross D, Regiani S. Science. 1986;234:200–203. doi: 10.1126/science.3018933. [DOI] [PubMed] [Google Scholar]
- 10.Agosti J M, Altman L C, Ayars G H, Loegering D A, Gleich G J, Klebanoff S J. J Allergy Clin Immunol. 1987;79:496–504. doi: 10.1016/0091-6749(87)90368-x. [DOI] [PubMed] [Google Scholar]
- 11.Mayeno A N, Curran A J, Roberts R L, Foote C S. J Biol Chem. 1989;264:5660–5668. [PubMed] [Google Scholar]
- 12.McCormick M L, Roeder T L, Railsback M A, Britigan B E. J Biol Chem. 1994;269:27914–27919. [PubMed] [Google Scholar]
- 13.Thomas E L, Bozeman P M, Jefferson M M, King C C. J Biol Chem. 1995;270:2906–2913. doi: 10.1074/jbc.270.7.2906. [DOI] [PubMed] [Google Scholar]
- 14.Wu W, Chen Y, d'Avignon A, Hazen S L. Biochemistry. 1999;38:3538–3548. doi: 10.1021/bi982401l. [DOI] [PubMed] [Google Scholar]
- 15.Holzbecher J, Ryan D E. Clin Biochem. 1980;13:277–278. doi: 10.1016/s0009-9120(80)80009-9. [DOI] [PubMed] [Google Scholar]
- 16.Ramsey P G, Martin T, Chi E, Klebanoff S J. J Immunol. 1982;128:415–420. [PubMed] [Google Scholar]
- 17.Slungaard A, Mahoney J R., Jr J Biol Chem. 1991;266:4903–4910. [PubMed] [Google Scholar]
- 18.Wu W, Samoszuk M K, Comhair S A A, Thomassen M J, Farver C F, Dweik R A, Kavuru M S, Erzurum S C, Hazen S L. J Clin Invest. 2000;105:1455–1463. doi: 10.1172/JCI9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosin M P, Anwar W A, Ward A J. Cancer Res Suppl. 1994;54:1929s–1933s. [PubMed] [Google Scholar]
- 20.Ishii A, Matsuoka H, Aji T, Ohta N, Arimoto S, Wataya Y, Hayatsu H. Mutat Res. 1994;305:273–281. doi: 10.1016/0027-5107(94)90247-x. [DOI] [PubMed] [Google Scholar]
- 21.Rosin M, Hofseth L J. In: Microbes and Malignancy. Parsonnet J, editor. New York: Oxford Univ. Press; 1999. pp. 313–345. [Google Scholar]
- 22.Halliwell B. Mutat Res. 1999;443:37–52. doi: 10.1016/s1383-5742(99)00009-5. [DOI] [PubMed] [Google Scholar]
- 23.Grisham M B, Jourd'heuil D, Wink D A. Aliment Pharmacol Ther, Suppl. 2000;114:3–9. doi: 10.1046/j.1365-2036.2000.014s1003.x. [DOI] [PubMed] [Google Scholar]
- 24.Jackson J H, Gajewski E, Schraufstatter I U, Hyslop P A, Fuciarelli A F, Cochrane C G, Dizdaroglu M. J Clin Invest. 1989;84:1644–1649. doi: 10.1172/JCI114342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wink D A, Kasprzak K S, Maragos C M, Elespuru R K, Misra M, Dunams T M, Cebula T A, Koch W H, Andrews A W, Allen J S, Keefer L K. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 26.Tamir S, Burney S, Tannenbaum S R. Chem Res Toxicol. 1996;9:821–827. doi: 10.1021/tx9600311. [DOI] [PubMed] [Google Scholar]
- 27.Byun J, Henderson J P, Mueller D M, Heinecke J W. Biochemistry. 1999;38:2590–2600. doi: 10.1021/bi9822980. [DOI] [PubMed] [Google Scholar]
- 28.Henderson J P, Byun J, Heinecke J W. J Biol Chem. 1999;274:33440–33448. doi: 10.1074/jbc.274.47.33440. [DOI] [PubMed] [Google Scholar]
- 29.van Dalen C J, Whitehouse M W, Winterbourn C C, Kettle A J. Biochem J. 1997;327:487–492. doi: 10.1042/bj3270487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maurer H. Disc Electrophoresis and Related Techniques of Polyacrylamide Gel Electrophoresis. New York: de Gruyter; 1971. [Google Scholar]
- 31.Derbyshire D H, Waters W A. J. Chem. Soc. 1950. 564–573. [Google Scholar]
- 32.Beers R J, Sizer I W. J Biol Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 33.Hazen S L, Hsu F F, Heinecke J W. J Biol Chem. 1996;271:1861–1867. doi: 10.1074/jbc.271.4.1861. [DOI] [PubMed] [Google Scholar]
- 34.Higashikubo R, Ragouzis M, Roti Roti J L. Cell Prolif. 1996;29:43–57. [PubMed] [Google Scholar]
- 35.Kanofsky J R, Hoogland H, Wever R, Weiss S J. J Biol Chem. 1988;263:9692–9696. [PubMed] [Google Scholar]
- 36.Kettle A J, Sangster D F, Gebicki J M, Winterbourn C C. Biochim Biophys Acta. 1988;956:58–62. doi: 10.1016/0167-4838(88)90297-x. [DOI] [PubMed] [Google Scholar]
- 37.Bryant R E, Rashad A L, Mazza J A, Hammond D. J Infect Dis. 1980;142:594–601. doi: 10.1093/infdis/142.4.594. [DOI] [PubMed] [Google Scholar]
- 38.Dubos R J. Lancet. 1955;269:1–5. doi: 10.1016/s0140-6736(55)93374-2. [DOI] [PubMed] [Google Scholar]
- 39.Iizasa T, Carson D A. Biochim Biophys Acta. 1986;888:249–251. doi: 10.1016/0167-4889(86)90027-3. [DOI] [PubMed] [Google Scholar]
- 40.Cadet J, Douki T, Ravanat J. Environ Health Perspect. 1997;105:1034–1039. doi: 10.1289/ehp.105-1470384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chaudhary A K, Nokubo M, Reddy G R, Yeola S N, Morrow J D, Blair I A, Marnett L J. Science. 1994;265:1580–1582. doi: 10.1126/science.8079172. [DOI] [PubMed] [Google Scholar]
- 42.Brandon M L, Mi L, Chaung W, Teebor G, Boorstein R J. Mutat Res. 2000;459:161–169. doi: 10.1016/s0921-8777(99)00061-0. [DOI] [PubMed] [Google Scholar]
- 43.Morris S M. Mutat Res. 1991;258:161–188. doi: 10.1016/0165-1110(91)90007-i. [DOI] [PubMed] [Google Scholar]
- 44.Maki H, Sekiguchi M. Nature (London) 1992;355:273–275. doi: 10.1038/355273a0. [DOI] [PubMed] [Google Scholar]
- 45.Yanofsky C, Cox E C, Horn V. Proc Natl Acad Sci USA. 1966;55:274–281. doi: 10.1073/pnas.55.2.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kriss J P, Maruyama Y, Tung L A, Bond S B, Revesz L. Cancer Res. 1962;23:260–268. [PubMed] [Google Scholar]
- 47.Boothman D A, Briggle T V, Greer S. Cancer Res. 1987;47:2354–2362. [PubMed] [Google Scholar]
- 48.Sowers L C, Goodman M F, Eritja R, Kaplan B, Fazakerley G V. J Mol Biol. 1989;205:437–447. doi: 10.1016/0022-2836(89)90353-7. [DOI] [PubMed] [Google Scholar]
- 49.Yu H, Eritja R, Bloom L B, Goodman M F. J Biol Chem. 1993;268:15935–15943. [PubMed] [Google Scholar]
- 50.Kaufman E R. Mutat Res. 1988;200:149–155. doi: 10.1016/0027-5107(88)90077-2. [DOI] [PubMed] [Google Scholar]
- 51.Albrich J M, McCarthy C A, Hurst J K. Proc Natl Acad Sci USA. 1981;78:210–214. doi: 10.1073/pnas.78.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moller A, Nordheim A, Kozlowski S A, Patel D J, Rich A. Biochemistry. 1984;23:54–62. doi: 10.1021/bi00296a009. [DOI] [PubMed] [Google Scholar]
- 53.Zemla J, Tarabek J. Antiviral Res. 1981;1:157–165. doi: 10.1016/0166-3542(81)90004-8. [DOI] [PubMed] [Google Scholar]
- 54.Dobersen M J, Jerkofsky M, Greer S. J Virol. 1976;20:478–486. doi: 10.1128/jvi.20.2.478-486.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X, Patel R, Melamed M R, Darzynkiewicz Z. Cell Prolif. 1994;27:307–319. doi: 10.1111/j.1365-2184.1994.tb01428.x. [DOI] [PubMed] [Google Scholar]
- 56.Sierakowska H, Shukla R R, Dominski Z, Kole R. Biochemistry. 1989;264:19185–19191. [PubMed] [Google Scholar]