

Abstract
Background and purpose:
Although GPR55 is potently activated by the endogenous lysophospholipid, L-α-lysophosphatidylinositol (LPI), it is also thought to be sensitive to a number of cannabinoid ligands, including the prototypic CB1 receptor antagonists AM251 and SR141716A (Rimonabant®). In this study we have used a range of functional assays to compare the pharmacological activity of selected cannabinoid ligands, AM251, AM281 and SR141716A with LPI in a HEK293 cell line engineered to stably express recombinant, human GPR55.
Experimental approach:
We evaluated Ca2+ signalling, stimulation of extracellular signal regulated kinase (ERK1/2) mitogen activated kinase MAP-kinases, induction of transcriptional regulators that are downstream of GPR55, including nuclear factor of activated T cells (NFAT), nuclear factor-κB (NF-κB) and cAMP response element binding protein (CREB), as well as receptor endocytosis. In addition, we assessed the suitability of a novel, label-free assay for GPR55 ligands that involves optical measurement of dynamic mass redistribution following receptor activation.
Key results:
GPR55 linked to a range of downstream signalling events and that the activity of GPR55 ligands was influenced by the functional assay employed, with differences in potency and efficacy observed.
Conclusions and implications:
Our data help to resolve some of the issues surrounding the pharmacology of cannabinoid ligands at GPR55 and highlight some differences in effector coupling associated with distinct GPR55 ligands.
Keywords: GPR55, GPCR, cannabinoid, LPI
Introduction
Recently, it has been suggested that the orphan G protein-coupled receptor, GPR55, is a novel lipid-sensing receptor that can be activated by certain cannabinoid ligands and L-α-lysophosphatidylinositol (LPI), of which 2-arachidonoyl-sn-glycero-3-phosphoinositol is the most active species (Johns et al., 2007; Oka et al., 2007; Lauckner et al., 2008; Waldeck-Weiermair et al., 2008; Henstridge et al., 2009; Kapur et al., 2009; Oka et al., 2009; Yin et al., 2009). There is evidence for endogenous GPR55 in endothelial cells (Waldeck-Weiermair et al., 2008) and microglia (Pietr et al., 2009) and this receptor may play an important role in inflammatory pain (Staton et al., 2008) and in the regulation of bone mass (Whyte et al., 2009). However, cannabinoid pharmacology at GPR55 and its coupling to downstream signalling pathways is at present controversial, possibly reflecting host and ligand-dependent effects (Brown and Robin, 2009; Ross, 2009). Although GPR55 is suggested to be activated by cannabinoid ligands, including the prototypic CB1 receptor antagonist AM251 (Ryberg et al., 2007; Henstridge et al., 2009; Kapur et al., 2009; Yin et al., 2009), this has been contested in a study measuring activation of extracellular signal regulated kinase (ERK) mitogen activated (MAP)-kinases (Oka et al., 2007). A more recent investigation of ERK MAP kinase signalling in U2OS cells stably expressing an epitope tagged, GPR55-vasopressin2 receptor chimera also failed to detect a response to cannabinoids, but did observe effects on receptor trafficking and beta-arrestin translocation (Kapur et al., 2009). In addition, the GPR55 activity of Rimonabant® (SR141716A), a CB1 receptor antagonist that has been used in the clinic for the treatment of obesity, is inconsistent. One study measuring intracellular Ca2+ signalling suggests that SR141716A is an antagonist at GPR55 (Lauckner et al., 2008), whereas other groups using beta-arrestin recruitment and a reporter gene assay suggest that it acts as an agonist (Kapur et al., 2009; Yin et al., 2009). GPR55 may be unique among GPCRs in that it is thought to signal primarily via activation of Gα12-family proteins and RhoA (Ryberg et al., 2007; Henstridge et al., 2009), although a link to Gαq has also been suggested (Lauckner et al., 2008; Waldeck-Weiermair et al., 2008). In HEK293 cells overexpressing recombinant GPR55, agonist binding promotes oscillatory Ca2+ signalling and activation of the nuclear factor of activated T cells (NFAT) family of transcription factors via an atypical, RhoA–dependent signalling cascade (Henstridge et al., 2009).
In this study we set out to define in more detail the ligand interactions and downstream signalling pathways of GPR55 using a range of different functional assays, including Ca2+ and ERK MAP-kinase signalling, activation of transcription factors and receptor internalization. We identify two new transcription factors that can be modulated by GPR55 activation and further evaluate the suitability of a novel, label-free methodology (Corning Epic®) for detecting GPR55 ligands.
Methods
Cell culture and transfection
HEK293-AD cells (hereafter termed HEK293; Invitrogen) were maintained in Dulbecco's modified Eagle's medium DMEM-F12 supplemented with 10% calf serum at 37°C and 5% CO2. For studies involving reporter gene and the dynamic mass redistribution (DMR) assay, cells were cultured in DMEM supplemented with 10% calf serum and 1% sodium pyruvate. Cell lines stably expressing 3xHA-GPR55 (GPR55-HEK293; see Henstridge et al., 2009) were maintained in G418 at 500 µg·mL–1. For experimental studies cells were plated onto poly-D-lysine-coated coverslips (10 µg·mL–1) and serum-starved for 12–48 h. Transient transfections were carried out using Lipofectamine 2000 according to the manufacturers instructions.
Ca2+ microfluorimetry
A digital epifluorescence imaging system (Perkin-Elmer, Emeryville, CA, USA) mounted on an Olympus BX50WI microscope was used to measure changes in intracellular Ca2+ concentrations, [Ca2+]i. Cells were incubated with the Ca2+ sensitive dye Fura-2-AM (6 µM; 40–60 min at 25°C) in HEPES-buffered saline (HBS; in mM: NaCl 135, HEPES 10, KCl 5, CaCl2 1.8, MgCl2 1.0, D-glucose 25, pH 7.4). Ratiometric images were collected at 5 s intervals with data derived from individual cells. The effects of ligands were quantified by measuring the mean change in fluorescence ratio. All n values represent data (number of cells) obtained from a minimum of three separate experiments derived from different passages or transfections.
Immunoblot analysis
Samples were separated on 12% polyacrylamide gels and transferred to nitrocellulose membranes. Following treatment with blocking solution (5% milk), membranes were incubated with polyclonal rabbit antibodies detecting ERK1/2, cAMP response element binding protein (CREB) or phoshorylated ERK1/2 (pERK1/2), at a dilution of 1:1000. The monoclonal antibody against pCREB was used at a dilution of 1:1000 and HRP-conjugated secondary antibodies at a dilution of 1:10 000. Detection was performed using enhanced chemoluminescence reagents. Densitometry analysis of bands was carried out using Photoshop software (Adobe Systems Incorporated, San Jose, CA, USA). Absolute band intensity values were generated by multiplying mean intensity levels above background by the pixel size of the band. Absolute intensity values of phosphorylated protein bands (pERK or pCREB) were then divided by those for total protein (tERK or tCREB) to generate a ratio. All ratio values were then normalized to that observed with 1 µM LPI, run in the same gel.
Reporter gene assays
GPR55-HEK293 cells (20 000 cells per well; DMEM, 10% FCS) were seeded in 96-well plates coated with 1% poly-D-lysine. Cells were transiently transfected with pNFAT-luciferase (Luc; 200 ng.well–1) or pNF-κB-Luc (50 ng.well–1) reporter plasmids (PathDetect cis-Reporters; Stratagene) using Lipofectamine 2000 (Invitrogen). Twenty four h later, cells were incubated with increasing amounts of LPI, AM251, AM281 and SR141716A for 6 h in serum-free OptiMEM medium at 37°C. Luciferase activity was visualized by using the Steadylite Plus Kit (Packard Instrument Company, Meriden, CT, USA) as previously described (Waldhoer et al., 2002; Henstridge et al., 2009). Luminescence was measured in a TopCounter (Top Count NXT; Packard) for 5 s. Luminescence values are given as relative light units (RLU).
NFκB-(p65)-EGFP translocation assay
GPR55-HEK293 cells were grown on coverslips to 70% confluency in DMEM supplemented with 10% FCS. Cells were transfected with 3 µg of the phosphorylated form of enhanced green fluorescent protein (EGFP)-p65 plasmid (Waldeck-Weiermair et al., 2008) and 12 h post transfection, cells were serum-starved in OPTIMEM for another 12 h. Cells were treated with the indicated concentrations of ligands for 15 min and the experiment was terminated by fixing cells with 3.7% formaldehyde. Fixed cells were washed twice with TBS (NaCl 135 mM, Tris-HCl 25 mM, CaCl2 1 mM, KCl 2.5 mM) and wet-mounted onto microscopy slides with Vectashield containing a DAPI dye to visualize nuclei (Roche). Images were taken with an Olympus inverted fluorescence microscope equipped with a Hamamatsu Orca CCD camera.
DMR assays
A beta version of the Corning® Epic® System (Corning, NY, USA) was used consisting of a temperature-control unit, an optical detection unit and an on-board robotic liquid handling device. Briefly, each well in the 384-well Epic® microplate contains a resonant waveguide grating biosensor. The system measures changes in the local index of refraction upon mass redistribution within the cell monolayer grown on the biosensor. Ligand-induced DMR in living cells is induced upon receptor stimulation and is manifested as a shift in the wavelength of light that is reflected from the sensor. The magnitude of this wavelength shift in picometres (pm) is proportional to the amount of DMR. Notably, GPCR activation is translated into signalling pathway-specific optical signatures. Further technical details and the assay principle have been provided earlier (Fang et al., 2006; 2007; Schröder et al., 2009). Forty eight h before the assay, naïve HEK293 cells or GPR55-HEK293 cells were seeded at a density of 7500 cells per well in 384-well Epic® sensor microplates with 30 µL growth medium (DMEM, 10% FCS) and cultured for 24 h (37°C, 5% CO2) to obtain confluent monolayers. For starvation, cell culture medium was removed by aspiration and replaced by 30 µL of starvation medium [Hank's buffered salt solution (HBSS) with 20 mM HEPES, pH 7.15] in which cells were then cultured for another 24 h (37°C, 5% CO2). Previous to the assay, cells were washed once with assay buffer [HBSS with 20 mM HEPES and 0.1% bovine serum albumin, fatty acid-free (BSAfaf), pH 7.15] and incubated in 30 µL per well of assay-buffer in the Epic® reader at 28°C. Hereafter, the sensor plate was scanned and a baseline optical signature was recorded. Ten µL of test compound dissolved in assay buffer was then transferred from the compound plate into the sensor plate and DMR responses were monitored for at least 3600 s.
Receptor endocytosis
GPR55-HEK293 cells grown on coverslips were incubated with a rabbit anti-HA antibody (1:1000) for 30–45 min at 37°C (in HBS) to label surface receptors (‘antibody feeding’). Subsequently, cells were treated with ligand for 60 min at 37°C, then fixed in 4% paraformaldehyde for a further 15 min. Pre-labelled receptors remaining at the cell surface were detected with an Alexa488-conjugated anti-rabbit secondary antibody (1:500; 30 min). Subsequently, cells were permeabilized with 0.2% Triton X-100 (15 min) and internalized receptors were visualized with Cy3 or Cy5-conjugated anti-rabbit secondary antibodies. Images obtained with Cy5 were pseudocoloured red using Zeiss Lasersharp software to aid visualization.
Data analysis
Dose–response curves and EC50 values were determined by nonlinear regression using Prism 4.02 (GraphPad Software Inc., CA, USA). For analysis of DMR data, the area under curve (AUC) values of DMR signals between the 1200 s and 3600 s time points with mean and standard error of the mean (SEM) were used to calculate agonist activities. AUC values were transformed into relative AUC to give equivalent baseline optical recordings for all dose–response curves from one assay plate. Data were then normalized and expressed as percent of maximum activation induced by a saturating concentration of LPI which was set 100%. The assays were performed in triplicate and figures are representative data of two to three independent experiments.
Statistical analyses were performed using analysis of variance (anova) for comparisons between multiple groups, followed by Bonferroni's post hoc analysis (using GraphPad Prism and Mirocal Origin software). P values <0.05 were considered to be significant.
Materials
Cell culture reagents, Alexa Fluor 488-tagged secondary antibodies and Lipofectamine 2000 were from Invitrogen (Paisley, UK). Rabbit anti-HA antibody was obtained from Abcam (Cambridge, UK). Cy3 and Cy5-tagged secondary antibodies were obtained from Jackson Immuno Research (West Grove, PA, USA). Monoclonal HA.11 antibody was obtained from Covance (Berkeley, CA, USA). Antibodies against pERK were obtained from Cell Signaling Technology (Danvers, MA, USA) and pCREB from Millipore (Upstate, Billerica, MA, USA). Horseradish peroxidase-conjugated secondary antibodies were purchased from Thermo Scientific (Pierce, Rockford, IL, USA). The pNFAT-Luc and pNF-kB-Luc reporter plasmids were obtained from Stratagene (La Jolla, CA, USA). SR141716A was a gift from Sanofi-Synthélabo Recherche (Montpellier, France). AM281 and AM251 were obtained from Tocris Cookson (Avonmouth, UK). L-α-lysophosphatidylinositol mixture (LPI; 58% C16, 42% C18), G418, poly-D-lysine, foetal calf serum (FCS), BSAfaf, HEPES and all other chemicals were obtained from Sigma-Aldrich (Dorset, UK or Taufkirchen, Germany).
Results
Effects of GPR55 ligands on Ca2+ signalling
We and others have shown that the activation of GPR55 by various ligands can promote the release of Ca2+ from intracellular stores (Oka et al., 2007; Lauckner et al., 2008; Henstridge et al., 2009). Here we have extended upon these findings to evaluate the effects of a series of well-described cannabinoid antagonists/inverse agonists on Ca2+ signalling in GPR55-HEK293 cells. Consistent with our previous work (Henstridge et al., 2009), a brief 5 min exposure of GPR55-HEK293 cells (Figure 1A) to LPI (300 nM, panel a) and AM251 (3 µM, panel b) resulted in the induction of oscillatory Ca2+ transients. This effect was concentration-dependent (Figure 1B), with EC50 values for LPI and AM251, shown in Table 1). Similar effects were observed with SR141716A (Figure 1A, panel c and 1B; Table 1), a close structural analogue of AM251 (see Figure S1). The related diarylpyrazole ligand AM281 was also able to promote Ca2+ signalling; however, it was much less potent, with responses observed over the range of 10–30 µM (Figure 1A, panel d and 1B; Table 1). No effects of any ligands on intracellular Ca2+ levels were observed in control HEK293 at the concentrations tested.
Figure 1.
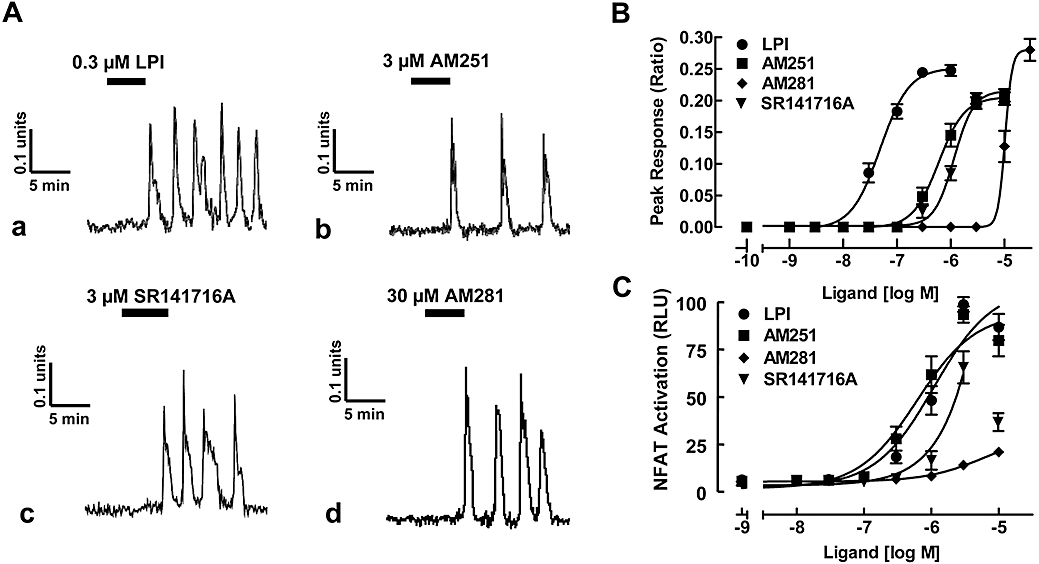
GPR55 ligands promote oscillatory Ca2+ transients and NFAT-activation in GPR55-HEK293 cells. (A) Representative Ca2+ transients in GPR55-HEK293 cells. (a) LPI (0.3 µM), (b) AM251 (3 µM), (c) SR141716A (3 µM) and (d) AM281 (30 µM) induce a sustained, oscillatory Ca2+ response in GPR55-HEK293 cells. Traces represent changes in fura-2 fluorescence ratio for individual cells. (B) Concentration–response curves for GPR55 ligands. LPI, AM251, SR141716A and AM281 all induced Ca2+ transients. Data are mean peak responses ± SEM of 40 cells derived from four independent experiments. (C) NFAT transcription factor activation in GPR55-expressing cells. GPR55-HEK293 cells were transfected with 200 ng of NFAT-luciferase-reporter plasmid and 24 h post transfection, cells were stimulated with increasing amounts of LPI, AM251, AM281 and SR141716A for 6 h in serum-free medium. Data are means ± SEM from one of four independent experiments performed in quadruplicate. Data were normalized and expressed as percent of maximum activation induced by a saturating concentration of LPI which was set as 100%. Note, for SR141716A, the concentration–response curve was fitted to values up to 3 µM. GPR55, G protein-coupled receptor 55; GPR55-HEK293, stable HEK293 cells expressing 3xHA-GPR55; LPI, L-α-lysophosphatidylinositol; NFAT, nuclear factor of activated T cells; RLU, relative light units.
Table 1.
Ligand potencies in various assays on GPR55-HEK cells (EC50 µM)
| Ligand | Ca2+microfluorimetry | NFAT-activation | ERK phosphorylation |
|---|---|---|---|
| LPI | 0.049 ± 0.004 | 1.10 ± 0.02 | 0.074 ± 0.013 |
| AM251 | 0.63 ± 0.11 | 1.13 ± 0.07 | 0.54 ± 0.15 |
| SR141716A | 1.14 ± 0.03 | >1 | 0.64 ± 0.25 |
| AM281 | 13.6 ± 3.30 | 13.1 ± 0.8 | >3 |
| CREB-phosphorylation | NF-κB activation | EPIC DMR | |
| LPI | 0.093 ± 0.01 | 1.9 ± 0.04 | 0.009 ± 0.0001 |
| AM251 | 0.43 ± 0.09 | >0.5 | 0.81 ± 0.14 |
| SR141716A | 0.48 ± 0.05 | >1 | 1.85 ± 0.1 |
| AM281 | >3 | 7.25 ± 0.23 | 4.1 ± 0.09 |
Data are means ± SEM (n= 2–5).
CREB, cAMP response element binding protein; DMR, dynamic mass redistribution; ERK, extracellular signal regulated kinase; GPR55-HEK293, stable HEK293 cells expressing 3xHA-GPR55; LPI, L-α-lysophosphatidylinositol; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor-κB.
As we have recently shown that the GPR55 agonist LPI can induce NFAT-activity in a concentration-dependent manner (Henstridge et al., 2009), we also compared the ability of the cannabinoid ligands to promote NFAT activation. LPI (Figure 1C) induced NFAT-luciferase activity and AM251 (Figure 1C) was as potent and efficacious as LPI (Table 1). High concentrations of SR141716A (>3 µM) appeared to suppress NFAT activation (Figure 1C), preventing an accurate determination of EC50 and Emax values. However, AM281 (Figure 1C) was clearly the least potent compound tested in activating NFAT (Table 1).
Activation of ERK1/2 MAP-kinase
Although GPR55 activation has been suggested to promote ERK1/2 signalling, this is controversial, having only been observed with LPI and not with cannabinoid ligands (Oka et al., 2007; Lauckner et al., 2008; Kapur et al., 2009). We therefore profiled the ability of various GPR55 ligands to activate ERK1/2 in GPR55-HEK293 and control HEK293 cells. In GPR55-HEK293 cells, LPI induced a marked, concentration-dependent increase in ERK1/2 phosphorylation (Figure 2A), with a peak response observed after 25 min stimulation (1 µM LPI; data not shown). No significant response was observed in control HEK293 cells at this time point (Figure 2A, HEK). In GPR55-HEK293 cells, threshold pERK responses were observed at 30 nM of LPI, with peak activation apparent at 300 nM LPI (Figure 2D) and, overall, with similar potency as already observed for Ca2+ signalling (Table 1).
Figure 2.
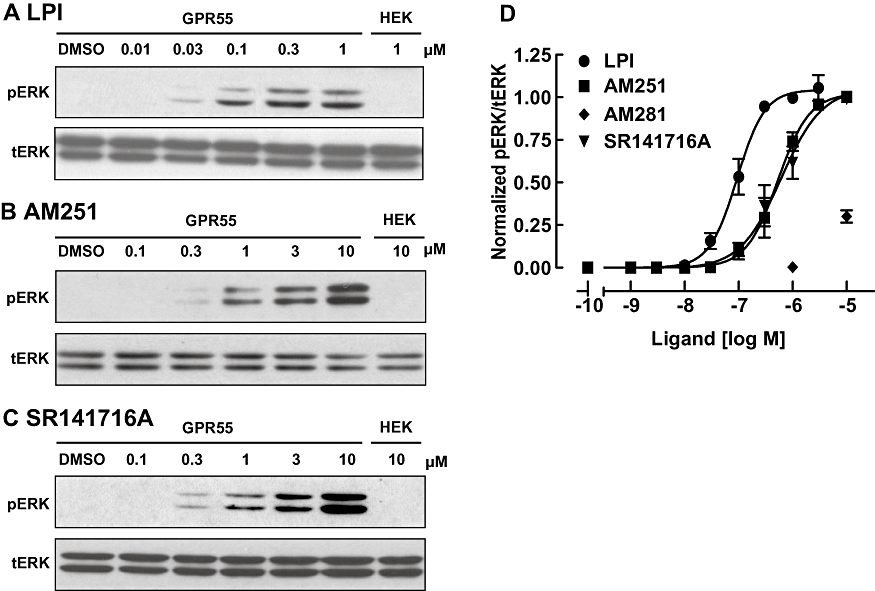
Activation of ERK1/2 MAP-kinase with GPR55 ligands. (A–C) Immunoblots showing ERK1/2 phosphorylation in response to increasing concentrations LPI (A), AM251 (B) and SR141716A (C) in GPR55-HEK293 cells. The absence of pERK responses in control HEK293 cells are illustrated on the right-hand panel of each blot. Blots for total ERK levels (tERK) are presented below the pERK bands and the immunoblots are representative of three separate experiments. (D) Concentration–response curves for GPR55 ligand induced ERK1/2 phosphorylation. LPI, AM251 and SR141716A induced ERK1/2 phosphorylation. Data points are also included for AM281 at 1 and 10 µM. Data are derived from densitometric analysis, showing the mean responses ± SEM (pERK/tERK ratio) normalized to the response of LPI at 1 µM run in the same blot. ERK1/2, extracellular signal regulated kinase 1/2; GPR55, G protein-coupled receptor 55; GPR55-HEK293, stable HEK293 cells expressing 3xHA-GPR55; LPI, L-α-lysophosphatidylinositol.
Next, we investigated the effects of AM251 and SR141716A on ERK1/2 signalling (Figure 2B–C). Interestingly, both cannabinoid ligands promoted ERK phosphorylation, with a similar efficacy to that observed with LPI. Both AM251 and SR141716A were slightly more potent than observed with the Ca2+ signalling assay (Figure 2D; Table 1). AM281 was clearly less potent at ERK activation than the other diarylpyrazole ligands, with effects only observed with concentrations >3 µM (Figure 2D). No ERK activation was observed in control HEK293 cells following treatment with any of these ligands (Figure 2A–C, HEK).
Activation of CREB transcription factor via GPR55
The CREB transcription factor activates the transcription of target genes in response to a diverse array of stimuli, including hormones and growth factors, that activate not only protein kinase A (PKA), but also a variety of other kinases such as ERK MAP-kinases and Ca2+/calmodulin-dependent protein kinases (CaMKs) (see Shaywitz and Greenberg, 1999). As LPI (Oka et al., 2007) and the cannabinoid antagonists/inverse agonists AM251, AM281 and SR141716A were able to activate the ERK MAP-kinase pathway (see Figure 2), we tested whether CREB, being a downstream target of ERK-MAP kinases and Ca2+ signalling, can also be activated by these ligands.
We investigated whether LPI, AM251, AM281 and SR141716A were capable of activating CREB using immunoblot detection of phospho-CREB (pCREB) (Figure 3). Treatment of GPR55-HEK293 cells with LPI induced a marked, concentration-dependent increase in CREB phosphorylation (Figure 3A), with a peak response observed after 25 min stimulation (1 µM LPI; data not shown). In GPR55-HEK293 cells, threshold pCREB responses were observed at 10 nM, with peak activation apparent in the range 0.3–1 µM LPI (Figure 3D; Table 1). Next, we investigated the effects of AM251 and SR141716A on CREB signalling (Figure 3B and 3C). Both these ligands exhibited a higher efficacy than LPI, with peak responses approximately 150–175% of the LPI response (Figure 3D). As with the pERK activation, both AM251 and SR141716A were more potent at inducing pCREB than triggering Ca2+ transients (Table 1). AM281 was also less potent at CREB activation than the other diarylpyrazole ligands, with effects observed with concentrations >3 µM (Figure 3D; Table 1). No CREB activation was observed in control HEK293 cells following treatment with these ligands (Figure 3A–C, HEK).
Figure 3.
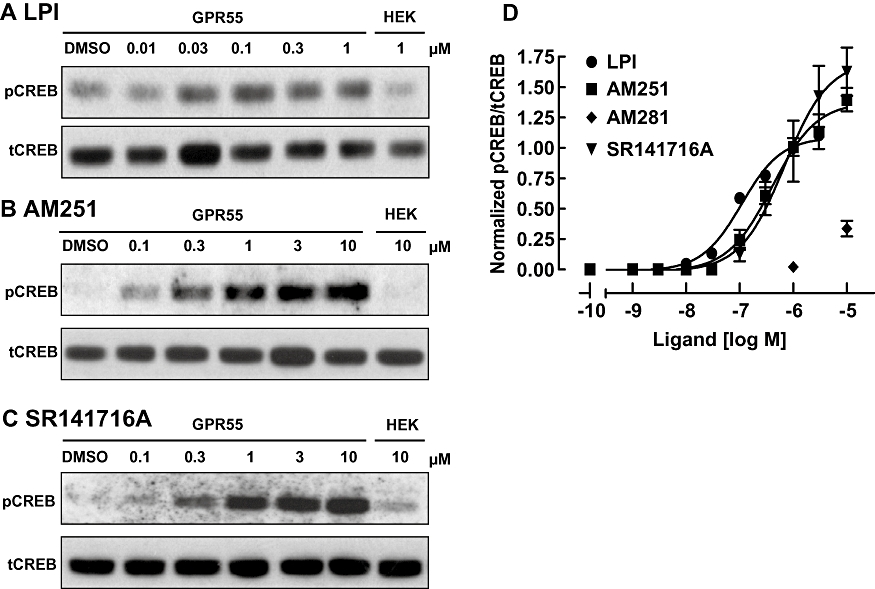
CREB phosphorylation with GPR55 ligands. (A) Immunoblot showing CREB phosphorylation (pCREB) in response to varying concentrations LPI (A), AM251 (B) and SR141716A (C) in GPR55-HEK293 cells. The absence of pCREB responses in control HEK293 cells are illustrated on the right-hand panel in each blot. Blots for total CREB levels (tCREB) are presented below the pCREB bands and the immunoblots are representative of three separate experiments. (D) Concentration–response curves for GPR55 ligand induced CREB phosphorylation. LPI, AM251 and SR141716A all induced pCREB activation. Data points are also included for AM281 at 1 and 10 µM. Data are derived from densitometric analysis, showing the mean responses ± SEM (pCREB/tCREB ratio) normalized to the response of LPI at 1 µM run in the same blot. CREB, cAMP response element binding protein; GPR55, G protein-coupled receptor 55; GPR55-HEK293, stable HEK293 cells expressing 3xHA-GPR55; LPI, L-α-lysophosphatidylinositol.
Activation of NF-κB transcription factor via GPR55
Recently, we have shown that the endogenous cannabinoid ligand anandamide can activate the transcription factor NF-κB in endothelial cells (Waldeck-Weiermair et al., 2008); however, we did not determine whether this was a GPR55-mediated effect. In addition, NF-κB transcription can be regulated by ERK MAP-kinases (Delfino and Walker, 1999). Hence, we tested whether LPI, AM251, AM281 and SR141716A can activate NF-κB transcription factor and nuclear translocation in GPR55-HEK293 cells.
As Figure 4A shows, LPI was as potent (Table 1) and efficacious in inducing NF-κB luciferase activity as NFAT activation (compare with Figure 1C). High concentrations of AM251 and SR141716A (>3 µM) appeared to suppress NF-κB activity, preventing accurate determinations of EC50 and Emax values. However, AM281 was clearly less potent (Figure 4A; Table 1).
Figure 4.
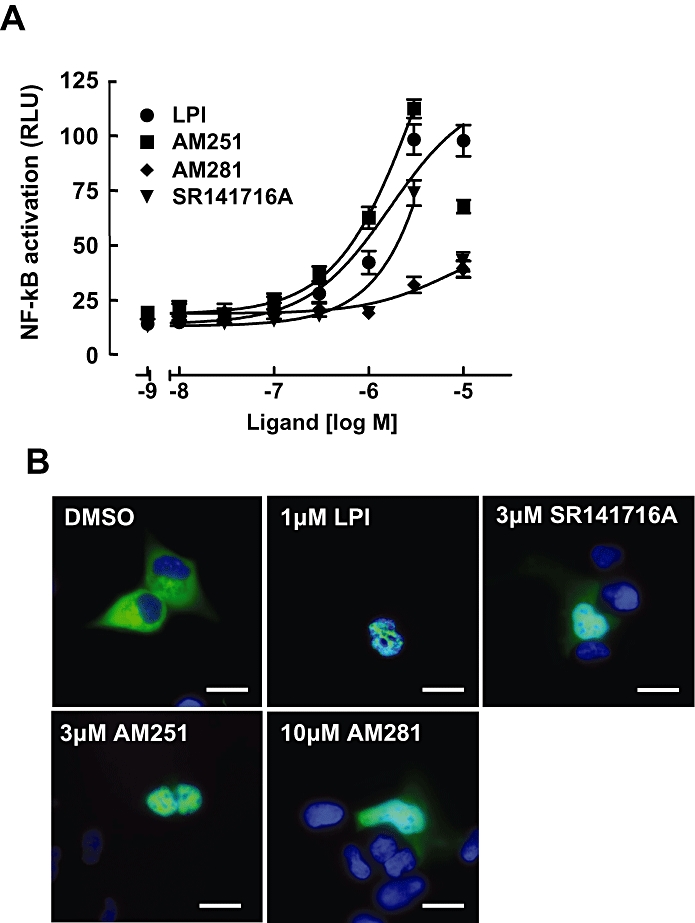
NF-κB transcription factor activation in GPR55 expressing cells. (A) GPR55-HEK293 cells were transfected with 200 ng of a NF-κB-luciferase-reporter. Twenty-four h post transfection, cells were stimulated with increasing amounts of LPI, AM251, AM281 and SR141716A for 6 h in serum-free medium. Data are means ± SEM from four independent experiments performed in quadruplicate. Data were normalized and expressed as percent of maximum activation induced by a saturating concentration (dose) of LPI which was set at 100%. Note, for SR141716A and AM251, the concentration–response curves were fitted to values up to 3 µM. (B) Nuclear translocation of p65-GFP upon ligand stimulation. The effect of 15 min treatment with 1 µM LPI, 3 µM AM251, 3 µM SR141716A and 10 µM AM281 on the translocation of an EGFP-tagged p65 subunit of NF-κB was visualized in GPR55-HEK293 cells. Cell nuclei were stained with DAPI (blue). Representative cells of three independent experiments are shown. Scale bars = 10 µM. GFP, green fluorescent protein; GPR55, G protein-coupled receptor 55; GPR55-HEK293, stable HEK293 cells expressing 3xHA-GPR55; LPI, L-α-lysophosphatidylinositol; NF-κB, nuclear factor-κB; RLU, relative light units.
Next, we tested whether these ligands could induce nuclear translocation of p65, the functional subunit of NF-κB, whose nuclear translocation upon stimulation can be monitored to report activation of this pathway, using EGFP-labelled p65 (Waldeck-Weiermair et al., 2008). While p65-EGFP was predominantly localized to the cytosol and excluded from the nucleus in cells treated with vehicle (DMSO) for 15 min (Figure 4B), cells treated with 1 µM LPI, 3 µM AM251, 3 µM of SR141716A or 10 µM AM281 all showed a redistribution of p65-EGFP (green) into the cell nucleus (overlay with DAPI, blue; Figure 4B). Cells treated with lower concentrations of LPI, AM251, AM281 or SR141716A did show sporadic p65-EGFP translocation to the nucleus, but to a lesser extent (data not shown).
Screening for GPR55 agonists using the label-free DMR assay
Given the difficulties associated with establishing efficient screening assays for GPR55, we evaluated an alternative – label-free and non-invasive – methodology based on optical detection of dynamic changes in cellular density following receptor activation (Fang et al., 2007; Schröder et al., 2009). Here, we employed the novel resonant wave guide grating biosensor technology (Corning Epic®) to resolve the real-time signalling capacity of GPR55 upon activation with the set of compounds identified in the assays above. In brief (see Schröder et al., 2009), activation of GPCRs is known to cause a translocation of multiple signalling molecules upon receptor stimulation; thus, the DMR of intracellular content is monitored as an optical signal. Typically, these signals, generated upon ligand addition, reflect the G protein coupling profile of the respective receptors, i.e. ‘optical signatures’ and are distinct for Gαi, Gαs, etc.-coupled receptors (Fang et al., 2007; Schröder et al., 2009).
The optical signals for increasing concentrations of LPI (0.01 nM to 10 µM) were recorded in GPR55-HEK293 cells (Figure 5A), and control HEK293 cells (Figure 5B). A concentration–response curve for LPI using the AUC values of the optical signal traces, illustrated in Figure 5A, is shown in Figure 5C and revealed an EC50 value in the low nanomolar range for LPI, thus appearing about 5-fold more potent than when measuring the Ca2+ assay (compare with Figure 1B; Table 1).
Figure 5.
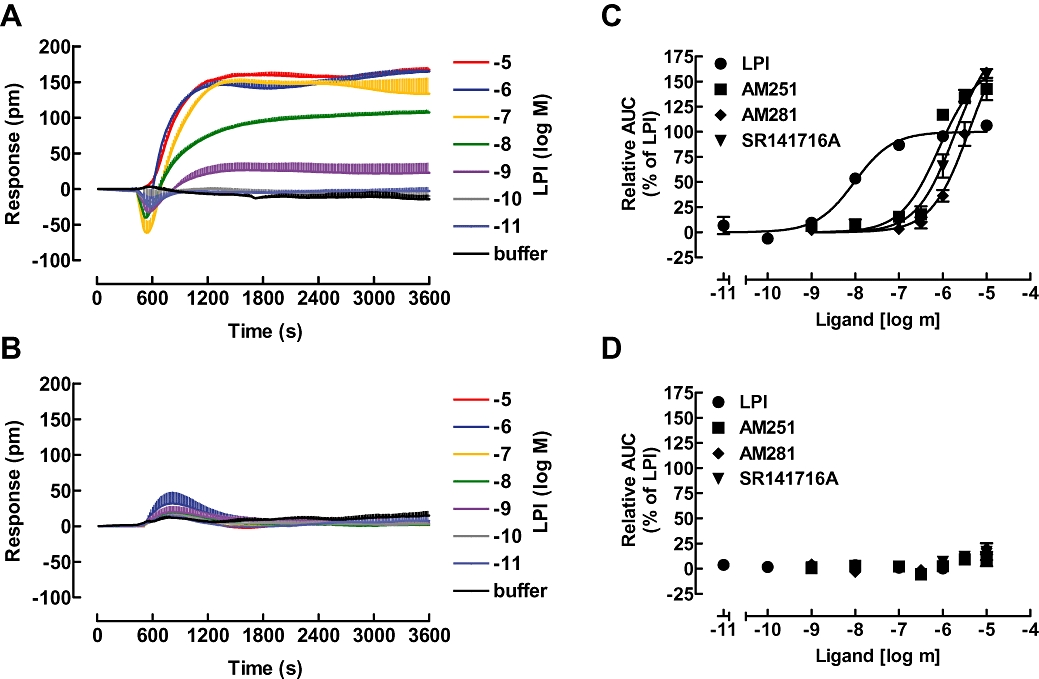
Dynamic mass redistribution (DMR) analysis of cellular responses to GPR55 ligands. GPR55-HEK293 cells (A) or HEK293 cells (B) were challenged with the indicated concentrations of the GPR55 agonist LPI and the resulting picometre-shifts of reflected light wavelength against the time [s] were monitored. Data shown are means ± SEM from a representative optical trace experiment carried out in triplicates. (C and D) Transformation of optical signatures into concentration response curves for the indicated GPR55 ligands in GPR55-HEK293 (C) or control HEK293 (D) cells using the AUC values between the 1200 and 3600 s time points. Data were normalized and expressed as percent of maximum activation induced by a saturating concentration of LPI (10 µM) which was set to 100%. Curves were normalized to the Emax of LPI (set to 100%). AUC, area under curve; GPR55, G protein-coupled receptor 55; GPR55-HEK293, stable HEK293 cells expressing 3xHA-GPR55; LPI, L-α-lysophosphatidylinositol.
Next, we tested the capacity of AM251, AM281 and SR141716A to elicit optical signals from GPR55-HEK293 (Figure 5C) and control HEK293 (Figure 5D) cells and calculated their respective potencies and efficacies (Table 1). All three ligands were approximately two log units less potent than LPI in this assay (Figure 5C), reflecting a rank order similar to those found for all other assays employed so far. However, using the Epic® real-time optical measurements, all compounds were potent, being equivalent to the most sensitive signalling assays readouts for detecting GPR55-mediated responses. In summary, this assay approach proved extremely suitable for non-invasively screening compounds on this Gα13-coupled GPCR.
Agonist-induced GPR55 receptor internalization
Lastly, we evaluated whether our candidate ligands were capable of inducing GPR55 internalization, using a modified antibody feeding protocol that distinguishes between cell surface and intracellular receptors (Coutts et al., 2001). Following antibody prelabelling of live GPR55-HEK293 cells for 30 min, cells were treated with the compounds and subsequently processed for cell surface and intracellular HA-immunolabelling (Figure 6). Analysis of HA-immunoreactivity using confocal microscopy showed that, under control conditions GPR55 was predominantly located on the cell surface (Figure 6, DMSO, green; Henstridge et al., 2009). However, following treatment with LPI (1 µM), SR141716A (3 µM) and AM251 (3 µM) for 60 min, a pronounced redistribution of GPR55 into intracellular vesicles was observed (red). Interestingly, only a modest effect on GPR55 internalization was seen with AM281 (30 µM).
Figure 6.
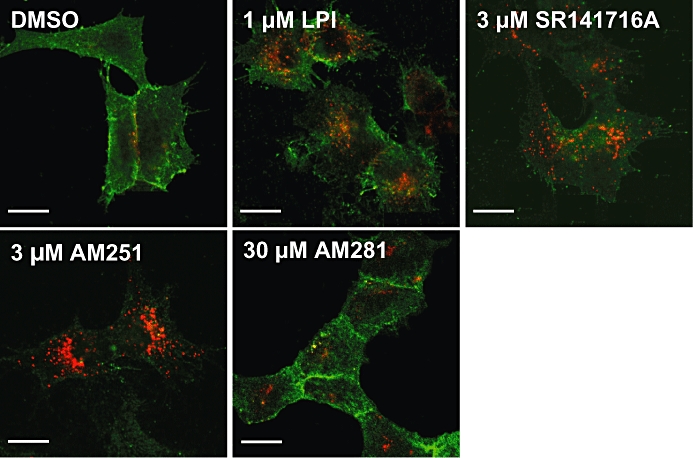
Agonist-induced internalization of GPR55. Confocal images of cell surface (green) and intracellular (red) 3xHA-GPR55 immunoreactivity. GPR55-HEK293 cells were pre-labelled for 30 min with anti-HA antibody, then treated for 1 h at 37°C with DMSO, 1 µM LPI, 3 µM SR141716A, 3 µM AM251 or 30 µM AM281. Cells were then processed for surface (green) and intracellular (red) HA-immunoreactivity. Control GPR55-HEK293 cells show primarily membrane localization of GPR55 (green) and very little internalized receptor (red). However, LPI, SR141716A and AM251 all induce marked endocytosis, represented by the strong red and weak green labelling. In contrast, AM281 induced very low levels of internalization. Scale bars = 20 µm. Representative cells from at least four independent experiments are shown. GPR55, G protein-coupled receptor 55; GPR55-HEK293, stable HEK293 cells expressing 3xHA-GPR55; LPI, L-α-lysophosphatidylinositol.
Discussion
GPR55 is a recently de-orphanized G protein-coupled receptor that can be activated by the endogenous lipid signalling molecule LPI and certain cannabinoid ligands. GPR55 is known to induce ERK1/2 phosphorylation and RhoA-dependent Ca2+ release from intracellular stores; however, the pharmacology of GPR55 is controversial. Here, we have identified new effects of GPR55 activation on transcription factors and profiled the activity of a variety of GPR55 ligands in different functional assays, which helps to rationalize some of the conflicting pharmacology reported for GPR55. In addition, we also evaluate a new label-free approach for evaluating GPR55 ligands and demonstrate its effectiveness for this purpose.
GPR55 signalling and pharmacology
In this study, we demonstrated that agonist interactions with GPR55 promoted the activation of a range of signalling pathways, including Ca2+ release, NFAT-, CREB- and NF-κB-transcription, ERK1/2 phosphorylation, Epic® traces and receptor internalization. It is becoming increasingly clear that the coupling efficiency of a GPCR to downstream effector systems can be selectively modulated by distinct ligands, a phenomenon sometimes referred to as ligand biased signalling (Kenakin, 2007; Galandrin et al., 2008). Although with the different GPR55 assays employed there were some notable differences in ligand efficacy, the rank order of potency for the ligands was consistent throughout this study. Thus, while there may be some evidence for agonist bias with GPR55, it has only a relatively modest impact on the observed pharmacology. The most marked difference was observed with the CREB assay, where LPI was clearly much less efficaceous than AM251 or SR141716A. An increasing number of studies have shown ERK MAP kinase activation with GPR55, both in recombinant systems (Oka et al., 2007; Kapur et al., 2009) and also in cells that express wild-type GPR55, including microglia (Pietr et al., 2009) and osteoclasts (Whyte et al., 2009). However, in some cases this is only observed with LPI and not with cannabinoid ligands (Oka et al., 2007; Kapur et al., 2009). This discrepancy may be due to a lack of concentration–response profiling with cannabinoid ligands in the different assays, as we find cannabinoids to have a similar efficacy as LPI for ERK activation, although they are less potent. In some of the reporter gene studies with SR141716A (NF-κB and NFAT assays) and AM251 (NF-κB assay) responses were suppressed at high ligand concentrations (>3 µM) preventing accurate determination of EC50 and Emax values. This may reflect non-specific effects and/or toxicity in these long-duration assays.
Cannabinoid activity at GPR55: implications for CB1 receptor antagonists
The widely used diarylpyrazole CB1 receptor antagonists/inverse agonists AM251 and SR141716A elicited a significant agonist response via GPR55 at concentrations typically encountered in experimental studies of CB1 receptor function. These data are consistent with previous studies reporting G protein activation with AM251 (Henstridge et al., 2009) and recently with beta-arrestin (Kapur et al., 2009; Yin et al., 2009) and pGL3-CRE-MRE-SRE-luc reporter assays (Yin et al., 2009) for both AM251 and SR141716A. The related compound, AM281 was a less potent agonist at GPR55, although its ability to bind to and antagonize the CB1 receptor is comparable with that of the other diarylpyrazole ligands (Lan et al., 1999). This suggests that the characteristics of the carboxamide group at position 3 of the diarylpyrazole backbone may be more important for ligand binding to GPR55 than for binding to the CB1 receptor, within this chemical series.
Although all the diarylpyrazole compounds induce GPR55 activity, it was generally observed that much higher concentrations than are typically used for CB1 receptor antagonsim [5–15 nM (Pertwee, 2005; Ryberg et al., 2007)] were required to elicit an effect. Thus, acute, physiological effects of low nM concentrations of these agents are likely to reflect CB1 receptor blockade. However, a role for GPR55 activation needs to be considered where micromolar concentrations of SR14171A and AM251 are used, a situation that is encountered with many functional/physiological studies of cannabinoid action. In addition, it is possible that long-term treatment with SR141716A (Rimonabant®) may have some impact on GPR55 function, especially if it accumulates in fatty tissues, such as the brain, and may therefore contribute to some of the biological activity and/or side-effects observed with this compound in humans.
Which GPR55 assay is best?
This study demonstrates that GPR55 mediates the activation of a plethora of downstream signalling pathways. However, evidence for agonist bias in their activation means that the pharmacology of GPR55 is influenced by the assay used to assess receptor function, and in some cases this could miss potential ligands. We therefore evaluated a novel label-free assay that should circumvent these problems, by allowing an integrated readout of GPR55 function. Indeed, using this approach, we were able to detect agonist activity for all GPR55 ligands tested. GPR55 pharmacology has also been assessed previously using beta-arrestin assays, and produced similar agonist profiles to the present study (Kapur et al., 2009; Yin et al., 2009). However, LPI was notably less potent with these assays, giving an EC50 values in the low µM range compared with the 9 nM value obtained from the Epic® real-time optical measurements.
Conclusion
GPR55 is thought to be sensitive to a range of cannabinoid ligands (Ryberg et al., 2007); however, there are a number of inconsistencies in the pharmacological profiles of cannabinoid ligands that interact with GPR55. In this study, we show that the activity of cannabinoid ligands at GPR55 is influenced by the assay used to assess receptor-mediated downstream signalling. Our findings help to resolve some of the issues surrounding the pharmacology of cannabinoid ligands at GPR55, and highlight differences in the effector coupling associated with distinct GPR55 ligands. Such activity is a basis for the therapeutic targeting of desirable signalling outcomes for GPR55.
Acknowledgments
This work was supported by TENOVUS SCOTLAND, the Royal Society, the MRC, the Caledonian Society and the Anonymous Trust (all to AJI) (a grant by) the Austrian Science Fund (FWF), the Jubiläumsfonds of the Austrian National Bank and the Lanyar Stiftung Graz (all to MW), a fellowship within the ‘Molecular Medicine Ph.D. program’ from the Medical University of Graz, Austria (NB) and a student's fellowship by the Austrian Government (JK). JLW was supported by funds provided by the state of California for medical research to the University of California, San Francisco and LM was supported by the Lundbeck Foundation, Denmark. We would like to thank Corning Life Sciences for their support on the Epic® System.
Glossary
Abbreviations:
- AM251
N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AM281
1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide
- CREB
cAMP response element binding protein
- ERK1/2
extracellular signal regulated kinase 1/2
- G13
Gα13 protein
- GFP
green fluorescent protein
- GPR55
G protein-coupled receptor 55
- GPR55-HEK293
stable HEK293 cells expressing 3xHA-GPR55
- HA
haemaglutinin
- HBS
HEPES-buffered saline
- LPI
L-α-lysophosphatidylinositol
- MAP
mitogen activated kinase
- NFAT
nuclear factor of activated T cells
- NF-κB
nuclear factor-κB
- PLC
phospholipase C
- SR141716A
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
Conflicts of interest
The authors declare no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Chemical structures of the compounds used in the study.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Brown AJ, Robin HC. Chapter 5. Is GPR55 an anandamide receptor? Vitam Horm. 2009;81:111–137. doi: 10.1016/S0083-6729(09)81005-4. [DOI] [PubMed] [Google Scholar]
- Coutts AA, Navi-Goffer S, Ross RA, Macewan DJ, Mackie K, Pertwee RG, et al. Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delfino F, Walker WH. Hormonal regulation of the NF-kappaB signalling pathway. Mol Cell Endocrinol. 1999;157:1–9. doi: 10.1016/s0303-7207(99)00127-6. [DOI] [PubMed] [Google Scholar]
- Fang Y, Ferrie AM, Fontaine NH, Mauro J, Balakrishnan J. Resonant waveguide grating biosensor for living cell sensing. Biophys J. 2006;91:1925–1940. doi: 10.1529/biophysj.105.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Li G, Ferrie AM. Non-invasive optical biosensor for assaying endogenous G protein-coupled receptors in adherent cells. J Pharmacol Toxicol Methods. 2007;55:314–322. doi: 10.1016/j.vascn.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpre G, Bonin H, Ogawa K, Gales C, Bouvier M. Conformational rearrangements and signalling cascades involved in ligand-biased mitogen-activated protein kinase signalling through the beta1-adrenergic receptor. Mol Pharmacol. 2008;74:162–172. doi: 10.1124/mol.107.043893. [DOI] [PubMed] [Google Scholar]
- Henstridge CM, Balenga NA, Ford LA, Ross RA, Waldhoer M, Irving AJ. The GPR55 ligand l-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signalling and NFAT activation. FASEB J. 2009;23:183–193. doi: 10.1096/fj.08-108670. [DOI] [PubMed] [Google Scholar]
- Johns DG, Behm DJ, Walker DJ, Ao Z, Shapland EM, Daniels DA, et al. The novel endocannabinoid receptor GPR55 is activated by atypical cannabinoids but does not mediate their vasodilator effects. Br J Pharmacol. 2007;152:825–831. doi: 10.1038/sj.bjp.0707419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Zhao P, Sharir H, Bai Y, Caron MG, Barak LS, et al. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J Biol Chem. 2009;284:29817–29827. doi: 10.1074/jbc.M109.050187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- Lan R, Gatley J, Lu Q, Fan P, Fernando SR, Volkow ND, et al. Design and synthesis of the CB1 selective cannabinoid antagonist AM281: a potential human SPECT ligand. AAPS PharmSci. 1999;1:E4. doi: 10.1208/ps010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci USA. 2008;105:2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka S, Nakajima K, Yamashita A, Kishimoto S, Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun. 2007;362:928–934. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- Oka S, Toshida T, Maruyama K, Nakajima K, Yamashita A, Sugiura T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: a possible natural ligand for GPR55. J Biochem. 2009;145:13–20. doi: 10.1093/jb/mvn136. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacological actions of cannabinoids. Handb Exp Pharmacol. 2005:1–51. doi: 10.1007/3-540-26573-2_1. [DOI] [PubMed] [Google Scholar]
- Pietr M, Kozela E, Levy R, Rimmerman N, Lin YH, Stella N, et al. Differential changes in GPR55 during microglial cell activation. FEBS Lett. 2009;583:2071–2076. doi: 10.1016/j.febslet.2009.05.028. [DOI] [PubMed] [Google Scholar]
- Ross RA. The enigmatic pharmacology of GPR55. Trends Pharmacol Sci. 2009;30:156–163. doi: 10.1016/j.tips.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder R, Merten N, Mathiesen JM, Martini L, Kruljac-Letunic A, Krop F, et al. The C-terminal tail of CRTH2 is a key molecular determinant that constrains Galphai and downstream signalling cascade activation. J Biol Chem. 2009;284:1324–1336. doi: 10.1074/jbc.M806867200. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Staton PC, Hatcher JP, Walker DJ, Morrison AD, Shapland EM, Hughes JP, et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain. 2008;139:225–236. doi: 10.1016/j.pain.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Waldeck-Weiermair M, Zoratti C, Osibow K, Balenga N, Goessnitzer E, Waldhoer M, et al. Integrin clustering enables anandamide-induced Ca2+ signalling in endothelial cells via GPR55 by protection against CB1-receptor-triggered repression. J Cell Sci. 2008;121:1704–1717. doi: 10.1242/jcs.020958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhoer M, Kledal TN, Farrell H, Schwartz TW. Murine cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signalling activities. J Virol. 2002;76:8161–8168. doi: 10.1128/JVI.76.16.8161-8168.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte LS, Ryberg E, Sims NA, Ridge SA, Mackie K, Greasley PJ, et al. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc Natl Acad Sci U S A. 2009;106:16511–16516. doi: 10.1073/pnas.0902743106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, et al. Lipid G-protein-coupled receptor ligand identification using beta-arrestin pathhunter assay. J Biol Chem. 2009;284:12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.