

Summary
Enhanced mesenchymal expression of FGF10 led to the formation of multifocal PIN or prostate cancer. Inhibition of epithelial FGFR1 signaling using DN FGFR1 led to reversal of the cancer phenotype. A subset of the FGF10-induced carcinoma was serially transplantable. Paracrine FGF10 led to an increase in epithelial androgen receptor and synergized with cell autonomous activated AKT. Our observations indicate that stromal FGF10 expression may facilitate the multifocal histology observed in prostate adenocarcinoma and suggest the FGF10/FGFR1 axis as a potential therapeutic target in treating hormone sensitive or refractory prostate cancer. We also show that transient exposure to a paracrine growth factor maybe sufficient for the initiation of oncogenic transformation.
Significance
Mechanisms underlying the multifocal nature of human prostate cancer are poorly understood. Using an in vivo reconstitution system that relies on epithelial-stromal interactions, we show that enhanced expression of mesenchymal FGF10, an essential gene for prostate development, led to the formation of well differentiated, multifocal prostate adenocarcinoma. Paracrine FGF10 signaling caused intrinsic changes in adjacent adult wild type epithelium, demonstrated by an increase in epithelial androgen receptor (AR) expression, and the development of hormone refractory disease. FGF10-induced PIN or adenocarcinoma could persist in an FGF10 low microenvironment, likely as a result of sustained epithelial changes that evade chronic dependence on this growth factor. We demonstrate a mechanism for the formation of multifocal prostate cancer.
Introduction
The multifocal nature of human prostate cancer is elusive and mechanisms accounting for this histologic presentation are poorly understood. It is not uncommon to identify areas of cancer adjacent to PIN and even normal prostate tubules in human prostatectomy specimens. This degree of heterogeneity has resulted in the Gleason scoring system, which grades the histologic severity of representative sections as a prognostic indicator of clinical behavior (Bostwick and Foster, 1999).
Heterogeneous genetic instability in the glandular epithelium due to telomerase shortening or infection with viruses such as BK, JC and a recently described retroviral isolate, are two mechanisms by which multifocal disease can occur (Das et al., 2004; Dong et al., 2007; Vukovic et al., 2003; Zambrano et al., 2002). Another widely proposed theory that may explain this multifocal heterogeneity is “field effect”, implicating global changes in the prostate that can subsequently give rise to independent polyclonal foci of disease (Harding and Theodorescu, 2000). Perturbations in the stroma may provide a mechanism by which a global cancer initiating microenvironment can be instigated (Harding and Theodorescu, 2000).
Interactions between stroma and epithelium are critical for development and alterations in this homeostatic balance can lead to neoplastic transformation (Kalluri and Zeisberg, 2006). Stromal cells are key regulators of adjacent epithelium in multiple epithelial tumor models such as mammary gland (Cheng et al., 2005), skin (Seftor et al., 2005), prostate (Bhowmick et al., 2004a; Hill et al., 2005) and stomach cancers (Bhowmick et al., 2004a). In the normal setting and in these cancer models, epithelial control is exerted by paracrine influences of the adjacent stroma. Paracrine factors stimulate growth and expand the adjacent epithelia by interacting with transforming growth factor β (TGF-beta), fibroblast growth factor (FGF), epidermal growth factor (EGF) and insulin like growth factor (IGF) family receptors (Bhowmick et al., 2004b).
The FGF receptor (FGFR) signaling cascade exemplifies the importance of epithelial and mesenchymal paracrine cross-talk through a diverse set of ligands and receptors that are compartmentalized in the stroma or epithelium (Powers et al., 2000). FGF10 is predominantly expressed in the mesenchyme of the developing prostate gland and is an essential gene for prostate development (Donjacour et al., 2003). In vitro data suggests that FGF10 has mitogenic actions on prostate epithelium and not stroma (Thomson and Cunha, 1999). FGF10 binds preferentially to the IIIb isoform of FGFR1 and FGFR2 (Kwabi-Addo et al., 2004; Lu et al., 1999). Both of these receptors are expressed in the normal prostate epithelium (Kwabi-Addo et al., 2004; Lin et al., 2007; Lu et al., 1999)
Several lines of evidence indicate that altered expression of the FGF/FGFR signaling axis may be important in prostate pathology. FGF10 transcripts have been detected in stroma derived from benign human prostatic hyperplasia specimens (Nakano et al., 1999). A thorough analysis of FGF10 expression is lacking in human prostate cancer. In the Dunning rat tumor model, FGF10 expression has been detected in the stroma and not epithelium of well differentiated tumors (Lu et al., 1999). Increased expression of FGFR1 is seen in both human prostate cancer and animal models, such as TRAMP tumors with progression to poorly differentiated adenocarcinoma (Huss et al., 2003; Sahadevan et al., 2007). The role of FGFR2 in prostate cancer is dependent on the expression of its specific isoform (Kwabi-Addo et al., 2004). Activation of the FGFR2 IIIb isoform, the binding partner for FGF10, is thought to have a role in maintaining prostatic epithelial homeostasis and over-expression of this receptor in neoplastic cells can restore differentiation (Feng et al., 1997; Matsubara et al., 1998).
Previous studies have investigated the role of FGFR1 and FGFR2 in the progression of prostate cancer (Freeman et al., 2003). In a transgenic mouse model, activation of FGFR1 led to formation of PIN, while activation of FGFR2 caused no discernable phenotype (Freeman et al., 2003). Autocrine epithelial over-expression of FGF7, also a stromal gene and a homologue of FGF10, in a transgenic animal model led to the formation of prostate hyperplasia after one year (Foster et al., 2002). These models utilized epithelial-based FGF expression and did not promote FGF signaling from the mesenchyme. As such, they do not examine the role of mesenchymal FGF on adjacent epithelial biology.
We set out to test whether enhanced expression of mesenchymal FGF10 would be sufficient to drive transformation of adult prostate epithelium. We hypothesized that the mitogenic paracrine effects of FGF10 could be regional and predominantly on the neighboring epithelial cells. To investigate the potential biologic effects of FGF10-mediated signaling in the development of prostate cancer we utilized a tissue recombination prostate regeneration system (Cunha and Lung, 1978), further modified into a dissociated single cell reconstitution method (Xin et al., 2003). In this model, adult dissociated prostate epithelial cells are combined with embryonic urogenital sinus mesenchyme (UGSM) and grafted under the kidney capsule of a SCID mouse resulting in formation of prostate gland like structures (Xin et al., 2003). This model allows for genetic manipulation of both compartments of the prostate gland, the epithelium and the mesenchyme, independently and, in contrast to previously published transgenic animal models, enables us to study the effects of paracrine factors on adult prostate epithelial cells.
We show that enhanced expression of mesenchymal FGF10 was sufficient for the formation of well-differentiated prostate carcinoma. The disease pattern observed was multifocal and similar to human prostate cancer when the number of FGF10 expressing UGSM cells was diluted. Inhibition of FGFR1 signaling with dominant negative (DN) FGFR1 could reverse the neoplastic phenotype even in the presence of excess mesenchymal FGF10. Paracrine FGF10 led to an increase in epithelial androgen receptor and, upon castration, FGF10 induced androgen independent survival and proliferation. FGF10-induced PIN or adenocarcinoma persisted in an FGF10 low microenvironment, with sustained activation of the AR machinery. Paracrine FGF10 synergized with cell autonomous activated AKT and resulted in the formation of poorly-differentiated adenocarcinoma.
Results
Paracrine signaling mediated by mesenchymal FGF10 is sufficient for the histologic transformation of the adjacent prostate epithelium
Retroviral constructs were designed to express GFP or FGF10 and GFP (supplementary figure 1A). UGSM was infected with either control vector (GFP) or FGF10-GFP retrovirus. Expression of secreted FGF10 was confirmed by western blot (supplementary figure 1B). FACS analysis confirmed greater than 90% infection of the UGSM cells (supplementary figure 1C).
Dissociated adult murine prostate cells (1×105) were combined with UGSM cells (1×105) infected either with control or FGF10 expressing vectors and engrafted under the kidney capsule of CB.17 SCID/SCID mice (Xin et al., 2003). After 8 weeks, mice were sacrificed and grafts were dissected off the kidney (figure 1A). The FGF10 expressing grafts weighed greater than twice the control grafts (figure 1A).
Figure1.
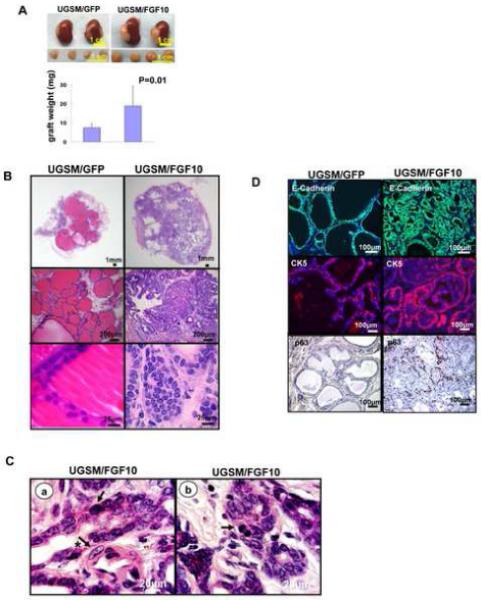
Paracrine FGF10 expression led to well-differentiated adenocarcinoma and an expansion of both luminal and basal cells.
A: Transilluminating image and weight (mean±SD) of regenerated tissue from wild type epithelia (1×105) combined with GFP or FGF10 expressing mesenchyme (1×105) placed in regeneration.
B: H&E analysis of FGF10 UGSM and GFP UGSM regenerated tissue.
C: High power (1000x) magnification of FGF10-induced adenocarcinoma demonstrating atypical (arrow), large nuclei (arrow and asterisk) in a and evidence of mitotic figures (arrow) in b.
D: Immunohistochemical analysis of the regenerated tissue using antibodies against E-cadherin, CK 5 and P63.
Histologic analysis of the control grafts revealed epithelial glands two layers thick with abundant luminal secretions (figure 1B). In contrast, FGF10 grafts showed an increased number of small glandular structures, one cell layer thick, containing cells with increased nuclear to cytoplasmic ratios, prominent multiple nucleoli, scattered apoptotic bodies, and occasional mitotic figures (figure 1B&1C). These features were diagnostic of well-differentiated prostate adenocarcinoma in 10/10 independent paracrine FGF10 regenerated grafts (figure 1B&1C) (Shappell et al., 2004). The commercially available antibodies for FGF10 can not detect this protein by immunohistochemistry. Therefore GFP faithfully expressed by an internal ribosomal entry site (IRES) in the FGF10 expressing constructs was localized in the mesenchymal compartment of regenerated grafts (supplemental figure 1D). Expression of GFP provided an indirect marker for over-expression of mesenchymal FGF10 (supplemental figure 1D). The contour of the epithelial cells was outlined by pancytokeratin staining (supplemental figure 1D). Enhanced expression of FGF10 was confirmed with western blot analysis in three independent FGF10 regenerated grafts (supplemental figure 1E).
To characterize the expanded epithelial cell population, immunohistochemistry was performed for epithelial markers cytokeratin 5 (CK5), CK8, p63, and E-cadherin (figure 1D and supplemental figure 2). Glands from FGF10 grafts expressed E-cadherin (figure 1D, top panel) and cytokeratin 8 (supplemental figure 2A) confirming the epithelial nature of these cells. We detected an approximately three fold expansion of p63 positive cells in FGF10 expressing grafts (figure 1D, bottom panel). We also observed a dramatic expansion of the luminal cells in FGF10 grafts compared to control (supplemental figure 2A). Paracrine FGF10 signaling resulted in the formation of a glandular adenocarcinoma in a background of prostatic hyperplasia. Previous models for prostate cancer lead to the formation of luminal or neuron-endocrine carcinoma (Freeman et al., 2003; Greenberg et al., 1995; Majumder et al., 2003; Wang et al., 2003). In our model we observed an expansion of luminal cells expressing CK8 (figure 2A) in addition to an expansion of basal cells expressing CK5/P63 (supplemental figure 2B and figure 1D bottom panel).
Figure 2.
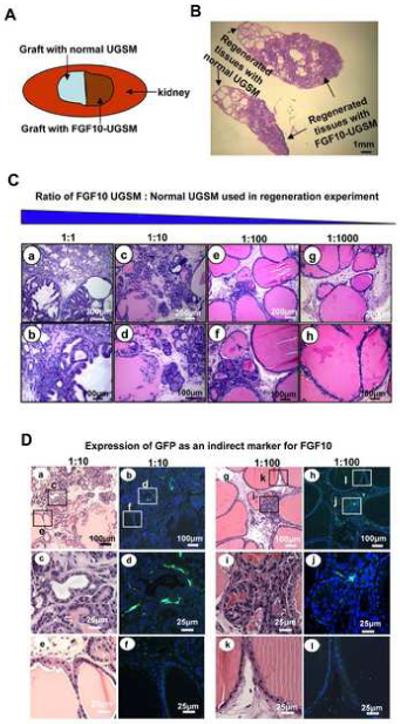
Paracrine FGF10 signaling promoted formation of multifocal prostate cancer resembling human PIN or prostate cancer, and exerted its effects with regional specificity.
A: Schematic representation of two adjacent grafts, one with FGF10 UGSM (1×105) and a second graft with normal UGSM (1×105) both combined with WT epithelium.
B: Histology of two adjacent grafts revealed normal prostate tubules on one side and well-differentiated prostate cancer on the other side, suggesting localized action of paracrine FGF10.
C: H&E analysis of regenerated tissue after a logarithmic reduction in the number of FGF10 UGSM revealed evidence of multifocal carcinoma. Cancer was seen in a&b, PIN adjacent to microinvasive carcinoma in c&d, multifocal PIN in e&f and predominantly normal histology was seen in g&h.
D: Expression of FGF10 indirectly detected by anti-GFP staining in areas of hyperplasia.
Paracrine FGF10 promotes localized transformation in the prostate regeneration system
We asked whether the transforming effects of FGF10 were local or extended beyond the source of FGF10 secreting cells. We placed one graft with FGF10 secreting UGSM adjacent to a second graft with normal UGSM cells (figure 2A). We then set up a dilution series of FGF10 secreting cells such that the number of FGF10 secreting UGSM were decreased in a logarithmic fashion by substituting normal UGSM (figure 2C). To measure the proliferation of regenerating epithelium at 6 weeks, mice were injected with bromodeoxyuridine (Brdu) i.p. (80 mg/kg) and sacrificed after two hours. Grafts were harvested and stained with Brdu antibody (figure 2B and supplemental figure 3). The Brdu labeled epithelia were counted and averaged in four independent high power fields on each end of the two adjacent grafts as well as the middle portion.
Two obviously distinct histologic zones were identified when an FGF10 secreting graft was placed adjacent to a normal graft (figure 2B). One end of the graft with normal UGSM showed normal prostatic tubules, while the opposite end with FGF10 secreting mesenchyme revealed well-differentiated adenocarcinoma (figure 2B). FGF10 led to a local increase in the number of proliferating epithelial cells, as shown by increased epithelial Brdu uptake in the FGF10 containing region (supplemental figure 3). The amount of Brdu uptake was 5 fold less on the control side compared to the cancer side of the graft, suggesting that the effects of FGF10 were localized (supplemental figure 3).
Decreasing the number of FGF10 secreting UGSM led to a less severe phenotype, suggesting that the transforming effects of paracrine FGF10 are dose dependent (figure 2C). In the presence of 50% FGF10 UGSM adenocarcinoma was noted, whereas progression to PIN adjacent to possibly micro invasive carcinoma (1:10) and PIN (1:100) were observed in FGF10 diluted grafts, culminating in normal prostatic epithelium when the FGF10 secreting mesenchymal cells were diluted by a factor of 1000. Microinvasive adenocarcinoma and hyperplastic tubules appeared adjacent to PIN lesions and normal prostate glands in 1:10 and 1:100 diluted samples. This disease pattern resembled the heterogeneous histologic appearance of human prostate cancer and PIN (figure 2C). To examine the location of FGF10 secreting UGSM cells, the expression of GFP was examined as an indirect marker for FGF10, in the diluted 1:10 and 1:100 specimens (figure 2D, a-l). We observed presence of GFP positive, hence FGF10 secreting, UGSM cells adjacent to areas of micro invasive carcinoma and PIN lesions (figure 2D, c&d, i&j). The expression of GFP was noted to be absent adjacent to normal regenerated tubules (figure 2D, e&f, k&l).
Our data indicates that regional PIN and multifocal adenocarcinoma could be recapitulated by altered adjacent cancer stromal cells, similar to the paracrine effects of FGF10 over-expressing mesenchyme.
Inhibition of FGFR1 signaling by DN FGFR1 reverts FGF10-induced adenocarcinoma
We hypothesized that blocking FGFR1 and/or FGFR2 signaling would lead to formation of normal tubules even in the presence of excess mesenchymal FGF10. To test our hypothesis lentiviral vectors were constructed to express DN FGFR1 and DN FGFR2, flag tagged for better detection on western blot (figure 3A) (Li et al., 1994). The DN FGFR1 and DN FGFR2 constructs encode proteins that are truncated down stream of the transmembrane domain and are signaling defective (Li et al., 1994). The lentiviral constructs express the red fluorescent protein (RFP) to mark the site of expression of the DN FGFR. Dissociated prostate epithelial cells were infected with either vector control, DN FGFR1, or DN FGFR2 and combined with equal numbers of FGF10 and WT UGSM and grafted for 6-8 weeks.
Figure 3.

FGF10 induces prostate cancer predominantly by activation of FGFR1.
A. The lentivirus FUCRW vector used for the expression of flag tagged DN FGFR1 and DN FGF R2. Western blot confirmed the expression of both dominant negative constructs.
B: Comparative analysis of graft weights (mean±SD) regenerated with control vector RFP, DN FGFR1-RFP or DN FGFR2-RFP and combined with FGF10 (5×104) added to WT (5×104) UGSM.
C: Immunofluorescent analysis of grafts regenerated with vector control RFP, DN FGFR1-RFP, or DN FGFR2-RFP. DN FGFR1-RFP infected tubules showed cells completely confined within simple glandular and tubular structures suggestive of normal prostate epithelium in contrast to RFP or DN FGFR2-RFP regenerated glands.
Expression of either DN construct resulted in formation of smaller grafts compared to vector control (Figure 3B). Fluorescence microscopy of the regenerated grafts was performed. The frequency of red tubules composed of DN FGFR1 or DN FGFR2 were significantly lower compared to vector control. Multiple sections of regenerated tissue were reviewed to identify relatively rare red tubules in the grafts containing DN FGFR1 or DN FGFR2. The decrease in the size and the rarity of red tubules in the DN regenerated tissue can be explained by the variation in the copy number and integration frequency of lentiviral DN constructs in the regenerating epithelia. High doses of either of the DN constructs in the single prostate epithelial cells could inhibit the regeneration into prostatic tubules and low copy numbers of the lentiviral vector could not be visualized by microscopy. Therefore the histologic data gathered in this experiment was on the group of cells expressing intermediate copy numbers of the DN constructs, visualized by the expression of RFP.
Regenerated red cells expressing DN FGFR1 were mostly confined within simple glandular and tubular structures resembling normal prostatic epithelial glands (figure 3C). These observations were confirmed in four independent experiments. The majority of control RFP or DN FGFR2 red cells were confined within clearly identifiable glands and tubular structures that showed tufting and extension into the glandular lumens, suggestive of epithelial hyperplasia and/or PIN (Figure 3C). The degree of tufting and hyper-cellularity noted in the DN FGFR2 regenerated glands was less compared to control RFP tissue in some regions. This finding suggests a partial blocking effect resulting from the DN FGFR2 expression, which may be due to non specific hetero-dimerization with FGFR1 (Dailey et al., 2005).
Quantitative PCR analysis showed that the expression level of all other predominant binding partners for FGFR1 (Zhang et al., 2006), in addition to FGF7 (a homologue of FGF10), were not altered significantly (table 2 in supplemental figure 4) when comparing WT to FGF10 UGSM. These findings rule out the possibility for an FGF10 induced FGF circuitry.
These data suggest that mesenchymal FGF10 predominantly exerted its effects through epithelial FGFR1 and validates previous data demonstrating that activation of FGFR1 plays an important role in neoplastic transformation of prostate epithelium (Freeman et al., 2003). Our data also demonstrates that inhibition of FGFR1 signaling in prostate epithelium reverses the neoplastic phenotype even in a high FGF10 microenvironment.
Enhanced paracrine mesenchymal FGF10 leads to an increase in epithelial androgen receptor
Multiple in vitro but no in vivo studies demonstrate the over-expression or activation of androgen receptor (AR) by fibroblast growth factor peptides. In vitro studies have shown that growth factors such as KGF can stimulate messenger RNA levels of AR in the absence of androgens (Planz et al., 2001). IGF-I, EGF, and KGF can activate AR in the absence of androgens, suggesting that these growth factors can directly activate the androgen signaling pathway (Culig et al., 1994).
To examine the effects of paracrine FGF10 on the expression of AR in our model, western blot analysis was performed on protein lysates obtained from regenerated grafts. Densitometry showed a roughly four fold increase in AR when comparing FGF10 to control regenerated grafts (figure 4A).
Figure 4.

Paracrine FGF10 resulted in an increase in epithelial AR
A: Western blot revealed a four-fold increase in expression of AR protein in FGF10 compared to control regenerated grafts.
B: Immunohistochemistry confirmed that paracrine FGF10 induced an increase in glandular epithelial AR compared to control regenerated tissue.
Immunohistochemistry was performed to localize the expression of AR. We determined that the majority of epithelial cells in the FGF10 grafts strongly expressed nuclear AR (figures 4B). The level of AR protein expression was higher in the glandular epithelium of FGF10 regenerated grafts compared to controls, as observed by the relative intensities of AR staining (figure 4B).
Similar to other steroid hormones, AR undergoes post-translational modifications that can culminate in an increase both in stability and activity of this receptor (Faus and Haendler, 2006). Signaling through growth factor receptors is one mechanism by which these post-translational modifications can be achieved (Reddy et al., 2006). Quantitative PCR revealed that mRNA levels of AR, obtained from epithelia exposed to paracrine FGF10, are not dramatically altered compared to control regenerated epithelium (data not shown). These results suggest that the FGF10-induced increase in epithelial AR maybe mediated at a post-translational level.
Paracrine FGF10 promotes androgen independent survival and proliferation of cancer cells
Signaling through growth factor receptors can promote androgen independent growth of prostate cancer cells by amplification of AR, an increase in the activation of existing AR, modulation of co-activators/co-repressors, and post translational modifications that stabilize AR (Feldman and Feldman, 2001).
To assess whether FGF10-induced adenocarcinoma survive following withdrawal of androgen, animals harboring grafts composed of FGF10 or control mesenchyme were castrated and followed for 6 weeks. Histologic analysis of the FGF10 grafts harvested from castrate animals revealed persistence of well differentiated prostate adenocarcinoma (figure 5A, a-c). In contrast, in the WT mesenchyme regenerated tissue, there was loss of many epithelia with a remaining, depleted-appearing glandular architecture (figure 5A, d-f). Our results suggest that the FGF10-induced prostate adenocarcinoma cells harbor survival mechanisms that allow proliferation and survival in castrate levels of androgen.
Figure 5.

Paracrine FGF10 promoted androgen independent survival of a subset of prostate adenocarcinoma
A: Histology of the FGF10 and WT mesenchyme regenerated grafts harvested from castrated animals. Contrary to normal WT regenerated grafts, persistence of adenocarcinoma was noted in all five FGF10 regenerated specimens.
B: Immunohistochemical localization of AR in FGF10 grafts obtained from intact and castrated animals. Castration led to partial cytoplasmic transport of AR in castrate (c&d) compared to intact grafts (a&b). Partial nuclear localization of AR persisted despite castration in the FGF10 regenerated tissue (c&d).
C: Comparative analysis of weight (mean±SD) of grafts harvested from intact and castrate animals.
Immunohistochemistry was performed to compare the localization and expression of AR in the intact vs. castrate FGF10 grafts (figure 5B). The glandular tissue of the intact FGF10 grafts expressed high levels of nuclear AR (figure 5B, a&b). Castration led to partial cytoplasmic translocation of AR in the FGF10 regenerated tissue (figure 5B, c&d). However, partial nuclear localization of AR persisted in the majority of the FGF10 glandular epithelium and there were foci that continued to harbor strong nuclear expression of AR (figure 5B c&d).
To assess the acute and chronic effects of androgen withdrawal on proliferation and apoptosis in FGF10 regenerated tissue, animals harboring FGF10 mesenchymal grafts were castrated and grafts were harvested at 1 and 6 weeks. Castrated grafts were half the size of grafts harvested from intact animals at 6 weeks (figure 5C). We quantified the proliferation index at 6 weeks, and rate of apoptosis at 1 week post-castration using Ki67 immunohistochemistry and TUNEL assays, respectively (supplemental figure 5A&B). We counted and averaged the Ki67 or TUNEL positive epithelia in five high-power microscopic fields. A decrease in proliferation measured by percentage of Ki67 positive cells (8% in intact vs. 4% in castrate), which did not reach statistical significance, and a statistical significant increase in apoptosis measured by percentage of TUNEL positive cells (0.8% in intact vs. 1.6% in castrate) was seen upon castration (supplemental figure 5A&B).
Decrease in the size of the grafts, increase in apoptosis and decrease in proliferation upon castration suggests that a subset of the FGF10 induced adenocarcinoma remain dependent on androgen hormone. Persistence of the remaining FGF10-induced prostate adenocarcinoma cells and Ki67 staining, suggests that paracrine FGF10 signaling can promote survival and proliferation of these cells in castrate levels of androgen. The dramatic increase in AR induced by paracrine FGF10 and subsequent hypersensitivity to residual castrate levels of androgens may be one survival strategy utilized by the surviving cancer cells after castration. Androgen independent activation of AR by paracrine FGF10 may be an alternate mechanism leading to androgen independent survival of a subset of the FGF10 induced adenocarcinoma.
A subset of FGF10-induced PIN or adenocarcinoma is serially transplantable
While chronic signaling through an oncogenic pathway is essential for maintaining transformation in some malignancies, cell autonomous alterations can be sufficient to bypass this continual dependence in other cancer models (Weinstein, 2002). We assessed whether transformation of epithelia exposed to FGF10 would evade or remain dependent on chronic paracrine signaling by this growth factor and asked whether the FGF10-induced adenocarcinoma was serially transplantable. Prostate epithelium from DS red transgenic animals were recombined with FGF10-GFP UGSM and placed in the regeneration system. DS red transgenic animals were utilized as they express the red fluorescent protein variant DS Red, providing a fluorescent marker for the epithelia. Grafts were harvested following 8 weeks of regeneration, minced, and digested into single cells.
Dilution of FGF10-UGSM cells by a factor of 1000 would lead to formation of normal tubules in this regeneration system (figure 2C). DS red dissociated single cells (2×104) harvested from FGF10 regenerated grafts were combined with WT epithelium (2×104) and an excess of WT mesenchyme such that the number of FGF10 expressing mesenchymal cells would be diluted by a factor of 1000. These cells were engrafted as a secondary tumor. After 8 weeks of regeneration, histologic analysis revealed areas of WT epithelium adjacent to hyperplastic foci of cells with uniform epithelial tufting, crowding, and nuclear atypia (supplemental figure 6A, a-c). Immunofluorescent microscopy revealed WT and red tubules forming double layered large tubules with normal architecture adjacent to red clusters of glands filled with several layers of tufted epithelial cells, reminiscent of PIN (supplemental figure 6A, d-f). This observation suggested that the FGF10-induced PIN could persist in an FGF10 low microenvironment.
DS red dissociated single cells harvested from FGF10 regenerated grafts were FACS sorted (R1), assuring exclusion of single or doublets of GFP-positive cells (R2+R3) (figure 6A). DS red sorted cells (3×104) were either recombined with WT epithelia (7×104) as helper cells, or engrafted alone with WT UGSM cells (105) as a secondary tumor in the regeneration system. Histologic analysis of grafts with added helper cells revealed multiple normal tubules, in addition to areas demonstrating cellular tufting, crowding and few foci with crowded microglandular structures containing cells with atypical nuclear features and higher nuclear to cytoplasmic ratios (figure 6A, a&b). Immunofluorescent microscopic evaluation of this regenerated tissue revealed formation of some double layered large red tubules resembling normal glands (figure 6B, a&b), in addition to other areas demonstrating small clusters of red tubules reminiscent of well differentiated adenocarcinoma (figure 6B, c&d).
Figure 6.
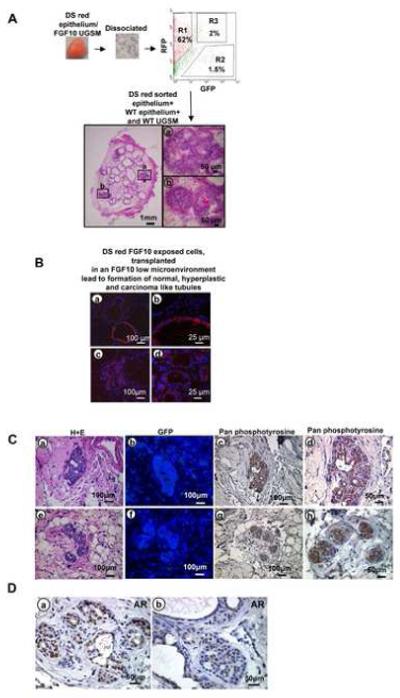
Serial transplantation of FGF10 induced adenocarcinoma
A: Schematic representation of transplantation experiments and histology of regenerated tubules.
B: Immunofluorescent analysis of regenerated tissue revealed formation of many WT and some red epithelial glands with a single cell layer resembling normal tubules (a&b), and multiple small red tubules adjacent to normal appearing WT glands reminiscent of well differentiated adenocarcinoma (c&d).
C: Histologic evaluation of transplanted tissue specimens revealed evidence of adenocarcinoma (a&e) and absence of contaminating FGF10 expressing stromal cells demonstrated by lack of GFP staining (b&f). The cancerous regenerated tissue revealed strong phosphotyrosine activation after withdrawal of retrovirally infected FGF10 UGSM cells (c&d, g&h).
D: Persistent strong nuclear localization of AR was observed in cancerous areas of the transplanted regenerated grafts (a&b).
Our findings suggest that an 8 week exposure to paracrine FGF10, may induce a subset of epithelial cells to continuously form abnormal epithelial structures, architecturally ranging from hyperplasia to well differentiated adenocarcinoma, even in low levels of this growth factor. To test whether the FGF10 induced adenocarcinoma is serially transplantable we dissociated secondary grafts into single cells and re-transplanted them into a tertiary graft (supplemental figure 6B). In these tertiary grafts, similar to the primary and secondary tumors, we observed abnormal epithelial structures architecturally ranging from epithelial hyperplasia to well differentiated adenocarcinoma, suggesting that a subset of FGF10-induced PIN/adenocarcinoma is serially transplantable (supplemental figure 6B). The rate of proliferation of the transplanted secondary and tertiary tumors was found to be similar to the primary FGF10 grafts and significantly more than normal regenerated tissue assessed and quantified by Ki67 staining (supplemental figure 7).
To ascertain possible mechanisms that could account for this sustained oncogenic phenotype, in low levels of FGF10, transplanted DS red regenerated tissue (without helper cells) were evaluated for phosphotyrosine activation in secondary and tertiary tumors. Immunohistochemical analysis of the transplanted tissue with pan-phosphotyrosine antibody revealed strong phosphotyrosine activation in cancerous regenerated glands (figure 6C c&d, g&h and supplemental figure 8B). In these histologic specimens the absence of GFP staining further confirmed the exclusion of exogenous growth factor expressing UGSM cells (figure 6C b&f). Regenerated tissue with normal UGSM were used as a negative control and cancerous tissue with FGF10 UGSM were used as positive control in assessing phosphotyrosine activity (supplemental figure 8B). Sustained nuclear localization of AR was seen in secondary and tertiary transplanted regenerated tissue, demonstrating sustained activation of the AR machinery (figure 6D a&b and supplemental figure 8A).
Cooperation between mesenchymal FGF10 and cell autonomous AKT leads to high-grade carcinoma
We sought to evaluate the biological consequences of combining two events in naïve prostate epithelial cells, “Outside-in” signaling through mesenchymal FGF10 and cell autonomous expression of activated AKT. We had previously shown that chronic activated epithelial AKT could lead to the formation of PIN while the combination of epithelial AKT and AR would lead to the formation of carcinoma (Xin et al., 2006). In this model, paracrine FGF10 signaling led to an increase in epithelial AR. We asked whether paracrine FGF10 signaling could synergize with cell autonomous activated AKT.
Prostate single cells were infected with control vector or activated AKT and combined with either FGF 10 expressing UGSM or vector control UGSM and placed in the regeneration system. Grafts were harvested after seven weeks, weighed (figure 7A) and the histology was evaluated (figure 7B, a-f). Addition of epithelial AKT to mesenchymal FGF 10 more than doubled the size of the grafts as compared to grafts with epithelial AKT alone (figures 7A). The combination of AKT and FGF10 resulted in grafts that were six times the weight of grafts with FGF10 alone (figures 7A). Histologic analysis of the 3/3 regenerated grafts revealed areas of poorly-differentiated adenocarcinoma with mesenchymal FGF10 and epithelial AKT, defined by sheets of atypical cells with almost complete loss of glandular architecture (figure 7B c&f, supplemental figure 9). Mesenchymal FGF10 led to the development of well-differentiated adenocarcinoma (figure 7B a&d) and epithelial AKT alone led to PIN defined by stratification of epithelial cells with nuclear atypia confined within glandular structures (figure 7B b&e). Expression of epithelial phospho- AKT was confirmed utilizing immunohistochemistry (figure 7B, h&i). Stromal expression of FGF10-GFP or GFP was confirmed by immunohistochemical detection of GFP (figure 7B, j-l). Our data demonstrates synergy between paracrine FGF10 signaling and cell autonomous AKT, implicating the importance of cooperation between stromal and epithelial perturbations in malignant transformation.
Figure 7.

Mesenchymal FGF10 synergized with cell autonomous AKT leading to a progressive cancer phenotype and a dramatic increase in epithelial AR
A: Transilluminating images and comparative weight (mean±SD) of grafts derived from WT epithelium with FGF10 UGSM, AKT epithelium with WT UGSM and AKT epithelium with FGF10 UGSM.
B: H&E staining for the regenerated tissue derived from WT epithelium with FGF10 UGSM (a&d), AKT epithelium with WT UGSM (b&e) and AKT epithelium with FGF10 UGSM (c&f). Immunohistochemical analysis of the expression phospho-AKT (g-i) and GFP (j-l).
C: Western blot analysis with AR antibody confirmed the increase in AR in the presence of mesenchymal FGF10. Vinculin used as loading control.
D: Immunofluorescence detection of AR revealed that FGF10 mesenchyme in combination with WT or AKT activated epithelium could lead to an increase in expression of epithelial AR compared to controls.
To ascertain the possible mechanisms for cooperation between paracrine mesenchymal FGF10 and cell autonomous AKT, regenerated grafts were lysed for western blot analysis (figure 7C). We had previously observed a four-fold increase in the expression of AR comparing FGF10 to control grafts (figure 4A). Similarly, densitometry on western blots showed roughly an eight-fold increase in AR protein when comparing grafts composed of epithelial AKT combined with mesenchymal FGF10 to grafts composed of epithelial AKT alone (figure 7C). Immunohistochemistry confirmed that AR was mainly over expressed in the epithelial compartment (figure 7D, a&b compared to c&d, e&f compared to g&h). These data indicate that paracrine FGF10 signaling leads to an increase in epithelial AR, and this secondary event can cooperate with activated epithelial AKT leading to a progressive cancer phenotype.
Discussion
Historically, essential growth factors have proven to be oncogenic in multiple tumor models (Kris et al., 1985). For example, platelet derived growth factor, an essential peptide for wound healing, was one of the first described growth factor oncogenes (Doolittle et al., 1983). We have demonstrated that enhanced expression of stromal FGF10, an essential gene for prostate development, is sufficient for the formation of multifocal adenocarcinoma concomitant with an increase in epithelial AR in an in vivo prostate cancer model.
Similar to FGF10, AR is essential for the development of the prostate (Cunha and Lung, 1978). We show that enhanced expression of stromal FGF10 is sufficient to increase levels of AR protein in both naïve and activated AKT prostate epithelium. Interestingly, in our study, expression of additional AR in WT epithelium utilizing lentiviral delivery vectors in combination with mesenchymal FGF10 led to no discernable histologic difference compared to mesenchymal FGF10 regenerated grafts (supplemental figure 10 A&B). Developmental studies utilizing in vitro organ culture systems of FGF10 null prostate demonstrate that addition of exogenous FGF10 can induce branching morphogenesis similar to the addition of testosterone, while addition of both FGF10 and testosterone do not provide further synergistic effects (Thomson and Cunha, 1999). Our findings concur with previous observations and suggest that paracrine FGF10 signaling can promote activation of AR. An increase in AR protein may be one mechanism by which this over-activation is achieved.
Amplification of the AR gene has been reported in up to 30% of human prostate cancers and is thought to be an important mechanism for formation of androgen independence (Chen et al., 2004; Feldman and Feldman, 2001). Perturbations in the growth factor regulatory loops are associated with progression to a more aggressive cancer phenotype leading to androgen independent metastatic disease (Culig et al., 2000; Culig et al., 2002; Scher et al., 1995). In our model, survival of a subset of the FGF10-induced prostate adenocarcinoma cells, concomitant with partial nuclear localization of AR in the castrate tumors, suggests activation of AR signaling axis by paracrine mesenchymal FGF10 in castrate levels of androgen.
Transformed cells, such as Bcr-Abl induced leukemia (Huettner et al., 2000) or ras induced melanoma (Chin et al., 1999), are found to be “addicted” to oncogenic signals, such that removal of the activating signal can lead to regression of the cancer phenotype (Weinstein, 2002). It is conceivable that a subset of cells exposed to an oncogene may undergo subsequent genetic alterations allowing them to evade chronic dependence on the initial growth promoting activated pathway (Weinstein, 2002). This phenomenon is seen in a sub-population of c-myc induced mammary carcinoma cells that can continue to grow in a myc independent manner after down regulation of this oncogene (Boxer et al., 2004). Formation of PIN or adenocarcinoma in transplanted epithelia, previously exposed to high levels of FGF10, suggests a possible “hit and run” mechanism for the paracrine FGF10 induced epithelial alterations. Persistence of pan-phosphotyrosine kinase activity and sustained nuclear localization of AR in this subset of transplanted cancerous tissue suggests intrinsic genetic alterations, resulting in survival and growth of this subpopulation of cells in low levels of FGF10. The non-genotropic activity of AR, has been linked to activation of several tyrosine kinases including the Src, PI3Kinase (phosphoinositide 3-kinase) and MAPK kinase (Mitogen-activated protein kinase) family receptors (Kousteni et al., 2001; Sun et al., 2003). Conversely, activation of protein tyrosine kinases can lead to activation of the AR (Feldman and Feldman, 2001). Either of these mechanisms, in conjunction or solely, could account for the sustained cancerous phenotype noted in the transplanted grafts.
Cancer progression can result from the accumulation of multiple cooperative genetic events, a concept that has been demonstrated in three recent prostate cancer models (Gao et al., 2006; Xin et al., 2006; Zhong et al., 2006). Cooperation between autocrine FGFR signaling and the PI3 Kinase pathway was shown in an in vivo transgenic animal model in which loss of PTEN and over-expression of FGF8b in the epithelium led to poorly differentiated adenocarcinoma (Zhong et al., 2006). We had previously shown synergy between epithelial AR and AKT resulting in adenocarcinoma that resists the effects of androgen ablation (Xin et al., 2006). Loss of PTEN and Nkx3.1 is another mechanism that can promote androgen independent prostate carcinoma (Gao et al., 2006). Our study emphasizes synergy between a paracrine growth factor, FGF10, and epithelial cell autonomous activated AKT. This model emphasizes the importance of stromal paracrine and epithelial cell autonomous signals leading to the development of poorly differentiated disease.
These data suggest the importance of targeting the growth factor receptor signaling axis either through inhibition of paracrine factors or direct inhibition of growth factor receptors, in conjunction with other therapeutic strategies, such as androgen ablation utilized in treating prostate cancer. Targeting growth factor signaling cascades similar to fibroblast growth factors may also delay emergence of androgen hormone independence which poses the largest clinical challenge in the treatment of prostate cancer.
Experimental procedures
Plasmids
cDNA encoding FGF10 was procured by RT-PCR, from NIH 3T3 cells, and cloned into the EcoRI site of MSCV-IRES-GFP (MSCV, murine stem cell virus; IRES, internal ribosomal entry site) (Hawley et al., 1994). DN FGFR1 and DN FGFR2 (Li et al., 1994) were flag tagged and sub cloned into the EcoRI cloning site of FU-CRW lentiviral expression vector. FU-CRW is a vector derived from FUGW (Xin et al., 2006). In the FU-CRW vector the GFP coding sequence of FUGW vector was substituted with a CMV driven RFP expression cassette. Myristoylated human AKT1 was sub cloned into EcoRI site of FU-CRW lentiviral expression vector.
Prostate Regeneration and prostate epithelial viral infections
The details of the regeneration process and viral infection of epithelial cells have been explained previously (Xin et al., 2003). Housing, maintenance and all surgical procedures were undertaken in compliance with the regulations of the Division of Laboratory Animal Medicine of the University of California, Los Angeles. All experimental procedures were approved by the Division of Laboratory Animal Medicine of the University of California, Los Angeles. Viral preparation procedures were performed under University of California, Los Angeles, safety regulations for lentivirus usage. Retrovirus and lentivirus were made as described previously (Wong et al., 2004; Xin et al., 2003; Xin et al., 2006)
Immunohistochemistry
Grafts were fixed in buffered formalin 10% and placed in 70% ethanol as described previously (Xin et al., 2003; Xin et al., 2006). 4 um sections were made and stained with hematoxylin and eosin (H+E) or with antibodies against CK5 (AF138, Abcam), P63 (sc-8431, Santa Cruz Biotechnology), E-Cadherin (610-182, Transduction), anti-AR (sc-816, Santa Cruz Biotechnology), P AKT (3787, Cell Signaling), GFP (mouse monoclonal clone 3C2-G2 produced in ON Witte labs), anti-Brdu (51-75512 Pharmingen), pan-phosphotyrosine (Upstate), CK8 (MMS-162P Covance), and Ki67 (Vector) antibodies. For visualization of CK5, E-cadherin, AR, pAKT biotinylated anti-mouse IgG (RO627, Vector biotechnology) and biotinylated anti-rabbit IgG (ab6720-1, Abcam) secondary antibodies were utilized. Histologic sections were visualized with fluorescent microscopy and counterstained with DAPI (Vector Laboratories).
Western Blot
Grafts and cells were manually lysed in RIPA buffer composed of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% SDS, 0.5% SD, 1% NP-40, 1 mM EDTA, 1 mM PMSF, 25 mM NaF, cocktail protease inhibitors (Roche) and phosphatase inhibitor I &2 (Sigma). Secreted FGF10 protein from UGSM cells 48 hour cultured media was collected and precipitated at a 4:1 ratio with 50% TCA (trichlor acetic acid), centrifuged, pelleted, washed with acetone, air dried and re suspended with 2x SDS lysis buffer. PH was adjusted with 10M NaOH. Protein lysates (40-100ug) were separated by SDS-PAGE followed by western blot analysis using AR (sc-816, Santa Cruz Biotechnology), FGF10 (sc-Santa Cruz Biotechnology), Vinculin (Sigma), anti-flag (Sigma) and Erk2 (sc-154, Santa Cruz Biotechnology) antibodies. Quantification of the western blot data was performed utilizing the NIH ImageJ program (http://rsb.info.nih.gov/ij/).
Quantitative RT-PCR
Quantitative RT-PCR was performed using an ABI 7700 Real Time PCR System. Primers are listed in the supplemental table 1 (supplemental figure 4). All primers span at least one intron. Standard curves of murine placenta cDNA were run for each primer pair, and the amount of target gene was normalized to the amount of actin.
Supplementary Material
Acknowledgements
We thank Ms. Barbara Anderson for her help in preparation of this manuscript and Ms. Donghui Cheng for her help in FACS sorting cells. S.M. has been supported by grants from NICHD-ABOG Reproductive Scientist Developmental Program 5K12HD00849-18 and NIH/National Cancer Institute, Clinical Scientist Training in Cancer Gene Medicine, K12-CA076905-09. L.X. is supported by a NIH Pathway to Independence Award 1K99CA125937 and a training grant from the California Institute for Regenerative Medicine. D.J.M. is funded by a National Institutes of Health (NIH) fellowship. H.W. is supported by NIH grant RO1 CA107166 and an Army Medical Research grant W81XWH-05-1-0542. M.A.T. is a Scholar of the Leukemia and Lymphoma Society and is supported by NIH grants R01CA90571, R01CA107300, and California Institute for Regenerative Medicine (CIRM) grant RS1-00313. O.N.W. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004a;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004b;432:332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostwick DG, Foster CS. Predictive factors in prostate cancer: current concepts from the 1999 College of American Pathologists Conference on Solid Tumor Prognostic Factors and the 1999 World Health Organization Second International Consultation on Prostate Cancer. Semin Urol Oncol. 1999;17:222–272. [PubMed] [Google Scholar]
- Boxer RB, Jang JW, Sintasath L, Chodosh LA. Lack of sustained regression of c-MYC-induced mammary adenocarcinomas following brief or prolonged MYC inactivation. Cancer Cell. 2004;6:577–586. doi: 10.1016/j.ccr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, Arteaga CL, Neilson EG, Hayward SW, Moses HL. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene. 2005;24:5053–5068. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O’Hagan R, Pantginis J, Zhou H, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Bartsch G, Klocker H. Androgen receptor--an update of mechanisms of action in prostate cancer. Urol Res. 2000;28:211–219. doi: 10.1007/s002400000111. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- Culig Z, Klocker H, Bartsch G, Hobisch A. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002;9:155–170. doi: 10.1677/erc.0.0090155. [DOI] [PubMed] [Google Scholar]
- Cunha GR, Lung B. The possible influence of temporal factors in androgenic responsiveness of urogenital tissue recombinants from wild-type and androgen-insensitive (Tfm) mice. J Exp Zool. 1978;205:181–193. doi: 10.1002/jez.1402050203. [DOI] [PubMed] [Google Scholar]
- Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:233–247. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Das D, Shah RB, Imperiale MJ. Detection and expression of human BK virus sequences in neoplastic prostate tissues. Oncogene. 2004;23:7031–7046. doi: 10.1038/sj.onc.1207920. [DOI] [PubMed] [Google Scholar]
- Dong B, Kim S, Hong S, Das Gupta J, Malathi K, Klein EA, Ganem D, Derisi JL, Chow SA, Silverman RH. An infectious retrovirus susceptible to an IFN antiviral pathway from human prostate tumors. Proc Natl Acad Sci U S A. 2007;104:1655–1660. doi: 10.1073/pnas.0610291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donjacour AA, Thomson AA, Cunha GR. FGF-10 plays an essential role in the growth of the fetal prostate. Dev Biol. 2003;261:39–54. doi: 10.1016/s0012-1606(03)00250-1. [DOI] [PubMed] [Google Scholar]
- Doolittle RF, Hunkapiller MW, Hood LE, Devare SG, Robbins KC, Aaronson SA, Antoniades HN. Simian sarcoma virus onc gene, v-sis, is derived from the gene (or genes) encoding a platelet-derived growth factor. Science. 1983;221:275–277. doi: 10.1126/science.6304883. [DOI] [PubMed] [Google Scholar]
- Faus H, Haendler B. Post-translational modifications of steroid receptors. Biomed Pharmacother. 2006;60:520–528. doi: 10.1016/j.biopha.2006.07.082. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- Feng S, Wang F, Matsubara A, Kan M, McKeehan WL. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res. 1997;57:5369–5378. [PubMed] [Google Scholar]
- Foster BA, Evangelou A, Gingrich JR, Kaplan PJ, DeMayo F, Greenberg NM. Enforced expression of FGF-7 promotes epithelial hyperplasia whereas a dominant negative FGFR2iiib promotes the emergence of neuroendocrine phenotype in prostate glands of transgenic mice. Differentiation. 2002;70:624–632. doi: 10.1046/j.1432-0436.2002.700915.x. [DOI] [PubMed] [Google Scholar]
- Freeman KW, Welm BE, Gangula RD, Rosen JM, Ittmann M, Greenberg NM, Spencer DM. Inducible prostate intraepithelial neoplasia with reversible hyperplasia in conditional FGFR1-expressing mice. Cancer Res. 2003;63:8256–8263. [PubMed] [Google Scholar]
- Gao H, Ouyang X, Banach-Petrosky WA, Gerald WL, Shen MM, Abate-Shen C. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc Natl Acad Sci U S A. 2006;103:14477–14482. doi: 10.1073/pnas.0606836103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO, Cunha GR, Donjacour AA, Matusik RJ, Rosen JM. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci U S A. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding MA, Theodorescu D. Prostate tumor progression and prognosis. interplay of tumor and host factors. Urol Oncol. 2000;5:258–264. doi: 10.1016/s1078-1439(00)00073-9. [DOI] [PubMed] [Google Scholar]
- Hawley RG, Lieu FH, Fong AZ, Hawley TS. Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1994;1:136–138. [PubMed] [Google Scholar]
- Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123:1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat Genet. 2000;24:57–60. doi: 10.1038/71691. [DOI] [PubMed] [Google Scholar]
- Huss WJ, Barrios RJ, Foster BA, Greenberg NM. Differential expression of specific FGF ligand and receptor isoforms during angiogenesis associated with prostate cancer progression. Prostate. 2003;54:8–16. doi: 10.1002/pros.10163. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- Kousteni S, Bellido T, Plotkin LI, O’Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- Kris RM, Libermann TA, Avivi A, Schlessinger J. Growth factors, growth-factor receptors and oncogenes. Nature Biotechnology. 1985;3:135–140. [Google Scholar]
- Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer. 2004;11:709–724. doi: 10.1677/erc.1.00535. [DOI] [PubMed] [Google Scholar]
- Li Y, Basilico C, Mansukhani A. Cell transformation by fibroblast growth factors can be suppressed by truncated fibroblast growth factor receptors. Mol Cell Biol. 1994;14:7660–7669. doi: 10.1128/mcb.14.11.7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Liu G, Zhang Y, Hu YP, Yu K, Lin C, McKeehan K, Xuan JW, Ornitz DM, Shen MM, et al. Fibroblast growth factor receptor 2 tyrosine kinase is required for prostatic morphogenesis and the acquisition of strict androgen dependency for adult tissue homeostasis. Development. 2007;134:723–734. doi: 10.1242/dev.02765. [DOI] [PubMed] [Google Scholar]
- Lu W, Luo Y, Kan M, McKeehan WL. Fibroblast growth factor-10. A second candidate stromal to epithelial cell andromedin in prostate. J Biol Chem. 1999;274:12827–12834. doi: 10.1074/jbc.274.18.12827. [DOI] [PubMed] [Google Scholar]
- Majumder PK, Yeh JJ, George DJ, Febbo PG, Kum J, Xue Q, Bikoff R, Ma H, Kantoff PW, Golub TR, et al. Prostate intraepithelial neoplasia induced by prostate restricted Akt activation: the MPAKT model. Proc Natl Acad Sci U S A. 2003;100:7841–7846. doi: 10.1073/pnas.1232229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara A, Kan M, Feng S, McKeehan WL. Inhibition of growth of malignant rat prostate tumor cells by restoration of fibroblast growth factor receptor 2. Cancer Res. 1998;58:1509–1514. [PubMed] [Google Scholar]
- Nakano K, Fukabori Y, Itoh N, Lu W, Kan M, McKeehan WL, Yamanaka H. Androgen-stimulated human prostate epithelial growth mediated by stromal-derived fibroblast growth factor-10. Endocr J. 1999;46:405–413. doi: 10.1507/endocrj.46.405. [DOI] [PubMed] [Google Scholar]
- Planz B, Wang Q, Kirley SD, Marberger M, McDougal WS. Regulation of keratinocyte growth factor receptor and androgen receptor in epithelial cells of the human prostate. J Urol. 2001;166:678–683. [PubMed] [Google Scholar]
- Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- Reddy GP, Barrack ER, Dou QP, Menon M, Pelley R, Sarkar FH, Sheng S. Regulatory processes affecting androgen receptor expression, stability, and function: potential targets to treat hormone-refractory prostate cancer. J Cell Biochem. 2006;98:1408–1423. doi: 10.1002/jcb.20927. [DOI] [PubMed] [Google Scholar]
- Sahadevan K, Darby S, Leung H, Mathers M, Robson C, Gnanapragasam V. Selective over-expression of fibroblast growth factor receptors 1 and 4 in clinical prostate cancer. J Pathol. 2007;213:82–90. doi: 10.1002/path.2205. [DOI] [PubMed] [Google Scholar]
- Scher HI, Sarkis A, Reuter V, Cohen D, Netto G, Petrylak D, Lianes P, Fuks Z, Mendelsohn J, Cordon-Cardo C. Changing pattern of expression of the epidermal growth factor receptor and transforming growth factor alpha in the progression of prostatic neoplasms. Clin Cancer Res. 1995;1:545–550. [PubMed] [Google Scholar]
- Seftor EA, Brown KM, Chin L, Kirschmann DA, Wheaton WW, Protopopov A, Feng B, Balagurunathan Y, Trent JM, Nickoloff BJ, et al. Epigenetic transdifferentiation of normal melanocytes by a metastatic melanoma microenvironment. Cancer Res. 2005;65:10164–10169. doi: 10.1158/0008-5472.CAN-05-2497. [DOI] [PubMed] [Google Scholar]
- Shappell SB, Thomas GV, Roberts RL, Herbert R, Ittmann MM, Rubin MA, Humphrey PA, Sundberg JP, Rozengurt N, Barrios R, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- Sun M, Yang L, Feldman RI, Sun XM, Bhalla KN, Jove R, Nicosia SV, Cheng JQ. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem. 2003;278:42992–43000. doi: 10.1074/jbc.M306295200. [DOI] [PubMed] [Google Scholar]
- Thomson AA, Cunha GR. Prostatic growth and development are regulated by FGF10. Development. 1999;126:3693–3701. doi: 10.1242/dev.126.16.3693. [DOI] [PubMed] [Google Scholar]
- Vukovic B, Park PC, Al-Maghrabi J, Beheshti B, Sweet J, Evans A, Trachtenberg J, Squire J. Evidence of multifocality of telomere erosion in high-grade prostatic intraepithelial neoplasia (HPIN) and concurrent carcinoma. Oncogene. 2003;22:1978–1987. doi: 10.1038/sj.onc.1206227. [DOI] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Wong S, McLaughlin J, Cheng D, Zhang C, Shokat KM, Witte ON. Sole BCR-ABL inhibition is insufficient to eliminate all myeloproliferative disorder cell populations. Proc Natl Acad Sci U S A. 2004;101:17456–17461. doi: 10.1073/pnas.0407061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Ide H, Kim Y, Dubey P, Witte ON. In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci U S A. 2003;100(Suppl 1):11896–11903. doi: 10.1073/pnas.1734139100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc Natl Acad Sci U S A. 2006;103:7789–7794. doi: 10.1073/pnas.0602567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano A, Kalantari M, Simoneau A, Jensen JL, Villarreal LP. Detection of human polyomaviruses and papillomaviruses in prostatic tissue reveals the prostate as a habitat for multiple viral infections. Prostate. 2002;53:263–276. doi: 10.1002/pros.10157. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Saribekyan G, Liao CP, Cohen MB, Roy-Burman P. Cooperation between FGF8b overexpression and PTEN deficiency in prostate tumorigenesis. Cancer Res. 2006;66:2188–2194. doi: 10.1158/0008-5472.CAN-05-3440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.