

Abstract
Despite investigations into mechanisms linking type 2 diabetes and cancer, there is a gap in knowledge about pharmacotherapy for diabetes in cancer patients. Epidemiological studies have shown that diabetic cancer patients on different antidiabetic treatments have different survival. The clinically relevant question is whether some antidiabetic pharmacotherapeutic agents promote cancer whereas others inhibit cancer progression. We investigated the hypothesis that various antidiabetic drugs had differential direct impact on cancer cells using four human cell lines (pancreatic cancer: MiaPaCa2, Panc-1; breast cancer: MCF7, HER18). We found that insulin and glucose promoted cancer cell proliferation and contributed to chemoresistance. Metformin and rosiglitazone suppressed cancer cell growth and induced apoptosis. Both drugs affected signalling in the protein kinases B (AKT)/mammalian target of rapamycin pathway; metformin activated adenosine monophosphate (AMP)-activated protein kinase whereas rosiglitazone increased chromosome ten level. Although high insulin and glucose concentrations promoted chemoresistance, the combination of metformin or rosiglitazone with gemcitabine or doxorubicin, resulted in an additional decrease in live cancer cells and increase in apoptosis. In contrast, exenatide did not have direct effect on cancer cells. In conclusion, different types of antidiabetic pharmacotherapy had a differential direct impact on cancer cells. This study provides experimental evidence to support further investigation of metformin and rosiglitazone as first-line therapies for type 2 diabetes in cancer patients.
Keywords: metformin, rosiglitazone, pancreatic cancer, breast cancer, type 2 diabetes mellitus, insulin, gemcitabine, doxorubicin
Introduction
Extensive epidemiological data suggest important roles of type 2 diabetes mellitus (DM2) in carcinogenesis [1–4] and prognosis [5]. Pancreatic cancer is well documented to be associated with DM2 [6–10], and up to 80% of pancreatic cancer patients have overt DM2 or impaired glucose tolerance [11]. Breast cancer is another prevalent cancer among many other cancers that are associated with DM2 [12], and up to 15% of breast cancer patients have DM2 (i.e. about 375,000 in the United States).
Overt DM2 is characterized by hyperglycaemia, hyperinsulinemia and high insulin-like growth factor-1 (IGF-1), and all three characteristics may promote cancer. As demonstrated by glycohaemoglobin levels in cancer patients, elevated average blood glucose may be associated with a higher proportion of patients with active cancer than patients in remission [13], suggesting that hyperglycaemia may promote cancer progression. Elevations in levels of glucose and free fatty acids in DM2 were also correlated with enhanced tumour growth both in vivo and in vitro[14]. Insulin was reported to stimulate proliferation and glucose utilization in pancreatic cancer cells; insulin augmented DNA synthesis mainly by mitogen-activated protein kinase activation and glucose uptake mainly by phosphoinositide 3-kinase activation and enhancement of glucose transporter-1 expression [15]. Furthermore, IGF-1 was found to prevent apoptosis of cancer cells induced by chemotherapeutic agents leading to chemoresistance in vitro[16]. Specific types of pharmacotherapy for DM2 may have different impacts on these mechanisms, thereby having different end results in terms of cancer progression.
Current medications for DM2 include thiazolidinediones (e.g. rosiglitazone), biguanides (e.g. metformin), sulfonylureas, meglitinides, α-glucosidase inhibitors, amylin analogues, dipeptidyl peptidase-4 inhibitors, incretin mimetics (e.g. exenatide) and insulin preparations. Although all these different classes of medications can lower blood glucose, the mechanisms of actions of these agents are different and they have different impacts on the circulating insulin levels. In addition, some antidiabetic drugs may have direct anti-tumour effects. For example, thiazolidinediones (agonists of peroxisome proliferator-activated receptor-γ) have been shown to suppress various types of cancer cells in cell culture and in animal models [17–19], and so has metformin [20–22], a biguanide that activates AMP-activated protein kinase (AMPK) and decreases signalling through mammalian target of rapamycin (mTOR) [23]. In a population-based cohort study [24], diabetic patients treated with sulfonylureas and/or insulin were more likely to die from cancer than patients treated with metformin. This study highlighted the differential impact of antidiabetic medications on cancer in diabetic cancer patients.
In this report, we investigated the differential impact of some antidiabetic medications on cancer cells. We examined the impact of insulin and glucose on cancer cell growth and chemoresistance, and the direct effects of some antidiabetic drugs on cell proliferation, apoptosis and chemosensitivity of human breast and pancreatic cancer cells. Our results provide key experimental evidence to guide future clinical investigation to establish the optimal pharmacotherapy for DM2 in cancer patients.
Materials and methods
Chemicals and reagents
All cell culture reagents and media were obtained from Invitrogen. Human insulin, D-Glucose and methylthiazolydiphenyl-tetrazolium bromide (MTT) were purchased from Sigma (St. Louis, MO, USA). Metformin was obtained from Alexis Biochemicals (San Diego, CA, USA) and dissolved in water prior to dilution in culture media. Rosiglitazone was from Cayman Chemicals (Ann Arbor, MI, USA) and dissolved in dimethylsulfoxide (DMSO) prior to dilution in culture media. Exenatide and gemcitabine injectable formulations were obtained from Lilly (Indianapolis, IN, USA). Anti-poly (ADP ribose) polymerase (PARP) was from BD Pharmingen (San Diego, CA, USA), and anti-B-Cell Lymphoma/Leukemia-2 (BCL-2) was from BD Transduction Laboratories (San Diego, CA, USA). Anti-phospho-AMPK was from Cell Signaling (Carlsbad, CA, USA). Anti-mTOR was from Strategic Diagnostics, Inc. (Newark, DW, USA). Anti-phospho-mTOR(Ser2448), anti-S6 ribosomal protein and anti-phospho-pS6 ribosomal protein (Ser 235/236), anti-Eukaryotic Translation Initiation, Factor 4E-Binding Protein 1 (4EBP1) and anti-p-4EBP1(Ser65) antibodies were from Cell Signaling. Anti-phosphatase and Tensin homologue deleted on chromosome ten (PTEN) was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA), and anti-α-tubulin was purchased from Sigma Aldrich (St. Louis, MO, USA). Horseradish peroxidase-conjugated anti-rabbit IgG and antimouse IgG were from Pierce Chemical (Rockford, IL, USA). Annexin V apoptosis detection kit and enhanced chemiluminescence were from BD Bioscience (San Jose, CA, USA).
Cell lines and culture conditions
MiaPaCa2, Panc-1 and MCF7 cells were originally obtained from ATCC (Manassas, VA, USA). HER18 cells are MCF7 cells stably overexpressing HER2. Cells were cultured in DMEM/F12 and supplemented with 10% foetal bovine serum (FBS) and 100 unit/ml penicillin G and 100 μg/ml streptomycin at 37°C and 5% CO2 in 10-cm-diameter dishes. Cells were subcultured by 0.25% Trypsin-ethylenediaminetetraacetic acid (EDTA) when they reached >80% confluence.
Cell proliferation assay
The cell proliferation was assessed by MTT assay. In brief, cells were cultured in a 96-well plate (Corning, Inc., Corning, NY, USA) at a density of 5000–10,000 cells per well in DMEM/F12 with 10% FBS. After 24 hrs incubation, various concentrations of insulin and glucose were added to DMEM without glucose (GIBCO, Carlsbad, CA, USA) with 0.5% FBS to replace the original DMEM/F12 (200 μl/well). After 72 hrs, 20 μl of MTT solution 5 mg/ml was added to each well. Plates were incubated in the dark for 2 hrs and then the formazan dye was dissolved in 100 μl DMSO after removing culture medium and MTT solution. The absorbance was measured at 570 nm using a microtitre plate reader (MRX Revelation, DYNEX Technologies, Chantilly, VA, USA).
Protein extraction and Western blot analysis
Cells were washed twice with PBS and lysed in 300 μl lysis buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P40, 0.5% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1 mM NaF, 1 mM sodium orthovanadate, 1 mg each of aprotinin, leupeptin and pepstatin per millilitre). Analyses were performed on 10% polyacrylamide gels with 5% polyacrylamide stacking gels. After semi-dry electrotransfer (Amersham Pharmacia Biotech, Piscataway, NJ, USA) of protein from SDS-PAGE to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA), the membranes were blocked with the buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween- 20, 5% Blotto (Bio-Rad, Hercules, CA, USA) at 4°C overnight and probed with antibodies of interest. Horseradish peroxidase-conjugated anti-rabbit IgG and antimouse IgG were used as secondary antibodies. Immunodetection was performed with the enhanced chemiluminescence reagent, and the images were recorded on X-ray films.
Flow cytometry of annexin V-fluorescein-isothiocyanate (FITC) and propidium iodine stained cells
Approximately 1 × 106 MiaPaCa2 cells were plated in each well in 6-well plate and incubated for 16 hrs. Then the cells were treated with either 20 mM metformin, 100 μM rosiglitazone, 0.5 μM gemcitabine or combinations for 48 hrs with DMEM/F12 and 10% FBS. The floating cells in the culture media were collected by centrifugation. Attached cells were rinsed with Ca2+ and Mg2+-free PBS and harvested after incubation with 0.25% Trypsin/ 0.21 mM EDTA in PBS for 10 min. Floating and attached cells were pooled and rinsed twice with PBS. Cells were resuspended in 1× binding buffer (Axxora Platform, San Diego, CA, USA) and incubated with FITC-conjugated annexin V for 15 min. at room temperature in the dark. Cells were washed twice with 1× binding buffer. Propidium iodine was added just before analysis of the samples. The samples were immediately analysed on fluorescence activated cell sorting (FACS) calibur flow cytometer (Becton Dickinson Immunocytometry System, Burlington, NC, USA) using the CellQuest Pro Software and WINMDI 2.9 software.
Statistical analysis
Significance (P < 0.05) was assessed by either a two-tailed Student’s t-test or a one-way ANOVA with post hoc intergroup comparisons using SPSS for Windows, version 12.0 (SPSS, Inc., Chicago, IL, USA). All data presented are the average of at least three independent experiments.
Results
Glucose and insulin promoted cell proliferation of cancer cells
DM2 is characterized by hyperinsulinemia and hyperglycaemia. MiaPaCa2 cells are known to express insulin and insulin-like growth factor receptors [25]. We showed that the proliferation of MiaPaCa2 cells was accelerated by high glucose and high insulin (Fig. 1). At a high glucose concentration of 4000 mg/l, MiaPaCa2 cells increased proliferation as the insulin concentration increased (Fig. 1A). At a high insulin concentration of 5 μg/ml, MiaPaCa2 cells increased proliferation as the glucose concentration increased, but at a normal insulin concentration of 0.0005 μg/ml, this effect of glucose was not so obvious (Fig. 1B). The growth promoting effect of glucose and insulin was confirmed using a different method, counting Trypan blue dye excluding cells under the microscope (Fig. 1C). These results indicate that high glucose and high insulin can accelerate the cell growth of cancer cells. Starting with the same number of cells in culture media containing different concentrations of glucose and insulin, the numbers of live cells after 3 days as measured by the MTT assay were plotted against insulin and glucose as 3D surfaces for two breast cancer cell lines (MCF7 and HER18) and two pancreatic cell lines (Panc-1 and MiaPaCa2) (Fig. 1D). These data show that insulin in general promotes cancer cell growth when the glucose concentration is ≥1000 mg/l, and that, with the exception of HER18, glucose promotes cancer cell growth when the insulin concentrations are low.
Fig 1.
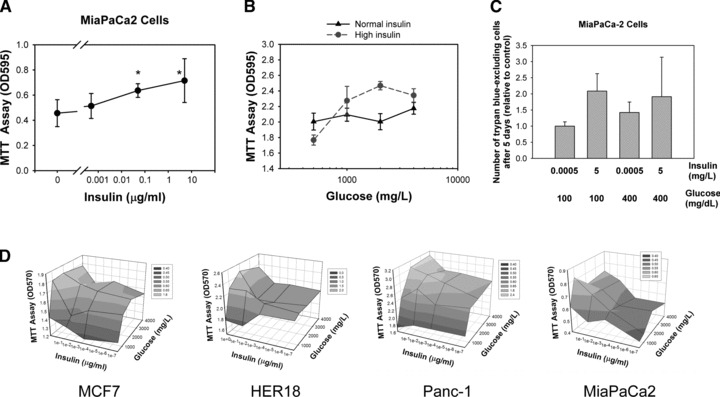
Glucose and insulin promote cancer cell proliferation. (A) Insulin stimulates cancer cell proliferation. MiaPaCa2 cells were cultured for 3 days in culture media containing different concentrations of insulin. The glucose concentration was fixed at 4000 mg/l. OD values in the MTT assay are plotted against the insulin concentration. Error bars represent 95% confidence intervals. The symbol ‘*’ denotes statistically significant difference (P < 0.05) in one-way ANOVA, post hoc intergroup comparison (Tukey’s test) with the control group in which no insulin was added. (B) The promoting effect of glucose on cancer cell proliferation is dependent on insulin. The impact of glucose on the OD of the MTT assay of MiaPaCa2 cells at normal (solid line and triangle) and high (dashed line and solid circle) insulin concentrations are plotted. (C) The ratio of the number of live cells as detected and counted by Trypan blue dye exclusion to that of control cells (first bar) are shown as a bar graph with insulin and glucose concentrations as labelled. Error bars – 95% confidence intervals. (D) OD values in the MTT assay of indicated cell lines are plotted along the z-axis versus the insulin (x-axis) and glucose (y-axis) concentrations. Grey-scale shading represents OD value according to the key.
Rosiglitazone and metformin suppressed the growth of cancer cells
To determine whether controlling the diabetic condition might affect the cell growth of pancreatic cancer cells, we used antidiabetic drugs to investigate the cell growth of pancreatic cancer. Therefore, we examined the impact of rosiglitazone, metformin and exenatide on the proliferation of MiaPaCa2 cells in culture media containing insulin and glucose concentrations mimicking four different stages in the natural history of DM2, i.e. normal state, pre-diabetes, overt diabetes and late diabetes (Fig. 2A) [26]. We found that metformin and rosiglitazone dose-dependently inhibited MiaPaCa2 cell proliferation in all four combinations of glucose and insulin tested (Fig. 2B and C). Interestingly, exenatide, an analogue of glucagon-like peptide-1, had no direct inhibitory effect on MiaPaCa2 cells at concentrations relevant to the treatment of DM2 (Fig. 2D). To further investigate the effects of these medications in other cancer cell types, breast cancer cell lines HER18 (Fig. 2E) and MCF7 (Fig. 2F) were treated with metformin (20 mM) and rosiglitazone (100 μM) in various combinations of glucose and insulin concentrations for 3 days followed by the MTT assay. The 3D plots showed that metformin and rosiglitazone decreased the number of live breast cancer cells in all the combinations of insulin and glucose concentrations tested. Metformin and rosiglitazone also decreased the number of live HER18 and MCF7 cells (MTT assay) in time- and dose-dependent manners whereas exenatide had no effect on these cells (data not shown). Together, these data indicated that metformin and rosiglitazone could directly inhibit the growth activity of human cancer cells whereas exenatide had no such an activity.
Fig 2.
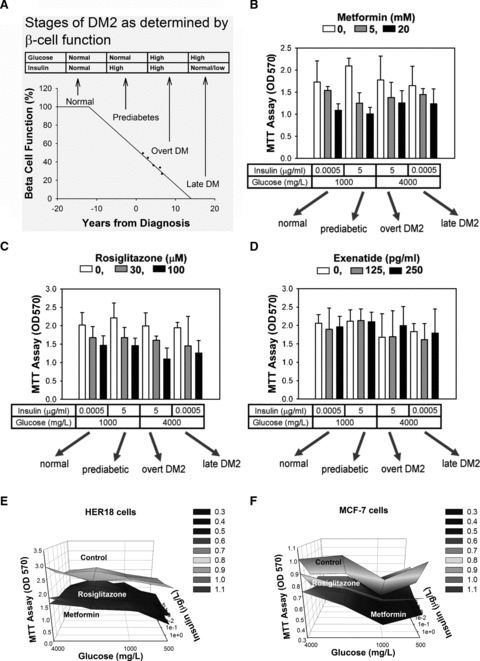
Rosiglitazone and metformin suppress proliferation of cancer cells. (A) The stages in the natural history of DM2 are determined by the loss of β cell function over time. The characteristics of glucose and insulin levels are listed in the table above the diagram, which has been redrawn based on data from the UK Prospective Diabetes Study 16 [26]. MiaPaCa2 cells were cultured for 3 days in media containing different concentrations of glucose and insulin, and were treated with different concentrations of metformin (B), rosiglitazone (C) and exenatide (D). The OD values in the MTT assay are plotted in the bar charts. The drug concentrations are as labelled by the grey-scale key. The glucose and insulin concentrations and the stages of DM2 that they represent are as labelled beneath the bar charts. Error bars represent 95% confidence intervals. The 3D surface demonstrated that the inhibitory effect of rosiglitazone (100 μM) and metformin (20 mM) in HER18 (E) and MCF7 (F) cells was present at all the insulin and glucose concentrations tested. Beginning with the same number of cells, the number of live cells were examined by the MTT assay after culturing for 3 days in DMEM without glucose + 1% FBS with glucose and insulin added at various concentrations.
Metformin and rosiglitazone induced apoptosis in cancer cells
To further investigate the mechanism behind the growth inhibitory effect of metformin and rosiglitazone, we determined if apoptosis is involved. Thus, we examined the ability of metformin and rosiglitazone to induce apoptosis of MiaPaCa2 and HER18 cells by FACS analysis. Cells treated with 20 mM metformin or 100 μM rosiglitazone were stained with FITC-annexin V and propidium iodide. Flow cytometry demonstrated that metformin- and rosiglitazone- treated cells significantly showed increases in cells with annexin V+ signals over control cells (Fig. 3A), indicating that metformin and rosiglitazone sensitized cells to apoptosis. The numbers of live (Trypan blue dye excluding) cells after metformin and rosiglitazone treatments were also accounted after Trypan blue staining (Fig. 3B). We found that both metformin and rosiglitazone significantly decreased the number of live MiaPaCa2 cells (one way-ANOVA, Tukey’s test: rosiglitazone versus control, P= 0.011; metformin versus control, P= 0.001). To further study their impact on apoptosis, we also examine the protein levels of BCL-2, an anti-apoptotic protein, and the specific cleavage of PARP, another apoptosis marker. We found that both metformin and rosiglitazone induced specific cleavage of PARP into an 85-kD fragment and decreased the protein level of BCL-2 in MiaPaCa2 cells (Fig. 3C). We also examined whether protein kinase B/AKT, a crucial regulator of oncogenic signals involved in cell survival, is regulated by the treatment of metformin and rosiglitazone. We found that rosiglitazone decreased the level of phospho-AKT, in parallel with increased PTEN, a known negative regulator of AKT activity. Although metformin had no direct impact on AKT activity, it induced the phosphorylation of AMPK (activated) (Fig. 3C), a positive regulator of p53, suggesting a positive impact on p53-mediated apoptosis. To investigate whether rosiglitazone and metformin affected mTOR signalling pathway further downstream, we evaluated the signalling through this pathway by assessing phosphorylation of mTOR kinase and its downstream targets S6 ribosomal protein and 4EBP1 in the rosiglitazone- and metformin-treated MiaPaCa2 cells. We found that both rosiglitazone and metformin decreased the level of phospho-mTOR kinase (Ser2448), but not total mTOR protein level (Fig. 3C). The expression of phospho-S6 (Ser235/236) and phospho-4EBP1 (Ser65) were also decreased by treatment of rosiglitazone and metformin in MiaPaCa2 cells. A positive value indicated an increase in level and a negative value indicated a decrease in level relative to the control cell sample. Thus, metformin and rosiglitazone promoted apoptosis in cancer cells and potentiated the apoptotic effect via AMPK and PTEN respectively, both regulating the AKT/mTOR signalling pathway.
Fig 3.
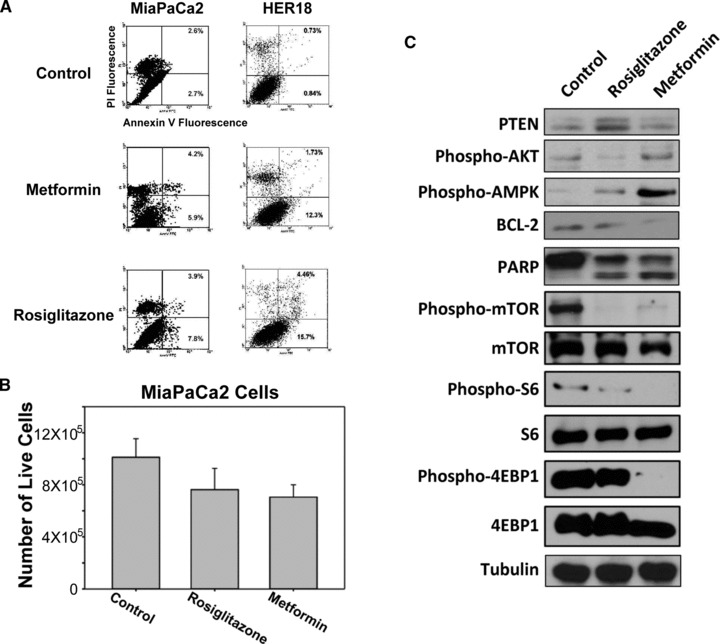
Metformin and rosiglitazone induce apoptosis in cancer cells. (A) MiaPaCa2 and HER18 cells were treated with or without the indicated drugs followed by FACS analysis. The vertical axis represents fluorescence due to PI, and the horizontal axis represents fluorescence due to FITC-annexin V binding. The two right quadrants indicate the percentage of apoptotic cells. (B) MiaPaCa2 cells were treated with metformin and rosiglitazone for 24 hrs. The numbers of live (dye-excluding) cells were counted using a haemocytometer after Trypan blue staining. The error bars represent 95% confidence intervals. (C) Immunoblots of MiaPaCa2 cell lysates after control, metformin and rosiglitazone treatments were shown. The antigens detected were labelled as indicated. Tubulin served as gel loading control.
High Insulin and high glucose induced chemoresistance of cancer cells
Doxorubicin, a topoisomerase II inhibitor, is a first-line chemotherapy drug for breast cancer, and gemcitabine, an analogue of pyrimidine, is a first-line chemotherapy drug for pancreatic cancer. Because DM2 is associated with poor outcome of cancer patients and our results showed that simulated hyperglycaemia and hyperinsulinemia promoted cancer growth, we next examined whether high insulin and glucose levels induced chemoresistance to doxorubicin in HER18 and MCF7 cells and chemoresistance to gemcitabine in MiaPaCa2 cells. The number of live cells in tissue culture was measured by the MTT assay as described above. In the presence of 1 μM of doxorubicin and 5 μg/ml of insulin, HER18 cells proliferated more quickly over 72 hrs in 2000 mg/l glucose. Likewise, MCF7 proliferated more in the presence of higher glucose concentrations, 2000 mg/l and 4000 mg/l, compared with lower glucose concentrations, 500 mg/l and 1000 mg/l (Fig. 4A), suggesting that high glucose compromised the growth inhibitory effect of doxorubicin. Further, in the presence of 1 μM doxorubicin and high glucose concentration (4000 mg/l), an increase in insulin concentration was associated with increased HER18 and MCF7 cell numbers (Fig. 4B), indicating that high insulin attenuated the inhibitory effect of gemcitabine. Similarly, in the presence of 0.1 μM gemcitabine and 5 μg/ml insulin, MiaPaCa2 cells proliferate more quickly in the presence of higher glucose concentrations, 2000 mg/l and 4000 mg/l, compared with lower glucose concentrations, 500 mg/l and 1000 mg/l (Fig. 4C), suggesting that high glucose compromised the growth inhibitory effect of gemcitabine. Likewise, in the presence of 0.1 μM gemcitabine and high glucose concentration (4000 mg/l), increase in insulin concentration was associated with increased MiaPaCa2 cell proliferation (Fig. 4D), indicating that high insulin attenuated the inhibitory effect of gemcitabine.
Fig 4.
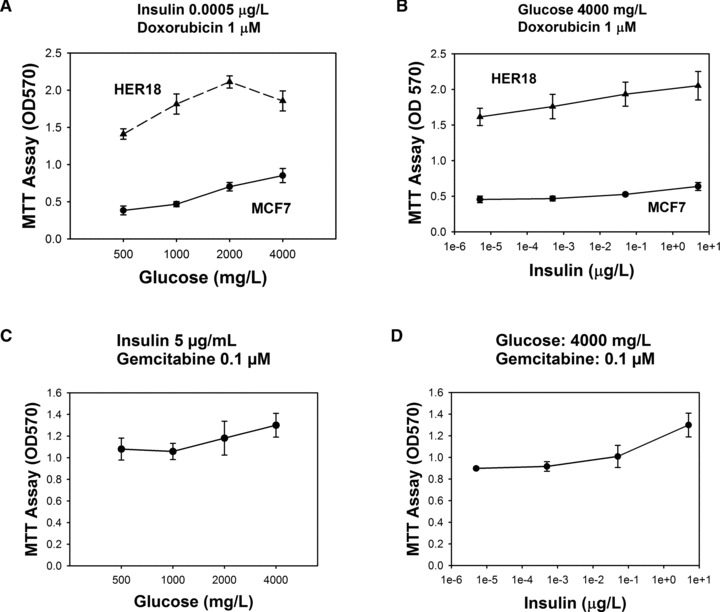
Insulin and glucose induce chemoresistance. Two indicated breast cancer cell lines were treated with doxorubicin. The OD values in the MTT assay are plotted against glucose (A) or insulin (B). The numbers of live MiaPaCa2 cells treated with gemcitabine as represented by the OD values in the MTT assay are plotted against glucose (C) or insulin (D). Error bars represent 95% confidence intervals.
The combination of gemcitabine with either rosiglitazone or metformin resulted in increased inhibition of pancreatic cancer cells
Because our results above showed that metformin and rosiglitazone activated signalling molecules that are already known to negatively regulate the AKT/mTOR pathway, we suggested that the combination of gemcitabine with either rosiglitazone or metformin could further increase inhibition of pancreatic cancer cells in four combinations of insulin and glucose concentrations mimicking different stages of DM2. As a negative control, we used the combination of exenatide with gemcitabine, which did not result in additional inhibition of MiaPaCa2 cells (Fig. 5A). Indeed, we showed that the combination of rosiglitazone and gemcitabine inhibited proliferation of MiaPaCa2 cells more than gemcitabine alone in four combinations of glucose and insulin (Fig. 5B). Similar results were obtained with metformin in combination with gemcitabine (Fig. 5C). Multiple linear regression analysis of the above MTT data using SPSS for Windows, version 12.0 (SPSS, Inc.), showed that glucose, insulin, gemcitabine, metformin and rosiglitazone were all statistically significant (P < 0.001) factors that determine the optical density (OD) values (i.e. the number of live cancer cells in the culture) whereas exenatide was not (P= 0.524) (i.e. changes in exenatide did not significantly affect the OD values). The above data were also used to evaluate antagonism, additivity and synergism by calculating the combination index (CalcuSyn, BioSoft, Cambridge, UK) based on the method of Chou and Talalay [27]. A combination index >1 indicates antagonism, = 1 indicates additivity and <1 indicates synergism. The data were presented as log (combination index) in Figure 5(D), and synergism (i.e. log [combination index] < 0] was observed for both combinations of gemcitabine with rosiglitazone and metformin in the four different combinations of glucose and insulin concentrations as the fractional effect was greater than approximately 0.5 (Fig. 5D).
Fig 5.
The combination of gemcitabine with rosiglitazone increases inhibition of pancreatic cancer cells. The gemcitabine dose–response curves of MiaPaCa2 cells in the presence of different concentrations of exenatide (A), metformin (B) and rosiglitazone (C) are shown as labelled. Data obtained with different combinations of insulin and glucose representing normal, pre-diabetes, overt diabetes and late diabetes are shown from left to right. (D) Evaluation of the combination of gemcitabine with metformin or rosiglitazone on pancreatic cancer cell proliferation. The combination index was evaluated using CalcuSyn for metformin + gemcitabine (triangles) and rosiglitazone + gemcitabine (circles). The log (combination index) – fractional effect plots of the data obtained with different combinations of insulin and glucose representing normal, pre-diabetes, overt diabetes and late diabetes are shown from left to right. The linear regression lines of the data are also shown.
To further examine the inhibitory effect of the combination of gemcitabine with either rosiglitazone or metformin on pancreatic cancer cells, we then analysed the amount of apoptotic cells under these combinations. MiaPaCa2 cells were treated for 48 hrs with gemcitabine 0.1 μM plus rosiglitazone 100 μM or metformin 20 mM, followed by FITC-annexin V staining for flow cytometry analysis. The combination of rosiglitazone and gemcitabine had the highest percentage of annexin V+ cells (14.9%+ 45.8%= 60.7%) in flow cytometry analysis compared with the combination of metformin and gemcitabine (16.9%+ 31.7%= 48.6%) or gemcitabine alone (14.2%+ 15.5%= 39.7%) (Fig. 6A). The data from multiple experiments were plotted as a bar graph with error bars representing 95% confidence intervals (Fig. 6B). Immunoblotting analysis also confirmed that the addition of either rosiglitazone or metformin further increased apoptosis as demonstrated by increased specific cleavage of PARP compared with gemcitabine alone (Fig. 6C). We also demonstrated the effects of rosiglitazone on increasing PTEN and reduced levels of phospho-AKT. Again, metformin increased the levels of phospho-AMPK. It is important to point out that gemcitabine alone did not alter the levels of PTEN, phospho-AKT or phospho-AMPK, suggesting the unique contribution from metformin or rosiglitazone when used in combination treatment. Together, these results indicate that high glucose and high insulin can reduce the inhibitory effect of gemcitabine, and that antidiabetic drugs including metformin and rosiglitazone can further increase the inhibitory effect as well as the apoptotic effect of gemcitabine in human pancreatic cancer.
Fig 6.
The combination of gemcitabine with rosiglitazone or metformin increases apoptosis of pancreatic cancer cells. (A) Representative scattergrams of FACS analysis of apoptotic cell death after indicated treatments are shown. The vertical axis represents fluorescence due to PI, and the horizontal axis represents fluorescence due to FITC-annexin V binding. The percentage of cells that are annexin V+ and PI– and the percentage of cells that are annexin V+ and PI+ are shown in the right panels. (B) The average percentages of annexin V+ cells after indicated treatments are plotted in the bar chart. The error bars represent 95% confidence intervals. (C) Immunoblots of cell lysates after indicated treatments are shown. The antigens detected are labelled to the left of each blot. Tubulin serves as gel loading control. (D) The diagram depicts the insulin signalling pathway connecting through AKT/mTOR signalling to cell proliferation. The possible mechanisms of action of rosiglitazone and metformin are indicated.
Discussion
Three major mechanisms have been postulated to explain the possible promoting impact of DM2 on cancer: hyperglycaemia, activation of the insulin signalling pathway and activation of the IGF signalling pathway. This report focused on the impacts of diabetic conditions and antidiabetic pharmacotherapy on cancer cells. First, we demonstrated that insulin promoted proliferation of cancer cells. Second, the impact of hyperglycaemia has not previously been well documented; in this report, we have defined the 3D relationship between cell proliferation, insulin, and glucose (Fig. 1D), demonstrating that high glucose does promote cancer cell growth but its effect is dependent on increased insulin levels. These results echo the Warburg phenomenon [28] in which activation of tyrosine kinase receptor signalling (for instance by insulin) can lead to increased glycolysis and high glucose concentrations can further facilitate glucose usage as fuel. On the other hand, evidence has been accumulating that some antidiabetic treatments inhibit growth of cancer cells. Biguanides and thiazolidinediones are widely used in diabetic patients, and both classes of drugs have direct inhibitory effects on cancer cells observed in vitro and in animal models [22, 29–37]. Our results showed that metformin and rosiglitazone inhibited cancer cells in combinations of glucose and insulin concentrations which attempted to mimic stages in the natural history of DM2 (normal, prediabetic, overt diabetic and late diabetic states) (Fig. 2). This is the first time that metformin and rosiglitazone have been evaluated for their cancer inhibiting effects in the context of DM2, and indeed we found that metformin activated AMPK, rosiglitazone up-regulated PTEN, and both promoted apoptosis in cancer cells (Fig. 3). These results may partially explain why DM2 can promote cancer progression, but more importantly, these results highlight the differential direct impact on cancer cell growth of different antidiabetic treatments: promotion of cancer cell growth by insulin preparations or insulin secretagogues versus inhibition of cancer cell growth by metformin and rosiglitazone.
In addition to direct impact on cancer cell growth in the absence of chemotherapy, another important issue in the context of antidiabetic pharmacotherapy pertains to chemoresistance. High insulin and glucose concentrations led to doxorubicin chemoresistance in breast cancer cells and chemoresistance to gemcitabine in pancreatic cancer cells. This resulted in higher numbers of live cancer cells than in control culture medium (Fig. 4). These results suggest that high circulating insulin levels and hyperglycaemia during chemotherapy may compromise the efficacy of chemotherapy. Whether insulin and glucose levels during chemotherapy are significant predictors of clinical response to chemotherapy in cancer patients with DM2 remains to be investigated clinically. Importantly, rosiglitazone or metformin increased the inhibitory effect of gemcitabine (Fig. 5B and C). In contrast, exenatide did not have such an effect (Fig. 5A). These results again highlight the differential direct impact on cancer cell chemoresistance of different antidiabetic treatments: promotion of chemoresistance by insulin preparations or insulin secretagogues versus additional inhibition of cancer cell growth by metformin and rosiglitazone versus a neutral effect of exenatide. Our future work will use a genetic mouse model of DM2 and pancreatic cancer to investigate whether DM2 influences pancreatic carcinogenesis and whether specific diabetic treatments including rosiglitazone and metformin can prevent pancreatic cancer or inhibit pancreatic cancer progression. Such animal model experiments will provide convincing evidence to support that specific diabetic treatments have beneficial impact on pancreatic cancer in vivo.
We investigated the mechanism by which rosiglitazone and metformin increased the inhibitory effect of gemcitabine by measuring apoptosis and signalling in key components of the AKT/mTOR pathway. Both metformin and rosiglitazone can further increase the percentage of apoptotic cells in gemcitabine-treated cancer cells (Fig. 6A and B). Increased apoptosis as indicated by specific cleavage of PARP was associated with inhibition of AKT/mTOR signalling by the effect of rosiglitazone on PTEN or the effect of metformin on AMPK (Fig. 6C). Here, we provide a working model for the action of these drugs on signal mediators in cancer cells in Figure 6(D).
Antidiabetic pharmacotherapy involves chronic administration of these drugs. A very small impact on cancer cell growth measured in the short duration of in vitro experiments may translate into a clinically meaningful impact in the long term. For example, a 2.5% decrease in the number of cancer cells by a drug compared with control observed in 3 days may become a 53.2% decrease in tumour burden in 3 months (i.e. 0.97530= 0.468). When comparing the differential impact of chronic antidiabetic medications on cancer, the opposite effects of insulin and metformin would make their difference stand out. We have also recently reported that metformin use is associated with reduced risk, and insulin or insulin secretagogue usage is associated with increased risk of pancreatic cancer in diabetic patients [22]. A population-based cohort study revealed an increased risk of cancer-related death in DM2 patients treated with sulfonylureas and insulin preparations than in those treated with metformin [24], and based on our results, this difference is very likely due to both detrimental effects of sulfonylureas/insulin preparations and the beneficial effect of metformin on cancer cell growth and chemoresistance. Recently, our colleagues have reported that diabetic patients with breast cancer receiving metformin and neoadjuvant chemotherapy have a higher complete response rate than do diabetics not receiving metformin [38]. This epidemiological evidence and our finding that metformin adds to the cytotoxic effect of doxorubicin corroborate each other. Nevertheless, epidemiological evidence for beneficial impacts of thiazolidinediones on cancer is still lacking.
There are no evidence-based guidelines in the current management of DM2 in patients with cancer, and current management guidelines for DM2 make no distinction for the coexistence of cancer [39]. Our results have brought into focus that there are differences among the currently available pharmacotherapy for DM2 in terms of their direct antineoplastic effects and effects on the circulating insulin levels. Future research is warranted to translate these findings into changes in clinical management strategies to benefit cancer patients in all stages of DM2.
Acknowledgments
This work was supported by an RO1 grant from the U. S. National Institute of Health (NIHRO1CA 089266) to M.H.L., Department of Defense, Breast Cancer Research Program (BCRP) of the Office of the Congressionally Directed Medical Research Programs (CDMRP) Synergistic Idea Development Award BC062166 to S.C.Y. and M.H.L., and a Promise grant from Susan G. Komen Foundation for Breast Cancer Research (KG081048, PI: S.C.Y.). The core facilities are supported by the Cancer Center Core Grant to The University of Texas M. D. Anderson Cancer Center (CA16672). G.V.-T. was supported in part, by a cancer prevention fellowship supported by the National Cancer Institute grant (R25T CA57730).
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Nilsen TI, Vatten LJ. Prospective study of colorectal cancer risk and physical activity, diabetes, blood glucose and BMI: exploring the hyperinsulinaemia hypothesis. Br J Cancer. 2001;84:417–22. doi: 10.1054/bjoc.2000.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verlato G, Zoppini G, Bonora E, et al. Mortality from site-specific malignancies in type 2 diabetic patients from Verona. Diabetes Care. 2003;26:1047–51. doi: 10.2337/diacare.26.4.1047. [DOI] [PubMed] [Google Scholar]
- 3.Richardson LC, Pollack LA. Therapy insight: influence of type 2 diabetes on the development, treatment and outcomes of cancer. Nat Clin Pract Oncol. 2005;2:48–53. doi: 10.1038/ncponc0062. [DOI] [PubMed] [Google Scholar]
- 4.Coughlin SS, Calle EE, Teras LR, et al. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol. 2004;159:1160–7. doi: 10.1093/aje/kwh161. [DOI] [PubMed] [Google Scholar]
- 5.van de Poll-Franse LV, Houterman S, Janssen-Heijnen ML, et al. Less aggressive treatment and worse overall survival in cancer patients with diabetes: a large population based analysis. Int J Cancer. 2007;120:1986–92. doi: 10.1002/ijc.22532. [DOI] [PubMed] [Google Scholar]
- 6.Lu XH, Wang L, Li H, et al. Establishment of risk model for pancreatic cancer in Chinese Han population. World J Gastroenterol. 2006;12:2229–34. doi: 10.3748/wjg.v12.i4.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuriki K, Hirose K, Tajima K. Diabetes and cancer risk for all and specific sites among Japanese men and women. Eur J Cancer Prev. 2007;16:83–9. doi: 10.1097/01.cej.0000228404.37858.40. [DOI] [PubMed] [Google Scholar]
- 8.Fisher WE. Diabetes: risk factor for the development of pancreatic cancer or manifestation of the disease. World J Surg. 2001;25:503–8. doi: 10.1007/s002680020344. [DOI] [PubMed] [Google Scholar]
- 9.Stolzenberg-Solomon RZ, Graubard BI, Chari S, et al. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA. 2005;294:2872–8. doi: 10.1001/jama.294.22.2872. [DOI] [PubMed] [Google Scholar]
- 10.Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA. 1995;273:1605–9. [PubMed] [Google Scholar]
- 11.Permert J, Ihse I, Jorfeldt L, et al. Pancreatic cancer is associated with impaired glucose metabolism. Eur J Surg. 1993;159:101–7. [PubMed] [Google Scholar]
- 12.Wolf I, Sadetzki S, Catane R, et al. Diabetes mellitus and breast cancer. Lancet Oncol. 2005;6:103–11. doi: 10.1016/S1470-2045(05)01736-5. [DOI] [PubMed] [Google Scholar]
- 13.Krone CA, Ely JT. Controlling hyperglycemia as an adjunct to cancer therapy. Integr Cancer Ther. 2005;4:25–31. doi: 10.1177/1534735404274167. [DOI] [PubMed] [Google Scholar]
- 14.Fisher WE, Boros LG, Schirmer WJ. Reversal of enhanced pancreatic cancer growth in diabetes by insulin. Surgery. 1995;118:453–7. doi: 10.1016/s0039-6060(05)80358-7. [DOI] [PubMed] [Google Scholar]
- 15.Ding XZ, Fehsenfeld DM, Murphy LO, et al. Physiological concentrations of insulin augment pancreatic cancer cell proliferation and glucose utilization by activating MAP kinase, PI3 kinase and enhancing GLUT-1 expression. Pancreas. 2000;21:310–20. doi: 10.1097/00006676-200010000-00014. [DOI] [PubMed] [Google Scholar]
- 16.Hudson CC, Liu M, Chiang GG, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–14. doi: 10.1128/MCB.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galli A, Mello T, Ceni E, et al. The potential of antidiabetic thiazolidinediones for anticancer therapy. Expert Opin Investig Drugs. 2006;15:1039–49. doi: 10.1517/13543784.15.9.1039. [DOI] [PubMed] [Google Scholar]
- 18.Blanquicett C, Roman J, Hart CM. Thiazolidinediones as anti-cancer agents. Cancer Ther. 2008;6:25–34. [PMC free article] [PubMed] [Google Scholar]
- 19.Wei S, Yang J, Lee SL, et al. PPARgamma-independent antitumor effects of thiazolidinediones. Cancer Lett. 2009;276:119–24. doi: 10.1016/j.canlet.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efferth T, Sauerbrey A, Steinbach D, et al. Analysis of single nucleotide polymorphism C3435T of the multidrug resistance gene MDR1 in acute lymphoblastic leukemia. Int J Oncol. 2003;23:509–17. [PubMed] [Google Scholar]
- 21.Hadad SM, Fleming S, Thompson AM. Targeting AMPK: a new therapeutic opportunity in breast cancer. Crit Rev Oncol/Hematol. 2008;67:1–7. doi: 10.1016/j.critrevonc.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 22.Hirsch HA, Iliopoulos D, Tsichlis PN, et al. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zakikhani M, Dowling R, Fantus IG, et al. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–73. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 24.Bowker SL, Majumdar SR, Veugelers P, et al. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–8. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 25.Fisher WE, Boros LG, O’Dorisio TM, et al. GI hormonal changes in diabetes influence pancreatic cancer growth. J Surg Res. 1995;58:754–8. doi: 10.1006/jsre.1995.1119. [DOI] [PubMed] [Google Scholar]
- 26.Ekelund S, Liminga G, Bjorkling F, et al. Early stimulation of acidification rate by novel cytotoxic pyridyl cyanoguanidines in human tumor cells: comparison with m-iodobenzylguanidine. Biochem Pharmacol. 2000;60:839–49. doi: 10.1016/s0006-2952(00)00382-8. [DOI] [PubMed] [Google Scholar]
- 27.UKPDS-Group. U.K. prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes. 1995;44:1249–58. [PubMed] [Google Scholar]
- 28.Yeung SJ, Pan J, Lee MH. Roles of p53, MYC and HIF-1 in regulating glycolysis – the seventh hallmark of cancer. Cell Mol Life Sci. 2008;65:3981–99. doi: 10.1007/s00018-008-8224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 30.Isakovic A, Harhaji L, Stevanovic D, et al. Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci. 2007;64:1290–302. doi: 10.1007/s00018-007-7080-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang LW, Li ZS, Zou DW, et al. Metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol. 2008;14:7192–8. doi: 10.3748/wjg.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fenner MH, Elstner E. Peroxisome proliferator-activated receptor-gamma ligands for the treatment of breast cancer. Expert Opin Investig Drugs. 2005;14:557–68. doi: 10.1517/13543784.14.6.557. [DOI] [PubMed] [Google Scholar]
- 33.Lyles BE, Akinyeke TO, Moss PE, et al. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARgamma-dependent and PPARgamma-independent pathways. Cell Cycle. 2009;8:268–77. doi: 10.4161/cc.8.2.7584. [DOI] [PubMed] [Google Scholar]
- 34.Dong YW, Wang XP, Wu K. Suppression of pancreatic carcinoma growth by activating peroxisome proliferator-activated receptor gamma involves angiogenesis inhibition. World J Gastroenterol. 2009;15:441–8. doi: 10.3748/wjg.15.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papageorgiou E, Pitulis N, Manoussakis M, et al. Rosiglitazone attenuates insulin-like growth factor 1 receptor survival signaling in PC-3 cells. Mol Med. 2008;14:403–11. doi: 10.2119/2008-00021.Papageorgiou. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cantini G, Lombardi A, Piscitelli E, et al. Rosiglitazone inhibits adrenocortical cancer cell proliferation by interfering with the IGF-IR intracellular signaling. PPAR Res. 2008;2008:904041. doi: 10.1155/2008/904041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teresi RE, Shaiu CW, Chen CS, et al. Increased PTEN expression due to transcriptional activation of PPARgamma by Lovastatin and Rosiglitazone. Int J Cancer. 2006;118:2390–8. doi: 10.1002/ijc.21799. [DOI] [PubMed] [Google Scholar]
- 38.Jiralerspong S, Palla SL, Giordano SH, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nathan DM, Buse JB, Davidson MB, et al. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2009;32:193–203. doi: 10.2337/dc08-9025. [DOI] [PMC free article] [PubMed] [Google Scholar]