

Abstract
Background and purpose:
‘Spice’ is an herbal blend primarily marketed in Europe as a mild hallucinogen with prominent cannabis-like effects and as a legal alternative to cannabis. However, a recent report identified a number of synthetic additives in samples of ‘Spice’. One of these, the indole derivative JWH018, is a ligand for the cannabinoid receptor 1 (CB1) cannabinoid receptor and inhibits cAMP production in CB1 receptor-expressing CHO cells. Other effects of JWH018 on CB1 receptor-mediated signalling are not known, particularly in neurons. Here we have evaluated the signalling pathways activated by JWH018 at CB1 receptors.
Experimental approach:
We investigated the effects of JWH018 on neurotransmission in cultured autaptic hippocampal neurons. We further analysed its activation of ERK1/2 mitogen activated protein kinase (MAPK) and internalization of CB1 receptors in HEK293 cells stably expressing this receptor.
Key results:
In cultured autaptic hippocampal neurons, JWH018 potently inhibited excitatory postsynaptic currents (IC50= 14.9 nM) in a concentration- and CB1 receptor-dependent manner. Furthermore, it increased ERK1/2 MAPK phosphorylation (EC50= 4.4 nM). We also found that JWH018 potently induced rapid and robust CB1 receptor internalization (EC50= 2.8 nM; t1/2= 17.3 min).
Conclusions and implications:
JWH018, a prominent component of several herbal preparations marketed for their psychoactivity, is a potent and effective CB1 receptor agonist that activates multiple CB1 receptor signalling pathways. Thus, it is likely that the subjective effects of ‘Spice’ are due to activation of cannabinoid CB1 receptors by JWH018, added to this herbal preparation.
Keywords: Spice, JWH018, cannabinoid, CB1 receptor, internalization, ERK1/2, MAPK, neurotransmission, marijuana, THC
Introduction
Cannabis sativa (cannabis, marijuana or hashish) is a widely used drug with well-known psychoactivity as well as potential medicinal value. Δ9-tetrahydrocannabinol (THC) has been identified as the principal psychoactive component of C. sativa, although it is only one of a number of bioactive phytocannabinoids found in the plant (Taura et al., 2007). The physiological effects of THC have been well described (Ameri et al., 1999; Howlett, 2002; Howlett et al., 2002; Costa, 2007; Pertwee, 2008).
The cannabinoid receptor 1 (CB1) cannabinoid receptor (nomenclature follows Alexander et al., 2008) has been identified as the receptor that mediates the behavioural effects of THC in animals (Monory et al., 2007) and likely does so in humans (Huestis et al., 2001). The CB1 receptor is predominately expressed in the CNS, particularly in areas such as the hippocampus, basal ganglia, cortex, amygdala and cerebellum – areas linked to behaviours affected by THC (Mackie, 2005). At the subcellular level, CB1 receptors are primarily found on axon terminals, a prime location to influence neurotransmission (Gulyas et al., 2004; Mackie, 2005; Nyiri et al., 2005; Yoshida et al., 2006; Matyas et al., 2007). The CB1 receptor is a G protein coupled receptor (GPCR) that couples to the Gi/o class of G proteins and as such, upon agonist activation leads to an inhibition of adenylyl cyclase and subsequent decrease in cellular cAMP levels. CB1 receptor activation also inhibits voltage gated calcium channels and activates inwardly rectifying potassium channels (Mackie et al., 1995; Twitchell et al., 1997; Howlett et al., 2002). Cumulatively, these effects on intracellular signalling result in reduced cellular excitability and, due to its proximity to synaptic terminals (Nyiri et al., 2005), in a reduction in the probability of neurotransmitter release (Shen et al., 1996). This ability to inhibit neurotransmission allows both exogenous cannabinoid agonists (such as THC) and endogenous cannabinoids (endocannabinoids) to have a profound impact on neuronal communication. As CB1 receptors are found on both glutamatergic and GABAergic terminals, their activation can suppress both inhibitory and excitatory synaptic transmission (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Chevaleyre et al., 2006; Straiker and Mackie, 2006).
CB1 receptor stimulation also results in activation of mitogen activated protein kinases (MAPKs), particularly extracellular signal-regulated kinase (p42/44 or ERK1/2) (Bouaboula et al., 1995; Daigle et al., 2008). MAPK activation results in phosphorylation of both nuclear transcription factors and other cytosolic targets that lead to changes in transcription, translation, cell motility, shape, proliferation, and differentiation (Derkinderen et al., 2003; Lefkowitz and Shenoy, 2005). Furthermore, in response to prolonged activation, CB1 receptor signalling is subject to regulation via receptor desensitization and internalization (Hsieh et al., 1999; Jin et al., 1999; Roche et al., 1999; Marchese et al., 2008). Desensitization is thought to result from phosphorylation of specific residues by GPCR kinases resulting in uncoupling of the receptor from G-protein signalling complexes. In contrast, internalization occurs via translocation of the receptor by endocytotic machinery to endosomes. Internalized receptors are subsequently recycled to the plasma membrane or degraded.
‘Spice’ is an herbal blend, marketed primarily in Europe for its cannabis-like effects and as an alternative to marijuana. A recent report used gas chromatography/mass spectrometry to analyse a number of different ‘Spice’ preparations as well as competing products from other manufacturers (Auwarter et al., 2009). Interestingly, these herbal ‘Spice’ blends contained diverse synthetic cannabinoid additives. Common among the different preparations was JWH018, a cannabinoid agonist from the aminoalkylindole family (Figure 1A). JWH018 has been shown to have a binding affinity for CB1 receptors in the low nanomolar range (∼9 nM) (Huffman et al., 1994; Showalter et al., 1996; Chin et al., 1999; Aung et al., 2000). In CB1 receptor expressing CHO cells, JWH018 inhibits forskolin-stimulated cAMP production with an EC50 of 14.7 nM with a maximal inhibition of 79% (Chin et al., 1999). Beyond this there has been no report of the effect of JWH018 on CB1 receptor-mediated cellular signalling. There has been a single report to date of the behavioural effects of JWH018 treatment. Wiley et al. (1998) found that JWH018 produced the tetrad of behaviours classically associated with cannabinoids (analgesia, catalepsy, hypomotility and hypothermia), having ED50 values ranging from a low of 0.09 mg·kg−1 for analgesia to a high of 1.47 mg·kg−1 for hypothermia in the rodent model, suggesting that JWH018 activated CB1 receptors in vivo.
Figure 1.
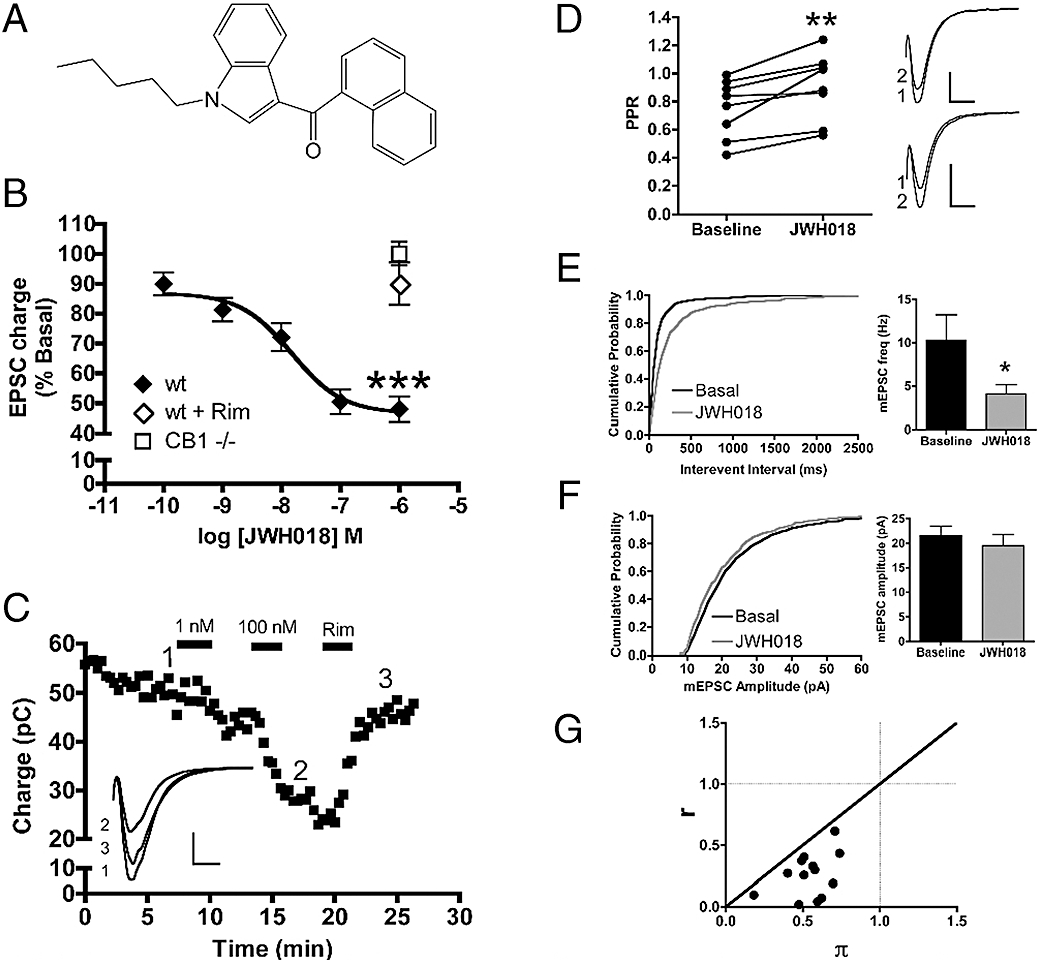
JWH018 decreases neurotransmitter release by activating presynaptic CB1 receptors. (A) Chemical structure of JWH018. (B) JWH018 concentration-dependently decreased EPSC charge (n= 8 to 14 for each concentration tested). This inhibitory effect was reversed by 1 µM rimonabant (n= 12) and was absent in CB1−/− neurons (n= 5). (C) A representative experiment showing a time course of EPSC inhibition by 1 nM JWH018 followed by 100 nM JWH018. 1 µM rimonabant reversed JWH018 inhibition. Inset shows representative traces for three indicated time points. JWH018 (100 nM) significantly increases the paired-pulse ratio (right: representative traces of paired pulses, numbers indicate order of pulses) (D) and decreases miniature EPSC frequency (E) without affecting miniature EPSC amplitude (F), indicative of decreasing the probability of neurotransmitter release by acting at a presynaptic site (n= 5 to 8 for each). Inset scale bars: 1 nA and 10 ms. (G) Coefficient of variation analysis demonstrating that r < π < 1, which is consistent with a presynaptic site for JWH018's action resulting in synaptic depression. Values are presented as mean ± SEM where applicable. *P < 0.05, **P < 0.01, ***P < 0.0001. CB1, cannabinoid receptor 1; EPSC, excitatory postsynaptic current.
Based on previous findings, we thought it was likely that JWH018 would act as an agonist in other CB1 receptor-mediated signalling pathways and sought to characterize its ability to function as such. We examined the effect of JWH018 on neurotransmission and ERK1/2 MAPK activation and its ability to produce CB1 receptor internalization. We found that JWH018 is both a potent and an efficacious CB1 receptor agonist in each of these areas, actions that likely explain the ability of ‘Spice’ preparations to produce marijuana-like effects.
Methods
Cell culture and transfection
All animal care and experimental procedures used in this study were approved by the Animal Care Committees of Indiana University and conform to the Guidelines of the National Institutes of Health on the Care and Use of Animals. Mouse (CD1 strain and GAD67-GFP) hippocampal neurons isolated from the CA1–CA3 region were cultured on micro-islands as previously described (Furshpan et al., 1976; Bekkers and Stevens, 1991). No significant differences were found between neurons isolated from either strain at any drug concentration tested, so the data from both strains were pooled. Neurons were obtained from animals (at postnatal day 0–2, killed via rapid decapitation without anaesthesia) and plated onto a feeder layer of hippocampal astrocytes that had been laid down previously (Levison and McCarthy, 1991). Cultures were grown in high-glucose (20 mM) minimum essential media containing 10% horse serum, without mitotic inhibitors and used for recordings after 8 days in culture and for no more than 3 h after removal from culture medium (Straiker and Mackie, 2005). All electrophysiological experiments were performed exclusively on excitatory neurons. All drugs were tested on cells from at least two different preparations.
Cell lines stably expressing CB1 receptors were made as previously described (Brown et al., 2002; Daigle et al., 2008). Stable clones, uniformly expressing CB1 receptors, were expanded and used for internalization and MAPK assays. Cells were grown in Dulbecco's modified Eagle's media with 10% fetal bovine serum and penicillin/streptomycin (GIBCO, Carlsbad, CA, USA) at 37°C in 5% CO2.
Electrophysiology
When a single neuron is grown on a small island of permissive substrate, it forms synapses – or ‘autapses’– onto itself. All experiments were performed on isolated autaptic neurons. Whole-cell, voltage-clamp recordings from autaptic neurons were carried out at room temperature using an Axopatch 200A amplifier (Axon Instruments, Burlingame, CA, USA). The extracellular solution contained (mM) NaCl 119, KCl 5, CaCl2 2, MgCl2 1, glucose 30 and HEPES 20. Continuous flow of solution through the bath chamber (2 mL·min−1) ensured rapid drug application and clearance. Drugs were typically prepared as a stock then diluted into extracellular solution at their final concentration and used on the same day. Recording pipettes of 1.8–4 MΩ were filled with solution containing (mM): potassium gluconate 121.5, KCl 17.5, NaCl 9, MgCl2 1, HEPES 10, EGTA 0.2, MgATP 2 and LiGTP 0.5. Access resistance was monitored and only cells with a stable access resistance were included for data analysis.
The membrane potential was held at −70 mV and excitatory postsynaptic currents (EPSCs) were evoked every 20 s by triggering an unclamped action current with a 1.0 ms depolarizing step. The resultant evoked waveform consisted of a brief stimulus artefact (i.e. a large downward spike representing inward sodium currents) followed by the slower EPSC. The size of the recorded EPSCs was calculated by integrating the evoked current to yield charge (in pC). Calculating the charge in this manner yields an indirect measure of the amount of neurotransmitter released while minimizing the effects of cable distortion on currents generated far from the site of the recording electrode (the soma). Data were acquired at a sampling rate of 5 kHz.
Depolarization suppression of excitation (DSE) is a process whereby depolarization of a neuron results in production of endocannabinoids and activation of presynaptic CB1 receptors with a subsequent transient decrease of glutamate release and EPSC amplitude. Cultured autaptic neurons are heterogeneous, with some expressing CB1 receptors and others not. The presence of DSE (which requires CB1 receptors) was used as a marker for neuronal cannabinoid sensitivity. DSE was induced as previously described (Straiker and Mackie, 2005). Recordings were primarily (see results) made from cells that exhibited DSE.
To determine the site of JWH018's action on neurotransmission, paired pulse ratio analysis, miniature EPSC (mEPSC) recordings and measurements of changes in the coefficient of variation (CV) were performed. Paired pulse ratios were calculated as the charge of the first of two pulses (60 ms interval) divided by the second. Ratios smaller than 1.0 were interpreted as paired-pulse depression. Analysis was performed only on neurons that displayed paired-pulse depression under basal conditions, indicating neurons with high probabilities of release. Ratios were calculated for each neuron under basal and drug-treated conditions.
mEPSC analysis was performed by measuring the frequency (Hz) and amplitude (pA) of mEPSC events under basal and drug-treated conditions. mEPSC events were analysed without knowledge of the treatments, and then the data from each condition were pooled. Plots of cumulative probability and graphs of mean mEPSC frequency and amplitude were made from this pooled data.
CV analysis was performed as described in Faber and Korn (1991) and Shen et al. (1996). Means and coefficients of variation were calculated from 6 to 20 sweeps from basal and drug-treated conditions. π and r were calculated for each individual experiment and means ± SEM were calculated for each. A presynaptic site of drug action leading to synaptic depression was deduced if r < π < 1.
MAPK and receptor internalization assays
MAPK activation was analysed as previously described (Daigle et al., 2008), with a few modifications. HEK293 cells stably expressing CB1 receptors were plated onto poly-D lysine-coated 96 well plates (Corning, Corning, NY) and allowed to grow to ∼95% confluency. The following day the growth media was removed and replaced with serum-free growth media and the cells were incubated overnight. The cells were washed once with HEPES buffered saline (HBS; 130 mM NaCl, 5.4 mM KCl, 1.8 mM MgCl2, and 10 mM HEPES, pH 7.5) containing 0.2 mg·mL−1 bovine serum albumin (BSA). Drug containing solutions were made in the HBS/BSA solution and added to the wells at appropriate time points. Following drug incubation, the wells were emptied and ice-cold 4% paraformaldehyde was added immediately to each well, and the plates were placed on ice for 15 min followed by 30 min at room temperature. The paraformaldehyde was then removed and at least 100 µL of ice-cold methanol was added to each well and the plate was incubated at −20°C for at least 20 min. For methanol incubation times shorter than an hour, an additional washing step was performed using phosphate-buffered saline (PBS; 137 mM NaCl, 10 mM NaH2PO4, 2.7 mM KCl, pH 7.4) containing 0.1% Triton-X 100 for 25 min. The methanol or PBS/Triton-X 100 was replaced with a blocking solution of Tris-buffered saline (TBS; 137 mM NaCl, 10 mM Tris, pH 7.4) containing 5 mg·mL−1 BSA and incubated for at least 1 h at room temperature. The blocking solution was then removed and replaced by a blocking solution containing rabbit anti-phospho-ERK1/2 MAPK antibody (1:200) (Cell Signaling Technologies Inc., Danvers, MA) and was allowed to shake overnight at 4°C or for 3 h at room temperature. The antibody solution was removed and the plates were washed five times with TBS containing 0.05% Tween-20 (TBST) for 5–15 min each time. A blocking solution containing an IRDye conjugated anti-rabbit IgG antibody [either donkey anti-rabbit IR800 (1:500 dilution) or goat anti-rabbit IgG IR680 (1:200) antibody (LI-COR Biosciences, Lincoln, NE)] was added and allowed to shake for 1 h at room temperature. The plates were then washed five times with TBST, 5–15 min each time, followed with a final quick single rinse in distilled water. The plates were patted dry and then scanned using a LI-COR Odyssey. Integrated intensities were used for each well. The amount of MAPK activation was calculated as the average integrated intensities of the drug-treated wells divided by the average integrated intensities of the untreated wells and are expressed as percentages.
The extent of CB1 receptor internalization was analysed as previously described in (Daigle et al., 2008). The extent of internalization was calculated as the average integrated intensities of the drug-treated wells divided by the average integrated intensities of the untreated wells and are expressed as percentages.
Data analysis
Data are reported as mean ± SEM (except EC50, IC50 and t1/2 data, which are reported as mean ± 95% CI). Non-linear regression was used to fit the concentration response curves and the time course of internalization. Paired Student's t-tests were used to evaluate the effect of drugs on paired pulse ratios and mEPSC data and unpaired Student's t-tests were used on all other comparison analyses. Statistical significance is indicated as follows: ***P < 0.0001, **P < 0.01, and *P < 0.05. All graphs and statistical analyses were generated using GraphPad Prism 4.0 software (Hearne Scientific Software, Chicago, IL, USA).
Materials
Drugs and reagents were purchased from Tocris Cookson (Ellisville, MO, USA), Cayman Chemical (Ann Arbor, MI, USA) or Sigma-Aldrich (St Louis, MO, USA). JWH018 was synthesized as described by Huffman et al. (1994). Heterozygote (CB1+/−) mice to establish our colony were generously provided by Dr Catherine Ledent (University of Brussels, Belgium; Reibaud et al., 1999). GAD67-GFP mice generated by Dr Yuchio Yanagawa (Gunman University, Gunma, Japan; Tamamaki et al., 2003) were provided by Dr Albert Berger (University of Washington, Seattle, WA, USA) and used with Dr Yanagawa's permission. Rimonabant (SR141716) was obtained from the National Institute of Drug Abuse drug supply.
Results
JWH018 decreases the probability of neurotransmitter release via CB1 receptor activation
In the CNS, CB1 receptors are predominately located on axon terminals. Upon agonist binding, CB1 receptor activation leads to a reduced probability of neurotransmitter release. To see whether JWH018 acts similarly to other CB1 receptor agonists in this regard, we recorded EPSCs from glutamatergic autaptic neuron cultures in the presence and absence of JWH018. Autaptic neuronal cultures are a well-characterized preparation that uses a single electrode to both stimulate and record EPSCs (Bekkers and Stevens, 1991). We found that JWH018 potently inhibited EPSCs (Figure 1B,C) in a concentration-dependent manner with a mean IC50 of 14.9 nM (4.8–45.9 nM; 95% CI) and a maximal inhibition of 48.0 ± 4.2% of control at 1 µM. As seen in Figure 1C, the inhibition by JWH018 was poorly reversed upon washout of the drug. However, the effect of JWH018 (1 µM) on EPSC charge was fully reversed by 1 µM rimonabant (Figure 1C), a CB1 antagonist, suggesting that the limited reversal is due to persistent receptor occupancy. The effect of JWH018 on EPSCs was examined in neurons that exhibited DSE, a transient reduction in EPSC size that results from depolarization. DSE is a well-described process that has been shown to be dependent on cannabinoid signalling molecules including CB1 receptors. We found that in neurons that did not exhibit DSE (and are likely to lack CB1 receptors); JWH018 had no effect on EPSC size (data not shown). To confirm that the effect observed with JWH018 was indeed due to action at CB1 receptors, we treated neurons from CB1 receptor null mice with 1 µM JWH018. In wild-type neurons, 1 µM JWH018 treatment reduced EPSC size (Figure 1B), whereas in the CB1 receptor null neurons, 1 µM JWH018 had no effect (Figure 1B). To establish that JWH018 was operating through presynaptic CB1 receptors to decrease the probability of neurotransmitter release, we performed paired-pulse analysis under baseline conditions and during JWH018 treatment. Figure 1D shows that 100 nM JWH018 treatment significantly increases the paired-pulse ratio (P= 0.0056), suggesting a presynaptic site of action. To further validate a presynaptic site of action we recorded mEPSCs before and after JWH018 treatment. Treatment of neurons with 100 nM JWH018 decreased mEPSC frequency (Figure 1E; P= 0.031) but did not significantly alter mEPSC amplitude (Figure 1F; P= 0.19). CV analysis further supported a presynaptic locus of JWH018's action (Figure 1G). 1 µM JWH018 treatment gave a mean value for r of 0.26 ± 0.049 and a mean π value of 0.54 ± 0.41. All of these measures are consistent with a presynaptic site of drug action resulting in synaptic depression. In summary, JWH018 reduced the probability of glutamate release in autaptic neurons by acting at presynaptic CB1 receptors in a concentration-dependent fashion.
JWH018 activated MAPK
In addition to modulating neurotransmission and cAMP levels, CB1 receptor signalling activates MAPK activity in cultured cells (Bouaboula et al., 1995; Daigle et al., 2008) and neurons (Derkinderen et al., 2003). This is conveniently detected by increased phosphorylation of ERK1/2. As JWH018 activated other CB1 receptor-mediated signalling pathways we hypothesized that JWH018 would also activate ERK1/2. HEK293 cells stably expressing CB1 receptors were treated with either JWH018 or WIN55,212, another well-characterized, efficacious CB1 receptor agonist. The time course of ERK1/2 MAPK activation for 100 nM of each drug was determined. Maximal activation occurred between 5 and 10 min of treatment with both drugs (Figure 2A). Interestingly, at both 5 and 7.5 min JWH018 was a more efficacious activator of ERK1/2 phosphorylation. A concentration-response analysis was performed at the 7.5-min time point (Figure 2B). JWH018 was more potent with an EC50 of 4.4 nM (1.6–12.5 nM) compared with WIN55,212, which had an EC50 of 69.9 nM (37.2–131.4 nM). The two agonists had similar maximal effects at 1 µM, although JWH018 was more potent than WIN55,212. In addition, to ensure that the MAPK activation observed from these drug treatments required CB1 receptors, the cells were treated with rimonabant and then agonist. Here, 1 µM rimonabant prevented the activation of MAPK by either 1 µM JWH018 or WIN 55,212 (JWH018: 97.1 ± 5.7%, WIN55,212: 86.8 ± 5.8%, NS vs. untreated). Thus, in addition to its inhibition of adenylyl cyclase and neurotransmission, JWH018 also serves as a potent agonist of CB1 receptor-mediated ERK1/2 MAPK activation.
Figure 2.
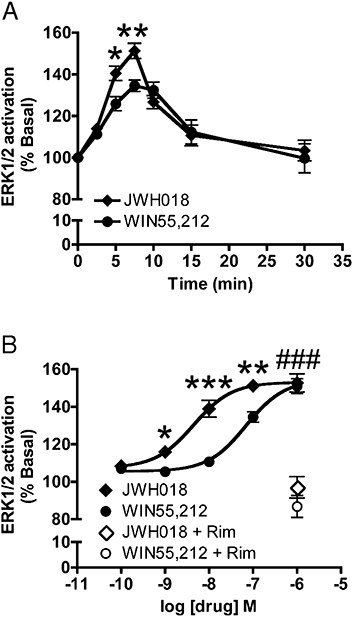
JWH018 activation of CB1 receptors stimulated ERK1/2 MAPK phosphorylation. (A) 100 nM JWH018 treatment of CB1 expressing HEK293 cells, transiently increased ERK1/2 phosphorylation, similar to the time course of 100 nM WIN55,212 activation (4–6 replicate samples from 7–15 independent experiments). (B) CB1 receptor expressing HEK293 cells were treated for 7.5 min with increasing concentrations of JWH018 and WIN55,212. JWH018 activated ERK1/2 in a concentration-dependent manner and was more potent than WIN55,212 (4–6 replicate samples from 8–15 independent experiments). 1 µM rimonabant reversed the effect of JWH018 and WIN55,212 on ERK1/2 activation. Values are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.0001: significantly different from WIN55,212 treatment. ###P < 0.0001: significantly different from rimonabant treatment. CB1, cannabinoid receptor 1; MAPK, mitogen activated protein kinase.
JWH018-induced robust CB1 receptor internalization
Following prolonged exposure to agonists, many GPCRs undergo internalization. This is thought to be a means whereby the cell can control its spatial and temporal response to receptor agonists (Drake et al., 2006; Marchese et al., 2008). CB1 receptor internalization has been described in response to numerous cannabinoid drugs (Hsieh et al., 1999; Coutts et al., 2001; Daigle et al., 2008). We hypothesized that based on previous experiments, if JWH018 was acting as a CB1 receptor agonist, long-term treatment of CB1 receptor expressing cells with JWH018 would lead to profound receptor internalization. Figure 3A shows the time course of CB1 receptor internalization in HEK293 cells stably expressing the receptor. After 3 h of exposure to 100 nM JWH018, 38.1 ± 3.1% of the CB1 receptors present at the plasma membrane under basal conditions remained on the cell surface (61.9% internalization). We again used an equal concentration of WIN55,212 as a control, in which 3 h of treatment led to 58.7 ± 3.2% internalization (NS vs. JWH018). JWH018 induced a much more rapid internalization than WIN55,212 with a t1/2 of 17.3 min (13.6–23.7 min) compared with 39.6 min (26.2–80.9 min) for WIN 55,212. Each drug caused similar maximal levels of internalization reaching plateaus of approximately 60% internalization (JWH018 = 40.7 ± 1.8% of basal levels; WIN55,212 = 41.1 ± 4.9% of basal levels). We performed concentration-response analysis of internalization using a 2 h treatment period (Figure 3B). JWH018 and WIN55,212 had similar maximal effects reaching 54.7 ± 3.4% and 56.0 ± 3.0% internalization at 1 µM concentrations respectively (NS). However, JWH018 was once again the more potent with an EC50 of 2.8 nM (1.2–6.3 nM) compared with WIN55,212, which had an EC50 of 19.4 nM (8.5–44.4 nM). 1 µM rimonabant prevented internalization by 1 µM of either drug (JWH018: 91.5 ± 3.8%, WIN55,212: 103.3 ± 3.3%, NS vs. untreated). Therefore, prolonged exposure to JWH018 leads to robust CB1 receptor internalization, as did treatment with WIN55,212, although JWH018 was more potent and caused faster internalization.
Figure 3.
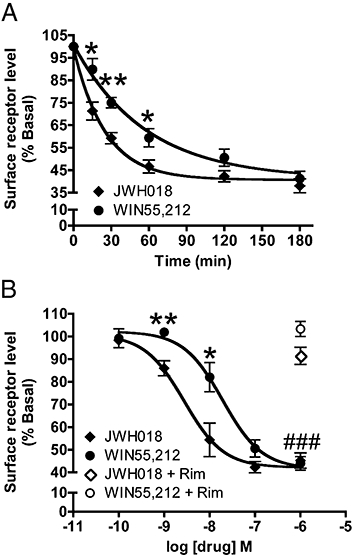
JWH018 induced CB1 receptor internalization. (A) In CB1 receptor expressing HEK293 cells, 100 nM JWH018 treatment resulted in robust receptor internalization that was quicker than that induced by 100 nM WIN55,212 (4–6 replicate samples from 5–10 independent experiments). (B) Following 2 h of exposure to increasing concentrations of each drug, CB1 receptors were internalized in a concentration-dependent manner with JWH018 being more potent than WIN55,212 (4–6 replicate samples from 5–10 independent experiments). 1 µM rimonabant, a CB1 receptor antagonist, prevented receptor internalization due to JWH018 or WIN55,212 treatment. Values are presented as mean ± SEM. *P < 0.05, **P < 0.01: significantly different from WIN55,212 treatment. ###P < 0.0001: significantly different from rimonabant treatment. CB1, cannabinoid receptor 1.
Discussion
JWH018 is a common synthetic additive found in diverse preparations of the herbal blend known as ‘Spice’ (Auwarter et al., 2009). JWH018 was first synthesized during an analysis of cannabimimetic indole structures that aimed to design new indoles with effects comparable with those of natural cannabinoids such as THC (Huffman et al., 1994). Relatively little characterization of this ligand has been performed. It has been reported to have a high affinity for CB1 receptor with a Ki of approximately 9 nM (Huffman et al., 1994; Showalter et al., 1996; Chin et al., 1999; Aung et al., 2000) and to inhibit adenylyl cyclase with an IC50 of 14.7 nM and a 79% maximal inhibition. Beyond these preliminary studies, there have been no other investigations of JWH018's effect on CB1 receptor-mediated signalling. In light of its presence in ‘Spice’ and its cannabinoid-like psychoactivity, we have examined the effects of this compound on neurotransmission, MAPK activity, and CB1 receptor internalization.
CB1 receptor activation may suppress neurotransmission and neuronal excitability (Kano et al., 2009). Here we have used autaptic excitatory hippocampal neurons as a well-characterized model system of cannabinoid-mediated effects on neurotransmission (Straiker and Mackie, 2005; 2007;). We found that JWH018 potently inhibits glutamate release in these neurons in a concentration-dependent fashion. The effect of JWH018 on synaptic transmission was due to its action at CB1 receptors as JWH018 had no effect on EPSC in neurons cultured from CB1 receptor knockout mice, and rimonabant, a CB1 receptor antagonist, blocked its effect in wild-type neurons. Furthermore, JWH018 is very likely acting at presynaptic CB1 receptors based on its ability to increase the paired-pulse ratio, to decrease the frequency of mEPSCs without affecting mEPSC amplitude and to increase the CV. Cannabinoid agonists sometimes do not decrease mEPSC frequency at glutamatergic synapses (Yamasaki et al., 2006). However, our results here are consistent with most reports that find cannabinoid suppression of mEPSC frequency (see Misner and Sullivan, 1999; Sullivan, 1999; Morisset and Urban, 2001; Robbe et al., 2001; Derbenev et al., 2004), and the difference may lie in the brain region, cell type or culture conditions used. The effect of JWH018 on neurotransmission is both potent and effective with an IC50 of 14.9 nM and a maximal inhibition to 48.0% of control at 1 µM. JWH018 exhibtied effects comparable with those of WIN55,212 (Straiker and Mackie, 2005). In light of the internalization data, JWH018's effect on neurotransmission may potentially be influenced by receptor desensitization or internalization. We cannot make any certain conclusions based on our data here as to whether desensitization had an effect on our recordings. Internalization is unlikely to play a role as little CB1 receptor internalization would occur at room temperature during the 20–30 min of recording. In summary, JWH018 inhibits synaptic transmission as a potent and efficacious CB1 receptor agonist.
Based on the earlier studies discussed above and our finding that JWH018 inhibited synaptic neurotransmission, it seemed probable that JWH018 would exhibit signalling effects similar to those of other CB1 receptor agonists. However, it was important to test this hypothesis as JWH018 may exhibit a different functional selectivity relative to other cannabinoid agonists (Urban et al., 2007). ERK1/2 MAPK activation is a typical consequence of CB1 receptor stimulation (Bouaboula et al., 1995; Daigle et al., 2008). We have demonstrated that as observed with other cannabinoid ligands such as WIN55,212, JWH018 also stimulated ERK1/2 MAPK activation in a concentration-dependent manner. This ERK1/2 activation had the typically observed rapid time course (Daigle et al., 2008) reaching a peak level of activation between 5 and 10 min. Comparing JWH018 with WIN55,212, we found that despite having a similar time course of activation, JWH018 was more potent with an EC50 of 4.4 nM compared with WIN55,212 with an EC50 of 69.9 nM. Both were similarly efficacious.
CB1 receptor internalization has been reported to occur in response to a number of different cannabinoid ligands in a number of cell types (Hsieh et al., 1999; Coutts et al., 2001; Daigle et al., 2008). Here we found that JWH018, consistent with its ability to act as a CB1 receptor agonist, produces robust CB1 receptor internalization that is rapid (t1/2= 17.3 min), potent (EC50= 2.8 nM) and efficacious (38.1% of basal surface levels at 3 h with 100 nM). We found it more potent and to induce internalization more rapidly than WIN55,212 (EC50= 19.4 nM and t1/2= 39.6 min). However, the two drugs are of similar efficacy (JWH018 – plateaus at 40.7% and WIN55,212 – plateaus at 41.1% of basal surface levels). Thus, prolonged engagement of CB1 receptors by JWH018, as with many other CB1 receptor agonists, leads to profound cellular adaptations that may serve to decrease cellular sensitivity to the drug. Interpretation of the effects of chronic JWH018 on behaviour must consider its ability to produce cellular desensitization and tolerance.
Previously we found that in autaptic neuronal cultures, THC did not inhibit EPSCs but rather, it antagonized inhibition by both WIN55,212 and 2-arachidonyl glycerol (2-AG) (Straiker and Mackie, 2005). Similar results have been reported in non-autaptic hippocampal cultures (Roloff and Thayer, 2009). In these systems, THC effectively acts as an antagonist with short-term treatment but desensitizes CB1 receptor signalling with long-term treatment. Since THC, which is the principal psychoactive component of marijuana, is a low-efficacy CB1 receptor agonist, we speculated that it prevents full CB1 receptor activation by the endocannabinoid 2-AG and mimics the effects of the low-efficacy endocannabinoid anandamide (Straiker and Mackie, 2005). This was a provocative hypothesis to explain the psychoactive effects of marijuana. However, in light of the findings from this study, this hypothesis must be revised. ‘Spice’ apparently produces marijuana-like psychoactivity when smoked (Auwarter et al., 2009). However, it has not been reported to contain THC but rather contains at least one potent and efficacious CB1 receptor agonist. Therefore, if the cognitive effects of ‘Spice’ are due to JWH018, our proposition that the psychoactivity of THC may in part be due to the antagonism of 2-AG activation of CB1 receptors requires rethinking. The effects we previously observed with THC may be unique to the cultured neuron preparation itself. In cultures, the number of CB1 receptors or their coupling may be limited, which will cause a low-efficacy agonist to act as an antagonist. In contrast, these factors may not be limiting for CB1 receptors expressed in brain, and a low-efficacy agonist such as THC may act as an agonist in vivo. Alternatively, as was suggested in a recent study, the difference may also lie in the firing rate of the neurons, which can influence a neuron's response to THC (Roloff and Thayer, 2009). Furthermore, it may be a possibility that there are other uncharacterized additives in ‘Spice’ that influence neurotransmission. Despite these uncertainties, it is clear that JWH018 potently inhibits neurotransmission, with an efficacy comparable with other synthetic cannabinoids such as WIN55,212.
Among the different preparations of ‘Spice’ that were analysed by Auwarter et al. (2009), JWH018 was a frequent additive. Here we have demonstrated that JWH018 treatment has cellular effects similar to those of other efficacious cannabinoid agonists such as WIN55,212. We have found that JWH018 is a more potent CB1 receptor agonist than WIN55,212, although of similar efficacy. This is consistent with the reports that ‘Spice’ has marijuana-like effects when smoked. While Auwarter et al. found that JWH018 was not the most abundant of the additives present in various spice preparations, its high potency suggests that it will produce behavioural effects in humans. The selectivity of JWH018 for CB1 receptors is low: JWH018 has a Ki of about 9 nM at CB1 receptors and a Ki of about 3 nM at CB2 receptors (Huffman et al., 1994; Chin et al., 1999; Aung et al., 2000). While the effects we observed are clearly due to CB1 receptor activation, the potential role of CB2 receptors in the effects of ‘Spice’ requires further study. This study has focused on JWH018; however, different preparations of ‘Spice’ apparently contain diverse synthetic additives such as a modified version of CP47,497 (extending the dimethylheptyl side chain to dimethyloctyl), a cannabinoid ligand, that may also act as agonists at CB1 receptors but so far remain uncharacterized and HU210 (Huffman et al., 2008; Auwarter et al., 2009). It is likely that these additional compounds might also contribute to the behavioural and subjective effects produced by smoking ‘Spice’, and their different pharmacologies might cause different preparations of ‘Spice’ to vary in their psychoactivity or health effects. Investigation into these additional synthetic additives requires further attention. Despite these caveats, we have shown that JWH018 has profound CB1 receptor-mediated effects on cellular signalling and neurotransmission, which are likely to have a significant impact on cognitive function. Thus, ‘Spice,’ which is marketed as a ‘natural’ herbal blend, is actually a vehicle of delivery for at least one very potent synthetic CB1 receptor agonist, and its presence is likely to account for the psychoactive effects produced when ‘Spice’ is smoked.
Acknowledgments
Supported by DA11322, DA21696, DA024122, DA009158 and DA003590.
Glossary
Abbreviations:
- CB1 receptor
cannabinoid receptor 1
- CP47,497
2-[(1R,3S)-3-hydroxycyclohexyl]-5-(2-methyloctan-2-yl)phenol
- EPSC
excitatory postsynaptic current
- ERK
extracellular signal regulated kinase
- GPCR
G protein coupled receptor
- JWH018
naphthalen-1-yl-(1-pentylindol-3-yl)methanone
- MAPK
mitogen activated protein kinase
- rimonabant
SR141716/5-(4-chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide
- THC
Δ9-tetrahydrocannabinol
- WIN55212-2
(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate
Conflict of interest
None.
References
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameri A, Wilhelm A, Simmet T. Effects of the endogeneous cannabinoid, anandamide, on neuronal activity in rat hippocampal slices. Br J Pharmacol. 1999;126:1831–1839. doi: 10.1038/sj.bjp.0702478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung MM, Griffin G, Huffman JW, Wu M, Keel C, Yang B, et al. Influence of the N-1 alkyl chain length of cannabimimetic indoles upon CB(1) and CB(2) receptor binding. Drug Alcohol Depend. 2000;60:133–140. doi: 10.1016/s0376-8716(99)00152-0. [DOI] [PubMed] [Google Scholar]
- Auwarter V, Dresen S, Weinmann W, Muller M, Putz M, Ferreiros N. ‘Spice’ and other herbal blends: harmless incense or cannabinoid designer drugs? J Mass Spectrom. 2009;44:832–837. doi: 10.1002/jms.1558. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, et al. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312:637–641. doi: 10.1042/bj3120637. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SM, Wager-Miller J, Mackie K. Cloning and molecular characterization of the rat CB2 cannabinoid receptor. Biochim Biophys Acta. 2002;1576:255–264. doi: 10.1016/s0167-4781(02)00341-x. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chin CN, Murphy JW, Huffman JW, Kendall DA. The third transmembrane helix of the cannabinoid receptor plays a role in the selectivity of aminoalkylindoles for CB2, peripheral cannabinoid receptor. J Pharmacol Exp Ther. 1999;291:837–844. [PubMed] [Google Scholar]
- Costa B. On the pharmacological properties of delta9-tetrahydrocannabinol (THC) Chem Biodivers. 2007;4:1664–1677. doi: 10.1002/cbdv.200790146. [DOI] [PubMed] [Google Scholar]
- Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, et al. Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Kearn CS, Mackie K. Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology. 2008;54:36–44. doi: 10.1016/j.neuropharm.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbenev AV, Stuart TC, Smith BN. Cannabinoids suppress synaptic input to neurones of the rat dorsal motor nucleus of the vagus nerve. J Physiol. 2004;559:923–938. doi: 10.1113/jphysiol.2004.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, et al. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci. 2003;23:2371–2382. doi: 10.1523/JNEUROSCI.23-06-02371.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake MT, Shenoy SK, Lefkowitz RJ. Trafficking of G protein-coupled receptors. Circ Res. 2006;99:570–582. doi: 10.1161/01.RES.0000242563.47507.ce. [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furshpan EJ, MacLeish PR, O'Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci USA. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, et al. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- Howlett AC. The cannabinoid receptors. Prostaglandins Other Lipid Mediat. 2002;68(69):619–631. doi: 10.1016/s0090-6980(02)00060-6. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Huestis MA, Gorelick DA, Heishman SJ, Preston KL, Nelson RA, Moolchan ET, et al. Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch Gen Psychiatry. 2001;58:322–328. doi: 10.1001/archpsyc.58.4.322. [DOI] [PubMed] [Google Scholar]
- Huffman JW, Dong D, Martin BR, Compton DR. Design, synthesis and pharmacology of cannabimimetic indoles. Bioorg Med Chem Lett. 1994;4:563–566. [Google Scholar]
- Huffman JW, Thompson AL, Wiley JL, Martin BR. Synthesis and pharmacology of 1-deoxy analogs of CP-47,497 and CP-55,940. Bioorg Med Chem. 2008;16:322–335. doi: 10.1016/j.bmc.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, et al. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci. 1999;19:3773–3780. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Levison SW, McCarthy KD. Characterization and partial purification of AIM: a plasma protein that induces rat cerebral type 2 astroglia from bipotential glial progenitors. J Neurochem. 1991;57:782–794. doi: 10.1111/j.1471-4159.1991.tb08220.x. [DOI] [PubMed] [Google Scholar]
- Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol. 2005;168:299–325. doi: 10.1007/3-540-26573-2_10. [DOI] [PubMed] [Google Scholar]
- Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matyas F, Watanabe M, Mackie K, Katona I, Freund TF. Molecular architecture of the cannabinoid signaling system in the core of the nucleus accumbens. Ideggyogy Sz. 2007;60:187–191. [PubMed] [Google Scholar]
- Misner DL, Sullivan JM. Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci. 1999;19:6795–6805. doi: 10.1523/JNEUROSCI.19-16-06795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monory K, Blaudzun H, Massa F, Kaiser N, Lemberger T, Schutz G, et al. Genetic dissection of behavioural and autonomic effects of delta(9)-tetrahydrocannabinol in mice. PLoS Biol. 2007;5:e269. doi: 10.1371/journal.pbio.0050269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisset V, Urban L. Cannabinoid-induced presynaptic inhibition of glutamatergic EPSCs in substantia gelatinosa neurons of the rat spinal cord. J Neurophysiol. 2001;86:40–48. doi: 10.1152/jn.2001.86.1.40. [DOI] [PubMed] [Google Scholar]
- Nyiri G, Cserep C, Szabadits E, Mackie K, Freund TF. CB1 cannabinoid receptors are enriched in the perisynaptic annulus and on preterminal segments of hippocampal GABAergic axons. Neuroscience. 2005;136:811–822. doi: 10.1016/j.neuroscience.2005.01.026. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199–215. doi: 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reibaud M, Obinu MC, Ledent C, Parmentier M, Bohme GA, Imperato A. Enhancement of memory in cannabinoid CBI receptor knock-out mice. Eur J Pharmacol. 1999;379:R1–R2. doi: 10.1016/s0014-2999(99)00496-3. [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche JP, Bounds S, Brown S, Mackie K. A mutation in the second transmembrane region of the CB1 receptor selectively disrupts G protein signaling and prevents receptor internalization. Mol Pharmacol. 1999;56:611–618. doi: 10.1124/mol.56.3.611. [DOI] [PubMed] [Google Scholar]
- Roloff AM, Thayer SA. Modulation of excitatory synaptic transmission by delta 9-tetrahydrocannabinol switches from agonist to antagonist depending on firing rate. Mol Pharmacol. 2009;75:892–900. doi: 10.1124/mol.108.051482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, Piser TM, Seybold VS, Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showalter VM, Compton DR, Martin BR, Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278:989–999. [PubMed] [Google Scholar]
- Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569:501–517. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Cannabinoids, electrophysiology, and retrograde messengers: challenges for the next 5 years. AAPS J. 2006;8:E272–E276. doi: 10.1007/BF02854897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Metabotropic suppression of excitation in murine autaptic hippocampal neurons. J Physiol. 2007;578:773–785. doi: 10.1113/jphysiol.2006.117499. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol. 1999;82:1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tamioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Taura F, Sirikantaramas S, Shoyama Y, Morimoto S. Phytocannabinoids in Cannabis sativa: recent studies on biosynthetic enzymes. Chem Biodivers. 2007;4:1649–1663. doi: 10.1002/cbdv.200790145. [DOI] [PubMed] [Google Scholar]
- Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Compton DR, Dai D, Lainton JA, Phillips M, Huffman JW, et al. Structure–activity relationships of indole- and pyrrole-derived cannabinoids. J Pharmacol Exp Ther. 1998;285:995–1004. [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Hashimoto K, Kano M. Miniature synaptic events elicited by presynaptic Ca2+ rise are selectively suppressed by cannabinoid receptor activation in cerebellar Purkinje cells. J Neurosci. 2006;26:86–95. doi: 10.1523/JNEUROSCI.2258-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, et al. Localization of diacylglycerol lipase-alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci. 2006;26:4740–4751. doi: 10.1523/JNEUROSCI.0054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]