

Abstract
The transient receptor potential V1 channel (vanilloid receptor, TRPV1) represents a promising therapeutic target for inflammatory pain and other conditions involving C-fiber sensory afferent neurons. Sensitivity of TRPV1 is known to be subject to modulation by numerous signaling pathways, in particular by phosphorylation, and we wished to determine whether TRPV1 structure-activity relations could be differentially affected. We demonstrate here that the structure activity relations of TRPV1, as determined by 45Ca2+ uptake, were substantially altered by treatment of the cells with cyclosporin A, an inhibitor of protein phosphatase 2B. Whereas the potency of resiniferatoxin for stimulation of 45Ca2+ was not altered by cyclosporin A treatment, the potencies of some other agonists were increased up to 8-fold. Among antagonists examined, potencies were reduced to a lesser extent, ranging from 1–2.5 fold. Finally, the efficacy of partial agonists was increased. In contrast to cyclosporin A, okadaic acid, an inhibitor of protein phosphatases 1 and 2A, had little effect on agonist potencies, and calyculin A, an inhibitor of protein phosphatases 1 and 2A but with somewhat different selectivity from that of okadaic acid, caused changes in structure activity relations distinct from those induced by cyclosporin A. Because phosphatase activity differentially modulates the structure activity relations of TRPV1 agonists and antagonists, our findings predict that it may be possible to design agonist and antagonists selective for TRPV1 in a specific regulatory environment. A further implication is that it may be desirable to tailor screening approaches for drug discovery to reflect the desired regulatory state of the targeted TRPV1.
Keywords: TRPV1, capsaicin, resiniferatoxin, vanilloid, cyclosporin A, protein phosphatase
Introduction
TRPV1, a member of the transient receptor potential channels, has emerged as a promising therapeutic target for a range of pathological conditions, including inflammatory and neuropathic pain, cystitis, and bladder hyperreflexia (Robbins, 2000). In addition to its prominent role in activation of C-fiber sensory neurons, TRPV1 functions as an integrator of nociceptive stimuli, including vanilloids such as capsaicin or resiniferatoxin, acidity, heat, and inflammatory mediators such as lipoxygenase products or, indirectly, bradykinin (Julius and Basbaum, 2001). Twin strategies being pursued for development of ligands targeted to TRPV1 are 1) direct antagonism and 2) desensitization/defunctionalization subsequent to agonist exposure (Szallasi and Blumberg, 1999). These two strategies have different mixes of advantages and disadvantages. Direct antagonism is readily reversible and limited to those responses of C-fiber neurons in which TRPV1 is directly involved (Krause et al., 2005). Desensitization/defunctionalization can have a broader effect, blocking other signaling pathways in the C-fiber sensory neurons, and be of long duration (Silva et al., 2002). Depending on the specific therapeutic application, the greater breadth and duration of desensitization/defunctionalization as compared to direct antagonism may be either advantageous or disadvantageous.
A critical aspect of TRPV1 regulation is its modulation by numerous signaling pathways (Cortright and Szallasi, 2004). Phosphatidylinositol-4,5-bisphosphate has been described as binding to and modulating TRPV1 (Chuang et al., 2001, Prescott and Julius, 2003, Stein et al., 2006). Kinases which phosphorylate and regulate TRPV1 include protein kinase A (Bhave et al., 2002), protein kinase C (Bhave et al., 2003), protein kinase D (Wang et al., 2004), and the calcium-calmodulin dependent protein kinase II (Jung et al., 2004). There is also evidence for phosphorylation of TRPV1 on tyrosine (Jin et al., 2004). The many surface receptors upstream of these widespread signaling elements thus should all influence TRPV1. Furthermore, since the state of phosphorylation of a protein depends on the balance between kinase activity and phosphatase activity, the activities of phosphatases such as the class 2B protein phosphatase calcineurin play an important part in TRPV1 regulation (Docherty et al., 1996; Cortright and Szallasi, 2004; Mohapatra and Nau, 2005). The signaling milieu in which TRPV1 is immersed will vary with local conditions, such as those prevailing at a site of inflammation. In addition, TRPV1 has now been described in a range of cell types other than C-fiber sensory neurons (Nagy et al., 2004), and these different cell types will differ in their internal signaling environment. These different signaling environments would be predicted to modulate the response of TRPV1 to drugs (Szallasi, 2006).
For capsaicin, it is established that the signaling environment modulates the response of TRPV1. For example, alteration in the phosphatidylinositol-4,5-bisphosphate binding site of TRPV1 shifted the EC50 of capsaicin from 0.42 μM to 3.0 μM (Prescott and Julius, 2003). Conversely, mutation of one of the proton sensing residues on TRPV1 (E600Q) sensitized the receptor to capsaicin, shifting the EC50 from 520 nM to 40 nM (Jordt et al., 2000). Similarly, it is clear that modulation of the signaling environment can change the pattern of response for compounds that are partial agonists/partial antagonists. We have demonstrated that the combination of elevated temperature, acidity, and phorbol ester treatment dramatically shifted the behavior of JYL-827 (N-[2-(3,4-dimethylbenzyl)-3-(pivaloyloxy)propyl]-N′-[4-(methylsulfonylamino)benzyl]thiourea) from 7% agonism to 89% agonism (Wang et al., 2003). Similarly, treatment with cyclosporin A converted anandamide from a partial to a complete agonist (Lizanecz et al., 2006).
In the present study, we have explored the extent to which modulation of TRPV1 may have differential effects on the potencies of different ligands. We examined the effect of cyclosporin A, which we have previously found to have a modest differential effect, sensitizing TRPV1 to anandamide in vivo (Lizanecz et al., 2006). We describe here that inhibition by cyclosporin A of TRPV1 dephosphorylation in CHO cells expressing rat TRPV1 caused a substantial, differential shift in structure activity relations of agonists and, albeit to a lesser extent, antagonists.
The conclusion that the potencies of agonist and antagonists are dependent on the context in which TRPV1 is present has important implications for drug discovery. It suggests that activated TRPV1 (e.g. by phosphorylation evoked by local release of inflammatory mediators) can be selectively targeted; consequently the possible side effects of TRPV1 modulation (like pain) can be dissected. The extent of such actual variation in ligand potency, of course, will await studies in animal models and the analysis of a broader array of ligand structures, but the TRPV1 ligands characterized here illustrate yet again the exciting opportunities afforded by TRPV1 as a therapeutic target.
Materials and Methods
Materials
Cyclosporin A was obtained from Biomol (Plymouth Meeting, PA). FK-506 was from Sigma-Aldrich (St. Louis, MO). Radioactive calcium (45Ca2+, specific activity 5 – 30 Ci/g) was purchased from MP Biomedicals (Irvine, CA). [20-3H] resiniferatoxin was from PerkinElmer (Boston, MA). Capsaicin, anandamide, calyculin A, and okadaic acid were from Sigma-Aldrich. RTX was from LC Laboratories (Waltham, MA). AMG9810 and SB366791 were from Tocris (Ellisville, MO). Evodiamine was supplied by Pharm-Rx Chemical Corporation (Denville, NJ, USA). Capsazepine was from Alexis (San Diego, CA). The syntheses of olvanil, DA-5018 (Jung et al., 1999), BCTC (Valenzano et al., 2003), and JYL-1511 (Wang et al., 2003) were by standard methods. The syntheses of CHK-679, HCR-211, HCR-331, MK-484, PSY-279, YHS-369 will be described elsewhere. The chemical names of these latter compounds are as follows. CHK-679: N-[2-(4-t-Butylbenzyl)-3-(pivaloyloxy)propyl]-2-[4-(2-aminoethoxy)-3-methoxyphenyl]acetamide; HCR-211: : N-(4-t-Butylphenyl)-N′-{4-[3-fluoro-4-(methylsulfonylamino)phenyl]-1-piperazyl}urea; HCR-331, N-[2-(4-t-Butylbenzyl)-3-(pivaloyloxy)propyl]-N′-(7-hydroxynaphthalen-1-yl)urea; MK-484: N-[(2R)-2-(4-t-Butylbenzyl)-3-(pivaloyloxy)propyl]-(2S)-2-[4-hydroxy-3-methoxyphenyl]propionamide; PSY-279, N-[2-(4-t-Butylbenzyl)-3-(pivaloyloxy)propyl]-2-(4-hydroxy-3-methoxyphenyl)acetamide; YHS-369, N-(3,4-Dichlorobenzyl)-N′-(7-hydroxynaphthalen-1-yl)urea; YS-282: N-[3,3-Bis-(4-fluorophenyl)propyl]-2-[3-fluoro-4-(methylsulfonylamino)phenyl)propionamide.
Cell Culture
The stable CHO-rTRPV1 cell clone (Tet-Off) was cultured in maintaining medium (Ham’s F-12 supplemented with 10% fetal bovine serum, 25 mM HEPES, 250 μg/ml Geneticin (G-418), and 1 ml/l tetracycline (all from Invitrogen, Carlsbad, CA)).
45Ca2+- Uptake
CHO-rTRPV1 cells were plated in 24-well plates, reaching 30 to 50% confluence in maintaining medium after 24 hours. The cells were then washed twice with Dulbecco’s phosphate buffered saline (DPBS; Invitrogen, Carlsbad, CA) to remove tetracycline, and fresh medium without tetracycline and geneticin were added to induce TRPV1 expression. Experiments were done approximately 48 hours after induction. The cells at this time were at least 90% confluent.
For 45Ca2+ uptake assays of agonists, the cells were pre-incubated for 15 minutes, with and without CsA or other phosphatase inhibitor, at 37° C in Dulbecco’s Modified Eagle’s Medium (DMEM) and 0.25 mg/ml bovine serum albumin (Sigma). After 15 minutes, DMEM containing 1 μCi/ml of 45Ca2+ together with capsaicin or the agonist being evaluated was added to the wells and subsequently incubated for an additional 5 minutes. After incubation, extracellular 45Ca2+ was removed by washing the cells two times with DPBS and then 400 μl of lysis buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, and 1% sodium deoxycholate) was added to lyse the cells. The cells were put on a shaker for at least 20 minutes. Aliquots of the cell lysate were counted in a liquid scintillation counter. Dose response curves were determined with quadruplicate points at each concentration of ligand in each experiment. All experiments were performed at least three times, unless otherwise indicated. The actual number of experiments is indicated in the table or figure legends. EC50 values were calculated from the dose response curves by fitting to the Hill equation using Origin 6.0 (Microcal Software, Northampton, MA).
For analysis of antagonists, a similar protocol was followed but the different concentrations of the antagonists were applied together with the specific TRPV1 agonist capsaicin (75 nM).
Intracellular calcium imaging
Cells (CHO-TRPV1 Tet-off) were plated on 25 mm round glass coverslips in maintaining medium (F-12 supplemented with 10% FBS, 25 mM HEPES, pH 7.4, 250 μg/ml geneticin, and 1 mg/L tetracycline). The next day, the medium was replaced by inducing medium (maintaining medium without tetracycline but with 1 mM sodium butyrate) to induce TRPV1 expression. Experiments were performed approximately 24 hours after induction. For fura-2 AM loading, the cells were incubated in DPBS containing calcium and magnesium (Invitrogen, Carlsbad, CA), 0.25 mg/ml BSA (Sigma, St. Louis, MO) and 1 μM fura-2 AM (Molecular Probes, Eugene, OR) for 90 minutes at room temperature. The cells were then kept in DPBS containing 0.25 mg/ml BSA and with or without 100 nM CsA at room temperature for 15 minutes prior to measurements. The fluorescence of individual cells was measured with an InCyt Im2 fluorescent imaging system (Intracellular Imaging Inc., Cincinnati, OH). The cells within a field were illuminated alternately at 340 and 380 nanometers. Emitted light >510 nm was measured. Data were analyzed with the Incyt 4.5 software and Microsoft Excel programs.
Statistical analysis
The statistical significance of the differences between means were calculated using an unpaired, two-tailed t-test.
Results
We and others have described that cyclosporin A treatment could increase the potency of agonists for stimulation of TRPV1 in various experimental models (Docherty et al., 1996, Mohaparta et al., 2005, Lizanecz et al., 2006), a result which is not unexpected in view of the sensitization of TRPV1 by various kinases (Galoyan, 2003).
To better evaluate the magnitude of the changes in sensitivity and the extent to which compounds differed in their responses, we examined a series of 8 TRPV1 agonists of diverse structures which spanned some 3 orders of magnitude in potency for stimulation of 45Ca2+ uptake by CHO cells heterologously expressing rat TRPV1 and showed full agonism. Cells were pretreated for 15 min with cyclosporin A before addition of agonist and 45Ca2+ uptake was measured with a 5 min incubation period. In no case did the cyclosporin A treatment cause a decrease in potency nor did it cause a reduction in the efficacy of these full agonists. Marked differences in the increase in potency were noted, however (Table 1). RTX was at one extreme, with potency the same as that of in the absence of cyclosporin A (p = 0.61, t-test), consistent with our previous results. All of the other seven agonists showed significantly enhanced potencies (p < 0.05 to p < 0.001, t-test). Two of the eight agonists showed enhancements of potency of between 5- and 8-fold. Capsaicin, for comparison, showed a 2.1-fold enhancement.
Table 1.
Effect of treatment with 100 nM cyclosporin A on the potencies of agonists for stimulation of 45Ca2+ uptake by CHO-rTRPV1 cells
| Agonist | Control * EC50, nM | + Cyclosporin A EC50, nM | Ratio Control/Cyclosporin A |
|---|---|---|---|
| Capsaicin | 94.8 ± 8.4 | 45.3 ± 4.8c | 2.1 |
| RTX | 1.49 ± 0.26 | 1.31 ± 0.19a | 1.1 |
| Evodiamine | 838 ± 41 | 441 ± 6.0d | 1.9 |
| DA-5018 | 381 ± 48 | 48.6 ± 5.8c | 7.8 |
| Olvanil | 193 ± 31 | 84 ± 18b | 2.3 |
| CHK-679 | 54.8 ± 9.0 | 10.7 ± 1.5c | 5.1 |
| PSY-279 | 5.49 ± 0.27 | 2.63 ± 0.13d | 2.1 |
| PPAHV | 157 ± 11 | 110.9 ± 3.3b | 1.4 |
Values represent the mean ± SEM of at least 3 independent experiments.
p > 0.05;
p < 0.05;
p< 0.01;
p < 0.001 (unpaired, 2-tailed t-test)
In these experiments, we used cyclosporin A at a concentration of 100 nM. In the case of the agonist DA-5018, we determined the EC50 of cyclosporin A for the enhancement of potency. The EC50 was 42.9 ± 7.0 nM (mean ± SEM, n = 6 experiments), consistent with the potency of cyclosporin A as a specific protein phosphatase 2B inhibitor (Figure 1). We also evaluated the maximal potential effect of cyclosporin A at 30 μM. This concentration of cyclosporin A by itself caused no increase in 45Ca2+ uptake under our assay conditions (3 experiments, data not shown). Treatment with 30 μM cyclosporin A induced a similar shift in structure activity relations but with increases of potency of up to 14-fold (3 or more independent experiments, data not shown).
Figure 1.
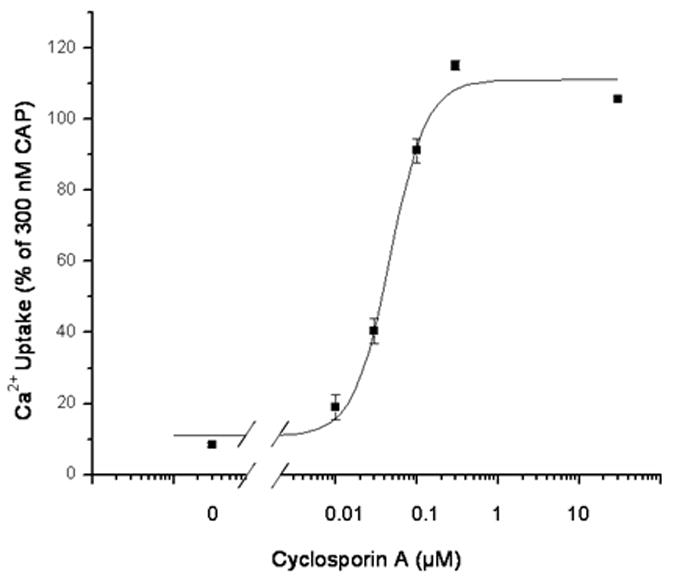
Potency of the protein phosphatase 2B inhibitor cyclosporin A for stimulating 45Ca2+ uptake in response to a suboptimal concentration of the agonist DA-5018. Cells were treated for 15 min in the absence or presence of the indicated concentrations of cyclosporin A. A suboptimal concentration of DA-5018 (100 nM) was then added and 45Ca2+ uptake was measured as described in methods. Results are of a single experiment. Bars, SEM of quadruplicate measurements in a single experiment. The experiments were repeated an additional 5 times with similar results.
To evaluate whether the apparent increased potency of agonists for 45Ca2+ uptake in the presence of cyclosporin A reflected a change in the kinetics of calcium uptake and retention leading to the enhanced uptake rather than a change in potency, we treated cells with increasing concentrations of DA-5018 in the presence or absence of cyclosporin A and measured response by monitoring the level of intracellular calcium as a function of time using the ratiometric calcium reporter dye Fura-2 (Figure 2). As can be seen, low concentrations of DA-5018 caused a gradually increasing level of intracellular calcium, similar to the pattern of response described for resiniferatoxin (Szallasi et al., 1999; Toth et al., 2005). At higher concentrations of DA-5018, intracellular calcium reached a plateau, showing little subsequent decrease over the 15 min time course of the experiment. Treatment with cyclosporin A (100 nM) caused an enhanced response at low DA-5018 concentrations and early times. Again, at higher DA-5018 concentrations and latter times the elevated level of intracellular calcium was maintained. We conclude that the increased potency of DA-5018 in the presence of cyclosporin A is not reflective of a change in the kinetics of increase or intracellular retention of calcium, independent of the increase in DA-5018 potency.
Figure 2.
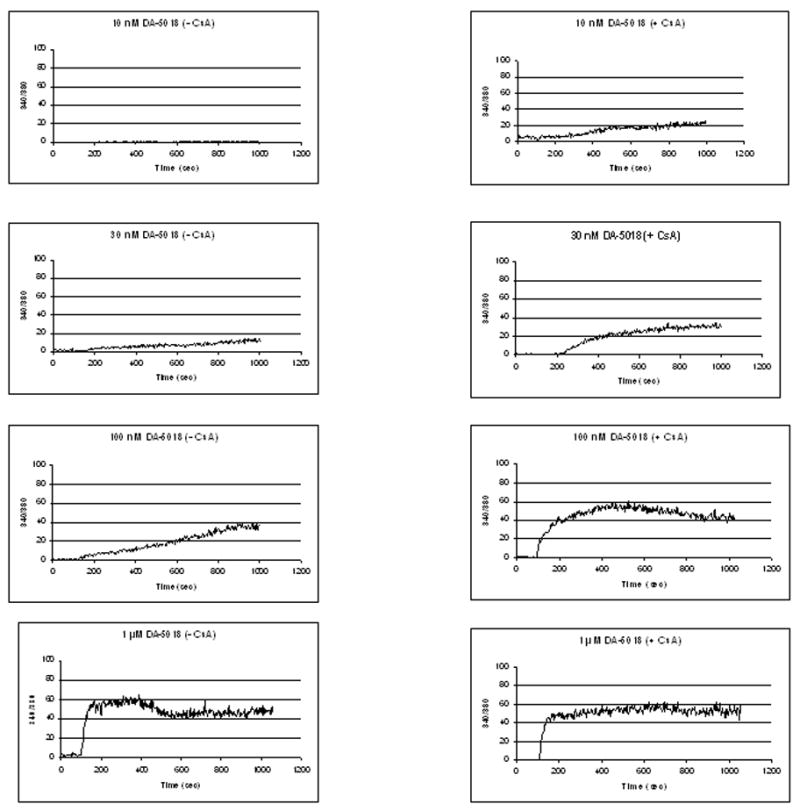
Effect of cyclosporin A on the time course of the response to increasing concentrations of DA-5018 as determined by calcium imaging. Cells were incubated in the absence or presence of 100 nM cyclosporin A for 15 min. DA-5018 at the indicated concentrations was then added. Intracellular calcium concentrations were monitored by Fura-2 imaging. Results are from a single, representative experiment. Each experiment was performed 3–5 times.
We next determined the effect of cyclosporin A treatment on the potencies of a series of 7 structurally diverse TRPV1 antagonists. This analysis was of particular importance, since there is very limited information about the sensitivity of TRPV1 antagonists to the phosphorylation state of TRPV1, in contrast to the data concerning capsaicin or the endogenous agonist anandamide. Cyclosporin A (100 nM) treatment caused either no change or a decrease in potency (increase in IC50), contrasting with the increases in potency for the TRPV1 agonists (Table 2). For 3 antagonists the changes in potency were not statistically significant (p > 0.05). For three of the others the decrease in potency was 1.4 – 1.7 fold, for the last it was 2.5-fold (p < 0.02 for each of the four compounds, t-test). We likewise evaluated the effect of cyclosporin A at 30 μM and obtained similar results (triplicate experiments, data not shown).
Table 2.
Effect of treatment with 100 nM cyclosporin A on the inhibition by antagonists of capsaicin stimulated 45Ca2+ uptake by CHO-rTRPV1 cells
| Antagonist | Control * IC50, nM | + Cyclosporin A IC50, nM | Ratio Control/Cyclosporin A(Cyclosporin A/Control) |
|---|---|---|---|
| BCTC | 0.35 ± 0.07 | 0.53 ± 0.12a | 0.66 (1.5) |
| HCR-331 | 4.62 ± 0.7 | 7.14 ± 0.7a | 0.65 (1.5) |
| YHS-369 | 17.6 ± 3.5 | 29.6 ± 4.0a | 0.6 (1.7) |
| AMG 9810 | 33.0 ± 0.6 | 50.2 ± 4.5b | 0.66 (1.5) |
| HCR-211 | 97.8 ± 9.0 | 137.2 ± 4.8b | 0.7 (1.4) |
| SB 366791 | 516 ± 60 | 1305 ± 57d | 0.4 (2.5) |
| Capsazepine | 574 ± 30 | 964 ± 61c | 0.6 (1.7) |
Values represent the mean ± SEM of at least 3 independent experiments.
p > 0.05;
p < 0.05;
p< 0.01;
p < 0.001 (unpaired, two tailed t-test)
Partial agonism represents a third pattern of response observed for some TRPV1 ligands. We examined the effect of cyclosporin A treatment (100 nM) on the behavior of the partial agonist JYL-1511. This compound was chosen for analysis since it had been shown to function competitively with capsaicin. As expected, cyclosporin A treatment increased the extent of agonism (Figure 3) (from 6.4 ± 0.67 % to 25.1 ± 5.9 % efficacy, n = 3 experiments). Similar results were seen with two other partial agonists (data not shown).
Figure 3.
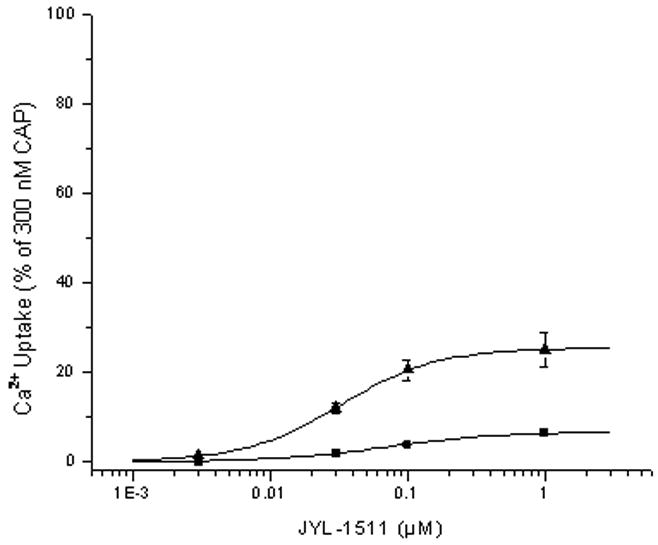
Effect of treatment with cyclosporin A on the efficacy of the partial agonist JYL-1511 for stimulation of 45Ca2+ uptake by CHO-rTRPV1 cells. Dose response curves were determined for stimulation of 45Ca2+ uptake by partial agonists in control cells (■) and those pretreated for 15 min with 100 nM cyclosporin A (▲). Stimulation was expressed relative to that by capsaicin (300 nM), likewise in the absence or presence of cyclosporin A. Data shown are average of three independent experiments. Error bars, SEM.
To assess the specificity of the action of cyclosporin A, we examined the effect of a structurally distinct protein phosphatase 2B inhibitor, FK506. In the absence of a TRPV1 agonist, FK506 treatment under our assay conditions caused no increase in 45Ca2+ uptake (triplicate experiments, data not shown). Like cyclosporin A, FK506 sensitized the cells to the agonist DA-5018 (Figure 4A). Its EC50 for potentiation was 147 ± 13 nM (mean ± SEM, n = 3 experiments).
Figure 4.
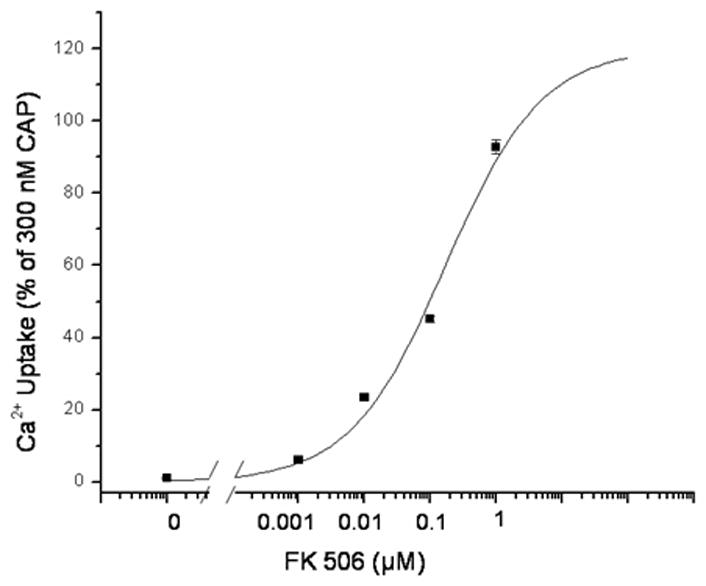
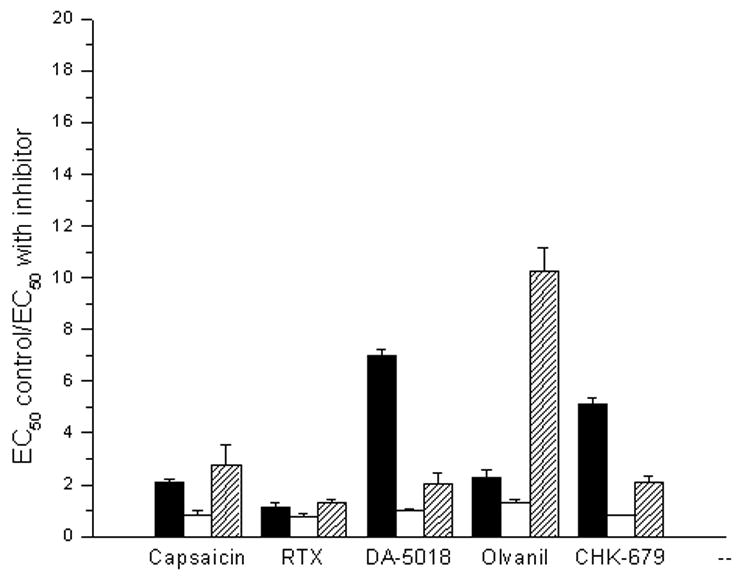
Analysis of other phosphatase inhibitors on the stimulation of calcium uptake by TRPV1 agonists. A. Potency of the protein phosphatase 2B inhibitor FK-506 for stimulating 45Ca2+ uptake in response to a suboptimal concentration of the agonist DA-5018. Cells were treated for 15 min in the absence or presence of the indicated concentrations of FK506. A suboptimal concentration of DA-5018 (100 nM) was then added and 45Ca2+ uptake was measured as described in methods. Results are of single experiments. Bars, SEM of quadruplicate measurements in a single experiment. The experiment was repeated an additional 2 times with similar results. B. Comparison of the effect of treatment with various protein phosphatase inhibitors on the potencies of agonists for stimulation of 45Ca2+ uptake by CHO-rTRPV1 cells. Cells were treated for 15 min with the protein phosphatase 2B inhibitor cyclosporin A (100 nM, filled bars) or with the protein phosphatase 1 and 2A inhibitors okadaic acid (300 nM, open bars) and calyculin A (100 nM, shaded bars). The EC50 values for stimulation of 45Ca2+ uptake by the various agonists were then determined and compared with their corresponding EC50 values for control cells not treated with a phosphatase inhibitor. Bars indicate the ratio of triplicate independent experiments. Error bars, ± SEM. All values for cyclosporin A treatment were significantly (p < 0.05, unpaired, two-tailed t-test) different from the control except for that of RTX. Okadaic acid induced no significant changes. All values for calyculin A treatment were significantly (p < 0.05, unpaired, two-tailed t-test) different from the control.
In contrast with the body of knowledge about calcineurin in the regulation of TRPV1 responsiveness, relatively little is known about the effect of other major protein phosphatases such as 1 and 2A. We therefore examined the effect of two protein phosphatase inhibitors (okadaic acid and calyculin A) with different selectivities for protein phosphatases 1 and 2A (Figure 4B). Okadaic acid had no significant effect (p > 0.05) on the potencies of the five TRPV1 agonists examined. Calyculin A had modest but significant (p < 0.05) effects (1.4- to 2.3-fold) on four of the five agonists but caused substantial sensitization (8.8-fold) for olvanil, thus yielding a profile of response markedly distinct from that of either cyclosporin A or okadaic acid. We conclude that not only are TRPV1 structure activity relations differentially modulated by phosphatase action but also the different phosphatases have differential effects.
Discussion
Our results clearly demonstrate the substantial, differential effect exerted by cyclosporin A treatment on TRPV1 agonist and antagonist potencies. Our first conclusion is that there is not some absolute potency of ligands for TRPV1, as determined by 45Ca2+ uptake. This conclusion had already been documented by multiple reports of different treatments sensitizing TRPV1 to capsaicin or anandamide (Tominaga et al., 1998; Di Marzo et al., 2002; Lizanecz et al., 2006), but our studies extend these conclusions to a diverse series of ligands, including full and partial agonists and also antagonists. Importantly, we show that sensitization does not occur in parallel for all agonists or antagonists. Rather, TRPV1 can be sensitized differentially to some structures relative to others. In case of the agonists, RTX was at one end of the spectrum of agonists, with negligible enhancement of potency, whereas DA-5018, for example, showed a 7.8-fold increase in potency. This shift in potencies changed the rank order of ligands. Thus, whereas the rank order of potencies was RTX > PSY-279 > CHK-679 > capsaicin > PPAHV > olvanil > DA-5018 > evodiamine under control conditions, it changed to RTX > PSY-279 > CHK-679 > capsaicin > DA5018 > olvanil > PPAHV > evodiamine in the presence of cyclosporin A. In the case of the competitive TRPV1 antagonists, cyclosporin A treatment did not increase but rather decreased the potency of most of the antagonists examined. Once again, appreciable shifts in IC50 were often observed, ranging up to 2.5-fold. Finally, we confirmed that the cyclosporin A treatment could enhance the agonist efficacy for the partial agonist JYL-1511, as we had described previously (Wang et al., 2003) for low pH, for elevated temperature, and for phorbol ester treatment.
Given the limited number of agonists examined in this study, it is premature to draw any firm conclusions about structural features of the ligands conferring sensitivity to the action of cyclosporin A. Nonetheless, it might be noted that DA-5018 and CHK-679 both have a 4-O-aminoethyl derivatized A region. Such a pattern of substitution has attracted attention previously because of improved metabolic stability and a potential slower rate of penetration (Wrigglesworth et al., 1996; Jung et al., 1999). A slow rate of penetration does not seem to be the relevant feature, however, since RTX, for example, shows no sensitization and would also be expected to penetrate slowly. Among agonists, those that have been shown to have limited pungency are of particular therapeutic interest. It is noteworthy that the potency of olvanil was substantially increased by calyculin A whereas that of RTX was not.
Phosphorylation represents a prominent mechanism for regulation of TRPV1 and we selected cyclosporin A as a well characterized modulator of phosphorylation, acting through inhibition of protein phosphatase 2B (Clipstone and Crabtree, 1992). We confirmed that FK-506, another protein phosphatase 2B inhibitor (Liu et al., 1991), similarly sensitized TRPV1, arguing that the effect we observed was not through a secondary mechanism unique to cyclosporin A (Zhang and Victor, 2000). Furthermore, the potencies of cyclosporin A and FK-506 for the enhancement of potency were in the range of their in vitro potencies as inhibitors of protein phosphatase 2B.
The effects of protein phosphatases 1 and 2A were also examined by the application of okadaic acid and calyculin A, two well known inhibitors of these enzymes (Ishihara et al., 1989). Okadaic acid was without substantial effects on the potencies of the compounds tested (potencies were 0.8–1.3 fold those in the absence of okadaic acid), whereas calyculin A enhanced the potency of olvanil by 8.8-fold. Previously, okadaic acid had been reported not to affect capsaicin-induced TRPV1 desensitization (Mohaptra and Nau, 2005). The different effects of these inhibitors are in accordance with previous findings, showing that okadaic acid is about two orders of magnitude more potent for inhibiting protein phosphatase 2A than 1 (Ishihara et al., 1989, Hori et al., 1991, Favre et al., 1997). Our data suggest that protein phosphatase 1 may contribute to the regulation of TRPV1 sensitivity to some (e.g. olvanil) but not to other TRPV1 agonists (e.g. DA-5018 and CHK-679). Interestingly, calyculin A showed a different pattern of sensitization than did the inhibitors of protein phosphatase 2B. These differences are clearly shown by three findings (i) the effects of RTX were not affected by any of the phosphatase inhibitors; (ii) calyculin A had a greater effect on the potency of olvanil than did cyclosporin A; conversely, (iii) cyclosporin A had an appreciably greater effect on the potencies of DA-5018 and CHK-679 than did calyculin A.
In any case, it was not our intention to link specific sites of phosphorylation on TRPV1 to specific patterns of structure activity relations. Such analysis will require rather different experimental strategies. Likewise, we recognize that effects of phosphorylation on calcium uptake will represent the potential contributions of multiple underlying mechanisms, not only phosphorylation of TRPV1. Rather, the goal of the present analysis was to focus on a key issue for drug discovery, namely whether TRPV1 selectivity for agonist and antagonist function was a constant or whether it depended on TRPV1 environment. Our data suggest that compounds can be designed to selectively target TRPV1 under conditions associated with increased/decreased protein phosphatase 1 or 2B activities. In addition, dephosphorylation of some phosphorylation sites in TRPV1 is phosphatase specific and with potential functional relevance.
It is important to recognize that potency of ligands for TRPV1 is not the only factor determining the impact of the ligand on intracellular calcium concentration over time. Toth et al. (2005), for example, highlighted differences in the rate of initial response to different ligands, probably reflecting the potential slow penetration of some compounds into cells (Lazar et al., 2006), differences in the kinetics of desensitization, and differences in the pattern of response of individual cells. Mohapatra and Nau (2005) have suggested an effect of cyclosporin A treatment on reduced desensitization in response to capsaicin. These factors do not appear to be responsible for the effects observed for cyclosporin A treatment on the response to DA-5018, for example, but emphasize that assays that are more sensitive to such effects might reveal yet larger differences in ligand response to TRPV1 environment.
For our analysis of antagonists, we measured their potencies for inhibition of TRPV1 stimulation by capsaicin. Low pH and temperature represent other classes of physiological stimulators for TRPV1, and our experiments do not address the effect of cyclosporin A on the structure activity relations of TRPV1 antagonists for these other physiological activators. Different structure activity relations have been suggested for antagonism of stimulation by capsaicin and by protons (Westaway, 2007), so it would be plausible that the effects of cyclosporin A might also differ. Once again, however, such studies could only broaden the scope of the dependence of ligand structure activity on the TRPV1 environment beyond that already shown here.
An on-going challenge will be to characterize in detail the actual states of regulation of TRPV1 in its physiological settings and under pathological conditions. It will then be informative to define the structure activity relations of TRPV1 under these various conditions. Although animal models naturally incorporate such modulation of the TRPV1 microenvironment, the greater ease of screening in cellular assays will provide enhanced efficiency of achieving this second order level of understanding of structure activity relations if the appropriate TRPV1 regulatory environment can be modeled in the cellular assays.
Although not the focus of our studies, a possible implication of the enhanced potency for agonists upon cyclosporin A treatment is that this enhanced potency might contribute to the development of pain reported as a side effect of cyclosporin A treatment (Bailie and Eisele, 1990; Ponticelli and Campise, 2005). TRPV1 is tonically activated in vivo, as shown by effects of antagonists on body temperature (Steiner et al., 2007; Gavva et al., 2007), so its sensitization by cyclosporin A might be expected to lead to further stimulation. Of course, this would depend on whether the endogenous agonist(s) behave comparably to most of the TRPV1 agonists studied here and are sensitized in response to cyclosporin A.
Acknowledgments
The research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by grants from the Basic Research Program of the Korean Science and Engineering Foundation (R01-2007-000-20052-0) and Research Institute of Pharmaceutical Sciences.
Nonstandard abbreviations
- TRPV1
Transient Receptor Potential channel type V1
- RTX
Resiniferatoxin
- CsA
Cyclosporin A
- FK-506
Tacrolimus
References
- Bailie GR, Eisele G. Cyclosporine-associated arthralgia. Clin Nephrol. 1990;33:256–267. [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th Protein Kinase C phosphorylation sensitizes but does not activate capsaicin receptor transient potential receptor vanilloid 1 (TRPV1) Proc Natl Acad Sci. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Clipstone NA, Crabtree GR. Identification of calcineurin as a key signaling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- Cortright DN, Szallasi Z. Biochemical pharmacology of the vanilloid receptor TRPV1. An update. Eur J Biochem. 2004;271:1814–1819. doi: 10.1111/j.1432-1033.2004.04082.x. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Blumberg PM, Szallasi A. Endovanilloid Signaling in Pain. Curr Opin Neurobiol. 2002;12:372–379. doi: 10.1016/s0959-4388(02)00340-9. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Bevan S, Boddeke HW. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurons from adult rats. Pflueg Arch Eur J Physiol. 1996;431:828–837. doi: 10.1007/s004240050074. [DOI] [PubMed] [Google Scholar]
- Favre B, Turowski P, Hemmings BA. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J Biol Chem. 1997;272:13856–13863. doi: 10.1074/jbc.272.21.13856. [DOI] [PubMed] [Google Scholar]
- Galoyan SM, Petruska JC, Mendell LM. Mechanisms of sensitization of the response of single dorsal root ganglion cells from adult rat to noxious heat. Eur J Neurosci. 2003;18:535–541. doi: 10.1046/j.1460-9568.2003.02775.x. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Bannon AW, Surapaneni S, Hovland DN, Jr, Lehto SG, Gore A, Juan T, Deng H, Han B, Klionsky L, Kuang R, Le A, Tamir R, Wang J, Youngblood B, Zhu D, Norman MH, Magal E, Treanor JJS, Louis JC. J Neurosci. 2007;27:3366–3374. doi: 10.1523/JNEUROSCI.4833-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori M, Magae J, Han YG, Hartshorne DJ, Karaki H. A novel protein phosphatase inhibitor, tautomycin. Effect on smooth muscle. FEBS Lett. 1991;285:145–148. doi: 10.1016/0014-5793(91)80745-o. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Martin BL, Brautigan DL, Karaki H, Ozaki H, Kato Y, Fusetani N, Watabe S, Hashimoto K, Uemura D, Hartshorne DJ. Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem Biophys Res Commun. 1989;159:871–877. doi: 10.1016/0006-291x(89)92189-x. [DOI] [PubMed] [Google Scholar]
- Jin X, Morsy N, Winston J, Pasricha PJ, Garrett K, Akbarali HI. Modulation of TRPV1 by nonreceptor tyrosine kinase, c-Src kinase. Am J Physiol Cell Physiol. 2004;287:C558–563. doi: 10.1152/ajpcell.00113.2004. [DOI] [PubMed] [Google Scholar]
- Jordt SE, Tominaga M, Julius D. Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci USA. 2000;97:8134–8139. doi: 10.1073/pnas.100129497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Jung J, Hwang SW, Kwak J, Lee SY, Kang CJ, Kim WB, Kim D, Oh U. Capsaicin binds to the intracellular domain of the capsaicin-activated ion channel. J Neurosci. 1999;19:529–538. doi: 10.1523/JNEUROSCI.19-02-00529.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U. Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem. 2004;279:7048–7054. doi: 10.1074/jbc.M311448200. [DOI] [PubMed] [Google Scholar]
- Krause JE, Chenard BL, Cortright DN. Transient receptor potential ion channels as targets for the discovery of pain therapeutics. Curr Opin Investig Drugs. 2005;6:48–57. [PubMed] [Google Scholar]
- Lazar J, Braun DC, Toth A, Wang Y, Pearce LV, Pavlyukovets VA, Blumberg PM, Garfield SH, Wincovitch S, Choi HK, Lee J. Kinetics of penetration influence the apparent potency of vanilloids on TRPV1. Mol Pharmacol. 2006;69:1166–1173. doi: 10.1124/mol.105.019158. [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Lizanecz E, Bagi Z, Pasztor ET, Papp Z, Edes I, Kedei N, Blumberg PM, Toth A. Phosphorylation-dependent desensitization by anandamide of vanilloid receptor-1 (TRPV1) function in rat skeletal muscle arterioles and in Chinese hamster ovary cells expressing TRPV1. Mol Pharmacol. 2006;69:1015–1023. doi: 10.1124/mol.105.015644. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C. Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem. 2005;280:13424–13432. doi: 10.1074/jbc.M410917200. [DOI] [PubMed] [Google Scholar]
- Ponticelli C, Campise MR. Neurological complications in kidney transplant recipients. J Nephrol. 2005;18:521–528. [PubMed] [Google Scholar]
- Nagy I, Santha P, Jancso G, Urban L. The role of the vanilloid (capsaicin) receptor (TRPV1) in physiology and pathology. Eur J Pharmacol. 2004;500:351–369. doi: 10.1016/j.ejphar.2004.07.037. [DOI] [PubMed] [Google Scholar]
- Prescott ED, Julius D. A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science. 2003;300:1284–1288. doi: 10.1126/science.1083646. [DOI] [PubMed] [Google Scholar]
- Robbins W. Clinical applications of capsaicinoids. Clin J Pain. 2000;16:S86–89. doi: 10.1097/00002508-200006001-00015. [DOI] [PubMed] [Google Scholar]
- Silva C, Ribeiro MJ, Cruz F. The effect Of intravesical resiniferatoxin in patients with idiopathic detrusor instability suggests that involuntary detrusor contractions are triggered by C-fiber input. J Urol. 2002;168:575–579. [PubMed] [Google Scholar]
- Stein AT, Ufret-vincenty CA, Hua L, Santan LF, Gordon SE. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J Gen Physiol. 2006;128:509–522. doi: 10.1085/jgp.200609576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner AA, Turek VF, Almeida MC, Burmeister JJ, Oliveira DL, Roberts JL, Bannon AW, Norman MH, Louis JC, Treanor JJS, Gavva NR, Romanovsky AA. Nonthermal activation of transient receptor potential vanilloid-1 channels in abdominal viscera tonically inhibits autonomic cold-defense effectors. J Neurosci. 2007;27:7459–7468. doi: 10.1523/JNEUROSCI.1483-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szallasi A. Small molecule vanilloid TRPV1 receptor antagonists approaching drug status: can they live up to the expectations? Naunyn-Schmiedeberg’s Arch Pharmacol. 2006;373:272–286. doi: 10.1007/s00210-006-0072-3. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM. Vanilloid (capsaicin) receptors and mechanisms. Pharmacol Rev. 1999;51:159–212. [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM, Annicelli LL, Krause JE, Cortright DN. The cloned rat vanilloid receptor VR1 mediates both R-type binding and C-type calcium response in dorsal root ganglion neurons. Mol Pharmacol. 1999;56:581–587. doi: 10.1124/mol.56.3.581. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Toth A, Wang Y, Kedei N, Tran R, Pearce LV, Kang SU, Jin MK, Choi HK, Lee J, Blumberg PM. Different vanilloid agonists cause different patterns of calcium response in CHO cells heterologously expressing rat TRPV1. Life Sci. 2005;76:2921–2932. doi: 10.1016/j.lfs.2004.10.056. [DOI] [PubMed] [Google Scholar]
- Valenzano KJ, Grant ER, Wu G, Hachicha M, Schmid L, Tafesse L, Sun Q, Rotshteyn Y, Francis J, Limberis J, Malik S, Whittemore ER, Hodges D. N-(4-tertiarybutylphenyl)-4-(3-chloropyridin-2-yl)tetrahydropyrazine-1(2H)-carboxamide (BCTC), a novel orally effective vanilloid receptor 1 antagonist with analgesic properties: In vitro characterization and pharmacokinetic properties. J Pharmacol Exp Therap. 2003;306:377–386. doi: 10.1124/jpet.102.045674. [DOI] [PubMed] [Google Scholar]
- Wang Y, Toth A, Tran R, Szabo T, Welter JD, Blumberg PM, Lee J, Kang SU, Lee J. High affinity partial agonists of the vanilloid receptor. Mol Pharmacol. 2003;64:325–333. doi: 10.1124/mol.64.2.325. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kedei N, Wang M, Wang QJ, Huppler AR, Toth A, Tran R, Blumberg PM. Interaction between protein kinase Cmu and the vanilloid receptor type 1. J Biol Chem. 2004;279:53674–53682. doi: 10.1074/jbc.M410331200. [DOI] [PubMed] [Google Scholar]
- Westaway SM. The potential of transient receptor potential vanilloid type 1 channel modulators for the treatment of pain. J Med Chem. 2007;50:2589–2596. doi: 10.1021/jm060637e. [DOI] [PubMed] [Google Scholar]
- Wrigglesworth R, Walpole CSJ, Bevan S, Campbell EA, Dray A, Hughes GA, James I, Masdin KJ, Winter J. Analogues of capsaicin with agonist activity as novel analgesic agents: structure-activity studies. 4. Potent, orally active analgesics. J Med Chem. 1996;39:4942–4951. doi: 10.1021/jm960512h. [DOI] [PubMed] [Google Scholar]
- Zhang W, Victor RG. Calcineurin inhibitors cause renal afferent activation in rats: a novel mechanism of cyclosporine-induced hypertension. Am J Hypertens. 2000;13:999–1004. doi: 10.1016/s0895-7061(00)00288-0. [DOI] [PubMed] [Google Scholar]