

Abstract
The two human CLC Cl− channels, ClC-Ka and ClC-Kb, are almost exclusively expressed in kidney and inner ear epithelia. Mutations in the genes coding for ClC-Kb and barttin, an essential CLC-K channel β subunit, lead to Bartter syndrome. We performed a biophysical analysis of the modulatory effect of extracellular Ca2+ and H+ on ClC-Ka and ClC-Kb in Xenopus oocytes. Currents increased with increasing [Ca2+]ext without full saturation up to 50 mM. However, in the absence of Ca2+, ClC-Ka currents were still 20% of currents in 10 mM [Ca2+]ext, demonstrating that Ca2+ is not strictly essential for opening. Vice versa, ClC-Ka and ClC-Kb were blocked by increasing [H+]ext with a practically complete block at pH 6. Ca2+ and H+ act as gating modifiers without changing the single-channel conductance. Dose–response analysis suggested that two protons are necessary to induce block with an apparent pK of ∼7.1. A simple four-state allosteric model described the modulation by Ca2+ assuming a 13-fold higher Ca2+ affinity of the open state compared with the closed state. The quantitative analysis suggested separate binding sites for Ca2+ and H+.
A mutagenic screen of a large number of extracellularly accessible amino acids identified a pair of acidic residues (E261 and D278 on the loop connecting helices I and J), which are close to each other but positioned on different subunits of the channel, as a likely candidate for forming an intersubunit Ca2+-binding site. Single mutants E261Q and D278N greatly diminished and the double mutant E261Q/D278N completely abolished modulation by Ca2+. Several mutations of a histidine residue (H497) that is homologous to a histidine that is responsible for H+ block in ClC-2 did not yield functional channels. However, the triple mutant E261Q/D278N/H497M completely eliminated H+ -induced current block. We have thus identified a protein region that is involved in binding these physiologically important ligands and that is likely undergoing conformational changes underlying the complex gating of CLC-K channels.
INTRODUCTION
In the kidney, transepithelial movement of ions and their relative reabsorption are guaranteed by the presence of channels and transporters along the nephron. In particular, Henle’s loop is the nephron segment in which a large fraction of NaCl and divalent cations are reabsorbed, maintaining the body’s salt equilibrium and fluid balance. In the thick ascending limb (TAL), Cl− is mainly taken up through the apical NKCC2 transporter, which works in concert with the Na-K-ATPase and the ROMK potassium channel, and is released from the basolateral membrane through ClC-Kb/barttin channels (Jentsch, 2005; Jentsch et al., 2005).
Human CLC-K channels belong to the CLC family of Cl− channels and transporters. ClC-Ka and ClC-Kb share 90% of identity in their primary structure and coassemble in the kidney and inner ear with the β subunit barttin (Uchida et al., 1993; Kieferle et al., 1994; Simon et al., 1997; Estévez et al., 2001; Jentsch, 2005; Jentsch et al., 2005). ClC-K1 (ClC-Ka) appears to be mainly expressed in the thin ascending limb of Henle’s loop of the nephron, whereas ClC-K2 (ClC-Kb) is restricted to the basolateral membranes of epithelial cells in the TAL, connecting tubule, distal convoluted tubule, and intercalated cells. Both isoforms are also localized in basolateral membranes of marginal cells of the stria vascularis and in dark cells of the vestibular organ, in which they play a role in the production of the endolymph (Rickheit et al., 2008). The role of CLC-K channels is stressed by the association of mutations in the genes coding for CLC-Ks and barttin to several kidney diseases. Mutations in ClC-Kb and barttin genes are associated with the renal disease called Bartter syndrome (type III and type IV, respectively), which is characterized by renal salt wasting due to an impaired NaCl reabsorption in the TAL (Simon et al., 1997). Mutations in the gene coding for barttin also cause deafness (Birkenhäger et al., 2001). Moreover, it has been shown that mice lacking the ClC-K1 channel exhibit nephrogenic diabetes insipidus (Matsumura et al., 1999). Yet, no human disease with mutations only in the gene coding for ClC-Ka has been described yet. However, simultaneous loss of function of both ClC-Ka and ClC-Kb leads to Bartter syndrome (type IV) with deafness, similar to the effect of barttin mutations (Schlingmann et al., 2004). This result supports the idea that ClC-Ka and ClC-Kb are functionally redundant in the inner ear, but not in the kidney.
In previous reports, it has been described that human CLC-K channels, like the rodent CLC-K1, are modulated by extracellular Ca2+ and protons in the millimolar range, and at physiological pH, respectively (Uchida et al., 1995; Estévez et al., 2001; Waldegger et al., 2002). In fact, both isoforms are enhanced by increasing [Ca2+]ext and blocked by increasing concentration of extracellular protons. Modulation of CLC proteins by pH is very common. This is not surprising for those CLC proteins that act as Cl−/H+ antiporters because H+ is one of the transported substrates (Friedrich et al., 1999; Accardi and Miller, 2004; Picollo and Pusch, 2005; Zifarelli and Pusch, 2009). However, most CLC channels are also strongly pH dependent because of the protonation/deprotonation of a conserved glutamate residue, the “gating glutamate,” that is involved in the opening of the channels (Hanke and Miller, 1983; Rychkov et al., 1996; Jordt and Jentsch, 1997; Saviane et al., 1999; Chen and Chen, 2001; Arreola et al., 2002; Dutzler et al., 2003; Traverso et al., 2006; Zifarelli et al., 2008; Niemeyer et al., 2009; Zifarelli and Pusch, 2010). However, in contrast with most CLC channels, in CLC-K channels a valine residue substitutes the gating glutamate. Hence, their pH dependence cannot be mediated by the same mechanism. Recently, for ClC-2 two distinct regulatory sites for protons have been identified (Niemeyer et al., 2009): protonation of the gating glutamate activates ClC-2 in a voltage-dependent manner, whereas protonation of an off-pore histidine residue leads to channel block. Conceivably, a similar mechanism might be responsible for the H+-induced block of CLC-K channels.
On the other hand, activation by extracellular Ca2+ is unique to CLC-K channels. The concentrations at which CLC-Ks are maximally sensitive to Ca2+ and H+ are close to the physiological values found in extracellular fluids. Therefore, the modulation by these agents is likely of physiological relevance, even though there is no direct evidence for this view.
Here, we performed a detailed biophysical analysis of this modulation. Moreover, we performed a mutagenic screen and identified a histidine residue (H497) that is responsible for H+-induced block as well as two residues (E261 and D278) that likely form an intersubunit Ca2+-binding site.
MATERIALS AND METHODS
Molecular biology
Mutations were introduced by recombinant PCR as described previously (Accardi and Pusch, 2003). cRNA was transcribed in vitro by SP6 RNA polymerase (Applied Biosystems) after linearization with MluI. ClC-Ka and its mutants were coexpressed with human barttin Y98A to enhance the expression (Estévez et al., 2001). The barttin construct was linearized with NotI, and RNA was transcribed using T7 RNA polymerase. All cDNA constructs were provided by T. Jentsch (Leibniz-Institut für Molekulare Pharmakologie, Berlin, Germany).
Electrophysiology
The cRNA of each construct was injected in defolliculated Xenopus oocytes. Oocytes were kept at 18°C in Barth’s solution containing (in mM) 84 NaCl, 1 KCl, 2.5 NaHCO3, 0.5 Ca(NO3)2, 0.6 CaCl2, and 7.5 Tris-HCl, pH 7.4, or in a solution containing (in mM) 90 NaCl, 10 HEPES, 2 KCl, 1 MgCl2, and 1 CaCl2, pH 7.5. Voltage clamp data were acquired 2–4 d after the injection at room temperature using a custom acquisition program (Gepulse) and a custom-built amplifier or a TEC-03X amplifier (npi electronic). The standard bath solution contained (in mM): 112 NaCl, 10 CaGluconate2, 1 MgSO4, and 10 HEPES, pH 7.3 (osmolarity: 235 mOsm). To study the effect of calcium on CLC-K currents, the concentration of CaGluconate2 of the standard solution was varied between 0.1 and 50 mM (the 50-mM Ca solution had an osmolarity of 313 mOsm). The somewhat elevated osmolarity of these solutions did not lead to the unspecific activation of endogenous currents in non-injected oocytes. HEPES was replaced by MES buffer in solutions having a pH <7. The holding potential was chosen close to the resting membrane potential (∼−30 mV). To evaluate the currents at different potentials, we used the following protocols of stimulation. For Figs. 1 and 2: after a prepulse to −140 mV for 200 ms, channels were stimulated with voltages ranging from −140 to 80 mV with 20-mV increments for 500 ms. Pulses ended with a tail to 60 mV for 200 ms. For all other figures: after a prepulse to −100 mV for 100 ms, channels were stimulated with voltages ranging from −140 to 80 mV with 20-mV increments for 200 ms. Pulses ended with a tail pulse to 60 mV for 100 ms.
Figure 1.

Ca2+ext dependence of ClC-Ka and ClC-Kb. (A) Typical currents of ClC-Ka evoked by the standard IV-pulse protocol at different Ca2+ concentrations as indicated, at pH 7.3. (B) Effect of [Ca2+]ext on ClC-Ka (circles; n = 9). Current at 60 mV acquired at different conditions was normalized to values measured in standard solution (I/I10 mM Ca2+, pH 7.3) and plotted versus [Ca2+]ext (used concentrations: 0.1, 0.5, 2, 5, 10, 20, 30, and 50 mM). The line represents the fit obtained using Eq. 2 as described in Results. (C) Superimposed traces measured at 60 mV at 5 mM [Ca2+]ext (black line) and at 20 mM [Ca2+]ext (gray line) measured from the same oocyte. Currents were scaled to the steady-state current at 60 mV. Horizontal bar, 200 ms. (D) Typical currents of ClC-Kb measured at different Ca2+ concentrations as indicated. (E) Effect of [Ca2+]ext (concentrations as in B) on ClC-Kb (squares; n = 5). Horizontal bars, 200 ms. Vertical bars, 5 µA.
Figure 2.

pHext dependence of ClC-Ka and ClC-Kb. (A) Voltage clamp traces of ClC-Ka measured at different pH as indicated, in 10 mM [Ca2+]ext. (B) Effect of pHext on ClC-Ka (circles; n = 7). Normalized currents were calculated as for Fig. 1. I/I10 mM Ca2+, pH 7.3, is plotted versus pH. The line represents the fit obtained using Eq. 1 as described in Results. (C) Superimposed traces measured at pH 7.0 (black line) and pH 8.0 (gray line) measured from the same oocyte. Currents were scaled to the steady-state current at 60 mV. Horizontal bar, 200 ms. (D) Traces of ClC-Kb measured at different pH as indicated, in 10 mM [Ca2+]ext. (E) Effect of pHext on ClC-Kb (squares; n = 4). Horizontal bars, 200 ms. Vertical bars, 5 µA.
To evaluate Ca2+ and pH effects on ClC-Ka wild type (WT) and all its mutants, the channels were stimulated with repetitive pulses to 60 mV for 200 ms applied at 1 Hz, and solutions with different pH and/or [Ca2+] were continuously applied until steady state was reached. Reversibility was always checked by returning to the standard solution. To estimate the contribution of endogenous currents, we applied a solution containing 100 mM NaI, 1 mM MgSO4, and 10 mM HEPES, pH 7.3, that specifically blocks currents carried by CLC-K, but not endogenous currents (Picollo et al., 2004).
The effect of calcium and pH was quantified by calculating the ratio between the mean current at 60 mV in the specific solution and in the standard bath solution (10 mM [Ca2+], pH 7.3). Leak currents (i.e., currents remaining in iodide) were subtracted. Error bars in figures indicate SEM.
Patch clamp experiments
For patch clamp recordings, the vitelline membrane of oocytes was mechanically removed during exposure to a hypertonic solution. The intracellular solution contained (in mM): 100 N-methyl-d-glucamine-Cl (NMDG-Cl), 2 MgCl2, 10 HEPES, and 2 EGTA, pH 7.3. The standard extracellular solution contained (in mM): 90 NMDG-Cl, 10 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.3. For some experiments, the pH was changed to 8 (keeping HEPES-buffer) or the Ca2+ concentration was changed to 2 or 1.8 mM CaCl2. For the 0 Cl− solution (Fig. 3), Cl− was exchanged by glutamate. Pipettes were pulled from aluminosilicate glass capillaries (Hilgenberg) and had resistances of 1–2 × 106 Ohm in the recording solutions. Currents were recorded at 50 kHz after filtering at 10 kHz with an eight-pole Bessel filter. For the recordings shown in Fig. 3, the outside-out configuration of the patch clamp technique was used, whereas for the noise analysis experiments shown in Fig. S1, the inside-out configuration was used (Hamill et al., 1981; Stühmer, 1998).
Figure 3.

Relaxation kinetics upon fast concentration jumps in outside-out patches. (A) Example recording of a patch held at 60 mV in which [Ca2+]ext was switched from 10 to 1.8 mM, and then back to 10 mM. Finally, the patch was exposed to a solution without Cl− (and without Ca2+). The three insets below show the transition regions on an expanded time scale (bar, 500 ms), superimposed with a single-exponential fit (red line). Time constants for the solution exchange (inset c) were on the order of 10 ms. (B) Average time constants (n > 3 patches) for the indicated transitions.
For noise analysis, 30–100 identical pulses to −100 or −140 mV were applied, and the mean response, I, was calculated. The variance, σ2, was calculated from the averaged squared difference of consecutive traces. Background variance at 0 mV was subtracted, and the variance-mean plot was constructed by binning as described by Heinemann and Conti (1992).
The binned variance-mean plot was fitted by σ2 = iI−I2/N, with the single-channel current, i, and the number of channels, N, as free parameters (Pusch et al., 1994). The single-channel conductance was calculated assuming a linear i-V relationship.
For the experiments shown in Fig. 3, a rapid solution exchanger (RSC-160; Biological Science Instruments) was used. In this instrument, three glass capillaries of 0.5-mm diameter are mounted on a rod that is rotated by a step motor. Full switching from one solution to the other requires ∼10 ms (see Fig. 3 A, inset c).
Data analysis
Currents measured at different [Ca2+]ext and pH were normalized to the steady-state current measured at 60 mV in the standard bath solution, i.e., 10 mM Ca2+, pH 7.3. Normalized currents were plotted versus [Ca2+]ext and pH.
Online supplemental material
In Fig. S1, we show example experiments for nonstationary noise analysis done for ClC-Ka at various pH and [Ca2+]ext conditions. In Fig. S2, we summarize the results of the effects of [Ca2+]ext on all mutants. In Fig. S3, we summarize the results of the effects of [pH]ext on all mutants. Figs. S1–S3 are available at http://www.jgp.org/cgi/content/full/jgp.201010455/DC1.
RESULTS
Effect of extracellular calcium and protons on ClC-Ka and ClC-Kb channels
CLC-K currents, measured in the standard solution containing 10 mM Ca2+, pH 7.3, show the following typical behavior: ClC-Ka partially deactivates at positive voltages and activates at negative voltages (Estévez et al., 2001; Picollo et al., 2004) (Fig. 1 A), whereas ClC-Kb activates at positive voltages and deactivates at negative voltages with slower kinetics compared with ClC-Ka (Estévez et al., 2001; Picollo et al., 2004) (Fig. 1 D).
In the first set of experiments, we applied solutions with different Ca2+ concentrations ranging from 0.1 to 50 mM Ca2+, keeping the pH fixed at 7.3. ClC-Ka–mediated currents increase with increasing [Ca2+]ext (Estévez et al., 2001), without saturation up to 50 mM (Fig. 1 A). Such high Ca2+ concentrations did not induce unspecific endogenous or leak currents in non-injected oocytes (not depicted). An important finding from a mechanistic point of view is that, at a Ca2+ concentration of 0.1 mM, ClC-Ka currents are still ∼20% of the currents measured in 10 mM [Ca2+]ext (Fig. 1 B). Preliminary experiments showed that reducing [Ca2+]ext to nominally zero did not further reduce currents compared with 0.1 mM [Ca2+]ext (not depicted). However, the development of endogenous currents under these conditions precluded a more systematic investigation. These results suggest that extracellular Ca2+ is not strictly essential for opening. As can be seen from the traces in Fig. 1 A, the kinetics—and thus the voltage dependence—are, to a first approximation unaffected by [Ca2+]ext. This is illustrated in more detail in Fig. 1 C, where currents recorded from the same oocyte at 5 mM [Ca2+]ext and at 20 mM [Ca2+]ext measured at 60 mV are superimposed and normalized to the steady-state current. ClC-Kb has a qualitatively very similar but slightly less pronounced dependence on [Ca2+]ext (Fig. 1, D and E).
Next, we investigated the pH sensitivity of ClC-Ka and ClC-Kb (Fig. 2). In these experiments, [Ca2+]ext was fixed at 10 mM. ClC-Ka–mediated currents decrease at acidic pH with an almost complete block at pH 6.0 (Fig. 2, A and B). Extracellular pH clearly alters the kinetics and the voltage dependence of gating, as demonstrated by the superposition of traces at pH 8.0 and at 7.0 measured at 60 mV (Fig. 2 C). In fact, protons slightly alter the kinetics of the current relaxation at positive voltages and significantly affect the activation at negative voltages. However, we did not study the effect of pH on the kinetics of gating in more detail, mainly because of the well-known problems of voltage clamp studies in Xenopus oocytes. ClC-Kb shows a similar behavior, but it appears to be more sensitive to proton block than ClC-Ka (Fig. 2, D and E), and for ClC-Kb, the effect of pH does not seem to saturate. This may be explained by a shift of the apparent pK of the effect toward more alkaline pH. Qualitatively, Ca2+ and protons have a similar effect on both CLC-K homologues. Therefore, we focused our attention on ClC-Ka because of its higher expression compared with ClC-Kb.
An important mechanistic question is whether Ca2+ and H+ exert their effect by a direct pore-occluding mechanism or if they modulate in an allosteric manner the open probability of the channel that is governed by one or more gating processes. A crude distinction between these two possibilities can be based on a kinetic analysis: allosteric gating regulation is expected to be “slow,” whereas a direct occluding effect is expected to be “fast.” Rapid perfusion is difficult to achieve in whole oocyte recordings. Therefore, we conducted outside-out patch recordings and used a rapid perfusion system to change the extracellular pH or Ca2+ concentration. A typical experiment is shown in Fig. 3 A, in which the current recorded at 60 mV from a patch containing many channels is displayed as a function of time. Upon reduction of external Ca2+ from 10 to 1.8 mM, currents decrease reversibly. To test the speed of the perfusion system, a solution without Cl− was applied at the end of the experiment. The insets in Fig. 3 A (a, b, and c) show the solution changes at an expanded time scale, superimposed with single-exponential fits (red lines). The speed of the solution exchange was on the order of 10 ms (Fig. 3 A, inset c), clearly much faster than the relaxations observed upon changing the Ca2+ concentration. Similarly slow relaxations were observed upon switching the extracellular pH from 7.3 to 8 (see Fig. 3 B). Average time constants are shown in Fig. 3 B for Ca2+ jumps and pH jumps. These kinetic experiments suggest that both Ca2+ and protons exert their effects by an allosteric modulation of the channel open probability.
To further exclude a direct effect on ion permeation, we performed nonstationary noise analysis to estimate the single-channel conductance. Typical experiments are shown in Fig. S1. From these experiments, no effect of Ca2+ or pH on the single-channel conductance could be detected (Table I).
Table I.
Single-channel conductance estimated from noise analysis
| Condition | Conductance (pS) | n |
| 10 mM Ca2+, pH 7.3 | 16.0 ± 1.0 | 5 |
| 2 mM Ca2+, pH 7.3 | 16.3 ± 0.7 | 3 |
| 10 mM Ca2+, pH 8 | 16.6 ± 0.5 | 5 |
To find out whether Ca2+ and H+ compete for common binding sites, we performed experiments mixing the concentrations of the two cations. First, we applied solutions with various [Ca2+]ext, keeping the pH fixed at a value of 8, a condition of maximal stimulation in the range of pH values tested (see Fig. 2 A). Interestingly, ClC-Ka currents still increase without saturation up to 50 mM Ca2+ (Fig. 4 A). Also at pH 6.5, a condition at which channel activity is very low (see Fig. 2 B), the effect of Ca2+ was still clearly evident (Fig. 4 B). At this pH, measurements at a [Ca2+]ext <2 mM are not reliable due to an increase of leak and endogenous currents after a long exposure to low pH and low Ca2+. Next, keeping [Ca2+]ext fixed at 0.5 mM, the pH dependence was very similar to that seen at [Ca2+]ext of 10 mM (Fig. 4 C). These results suggest that extracellular Ca2+ and extracellular protons act via independent mechanisms and binding sites. This hypothesis was further scrutinized by kinetic modeling.
Figure 4.

Ca2+ and pH effects on ClC-Ka at mixed conditions. (A and B) Effect of [Ca2+]ext on ClC-Ka measured at pHext 8 (A, n = 6) and pHext 6.5 (B, n = 4). Currents at 60 mV were normalized to values measured in 10 mM [Ca2+]ext and plotted versus [Ca2+]ext. (C) Effect of pHext on ClC-Ka in 0.5 mM Ca2+ (n = 3). Currents at 60 mV were normalized to values measured at pH 7.3 and plotted versus pHext. Lines represent the theoretical predictions obtained using Eqs. 1 and 2 as described in Results using the same parameters obtained from the fit in the standard conditions (i.e., pK = 7.11; KC = 19.6 mM, KO = 1.5 mM, and r0 = 434).
Modeling pH and Ca2+ modulation of ClC-Ka
Acidic pH practically completely blocks CLC-Ka. We therefore fitted the pH dependence by a simple Michaelis-Menten titration curve,
| (1) |
where pu is the probability of not being blocked by a proton, [H] the proton concentration, KH the apparent binding constant, and n the Hill coefficient. We define pK = −log10(KH) for convenience. As shown in Fig. 2 C, proton-induced block is voltage dependent. For simplicity, however, we concentrated our analysis on the proton block at 60 mV. The solid line in Fig. 2 B is the best fit of Eq. 1, with pK = 7.11 and n = 1.61. The Hill coefficient is significantly larger than one suggesting that more than one proton (most likely two) is necessary to block the channel.
Regarding the modeling of the Ca2+-induced potentiation, we have to consider that ClC-Ka channels are partially open even in the absence of Ca2+. Therefore, a simple two-state mechanism is inadequate. Instead, we are forced to invoke an allosteric model of Ca2+ modulation that incorporates an open non-Ca2+–bound state. Conceptually, the simplest allosteric model is composed of four states, as shown below.
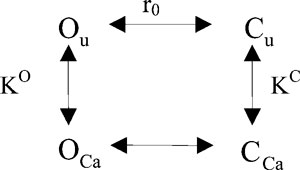 |
(SCHEME 1) |
Here, Ou is the open Ca2+-free state, and OCa is the open state bound to Ca2+. Cu and CCa refer similarly to the closed states. Ca2+ binding to the open state is governed by the dissociation constant KO, whereas Ca2+ binding to the closed state is described by the dissociation constant KC. r0 is the ratio between the probability of being in states Cu and Ou (r0 = p(Cu)/p(Ou)).
For Scheme 1, the open probability is given by the following equation:
| (2) |
The solid line in Fig. 1 B is the best fit of Eq. 2, resulting in KC = 19.6 mM, KO = 1.5 mM, and r0 = 434. The theoretical prediction of this model nicely fits the experimental data. The value obtained for r0 cannot be considered reliable. In fact, the model predictions are practically independent from the value of r0, as long as it is significantly >1. The precise determination of r0 would imply knowledge about the absolute open probability, clearly an impossible piece of information to be obtained from macroscopic measurements.
To further test if proton-induced block and Ca2+ potentiation are independent processes, we compared the experimental results obtained under “mixed conditions” (Fig. 4) with the predictions of the modeling obtained under standard conditions (Figs. 1 and 2). The lines in Fig. 4 were calculated using the same parameters obtained from the fits described above (i.e., pK = 7.11; KC = 19.6 mM, KO = 1.5 mM, and r0 = 434). Within the experimental error, these fits provide an adequate description of the data, consistent with the idea that Ca2+ and H+ act via independent mechanisms.
Thus, the model allows the following conclusions. First, two protons bind to the binding site for protons, and one Ca2+ binds to the binding site for Ca2+. Second, the analysis reveals that ClC-Ka has an affinity for Ca2+ that is 13-fold higher in the open state (KCaO = 1.5 mM) than in the closed state (KCaC = 19.6 mM), suggesting that Ca2+ promotes the open state of the channel. Proton block occurs with an apparent pK of ∼7.11. However, we would like to note that the two-electrode voltage clamp measurements present some technical limitations such as, for example, problems arising from the series resistance and difficulties distinguishing small currents (e.g., in low pH) from endogenous currents. Therefore, additional evidence to support the conclusions from the modeling is desirable.
Substitution of two externally accessible residues eliminates the sensitivity to Ca2+
The structure of the bacterial homologue EcClC-1 has been determined by x-ray crystallography (Dutzler et al., 2002). The bacterial EcClC-1 is significantly homologous to the eukaryotic CLCs (Maduke et al., 2000). In particular, they share the same transmembrane topology and several conserved regions. For this reason, we used the structure of the bacterial transporter as a guide to select and mutate all titratable residues of ClC-Ka that could be accessible from the extracellular side of the pore. In total, we chose and mutated 50 residues that are located between helices B and Q (see legend to Fig. S2). In general, charged residues (Glu, Asp, Arg, Lys, and His) were neutralized. Other titratable residues (Cys and Tyr) were changed with non-titratable residues. In cases where the mutations were not functional, we tried alternative and more conservative mutations. The extensive mutagenic screen is shown in Fig. 5 A, where we colored the mutated residues in light blue and in pink in the two different subunits. With the exception of H480 and E490, mutations of all other residues led to functionally active protein at least for one amino acid change. The majority of them displayed Cl− currents that rapidly deactivate at positive voltages and activate at negative voltages, similar to WT currents when measured in standard solution (not depicted). For each of these mutations, we studied the effect of extracellular Ca2+ and protons. The results of this screen are shown in Figs. S2 and S3, where normalized currents of the mutants are plotted versus the calcium concentrations and pH, respectively. Although several mutations slightly affected calcium sensitivity (Fig. S2), only E261Q and D278N had a drastically reduced calcium sensitivity (Fig. 6, B and C).
Figure 5.

Location of mutants mapped on the structure of ecClC-1. (A) A surface representation of the bacterial ecClC-1 (Protein Databank accession no. 1OTS) viewed from the extracellular side is shown. The two subunits that compose ecClC-1 are colored in gray and light gray, respectively. The residues corresponding to those selected for mutation are shown in pink and light blue in the two different subunits, respectively. The transparent surface also allows a glimpse of the internal mutated residues. The residues responsible for Ca2+ and H+ sensitivity are shown in different colors: yellow, E261 (corresponding to E235 of ecClC-1); red, D278 (corresponding to N250 of ecClC-1); green, H497 (corresponding to L421 of ecClC-1); the numbers of residues indicated in the figure correspond to those of ClC-Ka. (B) A zoom of a selected region is shown in cartoon representation. The three residues E235, N250, and L421 are highlighted as sticks and colored as in A. A hypothetical Ca2+ ion is shown as a light blue sphere between E261 and D278. (C) Alignments around the three residues responsible for Ca2+ and proton sensitivity are shown. E261, D278, H497, and the corresponding residues in other CLCs are in bold.
Figure 6.

Effect of [Ca2+]ext on ClC-Ka WT and its mutants E261Q, D278N, and E261Q/D278N. (A–D) The mean current, shown in color, at 60 mV plotted as a function of time. The type of solution applied is color coded as indicated in the middle inset. Breaks during the experiment are indicated with short dashed lines. Insets display representative current traces evoked by the standard IV-pulse protocol in standard solution. Horizontal bars, 100 ms; vertical bars, 3 µA. (E) Dose–response relationship of the modulation by Ca2+ of ClC-Ka WT (black circles; n = 16) and the mutants E261Q (yellow squares; n = 4), D278N (red triangles; n = 5), and E261Q/D278N (light blue rhombi; n = 5). Currents at 60 mV were normalized to values measured in standard solution and plotted versus [Ca2+]ext. Data for WT are different compared with Fig. 1 because they were obtained from different oocytes as control measurements in the mutagenic screen. The currents shown for the mutants D278N and E261Q/D278N are from oocytes with exceptionally large expression. On average, the current expression level measured at 60 mV were (in µA ± SD [no. of oocytes]): WT (2 d), 2.5 ± 1.4 (7); WT (3 d), 5.3 ± 1.9 (8); E261Q (1–2 d), 4.3 ± 0.9 (4); D278N (>3 d), 1.1 ± 0.9 (8); E261Q/D278N (>3 d), 2.2 ± 0.9 (7); E261Q/D278N/H497M (>3 d), 0.8 ± 0.3 (15); not injected, 0.17 ± 0.07 (8).
E261Q loses most of the Ca2+ sensitivity (Fig. 6, B and E). In fact, at 1 mM [Ca2+]ext, currents mediated by E261Q are >50% of the currents measured in 10 mM [Ca2+]ext, and when increasing [Ca2+]ext to 50 mM, the currents only slightly increase. For this mutant, the normalized current I([Ca2+]ext)/I(10 mM) varied less than twofold between 1 and 50 mM [Ca2+]ext, whereas for WT, the same ratio varied by ∼10-fold (Fig. 6, A and E).
Having identified E261 as being critical for [Ca2+]ext sensitivity, we selected and mutated all residues located nearby, but none of these mutations were significantly different from WT (Fig. S2).
The other mutation, D278N, is even less Ca2+ sensitive than E261Q (Fig. 6, C and E). In fact, currents mediated by D278N do not vary between 1 and 10 mM [Ca2+]ext, and they only slightly increase at 50 mM [Ca2+]ext (Fig. 6, C and E). These results lead us to suggest that E261 and D278 could be part of a binding site for calcium. In agreement with this hypothesis, the double mutant E261Q/D278N completely abolishes the calcium sensitivity (Fig. 6, D and E).
Residue E261 is conserved in both human CLC-K homologues and also in the rodent isoforms ClC-K1 and ClC-K2, but not in all CLCs. Residue D278 is conserved in all human and rodent CLC-Ks and quite conserved in other CLCs (Fig. 5 C). Both are located in the loop between helices I and J (Fig. 5 C). Based on the alignment of ClC-Ka with EcClC-1, we tentatively assigned the residue in EcClC-1 corresponding to D278 of ClC-Ka to N250 in EcClC-1 (Fig. 5 C). Because of a gap in the bacterial sequence, we could not uniquely align E261 (Fig. 5 C). Tentatively, we assigned the bacterial residue E235 as the one corresponding to E261 (Fig. 5 C). Keeping in mind these limits, we evidenced E235, in yellow, and N250, in red, in a surface representation of EcClC-1 (Fig. 5, A and B). These residues are quite far from each other in the primary sequence. Interestingly, in the 3-D structure of EcClC-1, E235 from one subunit of the homodimer and N250 from the other subunit are relatively close to each other (∼10 Å). Thus, based on the experimental evidence and the structure of EcClC-1, we conclude that E261 and D278 are excellent candidates for forming an intersubunit Ca2+-binding site.
Substitution of an external residue removes the sensitivity to protons
Another objective of our work was to identify the residues of ClC-Ka that are responsible for the sensitivity to protons. Therefore, we investigated the pH sensitivity of all the mutants prepared (Fig. S3). In particular, we paid attention to the residues involved in calcium sensitivity.
E261Q and D278N are still responsive to different pH. Nevertheless, at basic pH, both show a behavior different from WT. In fact, currents mediated by E261Q only slightly increase at pH 8, whereas those mediated by D278N even decrease in the same pH range (Fig. 7 B). These results might point to an indirect involvement of these residues in the binding of protons.
Figure 7.

Effect of [H+]ext on ClC-Ka WT and its mutants E261Q, D278N, E261Q/D278N, and E261Q/D278N/H497M. (A) The current at 60 mV, shown in color, of the triple mutant E261Q/D278N/H497M as a function of time (color code as in Fig. 6). Inset in A shows representative current traces of the mutant in standard solution. Horizontal bars, 100 ms; vertical bars, 1 µA. Capacitive transients have been blanked for clarity. (B) Dose–response relationship of the modulation by protons of ClC-Ka WT (black circles; n = 14) and the mutants E261Q (yellow squares; n = 4), D278N (red triangles; n = 5), E261Q/D278N (light blue rhombi; n = 5), and E261Q/D278N/E261Q (green hexagons; n = 5). Currents at 60 mV were normalized to values measured in standard solution and plotted versus pH. Data for WT are different compared with Fig. 2 because they were obtained from different oocytes as control measurements in the mutagenic screen.
Even though we selected and mutated all external titratable residues, we were not able to identify mutations that abolish the block induced at acidic pH effect. However, we could not obtain functional expression for several mutants of His-497, which corresponds to His-532 of the ClC-2 channel, where it has been shown to be responsible for H+-induced block (Niemeyer et al., 2009). His-497 is located at the N terminus of helix Q in a region that is conserved in most CLC proteins (Fig. 5 C). In ClC-2, the H532F mutation was specifically shown to eliminate H+ block. Unfortunately, the corresponding mutation in ClC-Ka (H497F) did not yield currents. In ClC-1, mutating the corresponding His to Ala was reported to yield normal channel function (Kürz et al., 1999). However, we could not obtain expression of the analogous H497A mutation. Among several other mutations (H497N, H497D, H497E, H497K, H497Y, and H497M), only H497M yielded very small currents that were, however, barely above background (not depicted). H497 corresponds to L421 in EcClC-1 (Fig. 5 C). We highlighted L421 in green in a surface representation of EcClC-1 (Fig. 5, A and B). Interestingly, H497 is localized relatively close to the amino acids E261 and D278, which form the acidic Ca2+-binding site (on the same subunit as D278).
The proximity of these three amino acids and their lower Ca2+ and H+ sensitivity than WT drove us to prepare the triple mutant E261Q/D278N/H497M. Fortunately, the triple mutant showed bigger currents than H497M, allowing us to investigate its pH sensitivity. The triple mutant E261Q/D278N/H497M completely eliminated H+-induced current block. In fact, currents did not vary significantly between pH 5 and 7.3 (Fig. 7, A and B). As expected, the triple mutant was also completely insensitive to [Ca2+]ext (Fig. 7 A).
DISCUSSION
Here, we performed for the first time a detailed analysis of the modulation of CLC-K channels by extracellular Ca2+ and protons. Both human homologues, ClC-Ka and ClC-Kb, are enhanced by increasing the extracellular Ca2+ concentration and are blocked by increasing the extracellular proton concentration. As shown previously (Estévez et al., 2001), ClC-Kb appears to be slightly more sensitive to pH compared with ClC-Ka. However, we focused on ClC-Ka because of its higher expression compared with ClC-Kb. The relatively slow relaxation kinetics upon stepwise changes of Ca2+ or pH in outside-out patch clamp recordings led us to conclude that both ions modulate CLC-K activity by altering the open probability of the channel. In fact, the absolute open probability of ClC-Ka and ClC-Kb in physiological conditions is significantly smaller than unity, allowing for a vast modulation of CLC-K–mediated membrane conductance by various ligands, including niflumic acid (NFA) (Zifarelli et al., 2010). Applying a two-state Michaelis-Menten model for H+ block revealed that the binding of two H+ is probably needed to induce channel closure, with an apparent pK of 7.1. A simple allosteric model for Ca2+ modulation was able to reproduce the concentration dependence of the Ca2+ modulation, assuming a dissociation constant of 19.6 mM for the closed state and 1.5 mM for the open state. The parameters obtained from the fits at standard conditions could also reasonably well describe H+-induced block at low [Ca2+] and Ca2+ modulation at more acidic and more alkaline pH, consistent with the idea that Ca2+ and H+ act via separate binding sites.
Next, using the structure of a bacterial CLC protein as a guide, we were able to identify the putative binding sites for both ions. Regarding the H+-binding site, we were aided by recent results obtained for the ClC-2 channel (Niemeyer et al., 2009). ClC-2 is regulated by external pH in a biphasic manner: slightly acidic pH values activate the channel, whereas strong acidic conditions block it (Jordt and Jentsch, 1997; Arreola et al., 2002; Niemeyer et al., 2009). Niemeyer et al. (2009) showed that the potentiation of ClC-2 by external protons is mediated by the protonation of the “gating glutamate” residue that is highly conserved in most CLC proteins. However, in CLC-K channels, this glutamate is exchanged by a not-titratable valine residue. In contrast, the H+-blocking site appears to be similar to the blocking site identified on the ClC-2 channel (Niemeyer et al., 2009). In ClC-2, protonation of His-532 appears to be responsible for H+-induced block, in that the H532F mutation was not blocked at acidic pH (Niemeyer et al., 2009). Unfortunately, most mutations of the corresponding His-497 residue in ClC-Ka resulted in nonfunctional channels. However, we could obtain reasonable currents of the triple mutant E261Q/D278N/H497M that were insensitive to pH in the range of 5 and 8, whereas the E261Q/D278N mutation was fully blocked at pH 5. Based on these results, we cannot completely rule out a certain interdependence of the pH and the Ca2+ effects because we could assert the importance of the His-497 residue for H+ block only in the context of a Ca2+-insensitive construct (i.e., E261Q/D278N). Nevertheless, these results also suggest that in ClC-K channels, protonation of this histidine residue at the beginning of helix Q is responsible for pH-induced block. As in ClC-2 (Arreola et al., 2002; Niemeyer et al., 2009), H+ block displays a functional stoichiometry of two; i.e., it appears that two H+ are necessary for closing the channel, further supporting the assumption of a similar mechanism of pH-induced block in the two channels. It has been speculated that H+ block acts on the “common gate” of the double-barreled channel. However, in the absence of single-channel data, such conclusions are difficult to draw conclusively. It will be interesting to find out if protonation of the corresponding histidine residue in other CLC channels, like ClC-0 or ClC-1, also affects channel function. Regulation by extracellular Ca2+ appears to be unique to CLC-K channels. Based on an exhaustive mutagenic screen, we could identify the most likely Ca2+-binding site. It is formed by two acidic residues (E261 and D278), both localized in the loop connecting helices I and J. This loop connects the two halves of one pseudosymmetric CLC monomer (Dutzler et al., 2002); i.e., helix I is the last helix of the first half, whereas helix J is the first helix of the second half, which is inserted oppositely into the membrane with respect to the first half. Sequence conservation in the I-J loop is relatively poor among distantly related CLCs, and the Escherichia coli sequence is considerably shorter than that of most eukaryotic CLCs, resulting in a certain ambiguity of assigning the residues of the E. coli EcClC-1 that correspond to E261 and D278 of ClC-Ka. Based on the alignment shown in Fig. 5, we identified E261-ClC-Ka with E235-ecClC-1 and D278-ClC-Ka with N250-ecClC-1. Interestingly, these two residues of one subunit of ecClC-1 are far from each other, but E235-ecClC-1 of one subunit and N250-ecClC-1 of the neighboring subunit are within ∼10 Å from each other. This distance is clearly too large to form a cation-binding site. However, because the I-J loop of ClC-K channels is considerably longer than that of ecClC-1, we speculate that in this channel, the two residues identified here, E261 and D278, form a millimolar affinity Ca2+-binding site between the two subunits. Consequently, two symmetrically located Ca2+-binding sites are present on each channel. The modeling of the functional data indicated that occupation of a single binding site is sufficient to promote channel activation. However, we believe that this issue deserves further detailed investigation, using, for example, heteromeric constructs in which only one of the subunits is manipulated.
Whereas D278 is rather conserved among the eukaryotic channel CLCs, E261 is not conserved in ClC-0, ClC-1, and ClC-5. Thus, it is not surprising that these CLCs are Ca2+ insensitive. It may be interesting to find out if Ca2+ sensitivity can be engineered into other CLC channels or transporters.
Recently, it was described that a mutation in the C-terminal cytosolic region of ClC-Kb (R538P) abolished its Ca2+ sensitivity, whereas the same mutation was without effect in ClC-Ka (Martinez and Maduke, 2008). This effect is likely caused indirectly by affecting the gating of ClC-Kb. Furthermore, ClC-Kb–mediated currents are rather small, such that unspecific effects cannot be fully excluded. In fact, in another recent report, the mutation R351W was suggested to abolish the Ca2+ and H+ sensitivity of ClC-Kb (Yu et al., 2010). However, in our hands, the similar mutation R351A in ClC-Ka was identical to WT (Figs. S2 and S3). Furthermore, the currents of the R351W mutant reported by Yu et al. (2010) were barely above background, casting serious doubts on the significance of this result.
Regarding the mechanism of the regulation of ClC-K channels by Ca2+ (and H+), three major questions have to be addressed in the future. First, what is the precise structure of the binding site? Second, what is the functional and structural nature of the conformational changes induced by Ca2+ and/or H+ binding? Lastly, what is the relationship of the modulation of CLC-K channels by Ca2+/H+ and the potentiation by NFA (Liantonio et al., 2006, 2008)? Regarding the latter point, we have previously identified a region on ClC-Ka where mutations strongly affect NFA sensitivity (Zifarelli et al., 2010). This region is distinct from the Ca2+-binding site identified here, and the less Ca2+-sensitive mutants E261Q and D278 do not affect the NFA sensitivity (Zifarelli et al., 2010). Nevertheless, it is not known whether Ca2+ (and H+) affects the same gating processes as does NFA.
A further important question is whether the regulation by Ca2+ and pH is of any physiological relevance. The normal plasma Ca2+ concentration ranges from 2.25 to 2.65 mM, 50% of which is in ionized form (i.e., the free concentration is ∼1.2–1.3 mM) (Vargas-Poussou et al., 2002). Several mechanisms regulate Ca2+ homeostasis. Bone, intestine, and kidney are the organs that determine the plasma Ca2+ level. Disorders in Ca2+ balance due to several diseases or pathological conditions are highly frequent (Houillier et al., 2006). Ca2+ is reabsorbed in the kidney, particularly in the proximal tubule (∼70%) and in the TAL through paracellular pathways (20%) (Frick and Bushinsky, 2003). The driving force for the paracellular flux of Ca2+ in the TAL is given by a transepithelial voltage gradient that is created by the basolateral secretion of Cl− and the apical secretion of K+ (Jeck et al., 2005). An impairment of Cl− transport leads to an abnormal Ca2+ absorption, implicating that Ca2+ reabsorption and CLC-K function are interrelated. Because CLC-Ks are modulated in the physiological range of Ca2+, and the interstitial Ca2+ level may vary in a wide range of conditions, we can speculate that this modulation is of physiological relevance. A critical test if Ca2+ regulation of CLC-K channels is physiologically relevant could be the analysis of animals that express Ca2+-insensitive CLC-K variants. In this respect, the single E261Q mutation has properties that are most favorable for a knock-in strategy because its overall properties are rather similar to the WT. In contrast, the D278N mutant and the E261Q/D278N double mutant show rather drastically reduced current levels.
The kidney is also critically involved in the maintenance of the acid-base balance. As for Ca2+, protons modulate CLC-Ks in the physiological range of pH. Unfortunately, the pH-insensitive mutation H497M shows a drastically reduced expression level. Thus, a direct test of the physiological relevance of the pH modulation using knock-in technology will be more difficult.
Lacking direct binding data, we can certainly not exclude that the mutations E261Q/D278N and H497M/E261Q/D278N abolish Ca2+ and H+ sensitivity of ClC-Ka in an indirect manner, for example, by prohibiting conformational changes associated with Ca2+ or H+ binding. However, we believe that our data provide rather strong evidence for a direct involvement of these residues in forming the respective Ca2+- and H+-binding sites because our mutational screen of externally accessible titratable or charged residues was exhaustive, without any other positive hits. Thus, if these mutations were not involved in Ca2+/H+ binding, the true binding site must be formed in an unconventional manner: Ca2+ would bind to uncharged residues, and H+ would bind to a non-titratable residue. We deem this possibility as unlikely.
In summary, we have identified two symmetrically repeated millimolar affinity Ca2+-binding sites on ClC-Ka that are localized at the dimer interface, each with contributions from both subunits. Our results provide a solid starting point for a detailed investigation of the precise structural basis and of the physiological relevance of the Ca2+ and pH sensitivity of ClC-K channels.
Acknowledgments
We thank T. Jentsch for providing all cDNA clones; Consuelo Murgia, Francesca Quartino, and Michela De Bellis for help in constructing some of the mutants; G. Gaggero for help in constructing the recording chamber; and Gianni Zifarelli for critical comments on the manuscript.
This work is supported by Telethon Italy (grant GGP08064) and the Italian “Ministero dell’Istruzione, dell’Università e della Ricerca” (grant MIUR PRIN 20078ZZMZW_002). The Compagnia San Paolo is gratefully acknowledged.
Christopher Miller served as editor.
Footnotes
Abbreviations used in this paper:
- NFA
- niflumic acid
- TAL
- thick ascending limb
- WT
- wild type
References
- Accardi A., Miller C. 2004. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 427:803–807 [DOI] [PubMed] [Google Scholar]
- Accardi A., Pusch M. 2003. Conformational changes in the pore of CLC-0. J. Gen. Physiol. 122:277–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arreola J., Begenisich T., Melvin J.E. 2002. Conformation-dependent regulation of inward rectifier chloride channel gating by extracellular protons. J. Physiol. 541:103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenhäger R., Otto E., Schürmann M.J., Vollmer M., Ruf E.M., Maier-Lutz I., Beekmann F., Fekete A., Omran H., Feldmann D., et al. 2001. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 29:310–314 [DOI] [PubMed] [Google Scholar]
- Chen M.F., Chen T.Y. 2001. Different fast-gate regulation by external Cl− and H+ of the muscle-type ClC chloride channels. J. Gen. Physiol. 118:23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R., Campbell E.B., Cadene M., Chait B.T., MacKinnon R. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294 [DOI] [PubMed] [Google Scholar]
- Dutzler R., Campbell E.B., MacKinnon R. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112 [DOI] [PubMed] [Google Scholar]
- Estévez R., Boettger T., Stein V., Birkenhäger R., Otto E., Hildebrandt F., Jentsch T.J. 2001. Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature. 414:558–561 [DOI] [PubMed] [Google Scholar]
- Frick K.K., Bushinsky D.A. 2003. Molecular mechanisms of primary hypercalciuria. J. Am. Soc. Nephrol. 14:1082–1095 [DOI] [PubMed] [Google Scholar]
- Friedrich T., Breiderhoff T., Jentsch T.J. 1999. Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J. Biol. Chem. 274:896–902 [DOI] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100 [DOI] [PubMed] [Google Scholar]
- Hanke W., Miller C. 1983. Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 82:25–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann S.H., Conti F. 1992. Nonstationary noise analysis and application to patch clamp recordings. Methods Enzymol. 207:131–148 [DOI] [PubMed] [Google Scholar]
- Houillier P., Froissart M., Maruani G., Blanchard A. 2006. What serum calcium can tell us and what it can’t. Nephrol. Dial. Transplant. 21:29–32 [DOI] [PubMed] [Google Scholar]
- Jeck N., Schlingmann K.P., Reinalter S.C., Kömhoff M., Peters M., Waldegger S., Seyberth H.W. 2005. Salt handling in the distal nephron: lessons learned from inherited human disorders. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288:R782–R795 [DOI] [PubMed] [Google Scholar]
- Jentsch T.J. 2005. Chloride transport in the kidney: lessons from human disease and knockout mice. J. Am. Soc. Nephrol. 16:1549–1561 [DOI] [PubMed] [Google Scholar]
- Jentsch T.J., Maritzen T., Zdebik A.A. 2005. Chloride channel diseases resulting from impaired transepithelial transport or vesicular function. J. Clin. Invest. 115:2039–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt S.E., Jentsch T.J. 1997. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 16:1582–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieferle S., Fong P., Bens M., Vandewalle A., Jentsch T.J. 1994. Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc. Natl. Acad. Sci. USA. 91:6943–6947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kürz L.L., Klink H., Jakob I., Kuchenbecker M., Benz S., Lehmann-Horn F., Rüdel R. 1999. Identification of three cysteines as targets for the Zn2+ blockade of the human skeletal muscle chloride channel. J. Biol. Chem. 274:11687–11692 [DOI] [PubMed] [Google Scholar]
- Liantonio A., Picollo A., Babini E., Carbonara G., Fracchiolla G., Loiodice F., Tortorella V., Pusch M., Camerino D.C. 2006. Activation and inhibition of kidney CLC-K chloride channels by fenamates. Mol. Pharmacol. 69:165–173 [DOI] [PubMed] [Google Scholar]
- Liantonio A., Picollo A., Carbonara G., Fracchiolla G., Tortorella P., Loiodice F., Laghezza A., Babini E., Zifarelli G., Pusch M., Camerino D.C. 2008. Molecular switch for CLC-K Cl− channel block/activation: optimal pharmacophoric requirements towards high-affinity ligands. Proc. Natl. Acad. Sci. USA. 105:1369–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduke M., Miller C., Mindell J.A. 2000. A decade of CLC chloride channels: structure, mechanism, and many unsettled questions. Annu. Rev. Biophys. Biomol. Struct. 29:411–438 [DOI] [PubMed] [Google Scholar]
- Martinez G.Q., Maduke M. 2008. A cytoplasmic domain mutation in ClC-Kb affects long-distance communication across the membrane. PLoS One. 3:e2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y., Uchida S., Kondo Y., Miyazaki H., Ko S.B., Hayama A., Morimoto T., Liu W., Arisawa M., Sasaki S., Marumo F. 1999. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat. Genet. 21:95–98 [DOI] [PubMed] [Google Scholar]
- Niemeyer M.I., Cid L.P., Yusef Y.R., Briones R., Sepúlveda F.V. 2009. Voltage-dependent and -independent titration of specific residues accounts for complex gating of a ClC chloride channel by extracellular protons. J. Physiol. 587:1387–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picollo A., Pusch M. 2005. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 436:420–423 [DOI] [PubMed] [Google Scholar]
- Picollo A., Liantonio A., Didonna M.P., Elia L., Camerino D.C., Pusch M. 2004. Molecular determinants of differential pore blocking of kidney CLC-K chloride channels. EMBO Rep. 5:584–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M., Steinmeyer K., Jentsch T.J. 1994. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys. J. 66:149–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickheit G., Maier H., Strenzke N., Andreescu C.E., De Zeeuw C.I., Muenscher A., Zdebik A.A., Jentsch T.J. 2008. Endocochlear potential depends on Cl− channels: mechanism underlying deafness in Bartter syndrome IV. EMBO J. 27:2907–2917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov G.Y., Pusch M., Astill D.S., Roberts M.L., Jentsch T.J., Bretag A.H. 1996. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J. Physiol. 497:423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C., Conti F., Pusch M. 1999. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 113:457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlingmann K.P., Konrad M., Jeck N., Waldegger P., Reinalter S.C., Holder M., Seyberth H.W., Waldegger S. 2004. Salt wasting and deafness resulting from mutations in two chloride channels. N. Engl. J. Med. 350:1314–1319 [DOI] [PubMed] [Google Scholar]
- Simon D.B., Bindra R.S., Mansfield T.A., Nelson-Williams C., Mendonca E., Stone R., Schurman S., Nayir A., Alpay H., Bakkaloglu A., et al. 1997. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 17:171–178 [DOI] [PubMed] [Google Scholar]
- Stühmer W. 1998. Electrophysiologic recordings from Xenopus oocytes. Methods Enzymol. 293:280–300 [DOI] [PubMed] [Google Scholar]
- Traverso S., Zifarelli G., Aiello R., Pusch M. 2006. Proton sensing of CLC-0 mutant E166D. J. Gen. Physiol. 127:51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida S., Sasaki S., Furukawa T., Hiraoka M., Imai T., Hirata Y., Marumo F. 1993. Molecular cloning of a chloride channel that is regulated by dehydration and expressed predominantly in kidney medulla. J. Biol. Chem. 268:3821–3824 [PubMed] [Google Scholar]
- Uchida S., Sasaki S., Nitta K., Uchida K., Horita S., Nihei H., Marumo F. 1995. Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J. Clin. Invest. 95:104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas-Poussou R., Huang C., Hulin P., Houillier P., Jeunemaître X., Paillard M., Planelles G., Déchaux M., Miller R.T., Antignac C. 2002. Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J. Am. Soc. Nephrol. 13:2259–2266 [DOI] [PubMed] [Google Scholar]
- Waldegger S., Jeck N., Barth P., Peters M., Vitzthum H., Wolf K., Kurtz A., Konrad M., Seyberth H.W. 2002. Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Arch. 444:411–418 [DOI] [PubMed] [Google Scholar]
- Yu Y., Xu C., Pan X., Ren H., Wang W., Meng X., Huang F., Chen N. 2010. Identification and functional analysis of novel mutations of the CLCNKB gene in Chinese patients with classic Bartter syndrome. Clin. Genet. 77:155–162 [DOI] [PubMed] [Google Scholar]
- Zifarelli G., Pusch M. 2009. Conversion of the 2 Cl(−)/1 H+ antiporter ClC-5 in a NO3(−)/H+ antiporter by a single point mutation. EMBO J. 28:175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zifarelli G., Pusch M. 2010. The role of protons in fast and slow gating of the Torpedo chloride channel ClC-0. Eur. Biophys. J. 39:869–875 [DOI] [PubMed] [Google Scholar]
- Zifarelli G., Murgia A.R., Soliani P., Pusch M. 2008. Intracellular proton regulation of ClC-0. J. Gen. Physiol. 132:185–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zifarelli G., Liantonio A., Gradogna A., Picollo A., Gramegna G., De Bellis M., Murgia A.R., Babini E., Camerino D.C., Pusch M. 2010. Identification of sites responsible for the potentiating effect of niflumic acid on ClC-Ka kidney chloride channels. Br. J. Pharmacol. 160:1652–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]