

Abstract
The current understanding of the molecular mechanisms involved in the bioinspired formation of silica structures laid foundation for investigating the potential of the S-layer protein SbpA from Lysinibacillus sphaericus CCM 2177 as catalyst, template and scaffold for the generation of novel silica architectures. SbpA reassembles into monomolecular lattices with square (p4) lattice symmetry and a lattice constant of 13.1 nm. Silica layers on the S-layer lattice were formed using tetramethoxysilane (TMOS) and visualized by transmission electron microscopy. In situ quartz crystal microbalance with dissipation monitoring (QCM-D) measurements showed the adsorption of silica in dependence on the presence of phosphate in the silicate solution and on the preceding chemical modification of the S-layer. An increased amount of precipitated silica could be observed when K2HPO4/KH2PO4 was present in the solution (pH 7.2). Further on, independent of the presence of phosphate the silica deposition was higher on S-layer lattices upon activation of their carboxyl groups with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) compared to native S-layers or EDC treated S-layers when the activated carboxyl groups were blocked with ethylene diamine (EDA). Fourier transform infrared attenuated total reflectance (FTIR-ATR) spectroscopy revealed the formation of an amorphous silica gel (SiO2)x·yH2O on the S-layer. The silica surface concentrations on the S-layer was 4 × 10−9 to 2 × 10−8 mol cm−2 depending on the modification of the protein layer and corresponded to 4–21 monolayers of SiO2.
Keywords: S-layer, Silica, Precipitation, Nanostructure, QCM-D, FTIR-ATR
1. Introduction
Currently there is much interest in the synthesis of inorganic materials using biomimetic approaches. One of the best examples is the synthesis of amorphous silica based on principles learned from nature. The current understanding of the key proteins (silicateins [1] and silaffins [2]), genes and molecular mechanisms involved in the bioinspired formation of silica structures laid foundation for investigating the potential of S-layer proteins and their self-assembly products as catalysts, templates, and scaffolds for the generation of novel silica architectures.
S-layers are the most commonly observed cell surface structures in prokaryotic organisms (bacteria and archaea) and have been optimized during billions of years of biological evolution[3-5]. S-layers exhibit oblique, square or hexagonal lattice symmetry. The unit cell dimensions are in the range of 3–30 nm and the thickness is in the range of 5–10 nm. They represent highly porous protein meshwork (30–70% porosity) with pores of uniform size and morphology in the 2–8 nm range. Native or recombinant S-layer proteins have the intrinsic capability to reassemble into monomolecular arrays either in suspension, at solid supports, the air–water interface or lipid films. In the present work the deposition of silica (SiO2)x·yH2O on the S-layer protein SbpA from Lysinibacillus sphaericus CCM 2177 was studied [6-8]. This S-layer protein reassembles with square (p4) lattice symmetry showing a lattice constant of 13.1 nm. The diameter of pores in the S-layer lattice is ~4.5 nm and the protein layer thickness is generally 7–8 nm [7]. A common feature of this S-layer is, with respect to the orientation on the bacterial cell, its smooth outer (extracellular) and more corrugated inner (intracellular) surface [7]. Functional groups on the surface are aligned in well-defined positions and orientations and act as binding sites for the fixation of various materials [8]. The outer surface does not have excess charges at physiological pH values (equimolar amount of carboxyl acid and amino groups), whereas the inner surface is net positively charged [8]. The isoelectric point (IEP) of the monomeric S-layer protein SbpA is 4.2 [8].
The natural analogue of the present study is the deposition of amorphous microporous gel-like or opal-like varieties of hydrated silica (SiO2)x·yH2O as observed in certain single- and multicellular organisms including diatoms, sponges or higher plants [1,2]. Various proteins and organic components form composites with silica. Thereby, the organic substances are acting as specific molecules in the biosilification process. In recent years, some constituents of the organic matrix of biosilificating organisms have been identified, e.g. the proteins of the silicatein family from marine sponges [1] and silaffins from diatoms [2]. In sponges (sponge classes: Demospongiae and Hexactinellida) hydrated amorphous silica is deposited around an axial organic filament composed of chitin or collagen [9,10]. These organic components allow, catalyzed by the enzyme silicatein, the precipitation of amorphous silica and hence the formation of lamellar silica layers in form of spicules [11]. The crucial moieties within the silicatein molecule that are involved in the catalytic mechanism are the hydroxyl groups of the serine residue and the imidazole nitrogen of the histidine residue in the active site of the enzyme [12]. Diatoms are able to accumulate silicon-containing molecular species in their cell walls to produce nanoscale spheres [13-15]. Silaffins and long-chain polyamines have a high affinity to silica and were identified as constituents of biosilica in diatoms. These organic components show to accelerate silica formation from a monosilicic acid solution in vitro whereas the presence of inorganic phosphate is required [2,16]. For polymerization and condensation of silicic acid interactions exist between amino groups of polyamine side chains and phosphorylated serine units with the silanol groups of silica.
Inspired by the process of biomineralization the S-layer protein SbpA, recrystallized on solid supports or in suspension, was used as organic template for the deposition of silica in form of thin layers. It was expected that the silica layer will replicate the topography of the S-layer. Carboxyl groups on the S-layer lattice were activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), and, for comparison, also subsequently blocked with ethylene diamine (EDA). The formation of the nanostructured silica layer was investigated by transmission electron microscopy (TEM), quartz crystal microbalance with dissipation monitoring (QCM-D) and Fourier transform infrared attenuated total reflectance (FTIR-ATR) spectroscopy.
2. Materials and methods
2.1. Isolation and recrystallization of the S-layer protein
The bacterial cell surface layer protein SbpA was isolated from L. sphaericus CCM 2177 (Czech Collection of Microorganisms). Growth, cell wall preparation and extraction of SbpA were performed as described previously [17,18]. The organism was grown in nutrient broth (NB 2, Oxoid, UK) and harvested at the late exponential growth phase by centrifugation. For extraction of S-layer protein from cell wall fragments intact cells were washed in 50 mM Tris/HCl buffer (pH 7.2), broken by sonication, centrifuged to remove non-open cells and cell content, and finally suspended in 5 volumes of the same buffer. To eliminate the cytoplasma constituent cell wall fragments were extracted in detergent solution (Triton-X-100) and washed for several times with water. The final solution was dialyzed against 1 l 50 mM Tris/HCl/10 mM CaCl2·H2O (pH 7.2) to get self-assembly products or against 1 l 2 M H2O/EDTA to get monomer solutions of the protein. Dialysis was performed for 20 min, 1 h, and 8 h at 4 °C, respectively.
For QCM-D and FTIR-ATR measurements the S-layer protein was mixed with buffer solution in a protein/buffer ratio of 0.1:1 ml and dropped on the gold or germanium surface, respectively. The buffer consisted of 0.5 mM Tris/HCl (pH 9) added to 10 mM CaCl2 and the protein concentration was 0.1 mg ml−1.
2.2. Fixation and modification of the S-layer protein
Glutaraldehyde was used in order to perform an intra- and intermolecular crosslinking of the amino groups of the S-layer protein. The crosslinking leads to a better stability and replication of the protein lattice structure. In addition, the S-layer was modified in some experiments with EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, Sigma–Aldrich) or EDC/EDA (EDA: ethylendiamine, Sigma–Aldrich). EDC can react in a first step with carboxyl groups of the protein to form highly reactive O-acylisourea intermediates [19]. This active surface species may then react in a second step with a nucleophile, such as EDA, in order to form an amide bond leading to a blocking of the carboxyl group.
2.3. Silification of the S-layer protein
A solution of silicic acid was freshly prepared by dissolving tetramethoxysilane Si(OCH3)4 (TMOS, Sigma–Aldrich) in 1 mM HCl to a final concentration of 1 M (hydrolysis). In the case of a phosphate solution, 800 μl K2HPO4/KH2PO4-buffer (pH 7.2) was added to this solution to get a final concentration of 0.1 M TMOS. For the non-phosphate solution, 800 μl KOH was added to adjust the pH of the acidic silicate solution to pH 7.2 yielding a final concentration of 0.1 M TMOS, too. After KOH addition the pH was monitored and no significant pH change was observed. The recrystallized S-layer was incubated with phosphate and non-phosphate containing solutions for varying times (1–50 min).
2.4. Transmission electron microscopy
S-layer samples were investigated with a CM12 and CM100 transmission electron microscope (TEM; FEI/Philips, Eindhoven, NL) operated at 80 keV. Self-assembly products were adsorbed for 20 min on a TEM grid (Cu) coated with pioloform and carbon. The S-layer lattice was subsequently stabilized by crosslinking with glutaraldehyde (2.5% in sodium cacodylate buffer, pH 7.2) for 15 min and washed two times with water for 5 min. Finally, the EM-grids with the attached S-layer self assembly products were incubated with the silicate solution, removed after 1–2 min and washed with water again for several times.
2.5. QCM-D measurements
QCM-D measurements were carried out using a Q-sense E4 instrument (Q-Sense AB, Gothenburg, S). Polished AT-cut piezoelectrical quartz crystals with gold electrodes (Q-Sense AB, Gothenburg, S) were used as substrates exhibiting a fundamental frequency f of 5 MHz. The adsorption of the material was monitored as a function of time by recording the change in frequency (Δf) and dissipation (ΔD). All measurements were done in a flow cell with a flow rate of 100 μl min−1 at a temperature of 21 ± 0.02 °C. The evaluation of the data was done with the QCM-D software QTools. For the interpretation and graphical presentation of the frequency and dissipation the averaged data of the third (15 MHz), fifth (25 MHz) and seventh (35 MHz) overtone signal were used. Viscosity and shear force were analyzed with the Voigt based viscoelastic model [20]. The mass, m, and thickness, d, of the adsorbed layers were calculated by the Sauerbrey equation m = (c·f)/n (c: 17.7 ng Hz−1 cm−2 using the fifth overtone (25 MHz) signal [21]. Depending on the adsorbed material, different layer densities were assumed for the calculations of the thickness d = m/ρ·A2 (ρ: S-layer 1.14 g cm−3, silica 2 g cm−3, A: area of the gold sensor) [22].
For the measurements a protein/buffer solution was spread in situ on a QCM-D crystal (crystallization time: 60 min). The S-layer was then washed with water for 5 min, fixed with glutaraldehyde and washed with water again prior to the subsequent silicate precipitation. In some experiments the S-layer was modified with 1 mM EDC or 1 mM EDC/EDA (incubation time: 10 min). The modified layer was washed with water to remove unbound molecules. A phosphate (pH 7.2, K2HPO4/KH2PO4) or non-phosphate (pH 7.2, KOH) containing TMOS solution was flowing across the protein layer (incubation time: 8 min). The silica layer was washed with water again to determine its stability.
2.6. Fourier transform infrared spectroscopy
Spectral analysis was performed using an FTIR-spectrometer (IFS 66, Bruker, Karlsruhe, D) equipped with a “lift-model” single-beam-sample-reference (SBSR) mirror attachment for ATR measurements (OPTISPEC, Neerach, CH). The instrument consisted of a hydrodynamic optimized and water-thermostat SBSR cell (flow-through cuvette) made of Delrin (OPTISPEC, Neerach, CH), with parallel and perpendicular polarized infrared light produced by an aluminium grid polarizer on a KRS-5 substrate (SPECAC, Orpington, U.K.) as well as a liquid-nitrogen cooled mercury–cadmium–telluride (MCT) detector (Bruker, Karlsruhe, D). A germanium trapezoid (54 mm × 30 mm × 2 mm, mean number of active total reflections N = 19.6, Komlas, Berlin, D) with an angle of incidence Θ = 45° was used as the multiple internal reflection element (MIRE). In SBSR measurements, the infrared beam passed alternatively through the upper reference (R) and the lower sample (S) compartment of a flow-through cuvette surrounding the MIRE. SBSR absorbance spectra were calculated from corresponding single channel spectra recorded with the infrared beam going to the two different compartments of the MIRE [23-26]. All measurements were done at 25 °C recording a spectral range of 700–4000 cm−1 with 4 cm−1 resolution. 100–1000 scans were accumulated to achieve the desired signal-to-noise ratio. Reference spectra were recorded with identical (phosphate) solutions as used for the sample spectra. The spectrometer software OPUS was used for the data evaluation. In addition, transmission measurements were done for evaluation of integral molar absorption coefficients. Solutions were analyzed in a CaF2 transmission cell using a 10 μm mylar spacer.
For the measurements the protein/phosphate solution was pumped in situ over the Ge-MIRE to crystallize the S-layer. The recrystallization was done in an alternation process of flowing solution (flow rate 50 μl min−1 for 5 min), stop of flow (for 30 min) and measurement (for 25 min). The recrystallized non-activated S-layer was washed with water several times before starting subsequent silicate adsorption. A phosphate (pH 7.2, K2HPO4/KH2PO4) containing TMOS solution was flowing on top of the protein layer (flow rate 50 μl min−1 for 45 min). The silica layer was washed with water again to check its stability.
3. Results
3.1. Transmission electron microscopy: ultrastructure of the silica layer
A TEM image of a self-assembled S-layer protein lattice almost completely covered by a silica gel layer is shown in Fig. 1. In this example, the silica layer was obtained by exposing the S-layer to a phosphate containing silicate solution. According to the investigation of silica formation by QCM-D and FTIR-ATR, time series studied by TEM showed that the S-layer was covered with silica within 1–2 min. Thus, a detailed replication of the S-layer lattice structure could only be achieved within the first minute of incubation with the silicate solution. In addition, according to the QCM-D and FTIR-ATR measurements, the observed contrast in the TEM images resembled the different layer thicknesses caused by the presence/absence of phosphate and the different chemical modifications of the S-layer protein (non-activated, EDC or EDC/EDA activated) (data only shown for presence of phosphate and EDC activated carboxyl groups on S-layer; Fig. 1).
Fig. 1.

Transmission electron microscopy image of an EDC-activated and silicified S-layer (bar: 300 nm).
3.2. QCM-D: adsorption process and stability of the protein layer
An increasing number of theoretical and experimental investigations with a quartz crystal microbalance with dissipation monitoring (QCM-D) working with liquids have opened the possibility for studying biomolecular assemblies, the adsorption of different materials on these assemblies and the quantitative interpretation of their viscoelastic properties. Thereby, the fixation of the adsorbed assemblies depends on the material of the sensor surface and on the type of surface modification [22,27].
In the present study, QCM-D measurements showed a decrease of the frequency adverse to an increase of the dissipation upon S-layer protein adsorption on the gold sensor (Table 1 and Fig. 2). The frequency decreased exponentially to a level of −87.9 Hz in the first 10 min and to a final level of −104.5 Hz within the next 50 min. The dissipation featured a characteristic initial maximum between 0 and 10 min followed by a small decrease and a small increase again. In detail, the dissipation increased to a level of 3.2 × 106 in the first 10 min and to a final level of 3.6 × 106. An averaged ΔD/Δf ratio of 0.04 was calculated. This indicates that the mass of the layer is relatively thin and rigid and is not deformed during the oscillatory motion of the gold crystal [28]. The observed frequency shift from approximately −80 to −100 Hz agrees with results of previously published studies, although Δf and ΔD are certainly affected by the individual preparation of the protein solution used in the experiments [22,27,29,30]. The mass uptake calculated by the Sauerbrey equation corresponded to 1546 ng cm−2 in the first 10 min and increased to 1850 ng cm−2 within the next 50 min. The calculated thickness (density: 1.14 g cm−3) was 13.5 nm (after 10 min) and finally 16.1 nm, indicating the formation of a protein double layer on the gold sensor with the outer surface exposed to the solution. Viscosity and shear force of the adsorbed protein were calculated by the Voigt model to 0.005 Pa s and 1.1 × 105 Pa. In comparison, the viscosity and shear forces of biomolecules and polymers are in the range 0.0005–0.01 Pa s and 100 Pa–1000 MPa, respectively [31].
Table 1.
QCM-D data (frequency change Δf, dissipation change ΔD, mass m, thickness d, viscosity V, shear force S) of the adsorbed S-layer after washing with water (±σ)
| n | Protein solution | |
|---|---|---|
| Frequency, f [Hz] | 3 | 104.7 ± 1.8 |
| 5 | 104.7 ± 1.0 | |
| 7 | 104.0 ± 1.5 | |
| Dissipation, D [×106] | 3 | 3.3 ± 0.2 |
| 5 | 3.7 ± 0.3 | |
| 7 | 3.8 ± 0.4 | |
| Mass, m [ng cm−2] | 5 | 1850 ± 40 |
| Thickness, d [nm] | 5 | 16.1 ± 0.7 |
| Viscosity, V [Pas] | 5 | 0.005 ± 0.0001 |
| Shear force, S [105 Pa] | 5 | 1.1 ± 0.2 |
Fig. 2.

QCM-D results (frequency and dissipation change) of the S-layer protein SbpA from Lysinibacillus sphaericus CCM 2177 adsorbed on a gold sensor and the adsorption of silica on that protein layer in a phosphate solution (pH 7.2, K2HPO4/KH2PO4). (a) EDC-activated S-layer, (b) EDC/EDA-activated S-layer and (c) non-activated S-layer.
3.3. QCM-D: adsorption process and stability of the silica layer
The silification of the S-layer depends on the phosphate concentration in the used silicate solution and the modification of the protein layer. Detailed results of the QCM-D measurements are presented in Tables 2 and 3 and Figs. 2 and 3. During the silification of the S-layer a decrease of the frequency and a strong increase of the dissipation were noted within the first 2 min when a phosphate containing silicate solutions (K2HPO4/KH2PO4) was used (Fig. 2). In this case additional frequency changes of −95.3 Hz (EDC-activated), −55.6 Hz (EDC/EDA-activated) and −43.9 Hz (non-activated) and dissipation changes of 23.8 × 106 (EDC-activated), 19.6 × 10 6 (EDC/EDA-activated) and 11.9 × 106 (non-activated) were measured. A change of the silica layer could be observed after a 5 min washing procedure. The frequency increased to a final level of −78.5 Hz (EDC-activated), −29.2 Hz (EDC/EDA-activated) and −23.8 Hz (non-activated) whereas the dissipation decreased to a final level of 17.0 × 106 (EDC-activated), 8.7 × 106 (EDC/EDA-activated) and 3.4 × 106 (non-activated), respectively. An averaged ΔD/Δf ratio of 0.22 (EDC-activated), 0.30 (EDC/EDA-activated) and 0.14 (non-activated) was calculated. The final adsorbed silicate mass was estimated by the Sauerbrey equation to 1355 ng cm−2 (EDC-activated), 502 ng cm−2 (EDC/EDA-activated) and 370 ng cm−2 (non-activated). If a silica layer density of 2.0 g cm−3 is assumed the layer thickness was calculated to 6.8 nm (EDC-activated), 2.5 nm (EDC/EDA-activated) and 1.9 nm (non-activated), respectively. The silica surface concentration was calculated using the molar masses of SiO2 and H4SiO4 (Table 4 [32]). Based on a dense package of SiO4 tetrahedra, the number of adsorbed SiO2 monolayers was 21 (EDC-activated), 8 (EDC/EDA-activated) and 5.5 (non-activated), respectively. Once comparing the calculated silica layer thickness, the possible inclusion of water and low amounts of potassium and phosphate ions should be noted, since they might affect the values slightly. When the protein layer was incubated with the phosphate containing silicate solution the viscosity decreased and the shear force increased. A washing process with water led to a desorption of unfixed silicate molecules, leading to an increase of the viscosity and a decrease of the shear force again. The final viscosity of the completely adsorbed layers (protein plus silica) was approximately 0.0037 Pa s (EDC-activated), 0.0033 Pa s (EDC/EDA-activated) and 0.0031 Pa s (non-activated), respectively. The shear force was 1.6 × 105 Pa (EDC-activated), 1.2 × 105 Pa (EDC/EDA-activated) and 1.4 × 105 Pa (non-activated). Therefore, the complete adsorbed silica layer had similar viscoelastic properties and shear forces as the underlying adsorbed protein layer.
Table 2.
QCM-D data of the silificated S-layer (K2HPO4/KH2PO4) after washing procedure with water (±σ)
| n | EDC | EDC/EDA | Non-activated | |
|---|---|---|---|---|
| Frequency, f [Hz] | 3 | 91.1 ± 1.9 | 33.9 ± 0.7 | 23.8 ± 0.5 |
| 5 | 76.3 ± 1.8 | 27.9 ± 0.8 | 23.8 ± 0.6 | |
| 7 | 68.0 ± 1.8 | 25.9 ± 0.7 | 23.8 ± 0.7 | |
| Dissipation, D [×106] | 3 | 19.7 ± 3.1 | 10.8 ± 1.6 | 3.7 ± 0.6 |
| 5 | 16.7 ± 1.5 | 8.5 ± 0.1 | 3.2 ± 0.3 | |
| 7 | 14.7 ± 0.4 | 6.9 ± 0.2 | 3.2 ± 0.1 | |
| Mass, m [ng cm−2] | 5 | 1355 ± 32 | 502 ± 14.2 | 370 ± 10.6 |
| Thickness, d [nm] | 5 | 6.8 ± 0.9 | 2.5 ± 0.4 | 1.9 ± 0.3 |
| Viscosity, V [Pas] | 5 | 0.0037 ± 0.0001 | 0.0033 ± 0.0001 | 0.0031 ± 0.0001 |
| Shear force, S [105 Pa] | 5 | 1.6 ± 0.3 | 1.2 ± 0.2 | 1.4 ± 0.2 |
Table 3.
QCM-D data of the silificated S-layer (KOH) after washing procedure with water (±σ)
| n | EDC | EDC/EDA | Non-activated | |
|---|---|---|---|---|
| Frequency, Δf [Hz] | 3 | 13.9 ± 0.3 | 8.6 ± 0.2 | 2.0 ± 0.0 |
| 5 | 13.1 ± 0.3 | 6.5 ± 0.2 | 2.5 ± 0.1 | |
| 7 | 12.0 ± 0.3 | 5.6 ± 0.1 | 1.1 ± 0.0 | |
| Dissipation, D [×106] | 3 | 0.3 ± 0.0 | 0.3 ± 0.0 | 0.1 ± 0.0 |
| 5 | 0.5 ± 0.1 | 0.6 ± 0.1 | 0.1 ± 0.1 | |
| 7 | 0.6 ± 0.0 | 0.3 ± 0.0 | 0.3 ± 0.0 | |
| Mass, m [ng cm−2] | 5 | 219 ± 5.3 | 142 ± 3.5 | 46 ± 1.8 |
| Thickness, d [nm] | 5 | 1.2 ± 0.2 | 0.6 ± 0.1 | 0.2 ± 0.1 |
| Viscosity, V [Pa s] | 5 | 0.0053 ± 0.0001 | 0.0049 ± 0.0001 | 0.0059 ± 0.0001 |
| Shear force, S [105 Pa] | 5 | 1.0 ± 0.2 | 1.4 ± 0.2 | 1.6 ± 0.2 |
Fig. 3.

From QCM-D studies calculated mass and thickness of adsorbed silica layers. Left: with phosphate solution (pH 7.2, K2HPO4/KH2PO4). Right: without phosphate solution (pH 7.2, KOH). The thickness was calculated with an assumed silica density of 2 g cm−3.
Table 4.
Theoretical and experimental surface concentrations Γ and number z of adsorbed silica layers calculated from QCM-D and FTIR-ATR data (±σ)
| QCM-D | |||||
|---|---|---|---|---|---|
| Mass, m [ng cm−2] |
Thickness, d [nm] |
Surface concentration, Γtheor [mol cm−2] (MSiO2 = 60.1 g mol−1) |
Surface concentration, Γtheor [mol cm−2] (MH4SiO4 = 96.1 g mol−1) |
Number zQCM-D of adsorbed Si—O—Si layers |
|
| pH 7.2, K2HPO4/KH2PO4 | |||||
| EDC | 1355 ± 32 | 6.8 ± 0.9 | 22.5 × 10−9 | 14.1 × 10−9 | 21.5 ± 0.5 |
| EDC/EDA | 502 ± 14.2 | 2.5 ± 0.4 | 8.35 × 10−9 | 5.22 × 10−9 | 8.0 ± 0.5 |
| Non-activated | 370 ± 10.6 | 1.9 ± 0.3 | 6.16 × 10−9 | 3.85 × 10−9 | 5.5 ± 0.5 |
| pH 7.2, KOH | |||||
| EDC | 219 ± 5.3 | 1.2 ± 0.2 | 3.64 × 10−9 | 2.28 × 10−9 | 4 |
| EDC/EDA | 142 ± 3.5 | 0.6 ± 0.1 | 2.36 × 10−9 | 1.48 × 10−9 | 2 |
| Non-activated | 46 ± 1.8 | 0.2 ± 0.1 | 0.77 × 10−9 | 0.48 × 10−9 | 1.0 |
| FTIR-ATR |
|||||
| Surface concentration, Γtheor [mol cm−2] |
Surface concentration, Γexp [mol cm−2] |
Number zQCM-D of adsorbed Si—O—Si layers |
|||
|
| |||||
| pH 7.2, K2HPO4/KH2PO4 | |||||
| Non-activated | 9.04 × 10−9 | 3.69 × 10−9 (⊥) | 4.0 ± 0.5 | ||
| 3.87 × 10−9 (II) | |||||
As basis for the surface concentration Γ a densely spherical package of SiO4 tetrahedra with a Si—O—Si distance of 0.3058 nm (rsphere = 0.1529 nm) was assumed.
QCM-D: surface concentration Γtheor = m/M, number of adsorbed Si—O—Si layers zQCM-D = d/2rSi—O (rSi—O = 0.1529 nm).
FTIR-ATR: surface concentration Γtheor = 4/(Na·a2) with a2: smallest unit cell (a2 = Π·r2, rSi—O = 0.1529 nm), Na: Avogadro constant, surface concentration Γmeas = A·d/(ε·N·ν·de) with A: integrated absorbance of the Si—O—Si band, d: thickness of the adsorbed layer, ε: molar extinction coefficient, N: middle number of intern active reflections, ν: number of functional groups/molecule, de: penetration depth. The molar extinction coefficient ε was calculated from transmission measurements of a (SiO2)x·yH2O gel with the help of the Lambert–Beersche law A = ε·c·dFringes.c: concentration of methanol, dFringes: cell thickness determined by “Fringes” [32], number of adsorbed Si—O—Si layers zFTIR = Γexp/Γtheor.
When silicate solutions without phosphate (KOH) were used, changes in frequency and dissipation were not as strong as in the case of silicate solutions with phosphate (K2HPO4/KH2PO4). For the adsorbed silica layer frequency changes of 21.5 Hz (EDC-activated), 16.5 Hz (EDC/EDA-activated) and 10.0 Hz (non-activated) and dissipation changes of 4.3 × 106 (EDC-activated), 4.6 × 106 (EDC/EDA-activated) and 3.6 × 106 (non-activated) could be determined. Nearly all adsorbed silica gel could be removed from the S-layer by a washing procedure with water. Thus, the frequency decreased to a final level of 13.0 Hz (EDC-activated), 6.9 Hz (EDC/EDA-activated) and 1.9 Hz (non-activated) whereas the dissipation increased to a final level of 0.5 × 106 (EDC-activated), 0.4 × 106 (EDC/EDA-activated) and 0.1 × 106 (non-activated), respectively. A ΔD/Δf ratio of 0.04 (EDC-activated), 0.09 (EDC/EDA-activated) and 0.02 (non-activated) was calculated. The use of a non-phosphate solution consequently led to a lower mass adsorption resulting in a lower thickness of the fixed silica layer compared to experiments with silicate solutions including phosphate (Fig. 3). Final adsorbed masses of the silica layers of 219 ng cm−2 (EDC-activated), 142 ng cm−2 (EDC/EDA-activated) and 46 ng cm−2 (non-activated) and thicknesses of 1.2 nm (EDC-activated), 0.6 nm (EDC/EDA-activated) and 0.2 nm (non-activated) were determined. The corresponding number of adsorbed SiO2 monolayers was 4 (EDC-activated), 2 (EDC/EDA-activated) and 1 (non-activated), respectively. The adsorption of silicate solutions without phosphate on the S-layer caused a decrease of the viscosity and an increase of the shear force. A subsequent flushing with water resulted in a decrease of the viscosity and an increase of the shear force. Finally, the viscosity of the complete adsorbed layers (protein plus silica) was 0.0053 Pa s (EDC-activated), 0.0049 Pa s (EDC/EDA-activated) and 0.0059 Pa s (non-activated) and the shear force was 1.0 × 105 Pa (EDC-activated), 1.4 × 105 Pa (EDC/EDA-activated) and 1.6 × 105 Pa (non-activated).
3.4. FTIR-ATR: adsorption process and stability of the silica layer
Attenuated total reflectance FTIR-ATR spectroscopy allows to record infrared spectra of biological specimen in an aqueous phase as a function of the time. Fig. 4a and b shows the FTIR-ATR absorption spectra of the non-activated recrystallized S-layer on the Ge-MIRE. Amide I (peptide C=O stretching) and amide II (peptide C—N stretching coupled to NH bending) absorption were found at 1597–1712 and 1482–1597 cm−1, respectively, where amide II peak appeared with a lower intensity. The amide I absorption frequency depends on the secondary protein structure (α-helical structure, β-structure) allowing to assess relative amounts of secondary structure parts [33-36]. Further IR-absorptions of the protein were observed at ~1460 cm−1 (δas(CH3), δ(CH2)), 1300–1420 cm −1 (νs(COO−)), 2844–2863 cm−1 (νs(CH2)), 2865–2888 cm−1 (νs(CH3)), 2900–2946 cm−1 (νas(CH2)) and 2900–2946 cm−1 (νas(CH2)). The washing procedure with water after stopping the reassembly of the S-layer protein led to no characteristic changing of the absorption protein bands. Fig. 4c and d shows the IR-absorption of silicate molecules bound on the S-layer. No shifting of the protein peaks could be observed. The main broaden asymmetric band at 1010–1300 cm−1 is assigned to the Si—O—Si stretching group. A smaller asymmetric band at 901–1002 cm−1 could be assigned to the Si—O stretching vibration of Si—OH groups [37,38]. In addition, three bands were measured at 920–940, 970–1010, and 1040–1120 cm−1, corresponding to the vibration modes of the used K2HPO4/KH2PO4 solutions. These bands interfered with the Si—O—Si and Si—O bands from the silicate molecules. A further strong band at 1005–1030 cm−1 appeared due to the formation of methanol during the hydrolysis of the TMOS solution. Both, the methanol band and the phosphate bands disappeared after washing with water (Fig. 4e and f). This procedure led to a decrease of the intensity of the Si—O—Si band and an explicit appearance of the Si—O band without a coincidence with the P—O bands.
Fig. 4.

FTIR-ATR absorption spectra (parallel and vertical polarized) of (a and b) crystallization of the S-layer on the Ge-MIRE, (c and d) silification of this S-layer, (e and f) the silicified S-layer after flushing with water. Left: Overview spectra, Right: silica spectral range.
In addition to qualitative information, the surface concentration of silica adsorbed on the protein layer could be calculated by quantification of the absorbance of the Si—O—Si band in both, parallel and vertical polarized light (following the approach of Wenzl et al. [40] and Fringeli [23]). Detailed information and results are listed in Table 4. The experimental surface concentration Γexp of the adsorbed silicate molecules on the S-layer was calculated to 3.7 × 10−8 mol cm−2 (parallel polarized Γpp) and 3.9 × 10−8 mol cm−2 (vertical polarized Γvp), respectively, by integration of the Si—O—Si band of the ATR measurements using the measured integral molar absorption coefficient ε(Si—O—Si) = 1.06 × 107 cm mol−1 (integration area 1010–1300 cm−1) of the transmission experiments. The dichroitic ratio Rexp (R = App/Avp) is 1.81 (theoretical expected value for a isotropic thin film Riso,heor = 1.90). The difference between the theoretical and experimental dichroitic ratio is close to the error in determining this value. Assuming the adsorbed silicate has a dense spherical packing, the theoretical surface concentration Γtheor for a monolayer is calculated to be 9.0 × 10−9 mol cm−2. A comparison between the experimental surface concentration Γexp and the theoretical surface concentration Γtheor for a monolayer indicate an adsorption of approximately 4 monolayers of silicate molecules or the formation of polymerized (SiO2)x·yH2O layers (Table 4). This agrees well with the surface concentration and the number of monolayers determined by QCM-D. In comparison the adsorption of the S-layer protein resulted in an experimental surface concentration Γexp of 3.5 × 10−12 mol cm−2 (parallel polarized Γpp) and 3.3 × 10−12 mol cm−2 (vertical polarized Γvp), respectively. This surface concentration corresponded to an adsorbed protein mass of 447 ng cm−2, which is less than 925 ng cm−2 (for a monolayer) determined by QCM-D. However, the results of both experiments are seen as consistent, as the adsorbed mass measured using QCM-D includes also water molecules hydrodynamically coupled to the deposited protein structure.
In addition the C—OH band at 1015 cm−1 was quantified to obtain information how many Si—OH bonds are formed from the start component TMOS (Si(OCH3)4). Therefore, transmission measurements were performed with different concentrations of methanol. The integral molar absorption coefficient ε of 1.28 × 106 cm mol−1 (integration area 1005–1030 cm−1) was calculated from a linear fit of the absorptions at the used concentrations. The bulk concentration cbulk of the methanol was estimated by integration of the C—OH band at 1015 cm−1 of the ATR measurements using the molar absorption coefficient ε of the transmission experiments. The calculated values are 3.8 × 10−4 mol cm−3 (cpp) for parallel respectively 4.0 × 10−4 mol cm−3 (cvp) for vertical polarized IR light. These results correspond to an average bulk concentration cbulk of the methanol of 3.9 × 10−4 mol cm−3. As the methanol concentration is 4.0 10−4 mol cm−3 (concentration of TMOS = 1.0 × 10−4 mol cm−3) all CH3 groups (100%) of TMOS were split off and reacted to form methanol in the bulk solution. This proofs the possibility that nearly all formed Si(OH)4-groups can polymerize to a Si—O—Si network and form a (SiO2)x·yH2O gel.
4. Discussion
4.1. Silica adsorption on the S-layer structure
Based on the microscopic studies presented in this work it can be stated that the pattern of bound silicate molecules resembled the lattice constant and symmetry of the S-layer template. The process of polycondensation of silicic acid seems to involve three stages through the adsorption on the S-layer: (1) homogeneous nucleation of silicate monomers and oligomers from the solution forming stable nuclei on the protein layer, (2) lateral growth of a silica network in all directions, and (3) fusion/aggregation of further molecules or polycondensates forming higher ordered structures including vertical growth of a silica network resulting in a corrugated silica film. Unfortunately, there are no direct indications where the first fixation of the silicate molecules (monomers, dimers, initial polymers) on the S-layer takes place (on cores, in proximity of the cores, in pores). In consideration to the fact that a silica network is formed, a first nucleation on the massive cores (major four-fold symmetry axis) can be assumed, which is followed by further fixation of silicate molecules on the arms (two-fold symmetry axis) and finally a coverage of the whole S-layer surface that comprises a closure of the pores, too. A model (Fig. 5) should illustrate the possibilities of a first silica fixation on the S-layer.
Fig. 5.
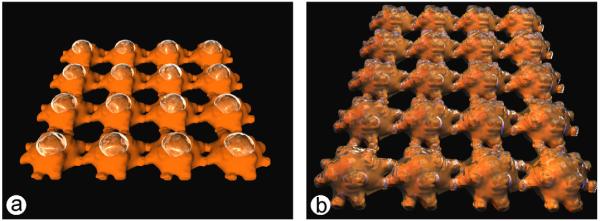
Models of the possible silification of the S-layer surface (a) homogeneous nucleation on the massive cores (major four-fold symmetry axis) and (b) coverage of the whole S-layer surface with pores initially left open.
4.2. Influence of phosphate molecules on silica adsorption
The present investigations demonstrate that the silification of the S-layer depends on different parameters, like the presence of phosphate in the silicate solution or the preceding modification of the protein layer. The results from QCM-D measurements indicate a higher silica adsorption in the presence of K2HPO4/KH2PO4 compared with non-phosphate containing solutions of the same pH (Fig. 3 and Table 4). The phosphate molecules seem to influence the interface of the organic S-layer and therefore the adsorption of silicate molecules. Possibly, phosphate molecules accelerate molecular deposition and homogeneous nucleation by increasing the extent of surface ionisation of the S-layer including the pore structure [41,42]. In addition, phosphate can act as regulator for the condensation of silicate molecules in solution and their adsorption on the protein layer. Thereby, the catalytic effect of the phosphate ions influences the surface ionisation of the next adsorbed silicate layers. This results in a decrease of the solubility of silica, which causes a higher polymerization rate including a higher deposition on the S-layer. A defined hydrogen-bonded network of silica adsorbed on the S-layer can be stabilized by balanced electrostatic interactions [41,43,44]. In nature, the silaffin1A component in diatoms has phosphorylated serine residues indicating that a high level of phosphorylation is essential for biological activity [45]. Non-phosphorylated silaffin1A precipitated significantly less silica, similar to the present silica adsorption on S-layers. In solutions without phosphate molecules the ionisation of the S-layer is smaller following a possible minimal catalytic effect and a reduced deposition of silica (Fig. 3 and Table 4). The outer surface of the protein layer does not have excess charges at physiological pH values (equimolar amount of carboxyl and amino groups) and is characterized by hydrophobic properties [46]. Consequently, the outer S-layer surface can function without phosphate molecules only as a weak catalyst for the adsorption of silicate, which could explain the lower deposition of silica.
4.3. Silica adsorption caused by S-layer functional groups
Generally, the extent of adsorption is affected by the number and type of functional groups on the protein molecules. Hydroxyl-, carboxyl-, amino-, and phosphoryl groups can be particularly effective in causing adsorption. In the case of the studied S-layer, there are large amounts of the hydrophilic amino acids threonine (13.9%), asparagine (8.2%), serine (5.6%), glutamic acid (5.2%), and aspartic acid (4.4%) and a smaller amount of basic amino acids, first of all lysine (6.1%). For the outer surface of the S-layer lattice 1.6 carboxyl groups per nm2 relevant for interactions with other protein molecules in solutions have been determined [47]. These carboxyl groups, in particular the amino acids glutamic acid (5.2%) and aspartic acid (4.4%), seem to be important for the fixation of silicate molecules. EDC treatment of the S-layer activates the carboxyl groups before the silica adsorption. The observed higher silica adsorption (Fig. 3 and Table 4) highlights the importance of carboxyl groups for the silification process. Attacking nucleophiles like oxygen atoms of phosphate as well as of silicate tetrahedra can interact with these carboxyl groups. Thus, a negative surface ionisation can be assumed, promoting the further adsorption of silica. A blocking of carboxyl groups through amino groups, like in the case of EDA/EDC modification of the S-layer, enhances the amount of amino groups (positive surface charges) on the protein surface at the expense of carboxyl groups (negative surface charges). Consequently, the lack of negatively charged groups yields a lower massadsorption of silica. Basic amino acids can also promote the hydrolysis and condensation of silicic acid [48]. However, in the case of this S-layer lattice negatively charged carboxyl groups seem to be more important compared to amino groups for the binding of silicate molecules and to design a complex network.
4.4. Molecular structure of adsorbed silica
FTIR-ATR measurements allow to identify the nature of the silica present on the protein surface and to determine its concentration. The broad absorption bands (Si—O—Si and Si—OH) are caused by several vibrations or interactions between atoms. In the case of the silicified S-layer, a polymerized (SiO2)x·yH2O gel is formed. The FTIR-ATR spectra of silica adsorbed on the S-layer are similar to natural biomineralized silica. Studies using FTIR spectroscopy have suggested that one of four oxygen atoms at the apices of the individual silica tetrahedra may be in the form of an OH group, indicating that biogenic silica is not as highly condensed as possibly assumed [49]. Other investigations showed that silicic acid molecules form preferentially a maximum number of siloxane (Si—O—Si) bonds and a minimum of uncondensed Si—OH groups during oligomerisation [41]. This results in the formation of three-dimensional infinite network structures with silanol group-saturated surfaces [50]. Quantitative results of the present FTIR-ATR measurements suggest that nearly all formed Si(OH)4-groups are polymerized to a (SiO2)x·yH2O gel with a Si—O—Si network. Therefore, the formed silica network adsorbed on the S-layer lattice seems to be linked to the bioorganic component, probably via Si—O−R+ bonds.
5. Conclusion
The S-layer protein SbpA of L. sphaericus CCM 2177 was used as organic template for the generation of nanostructured silica. It has been shown that this S-layer protein is capable to bind silicate and thus could be used for nanobiotechnological applications. TEM investigations showed the formation of a nanostructured silica network resembling the S-layer lattice. QCM-D measurements of silica adsorption demonstrated, that a certain amount of negatively charged sites, like phosphate molecules or activated carboxyl groups, significantly promote the deposition of silica on the S-layer. In contrast, the dominance of amino groups does not seem to be required for optimal catalysis of the silica formation on the protein layer. Investigations of the subsequent silification process using FTIR-ATR measurements showed the structure formation of an amorphous silica gel (SiO2)x·yH2O on the S-layer which was also supported by electron diffraction. A combined QCM-D and FTIR-ATR approach was successfully applied to calculate the silica surface concentration of the adsorbed silica layers.
Studying the formation of silicified S-layers may help to develop novel silicon-based materials. Advanced nanobiotechnological applications with such nanostructured framework materials will make use of their enhanced mechanical stability and optical properties.
Acknowledgments
This work was supported by AFOSR Biomimetics, Biomaterials and Biointerfacial Sciences Program (Agreement Award Nr. FA9550-06-1-0208) and by the Austrian Science Fund (FWF, project 20256-B11). We are grateful to Markus Gossmann for his assistance in the project modelling. We also thank Jacqueline Friedmann for her technical support and Norbert Hassler for his discussion of the FTIR-ATR data.
References
- [1].Shimizu K, Cha JH, Stucky GD, Morse DE. Proc. Natl. Acad. Sci. U.S.A. 1995;95:6234. doi: 10.1073/pnas.95.11.6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kröger N, Deutzmann R, Sumper M. Science. 1999;268:1129. doi: 10.1126/science.286.5442.1129. [DOI] [PubMed] [Google Scholar]
- [3].Sleytr UB, Messner P, Pum D, Sráa M. Angew. Chem. Int. Ed. 1999;38:1034. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1034::AID-ANIE1034>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- [4].Sleytr UB, Egelseer EM, Ilk N, Pum D, Schuster B. FEBS J. 2007;274:323. doi: 10.1111/j.1742-4658.2006.05606.x. [DOI] [PubMed] [Google Scholar]
- [5].Sleytr UB, Huber C, Ilk N, Pum D, Schuster B, Egelseer EM. FEMS Microbiol. Lett. 2007;267:131. doi: 10.1111/j.1574-6968.2006.00573.x. [DOI] [PubMed] [Google Scholar]
- [6].Pum D, Sleytr UB. Thin Solid Films. 1994;244:882. [Google Scholar]
- [7].Györvary ES, Stein O, Pum D, Sleytr UB. J. Microsc. 2003;212:300. doi: 10.1111/j.1365-2818.2003.01270.x. [DOI] [PubMed] [Google Scholar]
- [8].Györvary E, Schroedter A, Talapin DV, Weller H, Pum D, Sleytr UB. JNN. 2004;4:115. doi: 10.1166/jnn.2004.229. [DOI] [PubMed] [Google Scholar]
- [9].Schröder HC, Brandt D, Schloßmacher U, Wang X, Tahir MN, Tremel W, Belikov SI, Müller WEG. Naturwissenschaften. 2007;94:339. doi: 10.1007/s00114-006-0192-0. [DOI] [PubMed] [Google Scholar]
- [10].Ehrlich H, Krautter M, Hanke T, Simon P, Knieb C, Heinemann S, Worch H. J. Exp. Zool. Part B. 2007;308B:473. doi: 10.1002/jez.b.21174. [DOI] [PubMed] [Google Scholar]
- [11].Krasko A, Lorenz B, Batel R, Schröder HC, Müller IM, Müller WEG. Eur. J. Biochem. 2000;276:4878. doi: 10.1046/j.1432-1327.2000.01547.x. [DOI] [PubMed] [Google Scholar]
- [12].Zhou Y, Shimizu K, Cha JN, Stucky GD, Morse DE. Angew. Chem. 1999;111:826. doi: 10.1002/(SICI)1521-3773(19990315)38:6<779::AID-ANIE779>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- [13].Nakajima T, Volcani BE. Science. 1969;164:1400. doi: 10.1126/science.164.3886.1400. [DOI] [PubMed] [Google Scholar]
- [14].Hecky R, Mopper K, Kilham P, Degens T. Mar. Biol. 1973;19:323. [Google Scholar]
- [15].Swift D, Wheeler A. J. Phycol. 1992;28:202. [Google Scholar]
- [16].Kröger N, Deutzmann R, Bergsdorf C, Sumper M. PNAS. 2000;97:14133. doi: 10.1073/pnas.260496497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sleytr UB, Glauert AM. J. Bacteriol. 1976;126:869. doi: 10.1128/jb.126.2.869-882.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sleytr UB, Sara M, Kupcu Z, Messner P. Arch. Microbiol. 1986;146:19. doi: 10.1007/BF00690152. [DOI] [PubMed] [Google Scholar]
- [19].Williams A, Ibrahim IA. J. Am. Chem. Soc. 1981;103:7090. [Google Scholar]
- [20].Voinova MV, Rohdal M, Jonson M, Kasemo B. Phys. Scripta. 1999;59:391. [Google Scholar]
- [21].Sauerbrey G. Z. Phys. 1959;155:206. [Google Scholar]
- [22].Schuster B, Pum D, Sleytr UB. Biointerphases. 2008;3:FA3. doi: 10.1116/1.2889067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fringeli UP. In: Internal Reflection Spectroscopy. Mirabella FM, editor. Marcel Dekker; NY: 1992. p. 255. [Google Scholar]
- [24].Fringeli UP, Tranter GE, Holmes JC. In: Encyclopedia of Spectroscopy and Spectrometry. Lindon JC, editor. Academic Press; San Diego: 2000. p. 58. [Google Scholar]
- [25].Fringeli UP, Baurecht D, Bürgi T, Siam M, Reiter G, Schwarzott M, Brüesch P. In: Handbook of Thin Film Materials. Nalwa HS, editor. Vol. 2. Academic Press; San Diego: 2002. Chapter 4. [Google Scholar]
- [26].Baurecht D, Reiter G, Hassler N, Schwarzott M, Fringeli UP. CHIMIA. 2005;59:226. [Google Scholar]
- [27].Schuster B, Sleytr UB. In: Bioelectrochemistry Research Developments. Bernstein EM, editor. Nova Science Publishers Inc.; 2008. p. 105. [Google Scholar]
- [28].Glasmäster K, Larson C, Höök F, Kasemo B. J. Colloid Interface Sci. 2002;246:40. doi: 10.1006/jcis.2001.8060. [DOI] [PubMed] [Google Scholar]
- [29].Kepplinger C. Ph.D. Thesis. University of Natural Resources and Applied Life Sciences, Vienna; 2007. [Google Scholar]
- [30].Delcea M, Krastev R, Gutberlet T, Pum D, Sleytr UB, Toca-Herrera JL. Soft Matter. 2008;4:1414. doi: 10.1039/b719408k. [DOI] [PubMed] [Google Scholar]
- [31]. http://www.q-sense.com.
- [32].Heise G. IR-Sepktroskopie–Eine Einführung. VCH Weinheim; 1996. [Google Scholar]
- [33].Dong A, Huang P, Caughy WS. Biochemistry. 1990;29:3303. doi: 10.1021/bi00465a022. [DOI] [PubMed] [Google Scholar]
- [34].Surewisz WK, Mantsch HH, Chapman D. Biochemistry. 1993;32:389. doi: 10.1021/bi00053a001. [DOI] [PubMed] [Google Scholar]
- [35].Arrondo JL, Muga A, Castresana J, Goni FM. Prog. Biophys. Mol. Biol. 1993;59:23. doi: 10.1016/0079-6107(93)90006-6. [DOI] [PubMed] [Google Scholar]
- [36].Fahmy K, Merroun M, Pollmann K, Raff J, Savchuk O, Hennig C, Selenska-Pobell S. Biophys. J. 2006;91:996. doi: 10.1529/biophysj.105.079137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gendron-Badou A, Coradin T, Maquet J, Fröhlich F, Livage J. J. Non-Cryst. Solids. 2002;316:331. [Google Scholar]
- [38].Gelabert A, Pokrovsky OS, Schott J, Boudou A, Feurtet-Mazel A, Mielczarski J, Mielczarski E, Mesmer-Dudons N, Spalla O. Geochim. Cosmochim. Acta. 2004;68:4039. [Google Scholar]
- [40].Wenzl P, Fringeli M, Groette J, Fringeli UP. Langmuir. 1994;10:4253. [Google Scholar]
- [41].Iler RK. The Chemistry of Silica. Wiley; NY: 1979. [Google Scholar]
- [42].Weres O, Yee A, Tsao L. J. Colloid Interfaces Sci. 1981;84:379. [Google Scholar]
- [43].Mitzutani T, Nagase H, Fujiwara N, Ogoshi H. Bull. Chem. Soc. Jpn. 1998;71:2017. [Google Scholar]
- [44].Lutz K, Gröger C, Sumper M, Brunner E. Phys. Chem. Chem. Phys. 2005;7:2812. doi: 10.1039/b505945c. [DOI] [PubMed] [Google Scholar]
- [45].Kröger N, Lorenz S, Brunner E, Sumper M. Science. 2002;298:584. doi: 10.1126/science.1076221. [DOI] [PubMed] [Google Scholar]
- [46].Sara M, Egelseer EM, Huber C, Ilk N, Pleschberger M, Pum D, Sleytr UB. In: Microbial Bionanotechnology: Biological Self-Assembly Systems and Biopolymer-Based Nanostructures. Rehm B, editor. Horizon Scientific Press; Hethersett, Norwich, UK: 2006. p. 307. [Google Scholar]
- [47].Weigert S, Sara M. J. Membr. Sci. 1995;106:147. [Google Scholar]
- [48].Perry CC, Keeling-Tucker T. J. Biol. Inorg. Chem. 2000;5:537. doi: 10.1007/s007750000130. [DOI] [PubMed] [Google Scholar]
- [49].Frohlich F. Terra Nova. 1989;1:267. [Google Scholar]
- [50].Wirzing W, Fresenius Z. Anal. Chem. 1980;302:97. [Google Scholar]