

Abstract
The pontocerebellar hypoplasias (PCH) are a group of early-onset, autosomal recessive disorders resulting in abnormal growth and function of the brainstem and cerebellum. PCH type 2 (PCH2) is characterized by respiratory and feeding difficulties at birth, extrapyramidal dyskinesia, severe developmental impairment, progressive microcephaly and frequent death in childhood. Neuropathologic findings include diffuse cerebral gliosis with white matter changes, hypoplastic pons with depletion of neurons in the pontine nuclei, hypoplastic cerebellar hemispheres due to short cerebellar folia with poor branching, segmental loss of dentate, inferior olivary, and ventral pontine nuclei, and near absence of transverse pontine fibers with preservation of long fiber tracts and spinal anterior horn cells. On brain imaging, the cerebellar hemispheres appear very flat, and are more severely involved than the vermis. Most patients with PCH2 have mutations in TSEN54, with occasional mutations found in TSEN34 or TSEN2, genes that encode subunits of tRNA splicing endonuclease. Although this is a congenital disorder of pontocerebellar dysgenesis with fetal onset of neurodegeneration and symptoms at birth, prenatal imaging is unreliable in diagnosing this disorder in utero. We report on IVF dizygous twins with detailed prenatal imaging that failed to reveal any cerebellar abnormalities. Direct sequence analysis of TSEN54 showed homozygosity for c.919G>T, the common founder mutation in most PCH2 patients, and both parents were heterozygous for this mutation. We found no evidence of cerebellar dysgenesis on prenatal ultrasounds, but MRI tractography showed absence of pontine crossing fibers, a unique feature that might be useful for prenatal diagnosis of this condition.
Keywords: cerebellar hypoplasia, neurogenetics, prenatal diagnosis, neuroimaging, autosomal recessive, TSEN54 mutation, tRNA-splicing endonuclease, microcephaly, dyskinesia
INTRODUCTION
Several types of PCH have been described in the literature, all with variable neuropathologic characteristics and differing genetic loci. PCH2 is characterized by respiratory and feeding difficulties at birth, extrapyramidal dyskinesia, severe developmental impairment, progressive microcephaly and frequent death in childhood [Coppola et al., 2000; Chaves-Vischer et al., 2000; Steinlin et al., 2007]. At term, MRI imaging in PCH2 demonstrates mild brainstem hypoplasia and diffuse cerebellar hypoplasia with flattened cerebellar hemipheres that are more severely affected than the vermis, while second trimester brain ultrasounds are normal [Goasdoue et al., 2001]. Later in the first year, repeat brain-imaging shows more severe brainstem and cerebellar hypoplasia with cerebral atrophy, hence its designation as an inherited neurodegenerative disorder with fetal onset [Barth, 1993; Barth et al., 1995; Goasdoue et al, 2001; Steinlin et al., 2007; Barth et al., 2007]. PCH2 appears to be the most common cause of hemisphere predominant cerebellar hypoplasia. Neuropathology findings include diffuse cerebral gliosis and white matter changes, hypoplastic pons with depletion of neurons in the pontine nuclei, hypoplastic cerebellar hemispheres with short cerebellar folia with poor branching, segmental loss of dentate, inferior olivary, and ventral pontine nuclei, and near absence of transverse pontine fibers with sparing of anterior spinal horn cells [Barth et al., 2007]. In this report, we describe how MRI tractography demonstrated absence of cerebellar crossing fibers, which appears to be unique to PCH2.
Cerebellar foliar outgrowth begins at 25 weeks gestation in the human fetus, and the mature form of the inferior olivary nucleus does not become apparent until 22–27 weeks, with 5 out of 6 autopsies of PCH2 showing an immature folding pattern of this nucleus, and no patients showing a mature folding pattern of the dentate nucleus [Barth et al., 2007]. There are similar but more severe neuropathological changes in PCH4, such as much less foliar development, suggesting PCH2 and PCH4 are part of the same disease process, which initially affects the brainstem and cerebellum during the late second trimester [Barth et al., 2007; Leroy et al., 2007; Hevner, 2007]. Recently, mutations in several t-RNA-splicing endonuclease subunit genes have been found to be associated with PCH2, with a specific founder mutation 919G>T in TSEN54 causing most European cases of PCH2 and PCH4, suggesting that these disorders are allelic [Budde et al., 2008]. This gene is highly expressed in pontine neurons, as well as in cerebellar dentate and olivary nuclei during the late second trimester [Budde et al., 2008]. Interestingly, though PCH2 appears to be a congenital disorder of pontocerebellar neurodegeneration with fetal onset and neurological symptoms seen shortly after birth, clinically significant abnormalities are not routinely seen during the second trimester with conventional prenatal imaging [Goasdoue et al., 2001; Steinlin et al., 2007].
In this report we present prenatal and postnatal imaging of dizygous twins with PCH2. We confirm that at the present time, prenatal ultrasonography cannot reliably diagnose PCH2, especially early in gestation. Indeed, it appears that the characteristic imaging findings of pontocerebellar dysgenesis, supratentorial atrophy and delayed myelination found in this condition develop after 30 weeks gestation, resulting in progressive microcephaly, dystonia, and deterioration of neurologic function from birth. This is consistent with Barth’s classification of PCH2 as an inherited neurodegenerative disorder with late fetal onset.
CLINICAL REPORT
A 37-year-old gravida 1 para 0 woman was referred for prenatal diagnosis after successful IVF resulted in a twin pregnancy derived from 4 implanted embryos, which were conceived using the mother’s eggs and the 35-year-old father’s sperm. Ultrasonographic evaluations beginning at 16 weeks (from LMP) showed a normal dichorionic, diamniotic twin pregnancy. A detailed ultrasound evaluation at 20 weeks gestation revealed normal anatomy and no central nervous system abnormalities were seen (Fig 1). Transverse cerebellar diameter at 20 weeks was 2.1/2.1 cm (75th centile) and cisterna magna measurement was 0.7/0.7 cm (75th centile) for Twin A and B respectively. Routine maternal serum screening was normal, and no invasive testing, such as CVS or amniocentesis, was performed. Subsequent ultrasounds at 23, 25 and 29 weeks gestation continued to show normal growth in both twins. There was normal movement and fluid, and a normal-appearing posterior fossa, although no measurements were recorded. At 30 4/7 weeks, the mother presented to labor and delivery with spontaneous rupture of membranes. She underwent a course of antenatal corticosteroids and delivered the next day via cesarean section.
Figure 1.

Transaxial sections of Twin A (A) and Twin B (B) show cerebellar tissues to be within normal range at 20 weeks post-LMP. Note the normal proportion of the cisterna magna to the cerebellum. Measurements of the transverse cerebellar (TC) diameters at were 2.1/2.1cm (75th centile) and cisterna magna (CM) measurements were 0.7/0.7cm (75th centile) for Twin A (figure A) and Twin B (figure B) respectively.
At birth, Apgar scores were 1,6 and 7 for twin A, and 7 and 9 for twin B. Weights were 1510g and 1550g respectively (50th centile for gestational age). Both premature infants were admitted to the neonatal ICU. The infants manifested mild respiratory distress syndrome, poor feeding, apneas and intermittent bradycardia. The twins were normocephalic and not dysmorphic, with a head circumference of 28cm (50th centile) and 28cm (50th centile) and length of 41cm (50th centile) and 42cm (50th centile), respectively. Twin A was also found to have a mild left-sided hydronephrosis. Routine brain ultrasounds performed on Day of Life 4 (DOL#4) (31 weeks from LMP) and DOL#21 (34 weeks from LMP) were read as normal. Only on careful review, after the diagnosis of PCH2 was made, was the presence of subtle cerebellar hypoplasia appreciated (Fig 2). During their stay in the NICU, both infants remained extremely jittery. These jittery movements involved their arms and legs, and first appeared at around age 2 weeks of life (32 weeks from LMP). These small to medium amplitude, oscillatory movements of the limbs were initiated by stretching or moving their limbs and lasted variable periods of time from seconds to several minutes. These movements tended to be symmetric and were dampened or stopped by gentile tactile pressure and were generally not present at rest. This jitteriness persisted throughout the entire NICU stay and continued after discharge.
Figure 2.

Coronal sections from neonatal ultrasounds at 31 weeks (top) and 34 weeks (bottom) post-LMP in Twin A (left) and Twin B (right) reveal subtle cerebellar hypoplasia. Note empty cistern (marked with an arrow) in the lower half of the posterior fossa due to absent cerebellar tissue (remaining cerebellar hemispheres marked with asterisks).
EEG studies were performed on both infants at 10 weeks of life were normal. A CT scan performed on both twins at 9 weeks of life showed normal ventricles and cortical sulci, but underdeveloped cerebellar hemispheres with normal appearing vermi and posterior fossa fluid collections. Subsequent MRIs on both twins showed nearly identical results: hypoplastic cerebellar hemispheres with relative sparing of the vermis. The coronal views showed characteristic winging of the cerebellar hemispheres consistent with PCH2. The brainstem, especially the pons, of both patients also appeared hypoplastic with an empty lateral posterior fossa (Fig 3). Based on the neurologic, CT and MRI findings, and given that the findings in both dizygotic twins were nearly identical, a diagnosis of PCH2 was made. The Promega Powerplex16 kit was used to confirm that the twins were dizygotic.
Figure 3.
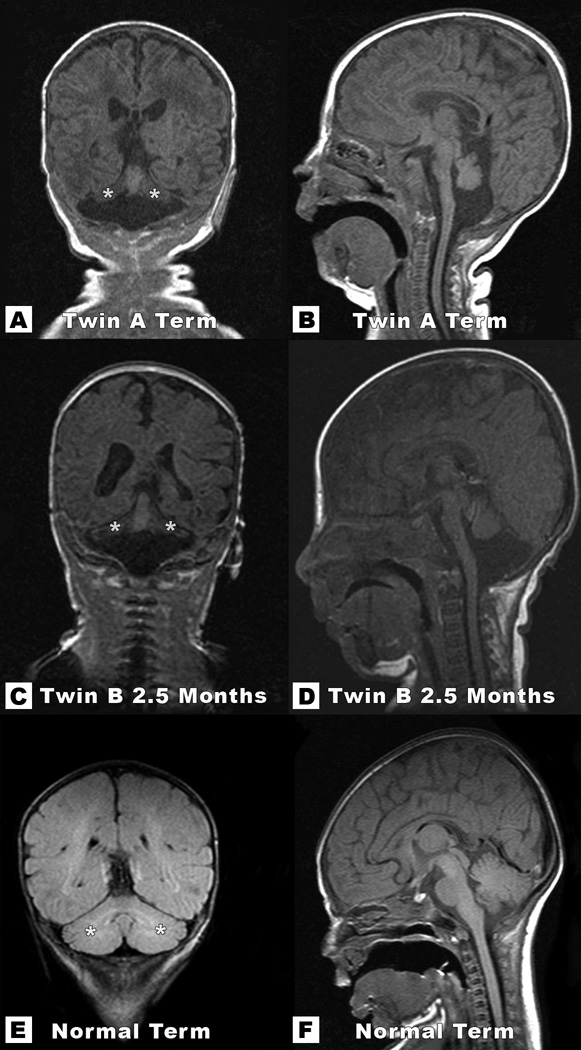
Coronal T1-weighted images of Twin A at 2.5 months of life (A and B) and Twin B at 5 months, 2.5 months corrected for prematurity (C and D) depict severe hypoplasia of both cerebellar hemispheres. Note the typical “winging” appearance of cerebellar hemispheres (marked with asterisks), which are compared with similar images in a term neonate (E and F). Mid sagittal T1-weighted images of each twin (Figures B and D) show significant volume loss of the pons and relative preservation of the vermis, with much better preservation of the cerebellar vermis than the cerebellar hemispheres.
After discharge, both twins became progressively microcephalic. Head circumference curves for both twins crossed percentiles downward from (50th centile) at birth to (<3rd centile) at 6 months. The head circumferences for the twins measured 38.5 cm and 39 cm at 6 months (-3 standard deviations for corrected age). Weights were 5.4 Kg and 5.8Kg respectively (10th centile for corrected age) and lengths were 62.5cm and 63cm respectively (both 25th centile). At 4 months their limb movements became more restless and less oscillatory. By 14 months, hypotonia and dyskinesia, with features of chorea, athetosis and dystonia, dominated their motor profile. Neither brother was able to sit, crawl or grasp objects. They made no attempts to reach for objects, though they tracked faces and appeared to see normally.
Since the differential diagnosis for cerebellar hypoplasia is quite varied, additional diagnostic testing was performed. Chromosomal microarray analysis of 1542 loci using 4685 BAC clones, which included the subtelomeres, pericentromeric regions and known genetic syndromes detected no abnormalities in Twin A. Joubert syndrome and congenital disorders of glycosylation were both considered, and an ophthalmologic evaluation and serum transferrin isoelectric focusing were both found to be normal. Once these preliminary tests were completed, based on their clinical and imaging findings, definitive testing for PCH2 was undertaken. Direct sequence analysis of TSEN54 showed homozygosity for a c.919G>T. This mutation results in an amino acid substitution of alanine for serine at codon 307 (A307S), and it has previously been described in a Northern European subset of patients with PCH2 [Budde et al., 2008]. As the parents of the twins were of Northern European decent, this c.919G>T mutation is consistent with the diagnosis of PCH2. Additionally, both parents were tested and found to be heterozygous for this mutation. As part of a research protocol at our institution, MRI tractography was performed during the MRI at age 2.5 months on Twin A and demonstrated absence of cerebellar crossing fibers (Fig 4), which appears to be unique to this disorder.
Figure 4.

Comparison of an age-matched control (right) with Twin A at 2.5 months (left). Fractional Anisotropy (FA) axial map of Twin A (Figure A) and coronal tractography (Figure C) outline attenuated corticospinal tracts and absent transverse fibers. FA of the age-matched control (Figure B) shows normal pontocerebellar transverse fibers traveling in X- axis depicted in red color. Coronal tractography map of a normal control (Figure D) outlines normal corticospinal fibers in blue and transverse fibers in red. Note the absent transverse pontine fibers with an axon stain in brainstem neuropathology from a 15-year-old patient with PCH2 (E), compared with normal transverse pontine fibers from an 8-year-old normal control (figures courtesy of Professor Peter G. Barth, Department of Neuropathology, Academic Medical Centre, University of Amsterdam. The Netherlands).
DISCUSSION
Pontocerebellar hypoplasia (PCH) represents a group of autosomal recessive neurodegenerative disorders that present with unique and overlapping clinical, pathological and molecular features. Due to the similar appearance of these disorders on imaging studies, their clinical, pathological and molecular differences are crucial for diagnosis (Table I). Initial classification of PCH distinguished type 1 and type 2 [Barth, 1993]. On imaging, PCH type 1 and 2 both exhibit cerebellar hypoplasia, particularly affecting the cerebellar hemispheres and sparing the vermis, as well as hypoplasia of the ventral pons. The cerebellar hemispheres of patients with PCH2 are often described as “bat wings” or “butterfly cerebellum”. Histologically, PCH1 is unique, consisting of loss of spinal anterior horn cells, similar to that seen in spinal muscular atrophy type 1. Clinically, patients with PCH1 present with polyhydramnios, joint contractures, neonatal hypotonia, arreflexia, poor respiratory effort, swallowing difficulty and early death. Recently, there have been reports of prolonged survival in PCH1 [Kalpana et al., 2009], and another recent report described an Ashkenazi Jewish family with PCH1 caused by a mutation in the VRK1 gene [Renbaum et al., 2009].
TABLE 1.
Types of Ponto-Cerebellar Hypoplasia (PCH): Clinical, Pathological and Molecular Differences.
| Clinical | Pathological | Gene | |
|---|---|---|---|
| PCH1 | Neonatal seizures, jerky movements, seizures, microcephaly at birth, early death |
Anterior horn cell degeneration, cerebellar hypoplasia with sparing of the vermis [Barth, 1993] |
VRK1 [Renbaum et al., 2009] |
| PCH2 | Hyperkinetic movements Mixed dyskinesia, spasticity, microcephaly after birth, longer survival |
Absent cerebellar crossing fibers, cerebellar hypoplasia with sparing of the vermis [Barth 1993] |
TSEN2, 34, 54 [Budde et al. 2008] |
| PCH3 | Hypotonia, hyper-reflexia, microcephaly, seizures, optic atrophy |
Hypoplasia of both cerebellar hemispheres and vermis without progressive encephalopathy |
Link 7q11-21 [Rajab et al., 2003] |
| PCH4 | Polyhydramnios, joint contractures, hypertonia early lethality |
Absent cerebellar crossing fibers, cerebellar hypoplasia with sparing of the vermis and C-shaped inferior olives. [Barth et al., 2007] |
TSEN54 [Budde et al., 2008] |
| PCH5 | Fetal seizures, cerebellar hypoplasia evident in second trimester (Patel et al., 2006) |
Vermis hypoplasia worse than cerebellar hemispheres |
Unknown |
| PCH6 | Severe cerebral atrophy, hypotonia, elevated lactate, variable multiple respiratory chain defects, edematous hands, feet |
Pontocerebellar atrophy affecting both cerebellum and vermis with progressive white matter loss |
RARS2 [Edvardson, et al. 2007] |
Most neonates with PCH2 are normocephalic at birth and develop progressive microcephaly [Coppola et al., 2000; Steinlin et al, 2007]. They occasionally have respiratory and sucking difficulties, some develop spasticity, and more than half go on to have a seizure disorder, which is initially difficult to distinguish from their movement disorder [Steinlin et al, 2007]. Patients with PCH2 are not dysmorphic, and unlike other types of pontocerebellar hypoplasia, they manifest early onset of hyperkinetic movements (jitteriness) with features of chorea, athetosis and some dystonia. This mixed dyskinesia is a distinguishing feature of PCH2. Remarkably, of the 24 patients reported by Steinlin et al. [2007], none of the prenatal ultrasounds performed on 14 women during the second half of their gestation showed any posterior fossa abnormality. One woman also had an MRI at 22 weeks, which was normal, Goasdoue et al. [2001] reported 5 patients with characteristic MRI findings for PCH2, all of which had normal prenatal ultrasounds between 22 and 25 weeks, while one of the 5 mothers had a subsequent ultrasound at 30 weeks, which showed cerebellar hypoplasia.
Recently, mutations in three of the four different subunits of the tRNA-splicing endonuclease complex were found in patients with PCH type 2 and 4 [Budde et al., 2008], confirming the long standing suspicion that these two types of pontocerebellar hypoplasia may represent varying severity along the same phenotypic spectrum. PCH4, also known as olivopontocerebellar hypoplasia, presents prenatally with polyhydramnios, joint contractures and neonatal hypertonia. The clinical course of PCH4 is significantly more severe and usually leads to early lethality, but both PCH2 and PCH4 are caused by mutations in TSEN54 (a non-catalytic subunit of tRNA splicing endonuclease). PCH2 can also be caused by mutations in TSEN2 and TSEN34 (catalytic subunits of tRNA splicing endonuclease) [Budde et al., 2008]. The cerebellar folia are less-developed than in PCH2, and the inferior olives are C-shaped in PCH4, which is consistent with the more rapid clinical course. PCH4 is usually caused by heterozygous mutations consisting of the common A307S missense mutation and a stopcodon. Since reduced function of TSEN54 causes the disease, two missense mutations are expected to generate a milder phenotype than a compound heterozygote, with a missense mutation together with a null allele [Budde et al., 2008]. Thus the genetic studies and the neuropathology findings suggest differing severity in allelic disorders [Barth et al., 2007; Leroy et al., 2007; Hevner, 2007; Budde et al., 2008].
PCH3 has been reported in a consanguineous family, which presented clinically with neonatal hypotonia, microcephaly, hyper-reflexia, seizure disorder, short stature, and unique optic atrophy [Rajab et al., 2003]. In this family, the locus for this gene was linked to chromosome 7q11-21. Durmaz et al. [2009] recently reported a second family with PCH3. At age 3 years, the index patient manifested seizures with two posterior epileptic foci on EEG, and MRI showed hypoplasia of the cerebellum, brainstem and cerebrum with associated hypoplasia of the corpus callosum and hearing loss and optic atrophy confirmed by auditory and visual evoked responses. Molecular analysis revealed a homozygous haplotype in the original critical region on 7q11.4 and narrowed the critical region from 20 Mb to 7 Mb [Durmaz et al., 2009]. These cases showed hypoplasia of both the cerebellar hemispheres and the vermis, without any progressive encephalopathy, suggesting that this disorder would be better classified as diffuse cerebellar hypoplasia rather than PCH, and also suggesting an alternate pathophysiology from PCH1 and PCH2/4.
PCH5 has also only been described in a single family with three affected siblings. This disorder presented with fetal onset of seizure-like activity, and uniquely, worse hypoplasia or atrophy of the cerebellar vermis than the hemispheres [Patel et al., 2006]. Due to fetal presentation and the occurrence of multiple affected siblings in one family, these authors were able to do extensive prenatal monitoring and delineate abnormal cerebellar development and progression during second and third trimesters. Autopsies at 20, 27, and 37 weeks gestation showed diffuse atrophy that was most marked for the cerebellum and brain stem structures. Prenatal ultrasounds at 16–18 weeks gestation demonstrated diminished cerebellar width, and this feature was used for prenatal diagnosis. Neuropathological abnormalities included dysplastic inferior olivary nuclei, absent or immature dentate nuclei, and cell paucity more marked for the cerebellar vermis than the hemispheres. In contrast to the hypoplasia of the cerebellar hemispheres seen in PCH type 2, the hypoplasia in PCH5 was more marked in the cerebellar vermis than the hemispheres. Based on these critical neuropathologic differences PCH5 clearly differs from PCH2/4.
PCH6 was recently described in three affected siblings of consanguineous family who presented with severe infantile encephalopathy with rapidly progressive pontocerebellar hypoplasia and multiple mitochondrial respiratory chain defects [Edvardson et al., 2007]. This is an autosomal recessive disorder resulting from homozygous mutations in one of the components of the nuclear encoded mitochondrial translation machinery (mitochondrial arginyl-transfer RNA (tRNA) synthase, or RARS2). These PCH6 patients presented with severe generalized hypotonia, elevated plasma and CSF lactate, and multiple mitochondrial respiratory chain defects due to a marked reduction of mitochondrial tRNA transcripts in the patient’s fibroblasts. The neonatal MRIs showed rapidly progressive pontocerebellar atrophy that affected both the cerebellum and the vermis, followed by parenchymal loss over the first few months of life that affected mostly the white matter, with relative sparing of the basal ganglia and cortex. The course was quite rapid leading to early demise during the first 2 years, but fetal brain ultrasonography at 20 weeks in the third affected sibling was normal. A second family with PCH6 was recently reported by Rankin et al. [2010].
This case of dizygous twins presented above exemplifies the difficulty in diagnosing PCH2 prenatally. The transverse cerebellar diameters in both twins were within standard observational limits at 20 weeks. Furthermore, ultrasound surveys at 23, 25 and 29 weeks did not show any obvious signs of hypoplasia in the cerebellar hemisheres or evidence of a mega cisterna magna. Postnatal brain ultrasounds performed at 31 weeks and 34 weeks post-LMP showed subtle loss of cerebellar tissue, which was not noted on the initial reports regarding these brain ultrasound findings. This underscores the problem of using prenatal sonographic imaging to identify abnormalities in the posterior fossa. Only at age 9 weeks (40 weeks post-LMP) did a CT scan demonstrate discernible cerebellar hypoplasia. Nomograms for transverse cerebellar diameters during the second trimester have been in clinical use for several decades [Smith, 1986], with a subsequent report of measurements of cerebellar foliation in prematurely born infants at 27 and 34 weeks [Korsten, et al., 2006]. Chavez et al. [2003] reported a fetal transcerebellar diameter nomogram with special emphasis on the third trimester because the cerebellar structures develop late in gestation making prenatal diagnosis of cerebellar hypoplasia extremely difficult. Despite development of such nomograms, prenatal diagnosis of PCH remains a subject of considerable debate, primarily because of the late development of cerebellar structures [Boltshauser, 2004: Kapur et al., 2009].
Cerebellar volume reduction can be a complication in extreme prematurity with very-low birth-weight infants born at less than 32 weeks of gestation, especially if the neonatal course is complicated by hemodynamic instability, supratentorial hemorrhagic lesions, infarcts, and/or sepsis [Messerschmidt et al., 2008]. Zelnik et al. [1996] reported monozygotic female twins with fetal-fetal transfusion in which the donor twin had pontocerebellar hypoplasia, while the recipient twin did not. Data provided by Snijers, et. al. [1994] suggests that in the case of infants with IUGR, transverse cerebellar diameters less than 2 standard deviations may suggest a poor perinatal outcome. Since this effect is most likely associated with decreased cerebellar perfusion, these findings suggest that not all cases of pontocerebellar hypoplasia have a genetic basis.
In the 30-week premature monozygotic twins with PCH2/4 reported by Chavez-Vischer et al. [2000], cranial ultrasound at 31 weeks showed cerebellar hypoplasia with a large cysterna magna, and neuropathology in both twins showed marked hypoplasia of the cerebellum and pons, moderate diffuse cerebral gliosis, depletion of neurons in pontine nuclei (especially the dentate and inferior olivary nuclei), absent transverse cereballar crossing fibers, and simplified cerebellar folia with normal spinal cord pathology. Ajibola, et al. [2009] reported a recurrence of pontocerebellar hypoplasia in a consanguineous family that was followed prenatally with measurements of fetal transcerebellar diameter (TCD) that did not fall significantly below the 5th centile until after 31 weeks gestation using curves developed by Chavez et al. [2003]. Based on previous reports, it appears that cerebellar degeneration in PCH2 is subtle and begins at the end of the second trimester. Even when PCH2 is known to be running in the family, second trimester ultrasonography appears to be normal.
Recently, diffusion tensor imaging (DTI) and tractography has been found to be a viable tool in evaluating the main projections and commissural pathways of human fetuses [Kasprian, et al. 2008]. Given the well-described loss of ventral pontine neurons and transverse pontine crossing fibers, we used this new technology to visualize the loss of these tracts. MRI tractography in one of these twins demonstrated absent crossing fibers at the level of the pons (Fig 4) at age 2.5 months (approximately 40 weeks post-LMP). Based on these findings, we feel that performing DTI and tractography in addition to conventional fetal MRI studies may have potential to assist in making this diagnosis prenatally, especially since absence of transverse pontine crossing fibers was previously documented in neuropathology from deceased monozygotic twins with PCH2/4 at 37 weeks post-conception [Chaves-Vischer et al., 2000]. Whether these findings can be reliably demonstrated prenatally remains to be proven. Until that time, secure prenatal diagnosis will require mutation analysis.
Since all types of pontocerebellar hypoplasia are inherited in an autosomal recessive fashion and result in devastating outcomes, prenatal diagnosis is crucial. Unfortunately, current prenatal second trimester imaging modalities appear to be inadequate to rule out a recurrence. We suggest that in the setting of a significant risk for recurrence of PCH2, a more detailed evaluation of the fetal brain be undertaken with transvaginal high-resolution ultrasound and MRI with DTI and tractography. In cases with a previous history of cerebellar hypoplasia, genetic evaluations should be undertaken with the hope that early prenatal diagnosis can be offered based on DNA findings.
ACKNOWLEDGMENTS
AHS and JMG appreciate support from SHARE's Childhood Disability Center, the Steven Spielberg Pediatric Research Center, the NIH/NICHD Program Project Grant (HD22657), and the Medical Genetics NIH/NIGMS Training Program Grant (5-T32-GM08243). This work was also partly supported by a Hersenstichting Grant (2009-81) to FB, and a NIH grant 1R01-NS050375 to WBD.
REFERENCES
- Ajibola AJ, Netzloff M, Samaraweera R, Omar SA. Two Cases of Pontocerebellar Hypoplasia: Ethical and Prenatal Diagnostic Dilemma. Am J Perinatol. 2009. 2009 Jul 30; doi: 10.1055/s-0029-1234030. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Barth PG, Aronica E, de Vries L, Nikkels PGJ, Scheper W, Hoozemans JJ, Poll-The BT, Troost D. Pontocerebellar hypoplasia type 2: a neuropathological update. Acta Neuropathol. 2007;114:373–386. doi: 10.1007/s00401-007-0263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth PG, Blennow G, Lenard HG, Begeer JH, van der Kley JM, Hanefeld F, Peters AC, Valk J. The syndrome of autosomal recessive pontocerebellar hypoplasia, microcephaly, and extrapyramidal dyskinesia (pontocerebellar hypoplasia type 2): compiled data from 10 pedigrees. Neurology. 1995;45:311–317. doi: 10.1212/wnl.45.2.311. [DOI] [PubMed] [Google Scholar]
- Barth PG. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993;15:411–422. doi: 10.1016/0387-7604(93)90080-r. [DOI] [PubMed] [Google Scholar]
- Boltshauser E. Cerebellum-small brain but large confusion: a review of selected cerebellar malformations and disruptions. Am J Med Genet A. 2004;126A:376–385. doi: 10.1002/ajmg.a.20662. [DOI] [PubMed] [Google Scholar]
- Budde BS, Namavar Y, Barth PG, Poll-The BT, Nürnberg G, Becker C, van Ruissen F, Weterman MA, Fluiter K, te Beek ET, Aronica E, van der Knaap MS, Höhne W, Toliat MR, Crow YJ, Steinling M, Voit T, Roelenso F, Brussel W, Brockmann K, Kyllerman M, Boltshauser E, Hammersen G, Willemsen M, Basel-Vanagaite L, Krägeloh-Mann I, de Vries LS, Sztriha L, Muntoni F, Ferrie CD, Battini R, Hennekam RC, Grillo E, Beemer FA, Stoets LM, Wollnik B, Nürnberg P, Baas F. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet. 2008;40:1113–1118. doi: 10.1038/ng.204. [DOI] [PubMed] [Google Scholar]
- Chaves-Vischer V, Pizzolato GP, Hanquinet S, Maret A, Bottani A, Haenggeli CA. Early fatal pontocerebellar hypoplasia in premature twin sisters. Eur J Paediatr Neurol. 2000;4:171–176. doi: 10.1053/ejpn.2000.0295. [DOI] [PubMed] [Google Scholar]
- Chavez MR, Ananth CV, Smulian JC, Lashley S, Kontopoulos EV, Vintzileos AM. Fetal transcerebellar diameter nomogram in singleton gestations with special emphasis in the third trimester: a comparison with previously published nomograms. Am J Obstet Gynecol. 2003;189:1021–1025. doi: 10.1067/s0002-9378(03)00894-9. [DOI] [PubMed] [Google Scholar]
- Coppola G, Muras I, Pascotto A. Pontocerebellar hypoplasia type 2 (PCH2): report of two siblings. Brain Dev. 2000;22:188–192. doi: 10.1016/s0387-7604(00)00093-0. [DOI] [PubMed] [Google Scholar]
- Durmaz B, Wollnik B, Cogulu O, Li Y, Tekgul H, Hazan F, Ozkinay F. Pontocerebellar hypoplasia type III (CLAM): Extended phenotype and novel molecular findings. J.Neurol. 2009;256:416–419. doi: 10.1007/s00415-009-0094-0. [DOI] [PubMed] [Google Scholar]
- Edvardson S, Shaag A, Kolesnikova O, Gomori JM, Tarassov I, Einbinder T, Saada A, Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goasdoue P, Rodriquez D, Moutard M-L, Robain O, Lalande G, Adamsbaum C. Pontocerebellar hypoplasia; definition of MR features. Pediatr Radiol. 2001;31:613–618. doi: 10.1007/s002470100507. [DOI] [PubMed] [Google Scholar]
- Hevner RF. Progress on pontocerebellar hypoplasia. Acta Neuropathol. 2007;114:401–402. doi: 10.1007/s00401-007-0282-x. [DOI] [PubMed] [Google Scholar]
- Kalpana D, Parvathy L, Ahamed SM, Iype M, Kunju MP. A mild variant of pontocerebellar hypoplasia type 1 in a 12-year-old Indian boy. Pediatr Neurol. 2009;40:302–305. doi: 10.1016/j.pediatrneurol.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Kapur RP, Mahoney BS, Finch L, Siebert JR. Normal and abnormal anatomy of the cerebellar vermis in midgestational human fetuses. Birth Defects Research (Part A) 2009;85:700–709. doi: 10.1002/bdra.20589. [DOI] [PubMed] [Google Scholar]
- Kasprian G. In utero tractography of fetal white matter development. Neuroimage. 2008;43:213–224. doi: 10.1016/j.neuroimage.2008.07.026. [DOI] [PubMed] [Google Scholar]
- Korsten A, Lequin M, Govaert P. Sonographic maturation of third-trimester cerebellar foliation after birth. Pediatric Research. 2006;59:695–699. doi: 10.1203/01.pdr.0000214991.07965.0f. [DOI] [PubMed] [Google Scholar]
- Leroy JG, Lyon G, Fallet C, Amiel J, De Praeter C, Van Den Broecke C, Vanhaesebrouck P. Congenital pontocerebellar atrophy and telencephalic defects in three siblings: a new subtype. Acta Neuropathol. 2007;114:387–399. doi: 10.1007/s00401-007-0248-z. [DOI] [PubMed] [Google Scholar]
- Messerschmidt A, Prayer D, Brugger PC, Boltshauser E, Zoder G, Sterniste W, Pollak A, Weber M, Birnbacher R. Preterm birth and disruptive cerebellar development: assessment of perinatal risk factors. Eur J Paediatr Neurol. 2008;12:455–460. doi: 10.1016/j.ejpn.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Patel MS, Becker LE, Toi A, Armstrong DL, Chitayat D. Severe, fetal-onset form of olivopontocerebellar hypoplasia in three sibs: PCH type 5? Am J Med Genet Part A. 2006;140A:594–603. doi: 10.1002/ajmg.a.31095. [DOI] [PubMed] [Google Scholar]
- Rajab A, Mochida GH, Hill A, Ganesh V, Bodell A, Riaz A, Grant PE, Shugart YY, Walsh CA. A novel form of pontocerebellar hypoplasia maps to chromosome 7q11-21. Neurology. 2003;60:1664–1667. doi: 10.1212/01.wnl.0000068548.58498.41. [DOI] [PubMed] [Google Scholar]
- Rankin J, Brown R, Dobyns W, Harington J, Patel J, Quinn M, Brown G. Pontocerebellar hypoplasia type 6: a British case with no respiratory chain defects in muscle. Am. J. Med. Genet. Part A. 2010 doi: 10.1002/ajmg.a.33531. In press. [DOI] [PubMed] [Google Scholar]
- Renbaum P, Kellerman E, Jaron R, Geiger D, Segel R, Lee M, King MC, Levy-Lahad E. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet. 2009;85:281–289. doi: 10.1016/j.ajhg.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders RJM, De Courcy-Wheeler RHB, Nicolaides KH. Intrauterine growth retardation and fetal transvers cerebellar diameter. Prenatal Diagnosis. 1994;14:1101–1105. doi: 10.1002/pd.1970141202. [DOI] [PubMed] [Google Scholar]
- Smith PA. Prenatal measurement of the fetal cerebellum and cisterna cerebellomedullaris by ultrasound. Prenatal Diagnosis. 1986;6:133–141. doi: 10.1002/pd.1970060209. [DOI] [PubMed] [Google Scholar]
- Steinlin M, Klein A, Haas-Lude K, Zafeiriou D, Strozzi S, Müller T, Gubser-Mercati D, Schmitt Mechelke T, Krägeloh-Mann I, Boltshauser E. Pontocerebellar hypoplasia type 2: variability in clinical and imaging findings. Eur J Paediatr Neurol. 2007;11:146–152. doi: 10.1016/j.ejpn.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Zelnik N, Dobyns WB, Forem SL, Kolodny EH. Congenital pontocerebellar atrophy in three patients: clinical, radiologic and etiologic considerations. Neuroradiology. 1996;38:684–687. doi: 10.1007/s002340050334. [DOI] [PubMed] [Google Scholar]