

Abstract
Background and purpose:
Cannabinoid receptor agonists reduce intestinal propulsion in rodents through the CB1 receptor. In addition to its antagonistic activity at this receptor, rimonabant (N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide) alone augments intestinal transit. Using rat and guinea-pig ileum MPLM (myenteric plexus-longitudinal muscle) preparations, we investigated whether the latter effect was through inverse agonism or antagonism of endocannabinoid agonist(s).
Experimental approach:
Inverse agonism was investigated by comparing the maximal enhancement of electrically evoked contractions of the MPLM by two CB1 receptor antagonists, AM 251 (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide) and O-2050 [(6aR,10aR)-3-(1-methanesulphonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6-H-dibenzo[b,d]pyran], with that produced by rimonabant. To reveal ongoing endocannabinoid activity, effects of inhibiting endocannabinoid hydrolysis by fatty acid amide hydrolase (FAAH) using AA-5HT (arachidonyl-5-hydroxytryptamine), PMSF (phenylmethylsulphonyl fluoride) or URB-597 (3′-carbamoyl-biphenyl-3-yl-cyclohexylcarbamate), or putative uptake using VDM-11 [(5Z,8Z,11Z,14Z)-N-(4-hydroxy-2-methylphenyl)-5,8,11,14-eicosatetraenamide] was evaluated.
Key results:
The presence of CB1 receptors was revealed by antagonism of exogenous anandamide, arachidonylethanolamide (AEA) and WIN 55,212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate] by rimonabant. The rank order of potentiation of contractions was AM 251 > rimonabant > O-2050. Neither the FAAH inhibitors nor VDM-11 affected electrically evoked contractions. Each FAAH inhibitor increased the potency of AEA but not WIN 55,212-2. VDM-11 did not alter the inhibitory effect of AEA.
Conclusions and implications:
The different levels of maximal potentiation of contractions by the CB1 receptor antagonists suggest inverse agonism. The potentiation of the action of AEA by the FAAH inhibitors showed that FAAH was present. The lack of effect of FAAH inhibitors and VDM-11 alone on electrically evoked contractions, and on the potency of exogenous AEA suggests that pharmacologically active endocannabinoids were not released and the endocannabinoid transporter was absent. Thus, the CB1 receptor antagonists behave as inverse agonists.
This article is part of a themed issue on Cannabinoids. To view the editorial for this themed issue visit http://dx.doi.org/10.1111/j.1476-5381.2010.00831.x
Keywords: cannabinoid, endocannabinoid tone, myenteric plexus, small intestine, fatty acid amide hydrolase, cannabinoid receptor agonists, cannabinoid receptor antagonists, cannabinoid receptor inverse agonists, inverse agonism
Introduction
Being a potent antagonist of the cannabinoid CB1 receptor, rimonabant (N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide) (pKB 8.00; Rinaldi-carmona et al., 1994) readily prevents or reverses the action of a range of agonists acting at this receptor in several in vivo and in vitro functional bioassays (Howlett et al., 2002). However, in addition to its antagonist activity, rimonabant and a number of other CB1 receptor antagonists, such as AM 251 (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide) (Gatley et al., 1997) and LY 320135 (Felder et al., 1998), alone elicit CB1 receptor-mediated effects that are invariably opposite in direction from those produced by cannabinoid receptor agonists (Pertwee, 2005). For example, while in vivo administration of cannabinoid receptor agonists into rodents delays intestinal transit, via a reduction in the propulsive function of the small intestine, rimonabant alone increases intestinal transit (Colombo et al., 1998; Izzo et al., 1999a,b; 2001;). Such antagonist-mediated changes have been attributed to the unmasking of a constitutive activity of the CB1 receptor via inverse agonism, or the antagonism of tonically released endocannabinoid agonists at this receptor. To date, rimonabant has only been conclusively demonstrated to behave as an inverse agonist in recombinant cell-based assays in which the CB1 receptor was overexpressed (Bouaboula et al., 1997; MacLennan et al., 1998). Therefore, it has appeared unlikely that such activity could occur in intact tissues in which receptor expression was likely to be at a natural physiological level.
Over the last decade, evidence has emerged to indicate that a functional endocannabinoid agonist tone may be present in the intestinal tract. This has been primarily because of the identification of anandamide, arachidonylethanolamide (AEA) and 2-arachidonylglycerol (2-AG, Izzo et al., 2003; Fegley et al., 2005; Guagnini et al., 2006) and the mechanisms for their inactivation by facilitated uptake, via a putative transporter protein (Mascolo et al., 2002), and enzymatic hydrolysis, such as by fatty acid amide hydrolase (FAAH, Katayama et al., 1997). Furthermore, the demonstration that in vivo administration of inhibitors of the putative uptake transporter or FAAH into rodents mimics the inhibitory effects of exogenously administered cannabinoid receptor agonists has added weight to a possible physiological inhibitory role of an endocannabinoid agonist tone on intestinal motility (Pinto et al., 2002; Capasso et al., 2005). Nevertheless, the intestinal prokinetic effect of rimonabant may not be attributed unequivocally to the presence of ongoing local enteric endocannabinoid agonist activity because the existence of a possible constitutive activity of the CB1 receptor in the small intestine has not been investigated.
Although in vivo bioassays have played an important role in characterizing the pharmacology of cannabinoids on intestinal motility, it has been difficult to assess whether locally released endocannabinoid agonists may modulate intestinal motility. To simplify studies, we have used the myenteric plexus-longitudinal muscle (MPLM) preparation of the rat and guinea-pig isolated ileum to elucidate the mechanism by which rimonabant augments small intestinal motility. Consistent with the inhibitory effect of cannabinoid receptor agonists on small intestinal transit in vivo, these in vitro bioassays exploit the ability of these agonists to attenuate twitch contractions evoked by electrical field stimulation (EFS) of the longitudinal smooth muscle, in a rimonabant-sensitive CB1 receptor-dependent manner, by reducing the release of acetylcholine (ACh) from the nerve terminals innervating the smooth muscle layer. Moreover, rimonabant alone, by increasing the release of ACh, augments the EFS-evoked twitch contractions of these tissues in a concentration-dependent manner (Coutts and Pertwee, 1997; Mang et al., 2001; Makwana et al., 2006).
Recently, a number of ligands that are thought to behave as pure CB1 receptor antagonists have been developed (Pertwee, 2005). Among these is O-2050 [(6aR,10aR)-3-(1-methanesulphonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6-H-dibenzo[b,d]pyran], a sulphonamide analogue of Δ9-tetrahydrocannabinol, which was reported to be devoid of agonist or inverse agonist activity on the EFS-evoked contractions of the mouse isolated vas deferens (Martin et al., 2002). Therefore, the availability of O-2050 offered the opportunity to elucidate the mechanism by which rimonabant alone produced its inherent effects. We also evaluated the effects of three inhibitors of FAAH, AA-5HT (arachidonyl-5-hydroxytryptamine), PMSF (phenylmethylsulphonyl fluoride) and URB-597 (3′-carbamoyl-biphenyl-3-yl-cyclohexylcarbamate), and an inhibitor of the putative endocannabinoid uptake transporter, VDM-11 [(5Z,8Z,11Z,14Z)-N-(4-hydroxy-2-methylphenyl)-5,8,11,14-eicosatetraenamide], as an approach to unmask an inhibitory endocannabinoid tone by limiting the inactivation of endocannabinoid agonists. Additionally, the effect of AA-5HT, PMSF, URB-597 and VDM-11 on the potency of exogenously applied AEA and WIN 55212-2, inhibitors of the EFS-evoked twitch contractions of the tissues, was examined. The latter cannabinoid receptor agonist was used because it is not a substrate for FAAH as it lacks an amide linkage, and in addition to its common use as an agonist, it displays stereospecific activity at the CB1 receptor in these tissues (Coutts and Pertwee, 1997; Makwana et al., 2006).
A preliminary account of some of the present data has been communicated to the British Pharmacological Society (Makwana et al., 2007).
Methods
Tissue preparation
Male Wistar rats (400–550 g) and Dunkin-Hartley or Heston-2 guinea-pigs of either sex (500–800 g) were used. All animals were bred at the Biological Services Unit of the University of Hertfordshire, Hatfield, UK, from stock originating at Charles River Laboratories UK (Margate, Kent, UK) and Harlan UK (Bicester, Oxford, UK) respectively. The animals were housed in rooms with a controlled temperature (22 ± 1°C), humidity (55 ± 10%) and 12 h light/dark cycle. Food and water were available ad libitum. Rats were killed by carbon dioxide (CO2) asphyxiation, while guinea-pigs were killed by cervical dislocation followed by exsanguination. All animal care and killing was conducted in accordance with requirements of the Animals (Scientific Procedures) Act 1986 and the University of Hertfordshire ethics committee.
A 15 cm ileum segment was excised from the small intestine of each animal and immersed in Krebs-Henseleit solution of the composition (in mM): NaCl 118.3, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, d-glucose 11.1, CaCl2 2.5, gassed with 95% O2 and 5% CO2 at room temperature (21 ± 4°C). A maximum of four MPLM strips were dissected from the ileal segment by the method of Paton and Zar (1968) after the first 5 cm length closest to the ileo–caecal junction had been discarded. Briefly, a 2 cm length of the ileum was flushed of its contents with Krebs-Henseleit solution and slipped over a glass rod of 5 mm diameter. After the mesentery had been trimmed, the longitudinal muscle with the adhered myenteric plexus was separated from the underlying circular muscle layer by gentle stroking with a cotton bud soaked with Krebs-Henseleit solution, starting at the mesenteric border and working along and around the circumference of whole ileum. Cotton sutures were tied at both ends of the MPLM strip, and the tissue was suspended in a 5 or 10 mL organ bath containing Krebs-Henseleit solution gassed with 95% O2 and 5% CO2 at 33°C. The experiments were carried out at 33°C in order to dampen down the irregular spontaneous activity of the tissues, particularly of the rat MPLM, to enable the evoked responses to be quantified easily.
Changes in tension of the MPLM strips was recorded isometrically in millinewtons (mN) using Dynamometer UF1 force transducers (Pioden Controls, Newport, Isle of White, UK) connected to either a MacLab Chart Version 3.5 data-acquisition system (AD Instruments, Chalgrove, Oxford, UK) on an Apple Macintosh computer (Apple Macintosh, http://www.apple.com/uk) or MX216 or MultiTrace-4 chart recorders (Lectromed, Hertfordshire, UK)
EFS experiments
Each MPLM strip was placed between a pair of platinum prong (10 mm length and 5 mm apart) electrodes connected to either Grass S11 or S88 stimulators (Grass Instruments, Slough, Berkshire, UK) or a Multistim D330 stimulator (Digitimer, Welwyn Garden City, Hertfordshire, UK).
After the tissue had been placed in the organ bath for 10 min, the Krebs-Henseleit solution was renewed once and each tissue was stretched by 5.0 mN and allowed to equilibrate for a further 50 min until EFS was commenced. The guinea-pig and rat MPLM strips were subjected to EFS over the entire duration of the experiment with single pulses of 0.5 ms duration repeated at a frequency of 0.1 and 0.05 Hz respectively. Each pulse was delivered at a voltage that was 10% greater than the voltage required to elicit maximal contractions.
No drugs were added until the amplitude of EFS-evoked contractions had become consistent for at least 30 min. The time taken for the amplitude of the contractions to become uniform was about 3–4 h. It was also noted that the amplitude of the contractions remained stable for a further 4 h without the need to renew the bathing Krebs-Henseleit solution. Cumulative concentration–response curves to the cannabinoid receptor agonists were constructed with a 20 min dosing interval. Only one concentration–response curve was constructed per tissue because of difficulties in washing the drug from the tissue with drug-free Krebs-Henseleit solution.
On each day, each MPLM strip from an animal was subjected to a different drug treatment, and the drug treatments were randomized between the organ baths. When competition studies were performed, rimonabant was added 20 min prior to the addition of a cannabinoid agonist whereas AA-5HT, PMSF, URB-597 or VDM-11 was added 15 min prior to the addition of the cannabinoid agonist. To exclude the possibility that rimonabant antagonized the inhibitory effect of the cannabinoid receptor agonists by virtue of its ability to enhance the EFS-evoked contractions, that is, through functional antagonism, the inhibitory effect of WIN 55,212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate] was compared after the contractions had been augmented with physostigmine, an acetyl cholinesterase inhibitor. This experiment was only performed on the guinea-pig MPLM because both rimonabant and physostigmine produced large and clearly defined enhancement of contractions in this tissue. Physostigmine was administered 20 min before the addition of WIN 55,212-2 at a concentration that potentiated the EFS-evoked contractions to a level nearly equal to that produced by rimonabant. All experiments were performed in parallel with vehicle-treated and time-matched controls.
Exogenous ACh stimulation
If any drug was found to alter the amplitude of the EFS-evoked contractions, its ability to produce a similar effect on contractions induced by exogenously applied ACh was investigated. A single high concentration of the drug, which had a maximally effect on the amplitude of EFS-evoked contractions, was administered between two ACh (10−10–10−5 M) cumulative concentration–response curves constructed 30 min apart. Each tissue acted as its own control. All experiments were performed in parallel with relevant vehicle-treated and time-matched controls.
Data analysis
The inhibition of the EFS-evoked contractions by a cannabinoid receptor agonist was quantified in percentage terms by calculating the reduction in the amplitude of the contractions after the addition of the agonist compared with the amplitude immediately before the first addition of the competing ligand or its vehicle. The enhancement of the contractions induced by certain ligands was quantified in percentage terms by calculating the increase in amplitude after each addition of the ligand compared with the amplitude immediately before the first addition of the same ligand.
Individual agonist concentration–response curves in the absence and presence of a competing ligand were fitted by non-linear regression to the four-parameter Hill equation (Equation 1), using GraphPad PRISM 4.0 for Windows (GraphPad Software, La Jolla, CA, USA);
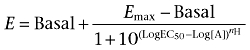 |
(Equation 1) |
where E denotes response, Log [A] the logarithm of the concentration of an agonist A, nH the midpoint slope of the curve, Log EC50 the logarithm of the midpoint location parameter along the concentration axis, and Emax and Basal the upper and lower asymptotes respectively.
The concentration–response data were plotted as the mean ± SEM, and n represents the number of preparations from different animals. Shifts of the agonist concentration–response curve by the presence of a competing ligand were quantified as the ratio of the agonist concentrations corresponding to the 50% equieffective response level. The concentration ratio for the rightward shift of the agonist curve was used to calculate the antagonist potency (pA2) using the Gaddum–Schild equation (Schild, 1949). The pA2 represents the negative logarithm of the concentration of the antagonist that produces a shift of the agonist concentration–response curve to the right by two linear units to give a concentration ratio of two.
The amplitude of the contractions to cumulative additions of ACh in the presence of a cannabinoid receptor ligand or its vehicle is expressed as a percentage of the maximal contraction to ACh (10−5 M), obtained from the initial ACh concentration–response curve constructed on each tissue. Where appropriate, shifts of an agonist concentration–response curve by the presence of a competing ligand were compared by a one-way anova followed by Dunnett's post hoc test for multiple comparisons or Student's unpaired t-test for comparisons of individual means. The probability P < 0.05 was taken to be statistically significant.
Drug and molecular target nomenclature conforms to British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2008).
Drugs and chemicals
ACh (acetylcholine chloride), atropine sulphate, physostigmine hemisulphate, hexamethonium bromide, PMSF were purchased from Sigma-Aldrich (Poole, Dorset, UK). AEA, AM 251, O-2050, TTX (tetrodotoxin), VDM-11 and WIN 55,212-2 were purchased from Tocris Biosciences (Avon, Bristol, UK). Rimonabant was a generous gift from Sanofi-Recherche, Montpellier, France. AA-5HT and URB-597 were purchased from Axxora Life Sciences, Bingham, Nottingham, UK. All other chemicals were purchased from Fisher Scientific, Loughborough, Leicestershire, UK. Atropine, physostigmine, hexamethonium, nicotine and TTX were dissolved in distilled water. All other drugs were prepared in 100% ethanol. The total volume of the solvents added to the organ baths did not exceed 1% of the bath volume. The presence of the solvents did not have a significant effect on the evoked responses of the tissues.
Results
The EFS responses of the rat and guinea-pig MPLM
Electrical field stimulation (EFS) of the MPLM from both species elicited a monophasic transient twitch contraction during each electrical pulse. The amplitudes of EFS-evoked contractions of the rat and guinea-pig MPLM before the addition of any drug were 4.02 ± 2.16 mN (n= 100) and 17.0 ± 7.45 mN (n= 100) respectively. The contractions of both MPLM tissues were abolished by treatment with either the voltage-gated Na+ channel blocker TTX (10−6 M, data not shown, n= 6) or the muscarinic ACh receptor antagonist atropine (10−6 M, data not shown, n= 6) but not with the nicotinic ACh receptor antagonist hexamethonium (10−4 M, data not shown, n= 6).
Effect of the AEA and WIN 55,212-2 in the absence and presence of rimonabant on the EFS-evoked contractions
Cumulative additions of AEA (Figure 1A and B) or WIN 55,212-2 (Figure 1C and D) resulted in a concentration-related reduction of the EFS-evoked contractions of both MPLM tissues with a similar Emax. Of the two agonists, WIN 55,212-2 was significantly (P < 0.05, unpaired t-test) more potent than AEA on both tissues. Non-linear regression analysis yielded a pEC50 of 7.67 ± 0.05 (n= 6) and 8.27 ± 0.06 (n= 6) for AEA and WIN 55,212-2 on the rat MPLM, respectively, and 6.81 ± 0.02 (n= 6) and 8.52 ± 0.03 (n= 6) for AEA and WIN 55,212-2 on the guinea-pig MPLM respectively. Unlike WIN 55,212-2, which showed similar potency on both MPLM tissues, AEA was significantly (P < 0.05, unpaired t-test) more potent, one logarithmic unit, on the rat MPLM. The inhibition of the EFS-evoked contractions of both tissues by each agonist was slow in onset with the maximal effect being achieved within 15 min of administration.
Figure 1.
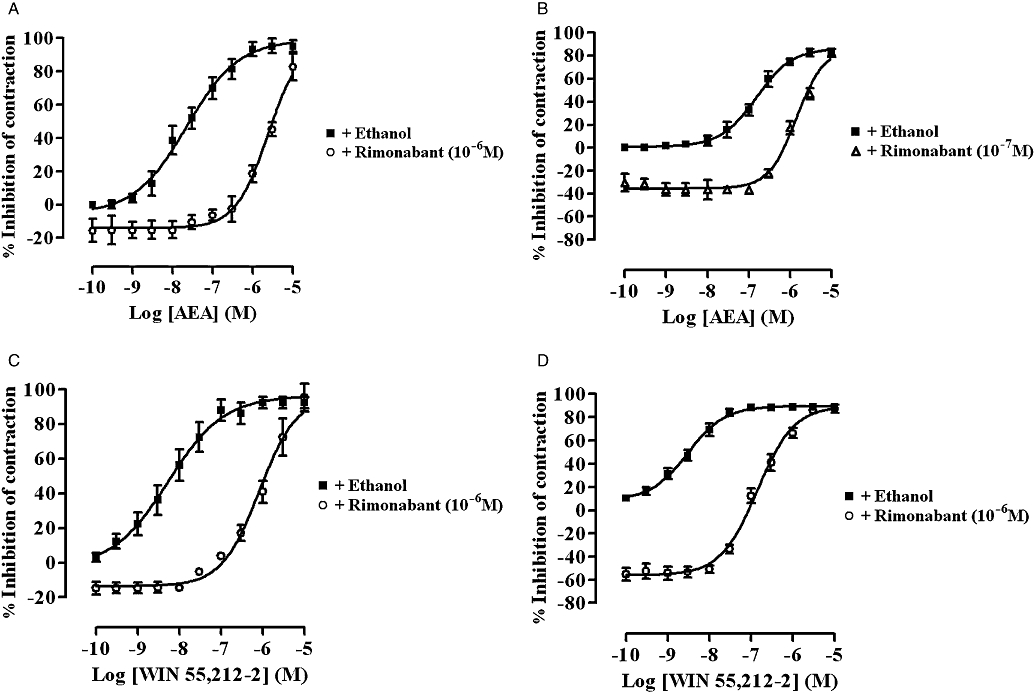
Concentration–response curves for the inhibition of the electrical field stimulation-evoked contractions of the rat (A and C) and guinea-pig (B and D) myenteric plexus-longitudinal muscle (MPLM) by anandamide, arachidonylethanolamide (AEA) (A and B) and WIN 55,212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate] (C and D) constructed in the presence of ethanol or rimonabant (N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide). The rat and guinea-pig MPLM were subjected to single electrical pulses of 0.5 ms duration, 110% supramaximal voltage at a frequency of 0.05 and 0.1 Hz respectively. Each curve was fitted by non-linear regression analysis. Each symbol represents the mean value of inhibition of the contractions expressed as a percentage reduction of the amplitude of the contractions measured immediately before the addition of any drug to the organ bath. Vertical lines indicate SEM, n= 6 for each curve. Rimonabant or ethanol was added 20 min before the first addition of an agonist.
A 20 min pretreatment of both tissues with rimonabant (10−7 or 10−6 M) resulted in a significant (P < 0.05, anova and Dunnett's test) dextral shift of the concentration–response curve of both agonists with no reduction in their Emax values (Figure 1). Figure 2 shows representative traces of the WIN 55,212-2-induced inhibition of the EFS-evoked contractions of the guinea-pig MPLM in the absence and presence of rimonabant. The pA2 values for rimonabant against AEA and WIN 55,212-2 were 7.68 and 8.21, respectively, on the rat MPLM and 7.90 and 7.97, respectively, on the guinea-pig MPLM.
Figure 2.
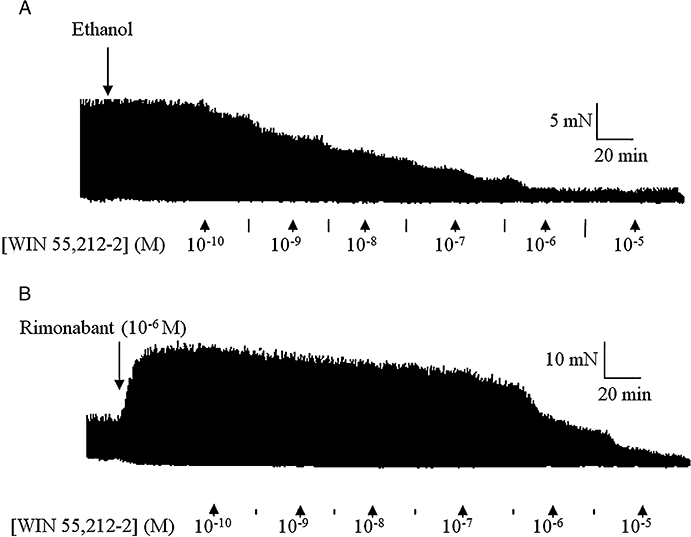
Representative traces of the inhibition of the electrical field stimulation (0.1 Hz frequency, 0.5 ms duration and 110% supramaximal voltage)-evoked contractions of the guinea-pig myenteric plexus-longitudinal muscle by WIN 55,212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate] in the presence of (A) ethanol and (B) rimonabant (N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide) (10−6 M). WIN 55,212-2 was added in half-logarithmic unit increments. Lines represent intermediate concentrations.
Effect of WIN 55,212-2 on the EFS-evoked contraction of the guinea-pig MPLM in the absence and presence of physostigmine
Physostigmine alone caused a concentration-related enhancement of the contractions of the guinea-pig MPLM up to 10−7 M; the enhancement induced by 10−7 M was 66.3 ± 12.3% (data not shown, n= 6). Further increases in concentration up to 3 × 10−6 M yielded a complex response, which consisted of a simultaneous progressive contraction of the muscle and an inhibition of the EFS-evoked contractions (recording not shown, n= 6). These responses were difficult to quantify. Using the enhancement to 10−7 M as the Emax, the pEC50 of physostigmine was 7.69 ± 0.01 (data not shown, n= 6). Treatment of the MPLM with 3 × 10−8 M physostigmine potentiated the EFS-evoked contractions by 48.7 ± 11.0% (n= 6), a level comparable to that produced by rimonabant (10−6 M). The presence of physostigmine at this concentration caused a small but non-significant (P > 0.05, anova and Dunnett's test) dextral shift of the WIN 55,212-2 concentration–response curve with no reduction in the Emax values (Figure 3).
Figure 3.
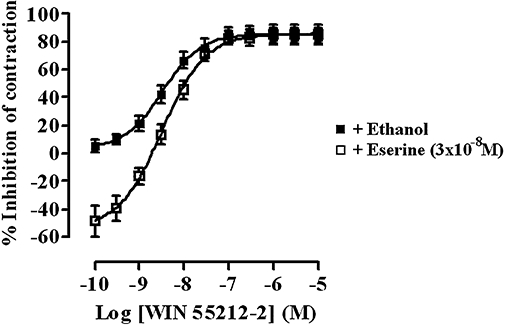
Concentration–response curves for the inhibition of the electrical field stimulation-evoked contractions of the guinea-pig myenteric plexus-longitudinal muscle (MPLM) by WIN 55,212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate] constructed in the absence and presence of physostigmine (eserine). The guinea-pig MPLM was subjected to single electrical pulses of 0.5 ms duration, 110% supramaximal voltage at a frequency of 0.05 and 0.1 Hz respectively. Each curve was fitted by non-linear regression analysis. Each symbol represents the mean value of inhibition of the contractions expressed as a percentage reduction of the amplitude of the contractions measured immediately before the addition of any drug to the organ bath. Vertical lines indicate SEM, n= 6 for each curve. Physostigmine was added 20 min before the first addition of WIN 55,212-2.
Effect of rimonabant, AM 251 or O-2050 alone on the EFS-evoked contractions
Figure 4 shows that rimonabant, AM 251 and O-2050 all caused a concentration-dependent enhancement of the EFS-evoked contractions of both the rat and guinea-pig MPLM. The EFS-evoked contractions in the presence of these cannabinoids were abolished by application of either TTX (10−6 M) or atropine (10−6 M). Figure 5 illustrates representative traces of the enhancement of the EFS-evoked contractions of both tissues by rimonabant and the inhibitory effect of atropine. All three cannabinoids produced different degrees of maximal enhancement of the EFS-evoked contractions in both tissues. The Emax values of all three antagonists were over 20 times (P < 0.05, unpaired t-test) greater in the guinea-pig MPLM than in the rat MPLM (Table 1). Nevertheless, the rank order of the Emax values were similar in both tissues that is, AM 251 > rimonabant > O-2050.
Figure 4.
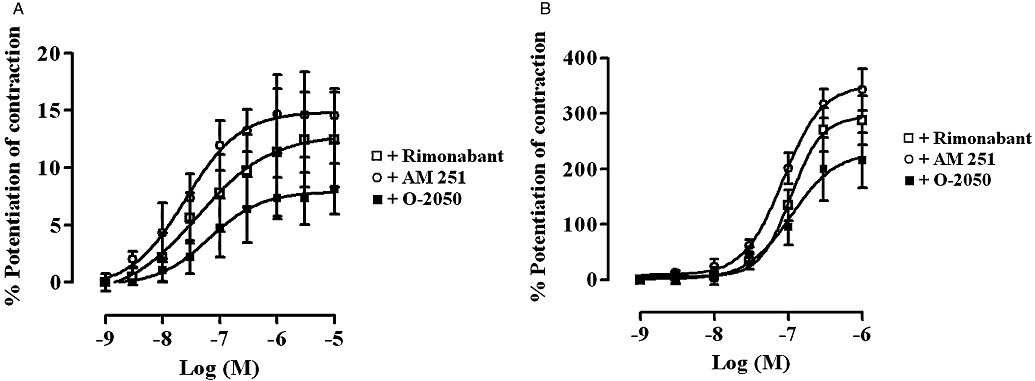
Concentration–response curves for the enhancement of the electrical field stimulation (EFS)-evoked contractions of the (A) rat and (B) guinea-pig myenteric plexus-longitudinal muscle (MPLM) by rimonabant (N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide), AM 251 (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide) and O-2050 [(6aR,10aR)-3-(1-methanesulphonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6-H-dibenzo[b,d]pyran]. The rat and guinea-pig MPLM were subjected to single electrical pulses of 0.5 ms duration, 110% supramaximal voltage at a frequency of 0.05 and 0.1 Hz respectively. Each curve was fitted by non-linear regression analysis. Each symbol represents the mean value for the increase in the EFS-evoked contractions expressed as a percentage of the amplitude of the contractions measured immediately before the addition of the same cannabinoid to the organ bath (n= 6). Vertical lines indicate the SEM.
Figure 5.
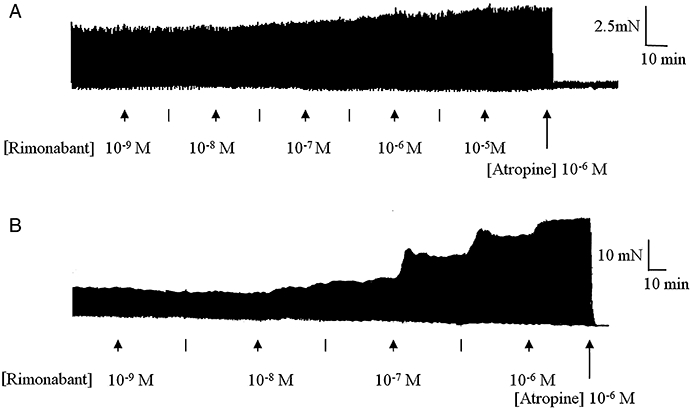
Representative traces of the electrical field stimulation (EFS)-evoked contractions of the (A) rat and (B) guinea-pig myenteric plexus-longitudinal muscle (MPLM) in the absence and presence of rimonabant (N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide), added in half-logarithmic unit increments. Lines represent intermediate concentrations. The rat and guinea-pig MPLM were subjected to single electrical pulses of 0.5 ms duration, 110% supramaximal voltage at a frequency of 0.05 and 0.1 Hz respectively. Note the inhibition of the rimonabant-enhanced EFS-evoked contractions by atropine (10−6 M).
Table 1.
A comparison of the potency (pEC50) and tissue maximal response (Emax) and the ratio of the Emax values of the enhancement of the EFS-evoked contractions of the rat and guinea-pig ileum MPLM by the CB1 receptor antagonist/inverse agonists rimonabant, AM 251 and O-2050
| Cannabinoid |
Rat MPLM |
Guinea-pig MPLM |
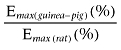 |
||
|---|---|---|---|---|---|
| pEC50 | Emax (%) | pEC50 | Emax (%) | ||
| Rimonabant | 7.46 ± 0.04 | 12.5 ± 4.1* | 6.97 ± 0.01 | 287.8 ± 43.9* | 23.1 |
| AM 251 | 7.60 ± 0.02 | 14.6 ± 2.3* | 7.07 ± 0.01 | 343.0 ± 37.5* | 23.5 |
| O-2050 | 7.19 ± 0.01 | 8.2 ± 2.2* | 6.96 ± 0.03 | 215.5 ± 50.2* | 26.4 |
spEC50 and Emax values were derived by non-linear regression analysis for the rat and guinea-pig MPLM strips stimulated electrically with single pulses of 0.5 ms duration, 110% supramaximal voltage at 0.05 and 0.1 Hz frequency respectively.
The asterisk represents a significant (P < 0.05, unpaired t-test) enhancement of the EFS-evoked contractions over base line contraction level in the absence of the cannabinoids. Where appropriate, values represent the mean ± SEM.
AM 251, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; EFS, electrical field stimulation; MPLM, myenteric plexus-longitudinal muscle; O-2050, (6aR,10aR)-3-(1-methanesulphonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6-H-dibenzo[b,d]pyran; rimonabant, N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide.
Because each set of curves in Figure 4 had different Emax, and the slopes were not parallel, a direct comparison between the pEC50 values of the three cannabinoids was not possible. However, a comparison between the concentrations corresponding to the 5% and 150% level of enhancement of the contractions of the rat and guinea-pig MPLM curves, respectively, indicated that the rank order of potency of the three antagonists on both tissues was similar that is, AM 251 > rimonabant > O-2050.
The time taken for each cannabinoid to enhance the EFS-evoked contractions was slow in both tissues, with the maximal enhancement at each concentration being achieved within 20 min of administration. Half-maximal potentiation of contraction by AM 251, O-2050 and rimonabant at 10−6 M was achieved after 4, 3.5 and 5.5 min, respectively, in the rat MPLM and 4, 4 and 5 min, respectively, in the guinea-pig MPLM.
Effect of AA-5HT, PMSF and URB-597 alone and on the inhibitory potency of AEA and WIN 55,212-2 on EFS-evoked contractions
Cumulative addition of AA-5HT (10−9–10−5 M), PMSF (10−6–10−4 M) or URB-597 (10−9–10−5 M) every 15 min had no effect on the basal tension or the EFS-evoked contractions of either the rat or guinea-pig MPLM (data not shown, n= 6 each). However, pre-incubation of both tissues for 15 min with AA-5HT (10−6 M), PMSF (10−4 M) or URB-597 (3 × 10−8 M) resulted in a significant (P < 0.05, anova and Dunnett's test) parallel leftward shift of the AEA concentration–response curve in both the rat and guinea-pig ileum MPLM with no change in the Emax or slope (Figure 6). The presence of these concentrations of AA-5HT, PMSF and URB-597 increased the potency of AEA by 2.0, 22.1 and 20.2 times, respectively, on the rat ileum MPLM and by 3.2, 32.4 and 48.8 times, respectively, on the guinea-pig ileum MPLM. By contrast, pretreatment of both MPLM tissues for 15 min with AA-5HT (10−6 M), PMSF (10−4 M) or URB-597 (3 × 10−8 M) had no effect on the location and Emax of the WIN 55,212-2 concentration–response curve (Figure 6).
Figure 6.
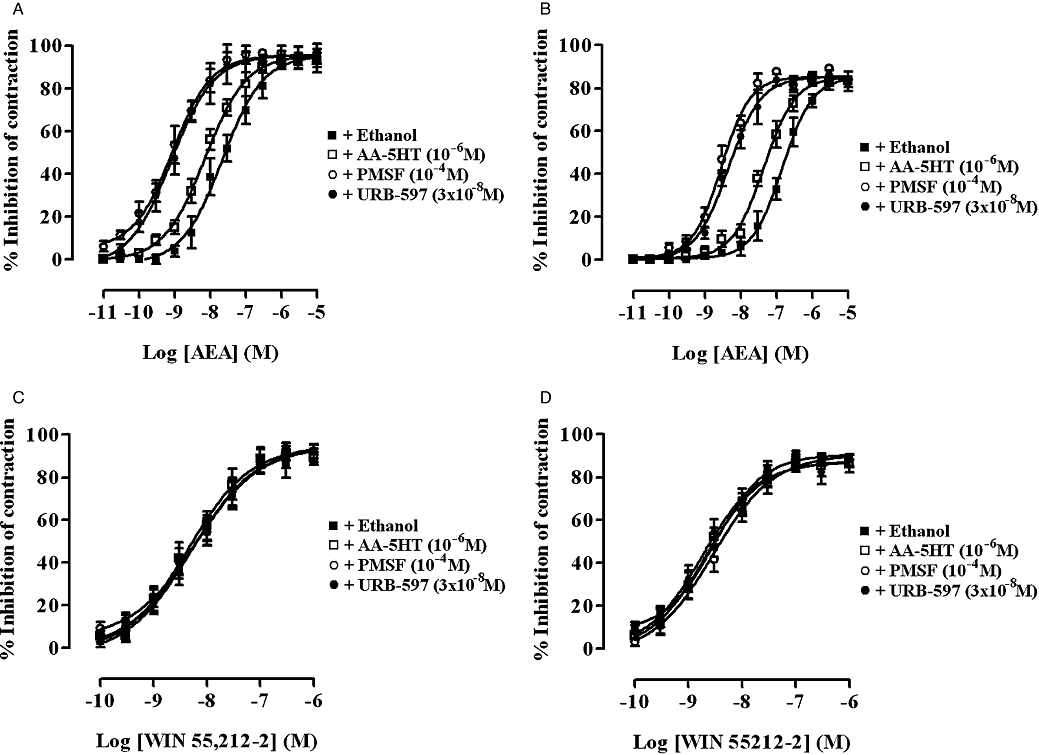
Concentration–response curves for the inhibition of the electrical field stimulation-evoked contractions of the rat (A and C) and guinea-pig (B and D) myenteric plexus-longitudinal muscle (MPLM) by anandamide, arachidonylethanolamide (AEA) (A and B) and WIN 55,212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate] (C and D), constructed in the presence of ethanol, AA-5HT (arachidonyl-5-hydroxytryptamine), PMSF (phenylmethylsulphonyl fluoride) or URB-597 (3′-carbamoyl-biphenyl-3-yl-cyclohexylcarbamate). The rat and guinea-pig MPLM were subjected to single electrical pulses of 0.5 ms duration, 110% supramaximal voltage at a frequency of 0.05 and 0.1 Hz respectively. Each curve was fitted by non-linear regression analysis. Each symbol represents the mean value of inhibition of the contractions expressed as a percentage reduction of the amplitude of the contraction measured immediately before the addition of any drug to the organ bath. Vertical lines indicate SEM, n= 6 for each curve. AA-5HT, PMSF or URB-597 or ethanol was added 15 min before the first addition of AEA and WIN 55,212-2.
Effect of VDM-11 alone and on the inhibitory potency of AEA on the EFS-evoked contractions
Cumulative addition of VDM-11 (10−9–10−5 M) every 15 min had no effect on basal tension or EFS-induced contractions of rat and guinea-pig ileum MPLM (data not shown, n= 6). Concentrations of VDM-11 higher than 10−5 M were not investigated. Pre-incubation of the tissues for 15 min with VDM-11 (10−5 M) did not cause a shift in the AEA concentration–response curve on either tissues (Figure 7).
Figure 7.
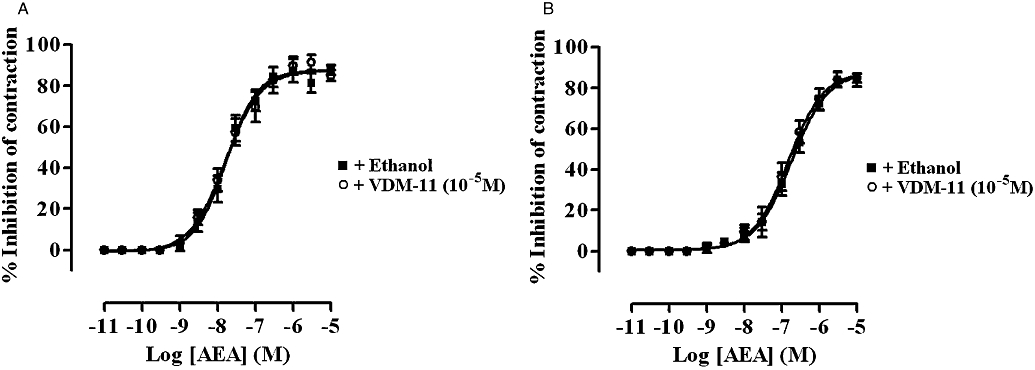
Concentration–response curves for the inhibition of the electrical field stimulation-evoked contractions of the rat (A) and guinea-pig (B) ileum myenteric plexus-longitudinal muscle (MPLM) by anandamide, arachidonylethanolamide (AEA) in the presence of ethanol or VDM-11 [(5Z,8Z,11Z,14Z)-N-(4-hydroxy-2-methylphenyl)-5,8,11,14-eicosatetraenamide]. The rat and guinea-pig MPLM were subjected to single electrical pulses of 0.5 ms duration, 110% supramaximal voltage at a frequency of 0.05 and 0.1 Hz respectively. Each curve was fitted by non-linear regression analysis. Each symbol represents the mean value of inhibition of contractions expressed as a percentage of the amplitude of the contraction measured immediately before the addition of any drug to the organ bath (n= 6). Vertical lines indicate the SEM. VDM-11 or ethanol was added 15 min before the first addition of AEA.
Effect of the cannabinoids on the basal tone and on contractions evoked by exogenously applied ACh
The basal tension and amplitude of the contractions of both the rat and guinea-pig MPLM elicited by cumulative addition of ACh (10−10–10−5 M) was not altered in the presence of AEA (10−5 M), WIN 55,212-2 (10−5 M), rimonabant (10−5 M), AM 251 (10−5 M) or O-2050 (10−5 M). Figure 8 shows the lack of effect of rimonabant (10−5 M), AM 251 (10−5 M) or O-2050 (10−5 M) on ACh-evoked contractions of the rat and guinea-pig MPLM.
Figure 8.
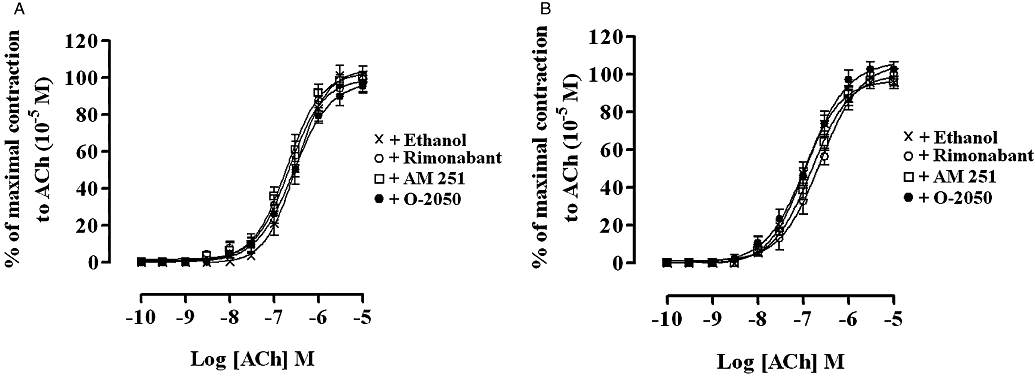
Concentration–response curves for the ACh-evoked contractions of the rat (A) and guinea-pig (B) ileum myenteric plexus-longitudinal muscle in the presence of ethanol, AM 251 (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide) (10−5 M), rimonabant (N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide) (10−5 M) or O-2050 [(6aR,10aR)-3-(1-methanesulphonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6-H-dibenzo[b,d]pyran] (10−5 M). Each curve was fitted by non-linear regression analysis. The amplitude of the contractions to ACh in the presence of ethanol or a cannabinoid was expressed as a percentage of the maximal contraction to ACh obtained at 10−5 M from an initial ACh concentration–response curve constructed on each tissue. Ethanol or the cannabinoid was added 30 min before constructing the second ACh concentration–response curve; n= 6 for all curves. All values represent mean ± SEM.
Discussion
Consistent with data from previous studies on these tissues (Coutts and Pertwee, 1997; Makwana et al., 2006), rimonabant antagonized the inhibitory effect of both AEA and WIN 55,212-2, and by itself potentiated the amplitude of the EFS-evoked contractions. These effects were shown to be mediated by the CB1 receptor; the pEC50 values of both agonists and their rank of order are in agreement with those reported previously and the surmountable antagonism by rimonabant yielded pA2 values, which are within the range reported at this receptor. There is also evidence that the CB1 receptors, through which both agonists probably reduce ACh release and so inhibit the EFS-evoked contractions (Coutts and Pertwee, 1997; Mang et al., 2001), are located presynaptically on postganglionic nerves of the myenteric plexus (Lynn and Herkenham, 1994; Coutts et al., 2002). The EFS-evoked contractions were inhibited after blockade of axonal conductance or the muscarinic ACh receptor, but not the nicotinic ACh receptor, and neither agonist had an effect on contractions to exogenous ACh.
The sensitivity of the EFS-evoked contractions augmented by rimonabant, AM 251 and O-2050 to TTX and atropine, and the lack of effect of these cannabinoids on contractions to exogenous ACh suggests that their effect was due to a presynaptic CB1 receptor-mediated increase in ACh release and not induced by sensitization of the muscle mACh receptor or an anti-ACh-esterase action. Although rimonabant up to 10−5 M appeared to act selectively through the CB1 receptor in the MPLM, it can cause non-CB1 receptor-mediated actions in the micromolar concentration range (White and Hiley, 1998). Although unlikely, it is possible that rimonabant also increases the amount of ACh released, and hence contraction, by blocking inhibitory autoreceptors of other mediators modulating ACh release.
Rimonabant produced an equal rightward shift of the WIN 55,212-2 curve but an unequal downward shift of the basal asymptote on both MPLM tissues. This implies that the antagonist activity of rimonabant is independent of its ability to enhance the EFS-evoked contractions, and therefore, the pA2 values were not overestimated by the latter property. This reasoning is supported by the finding that physostigmine increased the EFS-evoked contractions, albeit through a different mechanism involving an inhibition of ACh hydrolysis, but did not reduce the potency of WIN 55,212-2.
That all three cannabinoid antagonists were more potent but less efficacious on the rat MPLM could be ascribed to tissue-dependent factors, such as a low stimulus–response coupling capacity, a low tonic release of endocannabinoids (assuming the presence of an endocannabinoid tone) or a lower constitutive activity of the CB1 receptor (assuming inverse agonism).
The concentration–response curves for all three antagonists had different Emax values with respect to each other presumably because they behaved as inverse agonists in these tissues as the CB1 receptor was constitutively active. This is because the Emax of all three concentration–response curves should have been equal if the potentiation of the EFS-evoked contractions by each cannabinoid were due to a displacement of endocannabinoid agonists from the CB1 receptor, assuming that the amount of displacement was equal.
As the concentrations of all three antagonists were about three logarithmic units higher than their respective pKB values at the CB1 receptor, it is unlikely that tonically released endocannabinoid agonists bound to the CB1 receptor already occupied by the antagonist.
It is noteworthy that O-2050 also potentiated the EFS-evoked contractions of both MPLM tissues at concentrations previously shown to be devoid of activity on the EFS-evoked contractions of the mouse vas deferens (Martin et al., 2002). Although the reason for this discrepancy is not clear, this finding could be attributed to differences in the tissue, animal species, neurotransmitter released, stimulus–response coupling mechanism or a lower basal constitutive activity of the CB1 receptor in the MPLM. Previous studies have reported differences in the pharmacology of a number of cannabinoids in different bioassays. For example, AM 630, an analogue of WIN 55,212-2, behaved as an agonist in the guinea-pig MPLM (Pertwee et al., 1996), a competitive antagonist in the mouse vas deferens (Pertwee et al., 1995), an inverse agonist at the human CB1 receptor in CHO cells (Landsman et al., 1997), but was without inherent activity in the mouse urinary bladder (Pertwee and Fernando, 1996).
The termination of endocannabinoid signalling is regarded as a two-step process, involving the translocation of endocannabinoids released from the extracellular space into cells via a putative uptake transporter, followed by enzymatic hydrolysis, predominantly by FAAH. High levels of a number of endocannabinoids including AEA and 2-AG were detected in both the rat and guinea-pig ileum (Katayama et al., 1997; Valenti et al., 2005; Guagnini et al., 2006). Additionally, their protection from inactivation by cellular uptake or degradation by FAAH inhibited intestinal transit in mice (Pinto et al., 2002; Capasso et al., 2005). Therefore, an attempt was made to unmask the presence of a functional inhibitory endocannabinoid agonist tone in the MPLM using VDM-11, AA-5HT, PMSF and URB-597. VDM-11 up to a concentration almost equal to its pEC50, determined in the RBL-2H3 basophilic and C6 glioma cells (De petrocellis et al., 2000), did not inhibit the EFS-evoked contractions. Similarly, application of all three FAAH inhibitors by themselves, up to concentrations that completely inactivate FAAH and lack affinity for the CB1 receptor in vitro (Deutsch et al., 1997; Bisogno et al., 1998; Kathuria et al., 2003), were also without effect on the EFS-evoked contractions. While these results confirmed that VDM-11, AA-5HT, PMSF and URB-597 did not possess cannabinoid activity in these tissues, they suggested that pharmacologically active endocannabinoids or substrates of the putative uptake transporter and FAAH were not released in response to EFS. These findings support the notion that the CB1 receptor is constitutively active and that rimonabant, AM 251 and O-2050 are inverse agonists.
Because AA-5HT, PMSF and URB-597, but not VDM-11, potentiated the action of exogenous AEA in both MPLM tissues, it is possible that FAAH, unlike the putative uptake transporter, was present and functional in these tissues. The similar concentration ratios for the increase in potency of AEA by a given FAAH inhibitor in both MPLM preparations implies the presence of the same isoform of FAAH. The lack of an effect of all three FAAH inhibitors on the potency of WIN 55,212-2 confirms that this agonist is not a substrate of FAAH. This supports the deduction that FAAH was present in both tissues and the increase in potency of AEA was specific. These experiments also indicate that the FAAH inhibitors did not augment the sensitivity of the CB1 receptor to AEA.
Endocannabinoids, including AEA and 2-AG, are known not to be stored in vesicles in nerves following synthesis, but are synthesized and released on-demand in response to nerve stimuli (Di marzo et al., 2004). In vivo, PMSF (Mcvey et al., 2003) and AA-5HT (Capasso et al., 2005) delay small intestinal transit in rats and mice respectively. This effect is thought to be due to a potentiation of the inhibition induced by tonically released AEA and 2-AG because levels of these endocannabinoids were elevated in the small intestine. High levels of AEA and 2-AG were also measured in the guinea-pig MPLM (Guagnini et al., 2006). Hence, the levels of these endocannabinoids would be anticipated to be high in the MPLM during EFS. However, the lack of an inhibitory effect of the FAAH inhibitors on contractions to EFS suggests that the endogenous levels may not be sufficient to maintain a sustained activation of the CB1 receptor and may explain the antagonist-mediated potentiation of the contractions.
The effect of URB-597 on intestinal transit of rodents in vivo has not been studied, but this FAAH inhibitor, at concentrations that abolished FAAH activity, selectively elevated AEA levels in the brain without affecting the levels in the rat duodenum (Fegley et al., 2005). This suggests that the hydrolysis of AEA in the rat may occur by at least two enzymes, and that other endocannabinoids may modulate small intestinal transit in this species. It is possible that different enzymes might exist in different places, with FAAH not being strategically located to modulate endocannabinoid function at the cholinergic junction. Several enzymes including monoacylglycerol lipase and N-palmitoylethanolamine hydrolase have been implicated in inactivating endocannabinoids in the small intestine (Ueda et al., 2000; van der Stelt and Di Marzo, 2004). Because FAAH is capable of hydrolysing a broad spectrum of established and putative endocannabinoids, including 2-AG and palmitolyethanolamide, it is unlikely that these cannabinoids tonically activate the CB1 receptor in the MPLM.
Although the data available indicate that rimonabant augments intestinal motility by acting as an inverse agonist, it is conceivable that by using an isolated preparation, an endocannabinoid agonist tone was not detected because endocannabinoids may behave as hormones in vivo. For instance, endothelial cells of the mesenteric vasculature can synthesize AEA (Randall et al., 1996) and these vessels supply blood to the small intestine. Thus, endocannabinoid agonists synthesized in the mesenteric vasculature may tonically activate the CB1 receptor to inhibit enteric transmission via an endocrine hormonal effect. While these speculations may be valid in vivo, they do not appear to apply to the MPLM in vitro.
If it is assumed that rimonabant, AM 251 and O-2050 are inverse agonists in the MPLM, as the CB1 receptor was constitutively active, their ability to enhance EFS-evoked contractions may be explained using the theoretical ‘two state’ receptor model (Leff, 1995; Kenakin and Onaran, 2002). According to this model, the CB1 receptor exists in at least two interchangeable states, an active state, in which the receptor is coupled to the effector pathways to suppress ACh release, and an inactive sate, in which the receptor is not coupled to the effector pathways and thereby would increase ACh release. In the absence of any ligand, both conformations would exist at equilibrium; therefore, the CB1 receptor would be inhibiting ACh release. However, as inverse agonists, rimonabant AM 251 and O-2050 having preferential affinity for the inactive state would bind to this state and shift the equilibrium to reduce the number of receptors in the active conformation, thereby reversing the suppression of the ACh release.
Taken together, our results suggest that the ability of rimonabant, AM 251 and O-2050 to potentiate the EFS-evoked contraction of the rat and guinea-pig MPLM is through inverse agonism at the CB1 receptor and not due to antagonism of tonically released endocannabinoid agonists.
Glossary
Abbreviations:
- AA-5HT
arachidonyl-5-hydroxytryptamine
- AEA
anandamide, arachidonylethanolamide
- AM 251
N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- MPLM
myenteric plexus-longitudinal muscle
- O-2050
(6aR,10aR)-3-(1-methanesulphonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6-H-dibenzo[b,d]pyran
- PMSF
phenylmethylsulphonyl fluoride
- rimonabant
N-(piperidino)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-3-pyrazole-carboxyamide
- URB-597
3′-carbamoyl-biphenyl-3-yl-cyclohexylcarbamate
- VDM-11
(5Z,8Z,11Z,14Z)-N-(4-hydroxy-2-methylphenyl)-5,8,11,14-eicosatetraenamide
- WIN 55,212-2
(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)-pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate
Conflict of interest
The authors have no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels, 3rd edition. Br J Pharmacol. 2008;153:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Melck D, De Petrocellis L, Bobrov MYu, Gretskaya NM, Bezuglov VV, et al. Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Commun. 1998;248:515–522. doi: 10.1006/bbrc.1998.8874. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, et al. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J Biol Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- Capasso R, Matias I, Lutz B, Borrelli F, Capasso F, Marsicano G, et al. Fatty acid amide hydrolase controls mouse intestinal motility in vivo. Gastroenterology. 2005;129:941–951. doi: 10.1053/j.gastro.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Colombo G, Agabio R, Lobina C, Reali R, Gessa GL. Cannabinoid modulation of intestinal propulsion in mice. Eur J Pharmacol. 1998;344:67–69. doi: 10.1016/s0014-2999(97)01555-0. [DOI] [PubMed] [Google Scholar]
- Coutts AA, Pertwee RG. Inhibition by cannabinoid receptor agonists of acetylcholine release from the guinea-pig myenteric plexus. Br J Pharmacol. 1997;121:1557–1566. doi: 10.1038/sj.bjp.0701301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts AA, Irving AJ, Mackie K, Pertwee RG, Anavi-Goffer S. Localisation of cannabinoid CB(1) receptor immunoreactivity in the guinea pig and rat myenteric plexus. J Comp Neurol. 2002;448:410–422. doi: 10.1002/cne.10270. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000;483:52–56. doi: 10.1016/s0014-5793(00)02082-2. [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Lin S, Hill WA, Morse KL, Salehani D, Arreaza G, et al. Fatty acid sulfonyl fluorides inhibit anandamide metabolism and bind to the cannabinoid receptor. Biochem Biophys Res Commun. 1997;231:217–221. doi: 10.1006/bbrc.1997.6072. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, et al. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313:352–358. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Briley EM, Glass M, Mackie KP, Fahey KJ, et al. LY320135, a novel cannabinoid CB1 receptor antagonist, unmasks coupling of the CB1 receptor to stimulation of cAMP accumulation. J Pharmacol Exp Ther. 1998;284:291–297. [PubMed] [Google Scholar]
- Gatley SJ, Lan R, Pyatt B, Gifford AN, Volkow ND, Makriyannis A. Binding of the non-classical cannabinoid CP 55,940 and the diarylpyrazole AM251 to rodent brain cannabinoid receptors. Life Sci. 1997;61:PL191–PL197. doi: 10.1016/s0024-3205(97)00690-5. [DOI] [PubMed] [Google Scholar]
- Guagnini F, Cogliati P, Mukenge S, Ferla G, Croci T. Tolerance to cannabinoid response on the myenteric plexus of guinea-pig ileum and human small intestinal strips. Br J Pharmacol. 2006;148:1165–1173. doi: 10.1038/sj.bjp.0706813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, et al. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Mascolo N, Borrelli F, Capasso F. Defaecation, intestinal fluid accumulation and motility in rodents: implications of cannabinoid CB1 receptors. Naunyn Schmiedebergs Arch Pharmacol. 1999a;359:65–67. doi: 10.1007/pl00005325. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Mascolo N, Pinto L, Capasso R, Capasso F. The role of cannabinoid receptors in intestinal motility, defaecation and diarrhoea in rats. Eur J Pharmacol. 1999b;384:37–42. doi: 10.1016/s0014-2999(99)00673-1. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Fezza F, Capasso R, Bisogno T, Pinto L, Iuvone T, et al. Cannabinoid CB1-receptor mediated regulation of gastrointestinal motility in mice in a model of intestinal inflammation. Br J Pharmacol. 2001;134:563–570. doi: 10.1038/sj.bjp.0704293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Capasso F, Costagliola A, Bisogno T, Marsicano G, Ligresti A, et al. An endogenous cannabinoid tone attenuates cholera toxin-induced fluid accumulation in mice. Gastroenterology. 2003;125:765–774. doi: 10.1016/s0016-5085(03)00892-8. [DOI] [PubMed] [Google Scholar]
- Katayama K, Ueda N, Kurahashi Y, Suzuki H, Yamamoto S, Kato I. Distribution of anandamide amidohydrolase in rat tissues with special reference to small intestine. Biochim Biophys Acta. 1997;1347:212–218. doi: 10.1016/s0005-2760(97)00078-7. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Kenakin TP, Onaran O. The ligand paradox between affinity and efficacy: can you be there and not make a difference. Trends Pharmacol Sci. 2002;23:275–280. doi: 10.1016/s0165-6147(02)02036-9. [DOI] [PubMed] [Google Scholar]
- Landsman RS, Burkey TH, Consroe P, Roeske WR, Yamamura HI. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur J Pharmacol. 1997;334:R1–R2. doi: 10.1016/s0014-2999(97)01160-6. [DOI] [PubMed] [Google Scholar]
- Leff P. The two-state model of receptor activation. Trends Pharmacol Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- Lynn AB, Herkenham M. Localization of cannabinoid receptors and nonsaturable high-density cannabinoid binding sites in peripheral tissues of the rat: implications for receptor-mediated immune modulation by cannabinoids. J Pharmacol Exp Ther. 1994;268:1612–1623. [PubMed] [Google Scholar]
- MacLennan SJ, Reynen PH, Kwan J, Bonhaus DW. Evidence for inverse agonism of SR141716A at human recombinant cannabinoid CB1 and CB2 receptors. Br J Pharmacol. 1998;124:619–622. doi: 10.1038/sj.bjp.0701915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey D, Schmid P, Schmid H, Vigna S. Endocannabinoids induce ileitis in rats via the capsaicin receptor (VR1) J Pharmacol Exp Ther. 2003;304:713–722. doi: 10.1124/jpet.102.043893. [DOI] [PubMed] [Google Scholar]
- Makwana R, Molleman A, Parsons ME. The receptors mediating the inhibitory effects of cannabinoids on the electrically stimulated rat isolated myenteric plexus longitudinal muscle preparation are dependent on the frequency of stimulation. Symposium on the cannabinoids. 2006. Burlington, Vermont, International Cannabinoid Research Society: page 56.
- Makwana R, Molleman A, Parsons ME. Is there an endogenous cannabinoid tone in the guinea-pig and rat isolated ileum myenteric plexus-longitudinal muscle? Proceedings of the British Pharmacological Society. 2007. WWW document].URL http://www.pa2online.org/abstracts/Vol4Issue2abst094P.pdf.
- Mang CF, Erbelding D, Kilbinger H. Differential effects of anandamide on acetylcholine release in the guinea-pig ileum mediated via vanilloid and non-CB1 cannabinoid receptors. Br J Pharmacol. 2001;134:161–167. doi: 10.1038/sj.bjp.0704220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Stevenson LA, Pertwee RG, Breivogel CS, Williams W, Mahadevan A, et al. Agonists and silent antagonists in a series of cannabinoid sulfonamides. Symposium on the cannabinoids. 2002. Burlington, Vermont, International Cannabinoid Research Society, Page 2.
- Mascolo N, Izzo AA, Ligresti A, Costagliola A, Pinto L, Cascio MG, et al. The endocannabinoid system and the molecular basis of paralytic ileus in mice. FASEB J. 2002;16:1973–1975. doi: 10.1096/fj.02-0338fje. [DOI] [PubMed] [Google Scholar]
- Paton WDM, Zar AM. The origin of acetylcholine released from guinea-pig intestine and longitudinal muscle strips. J Physiol. 1968;194:13–33. doi: 10.1113/jphysiol.1968.sp008392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R, Griffin G, Fernando S, Li X, Hill A, Makriyannis A. AM 630, a competitive cannabinoid receptor antagonist. Life Sci. 1995;56:1949–1955. doi: 10.1016/0024-3205(95)00175-6. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005;76:1307–1324. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Pertwee RG, Fernando SR. Evidence for the presence of cannabinoid CB1 receptors in mouse urinary bladder. Br J Pharmacol. 1996;118:2053–2058. doi: 10.1111/j.1476-5381.1996.tb15643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Fernando SR, Nash JE, Coutts AA. Further evidence for the presence of cannabinoid CB1 receptors in guinea-pig small intestine. Br J Pharmacol. 1996;118:2199–2205. doi: 10.1111/j.1476-5381.1996.tb15663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto L, Izzo AA, Cascio MG, Bisogno T, Hospodar-Scott K, Brown DR, et al. Endocannabinoids as physiological regulators of colonic propulsion in mice. Gastroenterology. 2002;123:227–2234. doi: 10.1053/gast.2002.34242. [DOI] [PubMed] [Google Scholar]
- Randall MD, Alexander SPH, Bennett T, Boyd EA, Fry JR, Gardiner SM, et al. An endogenous cannabinoid as an endothelium-derived vasorelaxant. Biochem Biophys Res Commun. 1996;229:114–120. doi: 10.1006/bbrc.1996.1766. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- Schild HO. pAx and competitive drug antagonism. Br J Pharmacol. 1949;4:277–280. doi: 10.1111/j.1476-5381.1949.tb00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt M, Di Marzo V. Metabolic fate of endocannabinoids. Curr Neuropharm. 2004;2:37–48. [Google Scholar]
- Ueda N, Puffenbarger RA, Yamamoto S, Deutsch DG. The fatty acid amide hydrolase (FAAH) Chem Phys Lipids. 2000;108(1–2):107–121. doi: 10.1016/s0009-3084(00)00190-0. [DOI] [PubMed] [Google Scholar]
- Valenti M, Gianfrani C, Mukenge S, Scaglione G, D'Argenio G, Ferla G, et al. Involvement of endocannabinoids and palmitoylethanolamide in intestinal disorders with inflammatory complications: human studies. Symposium on the cannabinoids. 2005. Burlington, Vermont, International Cannabinoid Research Society: page 158.
- White R, Hiley CR. The actions of the cannabinoid receptor antagonist, SR 141716A, in the rat isolated mesenteric artery. Br J Pharmacol. 1998;125:689–696. doi: 10.1038/sj.bjp.0702127. [DOI] [PMC free article] [PubMed] [Google Scholar]