

Abstract
Background and purpose:
The CB1 cannabinoid receptor and the β2-adrenoceptor are G protein-coupled receptors (GPCRs) co-expressed in many tissues. The present study examined physical and functional interactions between these receptors in a heterologous expression system and in primary human ocular cells.
Experimental approach:
Physical interactions between CB1 receptors and β2-adrenoceptors were assessed using bioluminescence resonance energy transfer (BRET). Functional interactions between these receptors were evaluated by examining receptor trafficking, as well as extracellular signal-regulated kinase (ERK) and cyclic AMP response element binding protein (CREB) signalling.
Key results:
Physical interactions between CB1 receptors and β2-adrenoceptors were demonstrated using BRET. In human embryonic kidney (HEK) 293H cells, co-expression of β2-adrenoceptors tempered the constitutive activity and increased cell surface expression of CB1 receptors. Co-expression altered the signalling properties of CB1receptors, resulting in increased Gαi-dependent ERK phosphorylation, but decreased non-Gαi-mediated CREB phosphorylation. The CB1 receptor inverse agonist AM251 (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide) attenuated β2-adrenoceptor-pERK signalling in cells expressing both receptors, while the CB1 receptor neutral antagonist O-2050 ((6aR,10aR)-3-(1-methanesulfonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran) did not. The actions of AM251 and O-2050 were further examined in primary human trabecular meshwork (HTM) cells, which are ocular cells endogenously co-expressing CB1 receptors and β2-adrenoceptors. In HTM cells, as in HEK 293H cells, AM251 but not O-2050, altered the β2-adrenoceptor–pERK response.
Conclusion and implications:
A complex interaction was demonstrated between CB1 receptors and β2-adrenoceptors in HEK 293H cells. As similar functional interactions were also observed in HTM cells, such interactions may affect the pharmacology of these receptors in tissues where they are endogenously co-expressed.
This article is part of a themed issue on Cannabinoids. To view the editorial for this themed issue visit http://dx.doi.org/10.1111/j.1476-5381.2010.00831.x
Keywords: G protein coupled receptor, CB1 receptor, β2-adrenoceptor, bioluminescence resonance energy transfer, trabecular meshwork
Introduction
The CB1 cannabinoid receptor is a rhodopsin-like, family A, G protein-coupled receptor (GPCR) that is widely expressed both within the CNS and the periphery. The CB1 receptor (nomenclature follows Alexander et al., 2009) was originally described as a receptor for the primary psychotropic agent in the plant Cannabis sativa, Δ9-tetrahydrocannabinol (Matsuda et al., 1990), but has since been shown to also bind endogenous ligands including N-arachidonoylethanolamine or anandamide (AEA), and 2-arachidonoylglycerol (2-AG) (Devane et al., 1992; Mechoulam et al., 1995). CB1 receptors are involved in a wide range of biological functions both in the CNS and the periphery. Within the CNS, they are present presynaptically and act to inhibit neurotransmitter release, while in the periphery CB1 receptors are involved in the regulation of energy and metabolism, bone formation, embryo implantation, cardiovascular function and intraocular pressure (IOP) (Kunos et al., 2000; Wang et al., 2004; Szczesniak et al., 2006; Cota, 2007; Hashimotodani et al., 2007; Bab and Zimmer, 2008).
Functionally, CB1 receptors have been reported to couple primarily to Pertussis toxin (PTx)-sensitive Gi/o proteins to inhibit adenylyl cyclase and voltage-gated Ca2+ channels, while activating mitogen-activated protein kinases (Demuth and Molleman, 2006). However, it has recently been shown that CB1 receptors also couple to some degree with both Gs and Gq/11 proteins to activate adenylyl cyclase and increase intracellular Ca2+ respectively (Maneuf and Brotchie, 1997; Lauckner et al., 2005).
Like many other family A GPCRs, CB1 receptors physically interact with other GPCRs to form both homodimers, as well as heterodimers with the D2 dopamine receptor; the µ-, κ- and δ-opioid receptors; the orexin-1 receptor; and the A2A adenosine receptor (Wager-Miller et al., 2002; Kearn et al., 2005; Mackie, 2005; Ellis et al., 2006; Rios et al., 2006; Carriba et al., 2007). These interactions have been shown to influence many aspects of CB1 receptor function including ligand pharmacology, receptor trafficking and G protein coupling. Although, to date, these are the only GPCRs reported to physically interact with CB1receptors, given the biological significance and widespread distribution of this receptor it is likely that CB1 receptors interact with additional GPCRs.
The β2-adrenoceptor is also a rhodopsin-like, family A, GPCR that is widely expressed in several tissue and cell types. β2-adrenoceptors are predominantly coupled with Gs, but also to a lesser extent with Gi (Xiao et al., 1995). Like CB1receptors, β2-adrenoceptors have been shown to form homodimers and heterodimers with other family A GPCRs, including the β1- and β3-adrenoceptors, the prostaglandin EP1 receptor and the µ-opioid receptor (Hebert et al., 1996; Angers et al., 2000; Mcvey et al., 2001; Lavoie et al., 2002; Breit et al., 2004; Mcgraw et al., 2006). The tissue distribution of β2-adrenoceptors overlaps significantly with that of the CB1 receptors, including parts of the cardiovascular system, reproductive tract, brain, eye and bone (Jampel et al., 1987a; Wanaka et al., 1989; Tsou et al., 1998; Stamer et al., 2001; Wang et al., 2004; Pacher and Hasko, 2008). Despite this overlapping distribution, possible direct physical and functional interactions between CB1receptors and β2-adrenoceptors have not been examined. Instead, most studies on the interactions between the cannabinoid and adrenergic systems have focused on inhibition of noradrenergic neurotransmission by presynaptic CB1 receptors (Schlicker et al., 1997; Schultheiss et al., 2005; Pakdeechote et al., 2007; Tam et al., 2008).
One organ where interactions between CB1 receptors and β2-adrenoceptors may be of particular interest is the eye. Agonists targeting CB1 receptors and antagonists targeting β2-adrenoceptors in the eye are known clinically to decrease IOP (Hepler and Frank, 1971; Borthne, 1976; Pate et al., 1998; Mccarty et al., 2008). In fact, β2-adrenoceptor antagonists are a front-line treatment for glaucoma, a blinding eye disease for which the major risk factor is elevated IOP. In humans, IOP is maintained by the balance of aqueous humour production in the ciliary body epithelium and outflow through trabecular meshwork and uveoscleral pathways (Woodward and Gil, 2004). Interestingly, both ciliary epithelial cells and trabecular meshwork cells co-express CB1 receptors and β2-adrenoceptors, and these receptors have been implicated in the regulation of both aqueous humour production and outflow (Jampel et al., 1987b; Wax et al., 1989; Straiker et al., 1999; Stamer et al., 2001; Njie et al., 2006). Thus, these ocular cells provide an ideal model for studying endogenous interactions between these two receptors.
In the present study, novel physical and functional interactions between CB1 receptors and β2-adrenoceptors were identified in human embryonic kidney (HEK) 293H cells. These interactions were found to influence both signalling and trafficking of the two receptors. The functional consequences of this CB1/β2-adrenoceptors interaction were then further examined in primary human trabecular meshwork (HTM) cells. Together, our observations in HEK 293H and HTM cells suggest complex cell type-specific physical and functional interactions between CB1 receptors and β2-adrenoceptors that may be relevant to the cells that co-express these two receptors in vivo.
Methods
Constructs
Human CB1 cannabinoid receptor (CB1) carboxy-terminal GFP2 and Renilla luciferase (Rluc) constructs were generated by PCR; the CB1 sequence was amplified without its stop codon from the Rc/CMV-CB1 plasmid (from Tom Bonner, NIH, Bethesda, MD, USA) using forward (CGACGAATTCCAGCCTAATCAAAGACTGAGGTT) and reverse (TGACATGGATCCCACAGAGCCTCGGCAGAC) primers. The PCR product was digested with EcoRI and BamHI and inserted into the pGFP2-N3 and pRluc-N1 plasmids (PerkinElmer) to produce CB1-GFP2 and CB1-Rluc respectively. Constructs of human β2-adrenoceptors (β2AR-GFP2, or β2AR-Rluc) and of the human ether-a-go-go related gene (HERG-GFP2) were prepared as previously reported (Lavoie et al., 2002; Dupre et al., 2007). The human metabotropic glutamate receptor 6 RcCMV(mGLuR6) and mGluR6-GFP2 constructs were kind gifts from Dr Robert Duvoisin (Oregon Health, and Science University, Portland, OR, USA). The HA-tagged β2-adrenoceptor in pcDNA3.1/Zeo(−) (Invitrogen Canada Inc., Burlingon, ON, Canada), HA-β2AR(Zeo), was generated by inserting HA-β2AR into the EcoRI and HindIII sites of pcDNA3.1/Zeo(−). The neomycin resistant HA-β2AR(Neo) construct was generated by transferring the HA-β2AR sequence from HA-β2AR(Zeo) to pcDNA3.1(+) (Invitrogen Canada Inc.) using NheI and HindIII. To generate pTreHA-β2AR, the HA-β2AR sequence was cut out of HA-β2AR(Neo) using NotI and inserted into pTRE2hyg (Clontech Laboratories Inc., Mountain View, CA, USA). The pTet-ON plasmid was from Clontech.
Cell culture and transfection
Human embryonic kidney 293H cells (Invitrogen Canada Inc.) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Transfections were carried out using the Lipofectamine 2000 transfection reagent (Invitrogen Canada Inc.) according to the manufacturer's protocol. Stable cell lines for various expression constructs were generated by selection with the appropriate antibiotics. Primary HTM cells were obtained from ScienCell (Carlsbad, CA, USA) and maintained in DMEM with 10% FBS. Cell cultures were split every 3–4 days before reaching confluency and were passaged up to 10 times before their experimental use.
Generation of stable cell lines
CB1-GFP2 was transfected into HEK 293H cells and stably expressing clones were selected using the antibiotic Zeocin. These CB1-GFP2 cells were then transfected with the Tet-ON plasmid and selected using G418. Finally, cells expressing CB1-GFP2 and Tet-ON were transfected with pTreHA-β2AR and selected using hygromycin B to generate a cell line stably expressing CB1-GFP, Tet-ON and pTreHA-β2AR (CB1-GFP2/TreHA-β2AR cells). Using On-Cell Western, the addition of doxycycline (Dox) to CB1-GFP2/TreHA-β2AR cells was shown to produce a dose-dependent induction of HA-β2AR expression (data not shown). A cell line stably expressing both CB1-GFP2 and HA-β2AR was generated by transfecting HA-β2AR(Neo) into CB1-GFP2 cells and selecting using both Zeocin and G418. Cells only expressing HA-β2AR were generated using the HA-β2AR(Zeo) plasmid and selected using Zeocin.
Bioluminescence resonance energy transfer
Bioluminescence resonance energy transfer (BRET) experiments were carried out using a combination of the GFP2 BRET acceptor, and the DeepBlueC coelenterazine Rluc substrate (PerkinElmer, Waltham, MA, USA), as part of the previously described BRET2 technique (Ramsay et al., 2002). Cells were transfected with the GFP2 and Rluc constructs and cultured for 24–48 h before their use in BRET experiments. Cells were then washed twice with ice-cold phosphate-buffered saline (PBS) before being suspended in PBS supplemented with 1.0 g·L−1 glucose, 10 mg·L−1 benzamidine, 5 mg·L−1 leupeptin and 5 mg·L−1 soybean trypsin inhibitor (Roche Canada, Mississauga, ON, Canada). Cells were dispensed into a white 96-well plate and their GFP2 emission was measured using a FLx800 fluorescence plate reader (BioTek Instruments Inc., Winooski, VT, USA) with excitation and emission filters of 485/20 and 528/20 nm respectively. BRET measurements were carried out using a Luminoskan Ascent plate reader (Thermo Scientific, Waltham, MA, USA) immediately following the addition of DeepBlueC coelenterazine substrate (PerkinElmer, Waltham, MA, USA) to a final concentration of 5 µM. All BRET measurements were taken by setting the plate reader to make dual luminescent emission measurements using 510 and 405 nm filters with the integration time set to 10 s and the photomultiplier tube voltage set to 1200. BRET measurements were then converted to BRET efficiencies (BRETEff) according to a previously described method (James et al., 2006). Briefly, BRETEff values were calculated by normalizing the ratio of luminescent emissions at 510/405 nm for each sample to the minimum and maximum 510/405 nm emission ratios obtained using empty Rluc and GFP2-Rluc fusion constructs respectively.
Immunofluorescence and confocal microscopy
Cells expressing HA and/or GFP2 constructs or HTM cells were plated onto glass coverslips and maintained for 24–48 h. Cells were then treated as indicated in FBS-free DMEM before being fixed for 5 min in ice cold 100% methanol. After washing with PBS, cells were permeabilized with 100 µM digitonin, washed with PBS and blocked with 1% BSA in PBS. Cells were incubated with primary antibodies: monoclonal mouse anti-HA (Covance, Emeryville, CA, USA), or polyclonal mouse anti-β2AR (Abnova, Neihu District. Taipei City, Taiwan) and polyclonal rabbit anti-CB1 (Caymen Chemical, Ann Arbor, MI, USA) overnight at 4°C. After washing with PBS, coverslips were incubated with Cy3-conjugated anti-mouse IgG and/or fluorescein isothiocyanate-conjugated anti-rabbit IgG secondary antibodies for 1 h at room temperature. Coverslips were then washed again with PBS before being mounted on slides using Fluorsave reagent (Calbiochem, San Diego, CA, USA), and imaged using a Nikon Eclipse E800 microscope fitted with the D-Eclipse C1 confocal system (Nikon Canada Inc., Mississauga, ON, Canada). GFP2 and fluorescein isothiocyanate were imaged using a 488 nm air-cooled argon laser (Spectra-Physics Lasers Inc., Mountain View, CA, USA), while Cy3 was imaged with a 543 nm He-Ne laser (JDS Uniphase, Milpitas, CA, USA).
In-Cell Western blot analyses
Phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK) and cyclic AMP response element binding protein (CREB) was assessed using a modified In-Cell Western protocol, as previously reported (Mcintosh et al., 2007). Briefly, HEK 293H cells expressing CB1-GFP and/or HA-β2AR constructs or HTM cells were plated in 96-well plates and cultured to confluency. Cell culture media was then replaced with FBS-free DMEM and cells were maintained for 24 h prior to experiments. Cells were then treated as indicated for either 5 or 10 min in ERK experiments, or 30 min in CREB experiments, before being fixed for 1 h with 4% paraformaldehyde. Rabbit anti-phospho ERK1/2 and goat anti-total ERK2 primary antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) were used to assess ERK phosphorylation, while goat anti-phospho-CREB and rabbit anti-total CREB primary antibodies (Santa Cruz Biotechnology Inc.) were used to assess CREB phosphorylation. Secondary antibodies used were a IRDye800CW-conjugated donkey anti-rabbit IgG (Rockland Immunochemicals Inc., Gilbertsville, PA, USA) and an Alexa Fluor680-conjugated donkey anti-goat IgG (Invitrogen Canada Inc.). Plates were scanned to measure their fluorescent emission from the IRDye800CW- and Alexa Fluor680-conjugated antibodies using the Odyssey infrared imaging system (Li-Cor Biotechnology, Lincoln, NE, USA). In order to convert these data to relative pERK and pCREB values, background fluoresence was first determined and then subtracted using wells of the plate that received only the secondary antibodies. The ratio of the pERK/ERK2 or pCREB/total CREB signals (with background subtracted) were then determined for each well and normalized to the ratios obtained from the appropriate vehicle or untreated wells in order to obtain relative pERK or pCREB values. Experiments were in all cases repeated several times, and within each experiment, each condition was repeated in 2–8 wells. The total numbers of individual wells used are presented as the ‘n’ number in each figure.
On-Cell Western blot analyses
Cell surface expression of CB1-GFP2 and HA-β2AR was assessed using a modified In-Cell Western protocol utilizing non-permeablized cells (Miller, 2004). Cells were plated in 96-well plates and cultured for 24–48 h before experiments. Cells were treated as indicated in FBS-free DMEM and fixed in 4% paraformaldehyde. After washing with PBS, cells were blocked with 1% BSA in PBS at room temperature for 2 h. Primary antibodies directed at N-terminal epitopes of either the CB1-GFP2 or HA-β2AR constructs were polyclonal rabbit anti-CB1 (Cayman Chemical, Ann Arbor, MI, USA) or monoclonal anti-HA (Covance, Emeryville, CA, USA) antibodies respectively. Primary antibodies were diluted in 1% BSA in PBS and applied for 1 h at room temperature. After washing, cells were incubated in secondary antibodies: anti-rabbit IgG IRDye800CW and goat anti-mouse IgG Alexa Fluor680 (Invitrogen Canada Inc.), diluted in 1% BSA in PBS for 1 h at room temperature. Cells were washed with PBS then with distilled water before drying. Once dry, the plates were imaged using an Odyssey infrared imaging system (Li-Cor Biotechonology).
Statistical analysis and curve fitting
All data are presented as mean ± SEM. Statistical analysis and curve fitting of the data were performed using Graphpad Prism v.4 (GraphPad Software Inc., San Diego, CA, USA). To fit data to dose–response curves, vehicle treatments were plotted at a concentration equal to one and a half log units less than the lowest drug treatment concentration then data were fitted to a sigmoidal dose–response curve with variable slope. Statistical significance for curve fits was determined by the F-test comparing global fits, by t-test when comparing means of only two groups, or by one-way, two-way, or repeated measures analysis of variance (anova), as appropriate, when comparing the means of multiple treatment groups. Tukey's post hoc analysis was used to determine differences among groups for one-way anova, while Bonferroni's post hoc analysis was used for two-way anova. P < 0.05 was considered statistically significant.
Materials
Pertussis toxin, hygromycin B and G418 sulphate were from Calbiochem. (R)-(+)-WIN 55,212-2 mesylate ((R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate), AM251 (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide), AM630 (6-Iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)methanone), O-2050 ((6aR,10aR)-3-(1-methanesulfonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran), ICI 118,551 ((±)-erythro-(S*,S*)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride) and CGP 20712 (1-[2-((3-carbamoyl-4-hydroxy)phenoxy)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propan ol dihydrochloride) were from Tocris Bioscience (Ellisville, MO, USA). Zeocin and Opti-MEM were obtained from Invitrogen Canada Inc. FBS was from PAA laboratories Inc. (Etobicoke, ON, Canada). Restriction enzymes, DNA polymerases and other enzymes were from ferments Canada Inc. (Burlington, ON, Canada). All other chemicals and reagents were from Sigma-Aldrich Canada Ltd. (Oakville, ON, Canada).
Results
Physical interactions between CB1 receptors and β2-adrenoceptors in HEK 293H cells
Bioluminescence resonance energy transfer2 (BRET2) was used to demonstrate an interaction between CB1 receptors and the β2-adrenoceptors in HEK 293H cells. BRETEff was measured from cells co-transfected with either CB1-Rluc or β2AR-Rluc, and one of CB1-GFP2, β2AR-GFP2, HERG-GFP2 or mGluR6-GFP2 (Figure 1A). When co-expressed with CB1-Rluc, CB1-GFP2 and β2AR-GFP2 produced significantly increased BRETEff (P < 0.001) compared with either HERG-GFP2 or mGluR6-GFP2, two different membrane proteins not expected to interact with either CB1 receptors or β2-adrenoceptors. Similarly, when co-transfected with β2AR-Rluc both CB1-GFP2 and β2AR-GFP2 produced significantly increased BRETEff compared with the HERG-GFP2 (P < 0.001) and mGluR6-GFP2 (P < 0.01) controls. In all cases, GFP2 expression levels were equal to or less than those of the HERG-GFP2 negative control (data not shown). These data confirm previous reports of both CB1 receptor and β2-adrenoceptor homodimerization (Hebert et al., 1996; Angers et al., 2000; Wager-Miller et al., 2002), but also suggest a novel physical interaction between CB1 receptors and β2-adrenoceptors.
Figure 1.
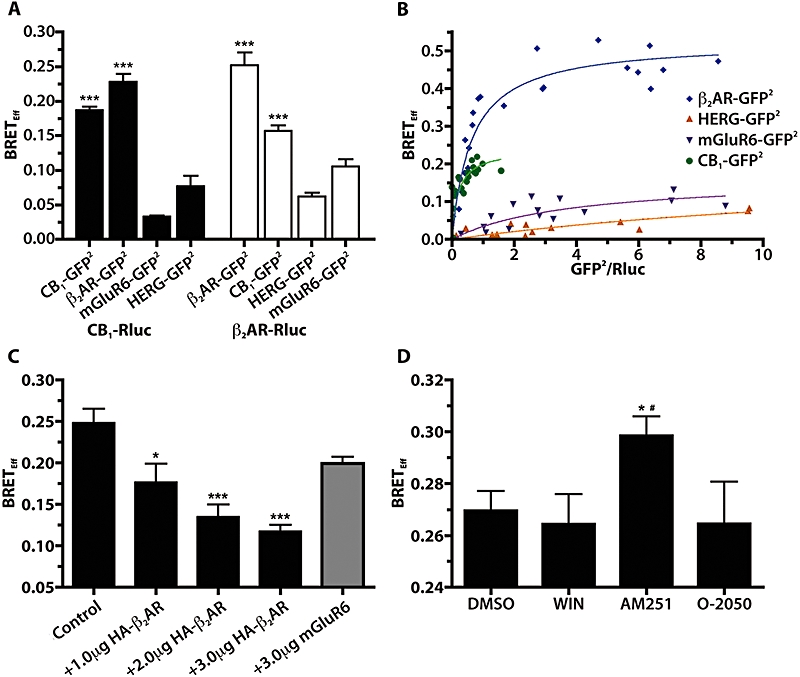
CB1 cannabinoid receptors physically interact with β2-adrenoceptors when expressed in 293H cells. (A) BRETEff values obtained from 293H cells transiently transfected with either CB1-Rluc or β2AR-Rluc and CB1-GFP2, β2AR-GFP2, mGluR6-GFP2 or HERG-GFP2. ***P < 0.001 compared with HERG-GFP2 controls; n= 4–9. (B) BRET saturation curves for CB1-Rluc with β2AR-GFP2, CB1-GFP2, HERG-GFP2 and mGluR6-GFP2. BRETEff is plotted against the ratio of GFP2 fluorescence (obtained by directly exciting GFP2) and Rluc emission and the data were fitted to a rectangular hyperbola. (C) BRETEff values from 293H cells transfected with a fixed amount of CB1-Rluc and β2AR-GFP2 (Control) and pcDNA or increasing amounts of HA-β2AR or mGluR6. *P < 0.05 and ***P < 0.001 compared with control column; n= 4–10. (D) BRETEff values obtained from cells transfected with CB1-Rluc and β2AR-GFP2 and treated for 15 min at room temperature with DMSO (0.05%), WIN (10 µM), AM251 (10 µM) or O-2050 (10 µM) prior to measuring BRETEff. *P < 0.05 compared with WIN; #P < 0.05 compared with O-2050; n= 9. AM251, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; BRET, bioluminescence resonance energy transfer; BRETEff, BRET efficiency; DMSO, dimethylsulphoxide; HERG, human ether-a-go-gorelated gene; mGluR6, metabotropic glutamate receptor 6; O-2050, (6aR,10aR)-3-(1-methanesulfonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran; Rluc, Renillaluciferase; WIN, WIN 55,212-2.
Bioluminescence resonance energy transfer saturation experiments were performed according to a previously described protocol (Roy et al., 2006). A fixed amount of the CB1-Rluc construct was co-transfected with increasing amounts of β2AR-GFP2, CB1-GFP2, HERG-GFP2 or mGluR6-GFP2. BRETEff values were plotted against the ratio of GFP2 fluorescent emission obtained by directly exciting GFP2 (measuring GFP2 emission in the absence of the Rluc coelenterazine substrate) to the Rluc bioluminescent emission and fitted to rectangular hyperbola curves (Figure 1B). Significantly different BRET50 values (P < 0.05) of 0.6 ± 0.1 and 0.19 ± 0.07, and BRETMax values (P < 0.001) of 0.53 ± 0.03 and 0.24 ± 0.03, were obtained from the saturation curves when β2AR-GFP2 and CB1-GFP2 were used as BRET acceptors respectively. These saturation curves demonstrate that there is a specific interaction between CB1 receptors and β2-adrenoceptors that is observed at low levels of receptor expression.
A BRET competition experiment was performed to demonstrate that the BRET signal between CB1-Rluc and β2AR-GFP2 could be reduced in a dose-dependent manner by co-transfection with HA-β2AR (Figure 1C). CB1-Rluc/β2AR-GFP2 BRETEff was significantly reduced by co-transfection with 1 µg of HA-β2AR cDNA (P < 0.05) and further reduced with 2 and 3 µg of HA-β2AR plasmid (P < 0.001), but was not significantly affected by 3 µg of RcCMVmGluR6 (P > 0.05) plasmid, indicating the specificity of the CB1/β2-adrenoceptors interaction.
The effect of various CB1 receptor ligands on the BRETEff for the CB1-Rluc/β2AR-GFP2 pair was then assessed (Figure 1D). The CB1 receptor inverse agonist AM251 resulted in a BRETEff that was significantly elevated compared with either the agonist WIN 55,212-2 (WIN) or the neutral antagonist O-2050 (P < 0.05). This finding suggests that AM251 modulates the CB1/β2-adrenoceptor heterodimer, either by altering the number of receptors interacting, or by altering the conformation of the dimer, such that it affects the orientation of the BRET donor and acceptor.
Functional interactions between CB1 receptors and β2-adrenoceptors in HEK 293H cells affect CB1 receptor constitutive activity
When CB1-GFP2 was stably expressed in HEK 293H cells a punctate pattern of internalized CB1 receptor distribution was observed in addition to a less intense pattern of receptors detected at the cell surface. Following addition of the CB1 receptor inverse agonist AM251 (10 µM, 24 h), CB1-GFP2 expression was redistributed to the cell surface (Figure 2A). Using On-Cell Western blots, a quantitative measure of the effect of AM251 (10 µM, 24 h) was obtained, confirming a significant (P < 0.001, 360%) increase in CB1-GFP2 cell surface expression in response to treatment with the inverse agonist (Figure 2B). These results demonstrate that CB1 receptors are constitutively active, resulting in a constitutive internalization of the receptor, which can be reversed by the inverse agonist AM251.
Figure 2.
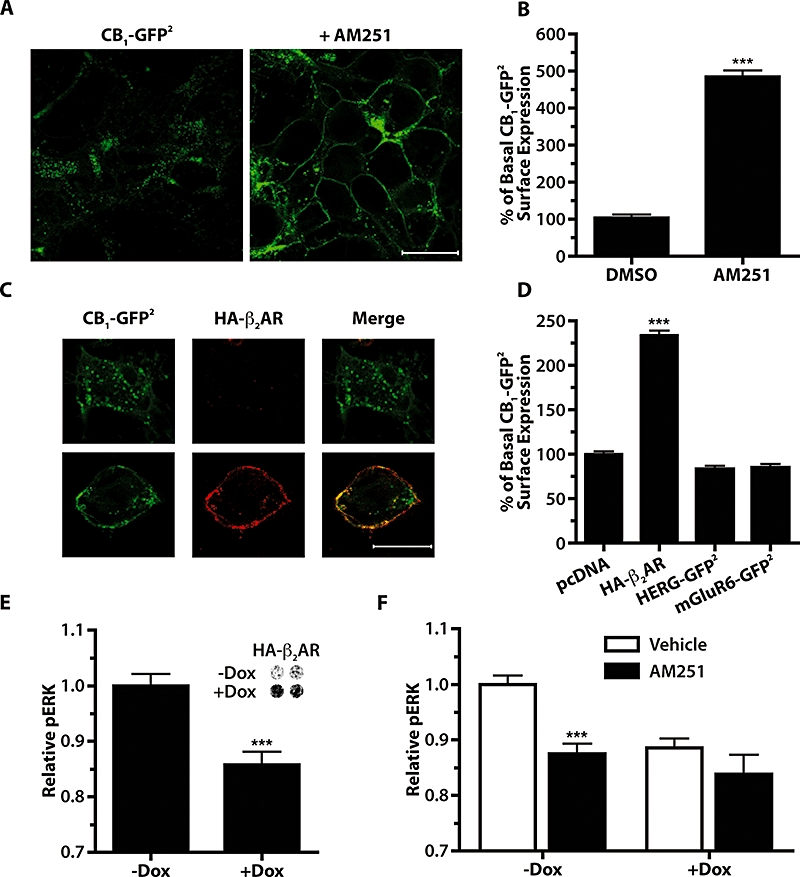
Co-expression of HA-β2AR reduces the constitutive activity of CB1-GFP2 in 293H cells. (A) Confocal images of 293H cells stably expressing CB1-GFP2 treated for 24 h with 0.05% DMSO vehicle (left panel) or 10 µM AM251 (right panel). Scale bar is 20 µm. (B) On-Cell Western quantitative measure of CB1 cell surface expression following 24 h AM251 treatment (10 µM) in 293H cells stably expressing CB1-GFP2. ***P < 0.001 compared with DMSO vehicle; n= 4–6. (C) Confocal images of 293H cells transfected with CB1-GFP2 and HA-β2AR. Left panels show GFP2 fluorescence, middle panels are anti-HA immunofluorescence utilizing a Cy3-conjugated secondary antibody, and the right panels are the merged images. Scale bar is 20 µm. (D) On-Cell Western quantitative measure of CB1-GFP2 cell surface expression in 293H cells stably expressing CB1-GFP2 and transiently transfected with pcDNA, HA-β2AR, HERG-GFP2 or mGluR6-GFP2. ***P < 0.001 compared with pcDNA transfected cells; n= 4–18. (E) Basal pERK levels in CB1-GFP2/TreHA-β2AR cells without and with Dox pretreatment (10 µg·mL−1, 24 h) to induce expression of HA-β2AR. ***P < 0.001; n= 20. Inset shows On-Cell Western using an anti-HA primary antibody to measure HA-β2 expression without or with Dox. (F) pERK levels in CB1-GFP2/TreHA-β2AR cells treated with 0.05% DMSO vehicle (open bars) or AM251 (1 µM, 10 min, solid bars) in cells without or with Dox pretreatment (10 µg·mL−1, 24 h). ***P < 0.001 compared with respective vehicle controls; n= 20–34. AM251, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; DMSO, dimethylsulphoxide; Dox, doxycycline; ERK, extracellular signal-regulated kinase; HERG, human ether-a-go-gorelated gene; mGluR6, metabotropic glutamate receptor 6.
Co-expression of CB1-GFP2 with HA-β2AR also resulted in increased localization of CB1-GFP2 at the cell surface. Immunofluorescence for the HA tag was carried out on HEK 293H cells transiently transfected with both CB1-GFP2 and HA-β2AR (Figure 2C). When cells were successfully transfected with both receptor constructs, CB1-GFP2 expression was largely observed at the cell surface (lower panels of Figure 2C), but when CB1-GFP2 was expressed alone, the distribution was consistent with an internalized receptor (upper panels of Figure 2C). In order to measure the effect that HA-β2AR co-expression had on CB1-GFP2 cell surface expression, On-Cell Western blots were used in CB1-GFP2 cells transiently transfected with HA-β2AR (Figure 2D). Transient expression of HA-β2AR in these cells resulted in a significant (P < 0.001, 130%) increase in CB1-GFP2 cell surface expression, while the expression of either HERG-GFP2 or mGluR6-GFP2 did not increase CB1-GFP2 surface expression.
To assess whether the increased CB1-GFP2 surface expression was the result of decreased constitutive activity of CB1receptors, basal CB1 pERK signalling was measured in CB1-GFP2/TreHA-β2AR cells treated with Dox to induce HA-β2AR expression (Figure 2E). Induction of HA-β2AR resulted in a significant decrease in the basal pERK level of these cells (P < 0.001). In order to show that this decrease in basal pERK was in fact the result of decreased constitutive activity of CB1 receptors, the ability of AM251 to reduce basal pERK level was then measured in these cells in the absence or presence of co-expressed HA-β2AR (Figure 2F). AM251 (1 µM, 10 min) significantly reduced basal pERK in the absence of HA-β2AR (P < 0.001), but had no significant effect on cells co-expressing HA-β2AR. These findings demonstrate that CB1 receptors are constitutively active in 293H cells, but that co-expression of β2-adrenoceptors attenuates this constitutive activity.
CB1 receptors and β2-adrenoceptors are co-internalized upon addition of a CB1 receptor or β2-adrenoceptor agonist
Co-transfection of CB1-GFP2 and HA-β2AR in HEK 293H cells resulted in expression of both receptors primarily at the cell membrane (Figure 3A). When these cells were exposed to the β2-adrenoceptor agonist, isoprenaline (10 µM, 30 min), internalization of not only HA-β2AR but also CB1-GFP2 was observed. In contrast, when cells were transfected with mGluR6-GFP2 and HA-β2AR and treated with isoprenaline, only HA-β2AR was internalized, while mGluR6-GFP2 remained at the cell surface. On-Cell Western analyses were used for a quantitative measure of the ability of isoprenaline to co-internalize CB1-GFP2 in cells stably expressing CB1-GFP2 and transiently transfected with HA-β2AR (Figure 3B). In the absence of HA-β2AR there was no effect on the cell surface distribution of CB1-GFP2 following treatment with isoprenaline. However, isoprenaline treatment of CB1-GFP2 cells transfected with HA-β2AR resulted in a significant decrease (P < 0.001, 49%) in CB1-GFP2 cell surface expression. As expected, HA-β2AR cell surface expression was also significantly decreased (P < 0.001) in CB1-GFP2 cells transfected with HA-β2AR following treatment with isoprenaline. These findings show that CB1-GFP2 is co-internalized with HA-β2AR following treatment with the β-adrenoceptor agonist, isoprenaline.
Figure 3.
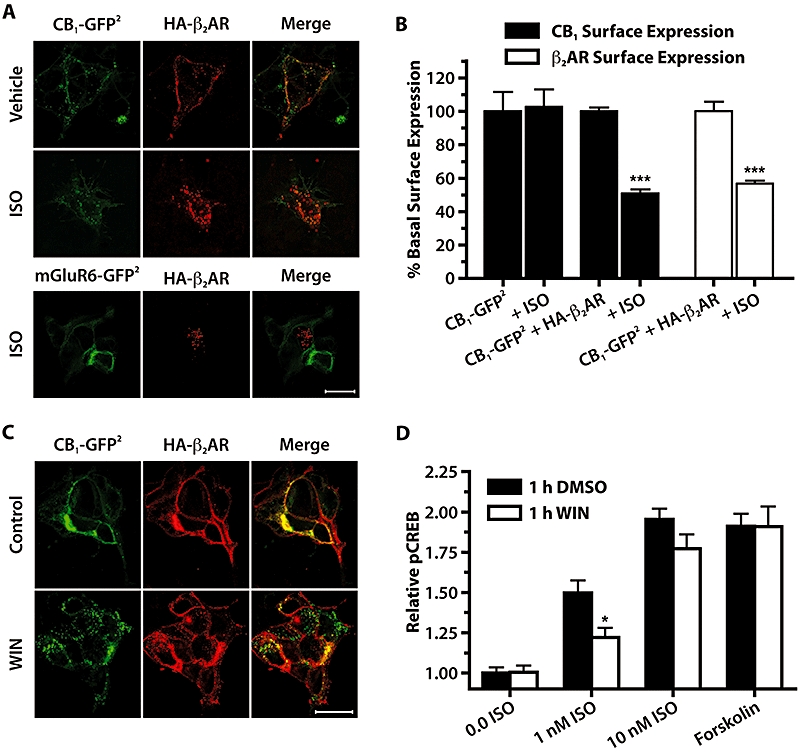
HA-β2AR and CB1-GFP2 are co-internalized when exposed to either isoprenaline (ISO) or WIN. (A) Confocal images of 293H cells transiently transfected with CB1-GFP2 or mGluR6-GFP2 and HA-β2AR treated for 30 min with vehicle or 10 µM isoprenaline. Left panels show GFP2 fluorescence, middle panels are anti-HA immunofluorescence utilizing a Cy3-conjugated secondary antibody, and right panels are merged images. Scale bar is 20 µm. (B) On-Cell Western quantitative measure of CB1-GFP2 or HA-β2AR cell surface expression in 293H cells stably expressing CB1-GFP2 and transiently transfected with either pcDNA (CB1-GFP2 bars) or HA-β2AR. Cells were treated with either H2O vehicle (labelled CB1-GFP2 + HA-β2) or isoprenaline (10 µM) for 30 min. ***P < 0.001 compared with CB1-GFP2 + HA-β2AR vehicle-treated groups; n= 6. (C) Confocal images of HEK 293H cells transiently transfected with CB1-GFP2 and HA-β2AR, upper panels are untreated controls, and lower panels are treated with WIN (10 µM, 30 min). Scale bar is 20 µm. (D) Relative pCREB levels in CB1-GFP2/TreHA-β2AR cells pretreated with Dox (10 µM, 24 h), treated for 1 h with either DMSO (0.05%) or WIN (10 µM), followed by 30 min treatment with isoprenaline (0–10 nM), or forskolin (10 µM). *P < 0.05 compared with respective 1 h DMSO treatments; n= 12–20. CREB, cyclic AMP response element binding protein; DMSO, dimethylsulphoxide; Dox, doxycycline; HEK, human embryonic kidney; mGluR6, metabotropic glutamate receptor 6; WIN, WIN 55,212-2.
Similar to the co-internalization observed when cells were co-transfected with CB1-GFP2 and HA-β2AR and treated with the CB1 receptor agonist WIN (10 µM, 30 min), internalization of both receptors was observed (Figure 3C). To further examine the functional significance of this co-internalization, the phospho-CREB (pCREB) signalling response to activation of β2-adrenoceptors was assessed in CB1-GFP2/TreHA-β2AR cells pretreated with Dox (10 µM, 24 h) to induce HA-β2AR expression, before being treated with WIN (10 µM, 1 h) to induce internalization (Figure 3D). In these cells, the ability of 1 nM isoprenaline to increase pCREB was attenuated by WIN treatment (P < 0.05), but the ability of 10 nM isoprenaline or 10 µM forskolin to increase pCREB were unaffected, indicating that the effect is not due to physiological antagonism of the pCREB signalling pathway. Taken together, these data demonstrate co-internalization of β2-adrenoceptors with CB1 receptors mediated by the agonist WIN, and that this co-internalization has functional consequences in the cross-desensitization of β2-adrenoceptors.
Co-expression of CB1 receptors and β2-adrenoceptors affects their ability to stimulate ERK and CREB phosphorylation
In-Cell Western analyses were used to assess levels of phospho-ERK (pERK) and pCREB in HEK 293H cells expressing CB1-GFP2 and/or HA-β2AR. When cells expressing CB1-GFP2 alone were treated with WIN, increases in both pERK (5 min exposure) and pCREB (30 min exposure) were observed. The WIN-dependent pERK response was sensitive to PTx, while the pCREB response was not (data not shown). pERK dose–response measurement following WIN treatment in CB1-GFP2/TreHA-β2AR cells that were not induced with Dox (i.e. no HA-β2AR expression) yielded a pEC50 of 6.85 ± 0.04, Emax of 1.74 ± 0.02 and a Hill coefficient of 1.07 ± 0.11 (Figure 4A). Pretreatment of these cells for 48 h with Dox to induce HA-β2AR expression, resulted in a significantly different WIN pERK dose–response curve (P < 0.001), with pEC50values of 6.66 ± 0.03, 1.91 ± 0.02 for Emax and a Hill coefficient of 1.64 ± 0.20. However, the PTx sensitivity of the WIN–pERK response was not affected by co-expression of HA-β2AR with CB1-GFP2 (data not shown). This demonstrates that co-expression of β2-adrenoceptors enhances CB1 receptor-dependent pERK signalling.
Figure 4.
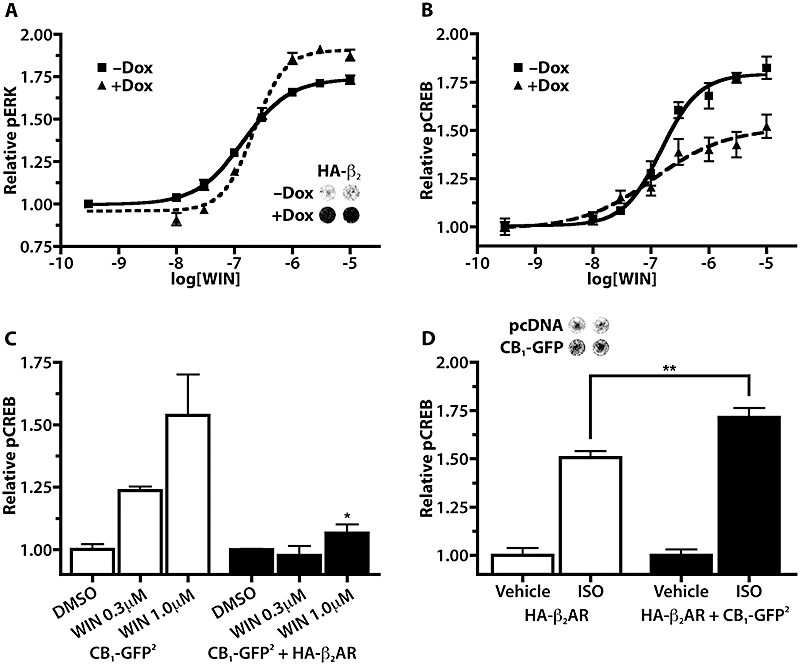
Co-expression of HA-β2AR and CB1-GFP2 alters receptor efficacy to activate ERK and CREB phosphorylation. (A) Dose–response curves for WIN pERK activation in CB1-GFP2/TreHA-β2AR cells pretreated for 48 h without or with Dox (10 µg·mL−1); n= 8. Inset shows On-Cell Western using an anti-HA primary antibody in CB1-GFP2/TreHA-β2AR cells without or with Dox (10 µg·mL−1, 48 h). (B) Dose–response curves for WIN pCREB activation in CB1-GFP2/Tet-ON/HA-β2AR cells pretreated for 48 h without or with Dox (10 µg·mL−1); n= 4–8. (C) WIN–pCREB responses in 293H cells stably expressing CB1-GFP2 alone or CB1-GFP2 and HA-β2AR. *P < 0.05 compared with 1.0 µM WIN in CB1-GFP2 cells; n= 3. (D) Isoprenaline–pCREB responses in 293H cells stably expressing HA-β2AR and transiently transfected with pcDNA (solid bars) or CB1-GFP2 (open bars). **P < 0.01; P= 7–8. Inset is On-Cell Western using anti-CB1 primary antibody of cells transfected with pcDNA or CB1-GFP2. CREB, cyclic AMP response element binding protein; Dox, doxycycline; ERK, extracellular signal-regulated kinase; WIN, WIN 55,212-2.
WIN treatment of CB1-GFP2/TreHA-β2AR cells that were not pretreated with Dox resulted in a pCREB dose–response curve with a pEC50 of 6.82 ± 0.08, Emax of 1.79 ± 0.03 and a Hill coefficient of 1.32 ± 0.29 (Figure 4B). Pretreatment of these cells with Dox (10 µg·mL−1, 48 h) resulted in a significantly different (P < 0.001) dose–response curve with values for pEC50of 6.92 ± 0.28, for Emax of 1.51 ± 0.07 and a Hill coefficient of 0.69 ± 0.33. To determine the long-term effect of co-expression of CB1 receptors and β2-adrenoceptors, a cell line stably expressing both CB1-GFP2 and HA-β2AR was employed. In these cells, the WIN-dependent pCREB response was nearly completely abolished at WIN concentrations of 0.3 and 1.0 µM, compared with cells expressing CB1receptors alone (Figure 4C), indicating that co-expression of β2-adrenoceptors inhibits the CB1 receptor-pCREB signalling pathway.
When cells stably expressing HA-β2AR were treated with isoprenaline, PTx-insensitive increases in both pCREB and pERK were observed (data not shown). In order to determine what influence co-expression of CB1-GFP2 had on the isoprenaline-stimulated pCREB and pERK responses, HA-β2AR cells were transfected with either pcDNA vector control or CB1-GFP2 48 h prior to treatment with isoprenaline. Cells transfected with CB1-GFP2 showed significantly greater (P < 0.01) isoprenaline-stimulated pCREB responses than those transfected with vector (Figure 4D), while there was no difference in the isoprenaline-stimulated pERK responses between cells transfected with pcDNA or CB1-GFP2 (data not shown). Again, the PTx insensitivity of the isoprenaline-induced pERK and pCREB responses was not affected by co-expression of CB1-GFP2 (data not shown).
Co-application of WIN and isoprenaline results in an additive response for pERK but not pCREB
Isoprenaline-stimulated pERK dose–response curves from CB1-GFP2/TreHA-β2AR cells pretreated with Dox to induce HA-β2AR expression were generated in the presence of dimethylsulphoxide (DMSO) vehicle, 0.1 and 0.3 µM WIN (Figure 5A). The curves generated in the presence of 0.1 and 0.3 µM WIN were both significantly different from the DMSO vehicle curve (P < 0.001). Similar pEC50 values of 7.57 ± 0.7, 7.48 ± 0.12 and 7.41 ± 0.14 were obtained for DMSO, 0.1 and 0.3 µM WIN respectively. Baseline pERK levels were increased from 0.91 ± 0.03 in DMSO-treated cells to 1.16 ± 0.04 and 1.30 ± 0.04 in 0.1 and 0.3 µM WIN-treated. Similarly, Emax values were also increased from 1.59 ± 0.04 in DMSO-treated cells to 1.84 ± 0.07 and 1.92 ± 0.07 in cells treated with 0.1 and 0.3 µM WIN respectively. This pattern of increased baseline pERK and Emax, but unchanged pEC50 is consistent with additive WIN- and isoprenaline-induced pERK responses. In contrast, when 1 µM WIN was co-applied with isoprenaline (1 and 10 nM) to these cells, the pCREB response was unchanged compared with isoprenaline application alone (Figure 5B). To determine if the pCREB response was saturated, 10 µM forskolin was co-applied with isoprenaline (1 and 10 nM); it was found that at 1 but not 10 nM isoprenaline, forskolin significantly further elevated pCREB levels over isoprenaline treatment alone (P < 0.001). This demonstrates that WIN and isoprenaline show additive pERK but not pCREB responses in cells co-expressing these two receptors.
Figure 5.
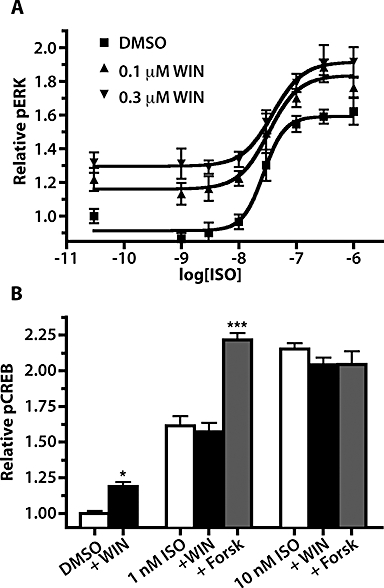
Co-application of isoprenaline (ISO) and WIN results in an additive increase in ERK phosphorylation. (A) Dose–response curves for pERK activation following co-exposure of isoprenaline and 0.05% DMSO vehicle, 0.1 µM WIN or 0.3 µM WIN in CB1-GFP2/TreHA-β2AR pretreated for 24 h with 10 µg·mL−1 Dox; n= 6. (B) pCREB responses in CB1-GFP2/Tet-ON/HA-β2AR cells pretreated for 24 h with Dox (10 µg·mL−1) then co-exposed to isoprenaline (0, 1 and 10 nM) and DMSO vehicle (0.05%), WIN (1.0 µM) or forskolin (10 µM). *P < 0.05, ***P < 0.001 compared with DMSO, or isoprenaline alone groups, respectively; n= 10–28. CREB, cyclic AMP response element binding protein; DMSO, dimethylsulphoxide; Dox, doxycycline; ERK, extracellular signal-regulated kinase; WIN, WIN 55,212-2.
The CB1 inverse agonist AM251 but not the neutral antagonist O-2050 inhibits the isoprenaline-stimulated pERK response
Dose–response curves for pERK were generated in CB1-GFP2 cells for the CB1 receptor agonist WIN, the inverse agonist AM251, and the neutral antagonist O-2050 (Figure 6A). AM251 produced a dose-dependent decrease in pERK from baseline to 0.78 ± 0.03 with a pEC50 of 8.2 ± 0.4, while O-2050 had no effect on basal pERK levels. Although both AM251 and O-2050 were capable of completely blocking the WIN–pERK response in these cells (data not shown), AM251 acts as an inverse agonist, while O-2050 acts as a neutral antagonist.
Figure 6.
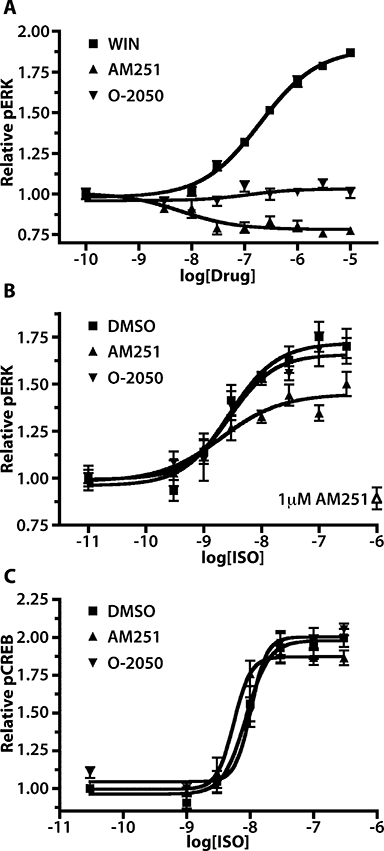
The CB1 inverse agonist AM251 but not the CB1 neutral antagonist O-2050 attenuates isoprenaline (ISO)-induced ERK phosphorylation. (A) pERK dose–response curves for WIN, AM251 and O-2050 in 293H cells stably expressing CB1-GFP2. WIN exposures were for 5 min, while AM251 and O-2050 exposures were for 15 min; n= 6–16. (B) Isoprenaline pERK dose–response curves for CB1-GFP2/Tet-ON/HA-β2AR cells pretreated for 24 h with Dox (10 µg·mL−1) and 15 min with 0.05 % DMSO (squares), AM251 (triangles) or O-2050 (inverted triangles); n= 9–12. Each curve is normalized to the pERK level in cells untreated with isoprenaline but pretreated with the DMSO, AM251 or O-2050. The effect of 20 min AM251 exposure on the basal pERK level in these cells is indicated by the open triangle. (C) Isoprenaline pCREB dose–response curves in CB1-GFP2/TreHA-β2AR cells pretreated for 24 h with Dox (10 µg·mL−1) and 15 min with 0.05% DMSO, AM251 or O-2050; n= 6–9. AM251, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; CREB, cyclic AMP response element binding protein; DMSO, dimethylsulphoxide; Dox, doxycycline; ERK, extracellular signal-regulated kinase; O-2050, (6aR,10aR)-3-(1-methanesulfonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran; WIN, WIN 55,212-2.
Isoprenaline-mediated pERK dose–response curves were produced in CB1-GFP2/TreHA-β2AR cells pretreated for 24 h with Dox to induce HA-β2AR expression then 15 min with either DMSO vehicle, AM251 or O-2050 (Figure 6B). Following DMSO pretreatment, the isoprenaline dose–response curve had a pEC50 of 8.57 ± 0.15 and an Emax of 1.71 ± 0.05. This dose–response curve was significantly altered by pretreatment with AM251 (P < 0.001) but not O-2050. AM251 pretreatment did not affect the pEC50 producing a value of 8.70 ± 0.28, but did result in a much lower Emax value of 1.45 ± 0.06. These findings demonstrate that the CB1 receptor inverse agonist AM251, but not the neutral antagonist O-2050 attenuates pERK signalling mediated by β2-adrenoceptors in HEK 293 cells.
Isoprenaline-mediated pCREB dose–response curves were also generated from CB1-GFP2/TreHA-β2AR cells pretreated for 24 h with Dox and 15 min with either DMSO vehicle, AM251 or O-2050 (Figure 6C). The isoprenaline pCREB dose–response curve with DMSO pretreatment had a pEC50 of 8.06 ± 0.07 and Emax of 1.98 ± 0.05. Neither pretreatment with AM251 or O-2050 significantly altered the isoprenaline pCREB dose–response curve in these cells. In addition, the β2-adrenoceptor inverse agonist timolol had no effect on CB1 receptor-mediated pERK or pCREB responses (data not shown).
Interactions between pERK signalling induced by CB1 receptors and β2-adrenoceptors in HTM cells
Immunofluorescence studies in HTM cells demonstrated clear labelling of endogenous CB1 receptors and β2-adrenoceptors (Figure 7A) when compared with secondary antibody-only controls (data not shown). CB1 receptor and β2-adrenoceptor expression was detected at the cell membrane in HTM cells, with co-localization of the two receptors apparent.
Figure 7.
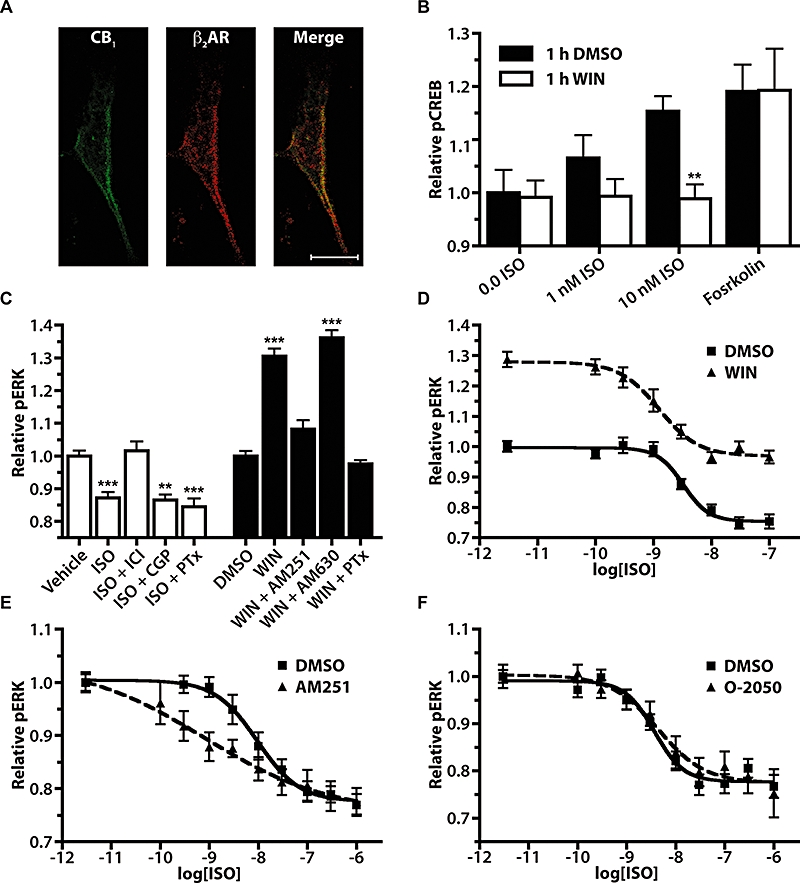
Interactions between CB1 receptor and β2-adrenoceptor pERK signalling in HTM cells. (A) Confocal immunofluorescence images of HTM cells using an anti-CB1 primary antibody with a FITC-conjugated secondary antibody (left panel), an anti-β2-adrenoceptor primary antibody with a Cy3-conjugated secondary antibody (middle panel) and the merged image (right panel). Scale bar is 20 µM. (B) Isoprenaline (ISO, 1 and 10 nM, 30 min) and forskolin (10 µM, 30 min) pCREB responses in HTM cells following 1 h DMSO or WIN (10 µM) pretreatment. **P < 0.01 compared with corresponding isoprenaline concentration pretreated with DMSO; n= 18–24. (C) pERK responses in HTM cells following a 10 min exposure to either aqueous vehicle and isoprenaline (100 nM) treatments, or to 0.05% DMSO vehicle and WIN (10 µM). Pretreatment with ICI (1 µM), CGP (1 µM), AM251 (1 µM) or AM630 (1 µM) was for 15 min and with PTx (100 ng·mL−1) for 24 h before isoprenaline or WIN application. PTx, ICI, CGP and AM251 bars are expressed as relative pERK level normalized to vehicle-treated cells that were also pretreated with the same antagonist or toxin to eliminate any affect these compounds may have had on the basal pERK levels. **P < 0.01 compared with vehicle, ***P < 0.001 compared with vehicle or DMSO; n= 8–27. (D) pERK dose–response in HTM cells following 10 min co-application of isoprenaline with 0.05% DMSO or 1 µM WIN; n= 13–16. (E) Isoprenaline pERK dose–response curves in HTM cells following 15 min pretreatment with either 0.05% DMSO or 1 µM AM251, followed by 5 min treatment with isoprenaline; n= 12–24. (F) Isoprenaline pERK dose–response curves in HTM cells following 15 min pretreatment with 0.05% DMSO or 1 µM O-2050 and 5 min exposure to isoprenaline; n= 12–16. AM251, N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide; CREB, cyclic AMP response element binding protein; DMSO, dimethylsulphoxide; ERK, extracellular signal-regulated kinase; HTM, human trabecular meshwork; O-2050, (6aR,10aR)-3-(1-methanesulfonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran; PTx, Pertussis toxin; WIN, WIN 55,212-2.
In order to assess the role CB1/β2-adrenoceptors heterodimerization might play in HTM cells, cross-desensitization of pCREB signalling induced by activation of β2-adrenoceptors, following treatment with the CB1 receptor agonist WIN was measured (Figure 7B). In these cells following a 1 h pretreatment with WIN (10 µM) the isoprenaline–pCREB response was significantly (P < 0.01) reduced. In contrast, WIN pretreatment had no effect on the forskolin-stimulated pCREB in these cells, indicating that the effect is not due to a non-specific effect on the pCREB signalling pathway by CB1 receptors. These findings indicate that, as in HEK 293 cells, the CB1 receptor agonist WIN produces a cross-desensitization of β2-adrenoceptors.
To demonstrate function of CB1 receptors and β2-adrenoceptors in HTM cells, the abilities of adrenergic and CB receptor ligands to affect pERK levels were examined using In-Cell Western analysis (Figure 7C). In these cells, isoprenaline treatment (100 nM) produced a significant decrease in basal pERK (P < 0.001) that was blocked by the selective β2-adrenoceptor antagonist ICI 118,551 (1 µM, P < 0.001), but not by the selective β1-adrenoceptor antagonist CGP 20712 (1 µM), or by PTx (100 ng·mL−1). In contrast, WIN treatment (10 µM) of HTM cells resulted in a significant increase in pERK (P < 0.001) that was blocked by the selective CB1 receptor inverse agonist AM251 (1 µM, P < 0.001) as well as by PTx (100 ng·mL−1, P < 0.001), but not by the selective CB2 receptor antagonist AM630 (1 µM). Treatment of HTM cells with AM251 did not alter basal pERK levels in HTM cells, suggesting CB1 receptors are not constitutively coupled to pERK signalling in these cells (data not shown). Together these findings demonstrate that both CB1 and β2AR are functionally expressed in HTM cells, and that activation of these receptors are capable of modulating pERK.
Isoprenaline-mediated pERK dose–response curves in HTM cells were generated in the presence of either DMSO vehicle or WIN (10 µM) (Figure 7D). The two isoprenaline dose–response curves were significantly different (P < 0.001) with values for pEC50 of 8.49 ± 0.08 and 8.91 ± 0.12; baseline pERK of 1.00 ± 0.01 and 1.28 ± 0.02; Emax of 0.75 ± 0.01 and 0.97 ± 0.02 for the curves in the presence of DMSO and WIN respectively. These data show that the WIN increase and the isoprenaline decrease in pERK inhibit each other to produce no net response when WIN and isoprenaline are applied together.
Isoprenaline-mediated pERK dose–response curves were generated in HTM cells following 15 min pretreatment with either DMSO vehicle, AM251 (1 µM),\ or O-2050 (1 µM) (Figure 7E and F). Pretreatment with AM251 resulted in a significantly altered isoprenaline dose–response curve in HTM cells (P < 0.001), which was unaffected by pretreatment with O-2050 (P > 0.05). Specifically, AM251 pretreatment resulted in an isoprenaline dose–response curve with a pEC50 of 9.10 ± 0.65 and a Hill coefficient of −0.34 ± 0.25, compared with the pEC50 of 8.02 ± 0.16 and Hill coefficient of −1.01 ± 0.32 for the DMSO vehicle-pretreated cells. These curves demonstrate that, as observed in HEK 293 cells, the CB1 receptor inverse agonist AM251 but not the CB1 receptor neutral antagonist O-2050 was capable of altering the pERK response to β2-adrenoceptors in HTM cells.
Discussion and conclusion
The ability of rhodopsin-like family A GPCRs to interact with each other as dimers or higher order oligomers has generated considerable interest in recent years. These complexes have received much attention, not only because they are formed by many different GPCRs, but also because they appear to influence nearly every aspect of GPCR function (Terrillon and Bouvier, 2004; Pfleger and Eidne, 2005). The findings presented here demonstrating that the CB1 receptor and β2-adrenoceptor BRET pair produce significantly increased BRETEff that is saturable and can be diluted by an untagged β2-adrenoceptor construct strongly suggests that CB1 receptors and β2-adrenoceptors can physically interact with each other. Similarly, the observed increased BRETEff values for CB1/CB1 receptor and β2/β2-adrenoceptor BRET pairs demonstrates that consistent with previous reports both of these receptors can also form homodimers in these cells (Hebert et al., 1996; Angers et al., 2000; Wager-Miller et al., 2002). In comparing the BRET saturation curves for the CB1 receptor homodimer with the CB1/β2-adrenoceptor heterodimer, it is apparent that the BRET50 value for the homodimer is lower than that of the heterodimer. This suggests that if these two receptors are expressed at similar levels, the CB1 receptor will preferentially form the homodimer over the heterodimer, although caution should be exercised in drawing such direct comparisons with two distinct BRET pairs (Mercier et al., 2002). In addition, the observation that treatment with the CB1 receptor inverse agonist AM251 altered BRETEff compared with either the agonist WIN or neutral antagonist O-2050, further supports the conclusion of a specific interaction, and implies that AM251 either facilitates increased interactions between the two receptors or alters the conformation of the heterodimer in such a way as to increase the BRETEff (Ayoub and Pfleger, 2009).
Functional interactions between CB1 receptors and β2-adrenoceptors were assessed by first examining the trafficking of these two receptors. When expressed in HEK 293 cells, CB1 receptors are highly constitutively active, resulting in constitutive internalization of the receptor (Leterrier et al., 2004; Ellis et al., 2006; Bohn, 2007). This is consistent with the punctate expression pattern observed when CB1-GFP2 was expressed in HEK 293H cells, which reverts to a cell surface expression pattern upon addition of the CB1 receptor inverse agonist AM251. In contrast, when β2-adrenoceptors are expressed in HEK 293 cells, they are predominantly localized to the cell surface (von Zastrow and Kobilka, 1992; Sunaguchi et al., 2003). This non-overlapping distribution of CB1-GFP2 and β2-adrenoceptors indicates that in order for these receptors to physically interact with each other, the subcellular expression of one receptor must be affected by the presence of the other. Our results demonstrate that when cells co-expressed CB1-GFP2 and HA-β2AR, CB1-GFP2 localization was indeed shifted towards the cell surface compared with when CB1-GFP2 was expressed alone, thus allowing for the physical interaction to take place. One possible mechanism for this would be that β2-adrenoceptors tempered the constitutive activity of CB1 receptors, as has been previously shown to occur in the β1/β2-adrenoceptor heterodimer (Zhu et al., 2005). This could explain why β2-adrenoceptors shift CB1 receptor expression towards the cell membrane, which does not seem to be the case when CB1 receptors interact with other GPCRs. For example, heterodimerization of CB1 receptors with the orexin-1 receptor did not affect the internalized localization of CB1 receptors, but instead altered the distribution of the orexin-1 receptor (Ellis et al., 2006). In addition, when CB1 receptors and another interacting GPCR, the µ-opioid receptor (Rios et al., 2006), were co-expressed neither CB1 nor µ-opioid receptor localization was affected, CB1 receptors remaining largely internalized while the µ-opioid receptors stayed at the cell surface (Ellis et al., 2006; Canals and Milligan, 2008).
Further support for the notion that CB1/β2-adrenoceptor dimerization attenuates CB1 receptor constitutive activity comes from the observation that basal pERK levels in CB1-GFP2/TreHA-β2AR cells were decreased following the induction of HA-β2AR expression, as was the ability of the CB1 receptor inverse agonist AM251 to decrease the basal pERK level. In addition, the fact that either CB1 or β2-adrenoceptor agonists were able to produce co-internalization of both receptors, a phenomenon reported for several other GPCR dimers (Terrillon and Bouvier, 2004), indirectly suggests that β2-adrenoceptors suppress constitutive activity of CB1 receptors. Specifically, as WIN produced co-internalization of β2-adrenoceptors and the constitutive trafficking of GPCRs generally follows a similar mechanism to agonist-induced receptor internalization (Leterrier et al., 2004; Marion et al., 2004), it should be expected that constitutive internalization of CB1 receptors would produce co-internalization of β2-adrenoceptors, as was the case when CB1 receptors were co-expressed with the orexin-1 receptor (Ellis et al., 2006). The fact that this was not observed, and that instead CB1 receptors were redistributed towards the cell surface, suggests that CB1 receptors are more likely to have a reduced constitutive internalization when β2-adrenoceptors are present.
G protein-coupled receptor heterodimerization may also influence the signalling pathways activated by the receptors present in the complex. In HEK 293H cells stably expressing CB1-GFP2 the cannabinoid agonist WIN produced both a PTx-sensitive increase in pERK and a PTx-insensitive increase in pCREB, indicating that CB1-GFP2 couples to both Gi/o and non-Gi/o pathways in these cells. Based on previous reports that in the presence of PTx, CB1 activates Gs to increase cAMP production (Maneuf and Brotchie, 1997; Jarrahian et al., 2004; Kearn et al., 2005), it is likely that the observed non-Gi/o pCREB–WIN response in these cells occurs via Gs. The fact that the induction of HA-β2AR expression in cells stably expressing CB1-GFP2 increased both the Emax and Hill coefficient of the WIN-mediated pERK dose–response, while decreasing the Emax and Hill coefficient of the WIN-mediated pCREB response suggests that HA-β2AR alters the G protein coupling preference of CB1 receptors. Specifically, the presence of β2-adrenoceptors shifts the Gi/o to Gs coupling ratio of CB1 receptors towards increased Gi/o, but decreased Gs coupling. GPCR heterodimerization has previously been shown to alter G protein coupling and similar signalling effects have previously been reported for both CB1 receptors as well as β2-adrenoceptors (Breit et al., 2004; Kearn et al., 2005; Mcgraw et al., 2006). It therefore is plausible that the physical interaction between CB1 receptors and β2-adrenoceptors directly affects CB1 receptor–G protein coupling, although it also cannot be ruled out that the shift in CB1 receptor coupling could be the result of Gs sequestration by β2-adrenoceptors (Vasquez and Lewis, 2003).
Similar to the changes observed in CB1 receptor signalling, β2-adrenoceptor signalling pathways were also affected by the presence of CB1 receptors. Co-expression of CB1 receptors and β2-adrenoceptors resulted in an increase in the isoprenaline-stimulated pCREB response with no change in the isoprenaline-mediated pERK response. The enhanced pCREB response is consistent with increased β2-adrenoceptor–Gs coupling that may result either directly from the physical interaction with CB1 receptors, or indirectly from decreased β2-adrenoceptor–Gi coupling caused by CB1 receptor–Gi sequestration (Vasquez and Lewis, 1999).
Co-application of WIN and isoprenaline to cells expressing both CB1-GFP2 and HA-β2AR resulted in an additive effect on pERK levels, but not on pCREB. The additive pERK response might be expected as WIN and isoprenaline activate pERK through different signalling pathways, WIN through PTx-sensitive Gi/o and isoprenaline through PTx-insensitive Gs or β-arrestin (Demuth and Molleman, 2006; Shenoy et al., 2006). However, the lack of an additive effect on pCREB is more interesting. Although activation of CB1 receptors has previously been shown to increase CREB phosphorylation (Casu et al., 2005), the underlying pathway has not been examined. Our observations suggest that the WIN-mediated pCREB response is not Gi/o-mediated, as it was insensitive to PTx, and that instead it is likely to be the result of CB1 receptor–Gs coupling. As β2-adrenoceptors also activates pCREB via Gs, the lack of an additive effect may be due to overlap in the two pathways. However, the fact that the pCREB response is not additive even at sub-maximal concentrations of isoprenaline suggests that instead it may arise from physiological antagonism resulting from CB1 receptor-induced activation of Gi, resulting in inhibition of adenylyl cyclase.
Another possible functional consequence of GPCR heterodimerization is altered receptor pharmacology of one or both receptors in the complex (Milligan, 2004; Terrillon and Bouvier, 2004). Our results demonstrate that the CB1 receptor inverse agonist AM251 inhibits β2-adrenoceptor-mediated pERK signalling, but had no effect on pCREB signalling, while the CB1 neutral antagonist O-2050 did not affect either β2-adrenoceptor-mediated pERK or pCREB activation. Recently, a similar result was reported where the CB1 receptor inverse agonist SR141716A enhanced pERK signalling of the µ-opioid receptor, while the neutral antagonist O-2050 had no effect (Canals and Milligan, 2008). In this study the result was attributed to the inverse agonist blocking constitutive activity of CB1 receptors and not to CB1/µ-opioid receptor heterodimerization because the two receptors were not detected in the same subcellular location. However, in the present study AM251 inhibited rather than enhanced the β2-adrenoceptor-mediated pERK response, suggesting that the AM251 effect is not simply the result of blocking CB1 receptor constitutive activity. Instead, our results are best explained by the concept that in a heterodimer, one GPCR can allosterically modulate the second receptor in the complex (Milligan and Smith, 2007). As an inverse agonist, such as AM251, will drive CB1 receptors from a constitutively active to an inactive state, it is conceivable that such a conformational change would, through heterodimerization, allosterically influence β2-adrenoceptors and their subsequent signalling. If so, it follows that the CB1 receptor neutral antagonist O-2050 should not alter the conformation of CB1 receptors, as a neutral antagonist shows no preference for either the active or inactive states of the receptor, and therefore should not influence β2-adrenoceptor signalling. Additional support for this conclusion may be drawn from the observation that AM251 treatment resulted in a change in the BRETEff compared with O-2050. Although alterations in BRETEff by ligands may be caused by changes in the number of receptors interacting as heterodimers, such ligand-mediated changes in BRETEff are now believed to be more representative of conformational changes within the heterodimer (Ayoub et al., 2002; Ayoub and Pfleger, 2009). Therefore, our observation that AM251 produces a change in BRETEff compared with O-2050 supports the notion that this inverse agonist is allosterically modulating the function of β2-adrenoceptors through the CB1/β2-adrenoceptor heterodimer.
CB1 receptors and β2-adrenoceptors are co-expressed in many tissues and cells, including parts of the cardiovascular system, female reproductive tract, brain, eye and bone (Jampel et al., 1987a; Wanaka et al., 1989; Tsou et al., 1998; Stamer et al., 2001; Wang et al., 2004; Pacher and Hasko, 2008). However, few studies have directly examined interactions between these two receptors in vivo. Instead, most work on interactions between the cannabinoid and adrenergic systems has focused on the presynaptic ability of CB receptors to inhibit noradrenergic neurotransmission (Schlicker et al., 1997; Schultheiss et al., 2005; Pakdeechote et al., 2007; Tam et al., 2008). We therefore examined the potential impact of a CB1/β2-adrenoceptor interaction on pERK signalling in primary HTM cells.
The trabecular meshwork represents the primary route of aqueous humour outflow in the human eye and as a result represents a key therapeutic target for the regulation of IOP and for the development of novel treatments for glaucoma (Kaufman et al., 1999; Ferrer, 2006). HTM cells have been shown to express β2-adrenoceptors, activation of which leads to increased cAMP levels and facilitates increased aqueous humour outflow (Jampel et al., 1987b; Erickson-Lamy and Nathanson, 1992). In addition, trabecular meshwork cells also express CB1 receptors, activation of which appears to increase aqueous humour outflow through activation of pERK (Straiker et al., 1999; Njie et al., 2006, Njie et al., 2008). Consistent with this previous work, our demonstration that HTM cells express both CB1 receptors and β2-adrenoceptors, that these receptors are functionally active in pERK signalling, and that they are co-localized at the cell membrane, suggests that any physical or functional interaction between these receptors may be relevant in HTM, and as a result in the regulation of aqueous humour outflow.
To determine if the co-internalization of CB1 receptors and β2-adrenoceptors observed in HEK 293H cells was also important to the function of these receptors in HTM cells, the ability of WIN to desensitize the isoprenaline-induced pCREB response in these cells was examined. As in HEK 293H cells, when HTM cells were pretreated with WIN, their isoprenaline–pCREB response was attenuated, while their response to forskolin was unaffected. Such cross-desensitization has previously been reported for other GPCR heterodimers (Pfeiffer et al., 2002) and supports the concept that CB1 receptors and β2-adrenoceptors form heterodimers in these cells, and further, that this heterodimer leads to cross-desensitization of β2-adrenoceptors.
To examine the functional interaction between CB1 receptors and β2-adrenoceptors in HTM cells, the ability of these receptors to affect pERK signalling was examined. In these cells, β2-adrenoceptor activation resulted in a PTx-insensitive decrease in pERK, while CB1 receptor activation led to a PTx-sensitive increase in pERK. As seen in the HEK 293H cells, co-application of the CB1 receptor and β2-adrenoceptor agonists to HTM cells produced an additive response; although given that the CB1 receptor response was positive while the β2-adrenoceptor response was negative in HTM cells, the overall results was net inhibitory. Also similar to that observed in HEK 293H cells, it was found that the CB1 receptor inverse agonist AM251 but not the neutral antagonist O-2050 altered the pERK response to the β2-adrenoceptor agonist isoprenaline. The combination of an increased pEC50, yet decreased Hill coefficient for the isoprenaline–pERK response when AM251 was present resulted in an increased ability of lower doses of isoprenaline to reduce pERK levels. Although this potentiation of the isoprenaline–pERK response by AM251 in HTM cells would seem to conflict with the inhibition AM251 observed in HEK 293H cells, as the HEK 293H–β2AR response was a positive pERK response, while the HTM–β2AR pERK response was negative it is conceivable that inhibition of a positive response and potentiation of a negative one could in fact be mediated by a similar mechanism.
Another interesting finding from our study is that the natures of ligand-induced effects differ depending on the cellular context. Other studies have demonstrated that both β2-adrenoceptor agonists and inverse agonists can activate the ERK pathway in HEK cells through multiple mechanisms (Azzi et al., 2003; Baker et al., 2003; Shenoy et al., 2006). However, to our knowledge, our findings in HTM cells are the first demonstration that a β2-adrenoceptor agonist can inhibit ERK phosphorylation, given the proper cellular context. Our observation that the CB1 receptor inverse agonist AM251 alters β2-adrenoceptor signalling in cells endogenously expressing both CB1 receptors and β2-adrenoceptors may be particularly relevant in vivo due to the interest in CB1 receptor inverse agonists for the clinical treatment of obesity, metabolic syndrome, type II diabetes and addiction (Xie et al., 2007; Le foll et al., 2008; Van diepen et al., 2008; Vemuri et al., 2008). Our results suggest that these CB1 receptor inverse agonists may exert their effects not only by directly blocking CB1 receptors, but also by indirectly affecting β2-adrenoceptor function in cells that co-express these receptors. In keeping with this, AM251 was found in one study to block the regional haemodynamic response to lipopolysaccharide in rats, a response that was attributed to β-adrenoceptor activation and was also blocked by the selective β2-adrenoceptor antagonist ICI 118,551 (Gardiner et al., 2005). In addition, the recent observation that CB1 receptors are expressed in murine cardiomyocytes (Mukhopadhyay et al., 2007) suggests that some of the cardiovascular effects of CB1 receptor inverse agonists (Van diepen et al., 2008) may be the result of indirect actions on β2-adrenoceptors in addition to the direct blockade of CB1 receptors.
The present study demonstrates a physical and functional interaction between CB1 receptors and β2-adrenoceptors. These results suggest a more complex picture than previously surmised in which interactions between the cannabinoid and the adrenergic systems were primarily attributed to CB1 receptor-mediated presynaptic inhibition of noradrenergic transmission. We suggest, in light of our findings and the wide biological and pharmacological importance of the β-adrenoceptor and endocannabinoid systems, a re-evaluation of the nature of previously reported functional interactions between the cannabinoid and β-adrenoceptor systems in cells and tissues that co-express these two receptors.
Acknowledgments
This study was supported by a Canadian Institutes of Health Research operating grant (MEMK) and by Natural Science and Engineering Council and Killam Trust student awards (BDH). TEH is a Chercheur National of the Fonds de la Recherche en Santé du Québec.
Glossary
Abbreviations:
- 2-AG
2-arachidonoylglycerol
- AEA
N-arachidonoylethanolamine, anandamide
- BRET
bioluminescence resonance energy transfer
- BRETEff
BRET efficiency
- CREB
cyclic AMP response element binding protein
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethylsulphoxide
- Dox
doxycycline
- ERK
extracellular signal-regulated kinase
- FBS
fetal bovine serum
- GPCR
G protein-coupled receptor
- HERG
human ether-a-go-go related gene
- HTM
human trabecular meshwork
- IOP
intraocular pressure
- mGluR6
metabotropic glutamate receptor 6
- PTx
Pertussis toxin
- Rluc
Renilla luciferase
- WIN
WIN 55,212-2
Statement of conflict of interests
The authors report no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, et al. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET) Proc Natl Acad Sci USA. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub MA, Pfleger KD. Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerization. Curr Opin Pharmacol. 2009;10:44–52. doi: 10.1016/j.coph.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Ayoub MA, Couturier C, Lucas-Meunier E, Angers S, Fossier P, Bouvier M, et al. Monitoring of ligand-independent dimerization and ligand-induced conformational changes of melatonin receptors in living cells by bioluminescence resonance energy transfer. J Biol Chem. 2002;277:21522–21528. doi: 10.1074/jbc.M200729200. [DOI] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bab I, Zimmer A. Cannabinoid receptors and the regulation of bone mass. Br J Pharmacol. 2008;153:182–188. doi: 10.1038/sj.bjp.0707593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol. 2003;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- Bohn LM. Constitutive trafficking – more than just running in circles? Mol Pharmacol. 2007;71:957–958. doi: 10.1124/mol.107.034223. [DOI] [PubMed] [Google Scholar]
- Borthne A. The treatment of glaucoma with propranolol (inderal). A clinical trial. Acta Ophthalmol (Copenh) 1976;54:291–300. doi: 10.1111/j.1755-3768.1976.tb01258.x. [DOI] [PubMed] [Google Scholar]
- Breit A, Lagace M, Bouvier M. Hetero-oligomerization between beta2- and beta3-adrenergic receptors generates a beta-adrenergic signaling unit with distinct functional properties. J Biol Chem. 2004;279:28756–28765. doi: 10.1074/jbc.M313310200. [DOI] [PubMed] [Google Scholar]
- Canals M, Milligan G. Constitutive activity of the cannabinoid CB1 receptor regulates the function of co-expressed mu opioid receptors. J Biol Chem. 2008;283:11424–11434. doi: 10.1074/jbc.M710300200. [DOI] [PubMed] [Google Scholar]
- Carriba P, Ortiz O, Patkar K, Justinova Z, Stroik J, Themann A, et al. Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacology. 2007;32:2249–2259. doi: 10.1038/sj.npp.1301375. [DOI] [PubMed] [Google Scholar]
- Casu MA, Pisu C, Sanna A, Tambaro S, Spada GP, Mongeau R, et al. Effect of delta9-tetrahydrocannabinol on phosphorylated CREB in rat cerebellum: an immunohistochemical study. Brain Res. 2005;1048:41–47. doi: 10.1016/j.brainres.2005.04.053. [DOI] [PubMed] [Google Scholar]
- Cota D. CB1 receptors: emerging evidence for central and peripheral mechanisms that regulate energy balance, metabolism, and cardiovascular health. Diabetes Metab Res Rev. 2007;23:507–517. doi: 10.1002/dmrr.764. [DOI] [PubMed] [Google Scholar]
- Demuth DG, Molleman A. Cannabinoid signalling. Life Sci. 2006;78:549–563. doi: 10.1016/j.lfs.2005.05.055. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Dupre DJ, Baragli A, Rebois RV, Ethier N, Hebert TE. Signalling complexes associated with adenylyl cyclase II are assembled during their biosynthesis. Cell Signal. 2007;19:481–489. doi: 10.1016/j.cellsig.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Ellis J, Pediani JD, Canals M, Milasta S, Milligan G. Orexin-1 receptor-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J Biol Chem. 2006;281:38812–38824. doi: 10.1074/jbc.M602494200. [DOI] [PubMed] [Google Scholar]
- Erickson-Lamy KA, Nathanson JA. Epinephrine increases facility of outflow and cyclic AMP content in the human eye in vitro. Invest Ophthalmol Vis Sci. 1992;33:2672–2678. [PubMed] [Google Scholar]
- Ferrer E. Trabecular meshwork as a new target for the treatment of glaucoma. Drug News Perspect. 2006;19:151–158. doi: 10.1358/dnp.2006.19.3.985929. [DOI] [PubMed] [Google Scholar]
- Gardiner SM, March JE, Kemp PA, Bennett T. Involvement of CB1-receptors and beta-adrenoceptors in the regional hemodynamic responses to lipopolysaccharide infusion in conscious rats. Am J Physiol Heart Circ Physiol. 2005;288:H2280–H2288. doi: 10.1152/ajpheart.00851.2004. [DOI] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Kano M. Endocannabinoids and synaptic function in the CNS. Neuroscientist. 2007;13:127–137. doi: 10.1177/1073858406296716. [DOI] [PubMed] [Google Scholar]
- Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, et al. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem. 1996;271:16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- Hepler RS, Frank IR. Marihuana smoking and intraocular pressure. JAMA. 1971;217:1392. [PubMed] [Google Scholar]
- James JR, Oliveira MI, Carmo AM, Iaboni A, Davis SJ. A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat Methods. 2006;3:1001–1006. doi: 10.1038/nmeth978. [DOI] [PubMed] [Google Scholar]
- Jampel HD, Lynch MG, Brown RH, Kuhar MJ, De Souza EB. Beta-adrenergic receptors in human trabecular meshwork. Identification and autoradiographic localization. Invest Ophthalmol Vis Sci. 1987a;28:772–779. [PubMed] [Google Scholar]
- Jampel HD, Lynch MG, Brown RH, Kuhar MJ, De Souza EB. Beta-adrenergic receptors in human trabecular meshwork. identification and autoradiographic localization. Invest Ophthalmol Vis Sci. 1987b;28:772–779. [PubMed] [Google Scholar]
- Jarrahian A, Watts VJ, Barker EL. D2 dopamine receptors modulate galpha-subunit coupling of the CB1 cannabinoid receptor. J Pharmacol Exp Ther. 2004;308:880–886. doi: 10.1124/jpet.103.057620. [DOI] [PubMed] [Google Scholar]
- Kaufman PL, Gabelt B, Tian B, Liu X. Advances in glaucoma diagnosis and therapy for the next millennium: new drugs for trabecular and uveoscleral outflow. Semin Ophthalmol. 1999;14:130–143. doi: 10.3109/08820539909061466. [DOI] [PubMed] [Google Scholar]
- Kearn CS, Blake-Palmer K, Daniel E, Mackie K, Glass M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Mol Pharmacol. 2005;67:1697–1704. doi: 10.1124/mol.104.006882. [DOI] [PubMed] [Google Scholar]
- Kunos G, Jarai Z, Batkai S, Goparaju SK, Ishac EJ, Liu J, et al. Endocannabinoids as cardiovascular modulators. Chem Phys Lipids. 2000;108:159–168. doi: 10.1016/s0009-3084(00)00194-8. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN 55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci USA. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie C, Mercier JF, Salahpour A, Umapathy D, Breit A, Villeneuve LR, et al. Beta 1/beta 2-adrenergic receptor heterodimerization regulates beta 2-adrenergic receptor internalization and ERK signaling efficacy. J Biol Chem. 2002;277:35402–35410. doi: 10.1074/jbc.M204163200. [DOI] [PubMed] [Google Scholar]
- Le Foll B, Forget B, Aubin HJ, Goldberg SR. Blocking cannabinoid CB1 receptors for the treatment of nicotine dependence: insights from pre-clinical and clinical studies. Addict Biol. 2008;13:239–252. doi: 10.1111/j.1369-1600.2008.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. Constitutive endocytic cycle of the CB1 cannabinoid receptor. J Biol Chem. 2004;279:36013–36021. doi: 10.1074/jbc.M403990200. [DOI] [PubMed] [Google Scholar]
- Mccarty CA, Burmester JK, Mukesh BN, Patchett RB, Wilke RA. Intraocular pressure response to topical beta-blockers associated with an ADRB2 single-nucleotide polymorphism. Arch Ophthalmol. 2008;126:959–963. doi: 10.1001/archopht.126.7.959. [DOI] [PubMed] [Google Scholar]
- Mcgraw DW, Mihlbachler KA, Schwarb MR, Rahman FF, Small KM, Almoosa KF, et al. Airway smooth muscle prostaglandin-EP1 receptors directly modulate beta2-adrenergic receptors within a unique heterodimeric complex. J Clin Invest. 2006;116:1400–1409. doi: 10.1172/JCI25840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcintosh BT, Hudson B, Yegorova S, Jollimore CA, Kelly ME. Agonist-dependent cannabinoid receptor signalling in human trabecular meshwork cells. Br J Pharmacol. 2007;152:1111–1120. doi: 10.1038/sj.bjp.0707495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K. Cannabinoid receptor homo- and heterodimerization. Life Sci. 2005;77:1667–1673. doi: 10.1016/j.lfs.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Mcvey M, Ramsay D, Kellett E, Rees S, Wilson S, Pope AJ, et al. Monitoring receptor oligomerization using time-resolved fluorescence resonance energy transfer and bioluminescence resonance energy transfer. the human delta-opioid receptor displays constitutive oligomerization at the cell surface, which is not regulated by receptor occupancy. J Biol Chem. 2001;276:14092–14099. doi: 10.1074/jbc.M008902200. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Brotchie JM. Paradoxical action of the cannabinoid WIN 55,212-2 in stimulated and basal cyclic AMP accumulation in rat globus pallidus slices. Br J Pharmacol. 1997;120:1397–1398. doi: 10.1038/sj.bjp.0701101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion S, Weiner DM, Caron MG. RNA editing induces variation in desensitization and trafficking of 5-hydroxytryptamine 2c receptor isoforms. J Biol Chem. 2004;279:2945–2954. doi: 10.1074/jbc.M308742200. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M. Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem. 2002;277:44925–44931. doi: 10.1074/jbc.M205767200. [DOI] [PubMed] [Google Scholar]
- Miller JW. Tracking G protein-coupled receptor trafficking using odyssey imaging. Li-Cor Bioscience. 2004 Available at http://www.licor.com/bio/PDF/Miller_GPCR.pdf (accessed August 2008). [Google Scholar]
- Milligan G. G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol. 2004;66:1–7. doi: 10.1124/mol.104.000497.. [DOI] [PubMed] [Google Scholar]
- Milligan G, Smith NJ. Allosteric modulation of heterodimeric G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:615–620. doi: 10.1016/j.tips.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Batkai S, Rajesh M, Czifra N, Harvey-White J, Hasko G, et al. Pharmacological inhibition of CB1 cannabinoid receptor protects against doxorubicin-induced cardiotoxicity. J Am Coll Cardiol. 2007;50:528–536. doi: 10.1016/j.jacc.2007.03.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njie YF, Kumar A, Qiao Z, Zhong L, Song ZH. Noladin ether acts on trabecular meshwork cannabinoid (CB1) receptors to enhance aqueous humor outflow facility. Invest Ophthalmol Vis Sci. 2006;47:1999–2005. doi: 10.1167/iovs.05-0729. [DOI] [PubMed] [Google Scholar]
- Njie YF, He F, Qiao Z, Song ZH. Aqueous humor outflow effects of 2-arachidonylglycerol. Exp Eye Res. 2008;87:106–114. doi: 10.1016/j.exer.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Pacher P, Hasko G. Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br J Pharmacol. 2008;153:252–262. doi: 10.1038/sj.bjp.0707582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakdeechote P, Dunn WR, Ralevic V. Cannabinoids inhibit noradrenergic and purinergic sympathetic cotransmission in the rat isolated mesenteric arterial bed. Br J Pharmacol. 2007;152:725–733. doi: 10.1038/sj.bjp.0707397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pate DW, Jarvinen K, Urtti A, Mahadevan V, Jarvinen T. Effect of the CB1 receptor antagonist, SR141716A, on cannabinoid-induced ocular hypotension in normotensive rabbits. Life Sci. 1998;63:2181–2188. doi: 10.1016/s0024-3205(98)00499-8. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M, Koch T, Schroder H, Laugsch M, Hollt V, Schulz S. Heterodimerization of somatostatin and opioid receptors cross-modulates phosphorylation, internalization, and desensitization. J Biol Chem. 2002;277:19762–19772. doi: 10.1074/jbc.M110373200. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Eidne KA. Monitoring the formation of dynamic G-protein-coupled receptor-protein complexes in living cells. Biochem J. 2005;385:625–637. doi: 10.1042/BJ20041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay D, Kellett E, Mcvey M, Rees S, Milligan G. Homo- and hetero-oligomeric interactions between G-protein-coupled receptors in living cells monitored by two variants of bioluminescence resonance energy transfer (BRET): hetero-oligomers between receptor subtypes form more efficiently than between less closely related sequences. Biochem J. 2002;365:429–440. doi: 10.1042/BJ20020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios C, Gomes I, Devi LA. Mu opioid and CB1 cannabinoid receptor interactions: reciprocal inhibition of receptor signaling and neuritogenesis. Br J Pharmacol. 2006;148:387–395. doi: 10.1038/sj.bjp.0706757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy AA, Baragli A, Bernstein LS, Hepler JR, Hebert TE, Chidiac P. RGS2 interacts with gs and adenylyl cyclase in living cells. Cell Signal. 2006;18:336–348. doi: 10.1016/j.cellsig.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Timm J, Zentner J, Gothert M. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn Schmiedebergs Arch Pharmacol. 1997;356:583–589. doi: 10.1007/pl00005093. [DOI] [PubMed] [Google Scholar]
- Schultheiss T, Flau K, Kathmann M, Gothert M, Schlicker E. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in guinea-pig vessels, but not in rat and mouse aorta. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:139–146. doi: 10.1007/s00210-005-0007-4. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, et al. Beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Stamer WD, Golightly SF, Hosohata Y, Ryan EP, Porter AC, Varga E, et al. Cannabinoid CB(1) receptor expression, activation and detection of endogenous ligand in trabecular meshwork and ciliary process tissues. Eur J Pharmacol. 2001;431:277–286. doi: 10.1016/s0014-2999(01)01438-8. [DOI] [PubMed] [Google Scholar]
- Straiker AJ, Maguire G, Mackie K, Lindsey J. Localization of cannabinoid CB1 receptors in the human anterior eye and retina. Invest Ophthalmol Vis Sci. 1999;40:2442–2448. [PubMed] [Google Scholar]
- Sunaguchi M, Nishi M, Mizobe T, Kawata M. Real-time imaging of green fluorescent protein-tagged beta 2-adrenergic receptor distribution in living cells. Brain Res. 2003;984:21–32. doi: 10.1016/s0006-8993(03)03004-x. [DOI] [PubMed] [Google Scholar]
- Szczesniak AM, Kelly ME, Whynot S, Shek PN, Hung O. Ocular hypotensive effects of an intratracheally delivered liposomal delta9-tetrahydrocannabinol preparation in rats. J Ocul Pharmacol Ther. 2006;22:160–167. doi: 10.1089/jop.2006.22.160. [DOI] [PubMed] [Google Scholar]
- Tam J, Trembovler V, Di Marzo V, Petrosino S, Leo G, Alexandrovich A, et al. The cannabinoid CB1 receptor regulates bone formation by modulating adrenergic signaling. FASEB J. 2008;22:285–294. doi: 10.1096/fj.06-7957com. [DOI] [PubMed] [Google Scholar]
- Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Van Diepen H, Schlicker E, Michel MC. Prejunctional and peripheral effects of the cannabinoid CB(1) receptor inverse agonist rimonabant (SR 141716) Naunyn Schmiedebergs Arch Pharmacol. 2008;378:345–369. doi: 10.1007/s00210-008-0327-2. [DOI] [PubMed] [Google Scholar]
- Vasquez C, Lewis DL. The CB1 cannabinoid receptor can sequester G-proteins, making them unavailable to couple to other receptors. J Neurosci. 1999;19:9271–9280. doi: 10.1523/JNEUROSCI.19-21-09271.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez C, Lewis DL. The beta2-adrenergic receptor specifically sequesters gs but signals through both gs and Gi/o in rat sympathetic neurons. Neuroscience. 2003;118:603–610. doi: 10.1016/s0306-4522(03)00024-1. [DOI] [PubMed] [Google Scholar]
- Vemuri VK, Janero DR, Makriyannis A. Pharmacotherapeutic targeting of the endocannabinoid signaling system: drugs for obesity and the metabolic syndrome. Physiol Behav. 2008;93:671–686. doi: 10.1016/j.physbeh.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager-Miller J, Westenbroek R, Mackie K. Dimerization of G protein-coupled receptors: CB1 cannabinoid receptors as an example. Chem Phys Lipids. 2002;121:83–89. doi: 10.1016/s0009-3084(02)00151-2. [DOI] [PubMed] [Google Scholar]
- Wanaka A, Kiyama H, Murakami T, Matsumoto M, Kamada T, Malbon CC, et al. Immunocytochemical localization of beta-adrenergic receptors in the rat brain. Brain Res. 1989;485:125–140. doi: 10.1016/0006-8993(89)90674-4. [DOI] [PubMed] [Google Scholar]
- Wang H, Guo Y, Wang D, Kingsley PJ, Marnett LJ, Das SK, et al. Aberrant cannabinoid signaling impairs oviductal transport of embryos. Nat Med. 2004;10:1074–1080. doi: 10.1038/nm1104. [DOI] [PubMed] [Google Scholar]
- Wax MB, Molinoff PB, Alvarado J, Polansky J. Characterization of beta-adrenergic receptors in cultured human trabecular cells and in human trabecular meshwork. Invest Ophthalmol Vis Sci. 1989;30:51–57. [PubMed] [Google Scholar]
- Woodward DF, Gil DW. The inflow and outflow of anti-glaucoma drugs. Trends Pharmacol Sci. 2004;25:238–241. doi: 10.1016/j.tips.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Ji X, Lakatta EG. Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol. 1995;47:322–329. [PubMed] [Google Scholar]
- Xie S, Furjanic MA, Ferrara JJ, Mcandrew NR, Ardino EL, Ngondara A, et al. The endocannabinoid system and rimonabant: a new drug with a novel mechanism of action involving cannabinoid CB1 receptor antagonism – or inverse agonism – as potential obesity treatment and other therapeutic use. J Clin Pharm Ther. 2007;32:209–231. doi: 10.1111/j.1365-2710.2007.00817.x. [DOI] [PubMed] [Google Scholar]
- von Zastrow M, Kobilka BK. Ligand-regulated internalization and recycling of human beta 2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J Biol Chem. 1992;267:3530–3538. [PubMed] [Google Scholar]
- Zhu WZ, Chakir K, Zhang S, Yang D, Lavoie C, Bouvier M, et al. Heterodimerization of beta1- and beta2-adrenergic receptor subtypes optimizes beta-adrenergic modulation of cardiac contractility. Circ Res. 2005;97:244–251. doi: 10.1161/01.RES.0000176764.38934.86. [DOI] [PubMed] [Google Scholar]