

Abstract
Background and purpose:
Methamphetamine (METH) is a psychostimulant amphetamine that causes long-term dopaminergic neurotoxicity in mice. Hypodopaminergic states have been demonstrated to increase voluntary ethanol (EtOH) consumption and preference. In addition, the endocannabinoid system has been demonstrated to modulate EtOH drinking behaviour. Thus, we investigated EtOH consumption in METH-lesioned animals and the role of cannabinoid (CB) signalling in this EtOH drinking.
Experimental approach:
Mice were treated with a neurotoxic regimen of METH, and 7 days later exposed to increasing concentrations of drinking solutions of EtOH (3, 6, 10 and 20%). Seven days after neurotoxic METH, the following biochemical determinations were carried out in limbic forebrain: CB1 receptor density and stimulated activity, 2-arachidonoyl glycerol (2-AG) and monoacylglycerol lipase (MAGL) activity, dopamine levels and dopamine transporter density.
Key results:
EtOH consumption and preference were increased in METH-treated mice. Seven days after METH, a time at which both dopamine levels and density of dopamine transporters in limbic forebrain were decreased, CB1 receptor density and activity were unaltered, but 2-AG levels were increased. At this same time-point, MAGL activity was reduced. The CB1 receptor antagonist AM251 prevented the METH-induced increase in EtOH consumption and preference, while N-arachidonoyl maleimide, an inhibitor of MAGL, increased EtOH consumption and preference in both saline- and METH-treated mice.
Conclusions and implications:
An increase in endocannabinoid tone may be involved in the increased consumption of and preference for EtOH displayed by METH-lesioned mice as blockade of the CB1 receptor decreased EtOH-seeking behaviours, whereas the MAGL inhibitor increased EtOH consumption.
This article is part of a themed issue on Cannabinoids. To view the editorial for this themed issue visit http://dx.doi.org/10.1111/j.1476-5381.2010.00831.x
Keywords: methamphetamine, ethanol consumption and preference, dopamine neurotoxicity, cannabinoid 1 receptor, 2-arachidonoyl glycerol, monoacylglycerol lipase, fatty acid amide hydrolase
Introduction
Methamphetamine (METH) is a dimethyl substituted phenylethylamine, with high abuse potential and is increasingly being manufactured and used illicitly. The United Nations Office on Drugs and Crime (UNODC) conservatively estimates that there are between 15 and 16 million METH users worldwide, a figure similar to those for heroin and cocaine (UNODC, 2008). Repeated administration of METH to mice produces a neurodegeneration of dopamine axons and terminals in the striatum and cell body loss in the substantia nigra (Sonsalla et al., 1996; Hirata and Cadet, 1997). Damage has been demonstrated both histologically (O'Callaghan and Miller, 1994; Granado et al., 2009) and biochemically (Sonsalla et al., 1989; Mann et al., 1997; Hogan et al., 2000; Itzhak et al., 2000; Staal et al., 2000), and is reflected as a substantial decrease in the concentration of dopamine and its metabolites, a loss in the density of plasmalemmal and vesicular dopamine transporters and a decrease in tyrosine hydroxylase activity.
3,4-Methylenedioxymethamphetamine (MDMA) is another amphetamine derivative, which exerts similar and selective persistent neurochemical effects to METH in the brain mouse. We recently reported that mice exposed to a neurotoxic dose of MDMA exhibit higher consumption of and preference for ethanol (EtOH) compared with saline-treated mice (Izco et al., 2007). Differences between saline- and MDMA-treated animals did not appear to be secondary to changes in acute EtOH clearance, and did not reflect a more global change in taste preferences or a need to consume more calories. Administration of the full D1 agonist, SKF81297, is sufficient to partially reduce EtOH consumption in MDMA-lesioned mice, suggesting that the impairment is caused, at least in part, by a reduction in the stimulation of D1 receptors subsequent to a deficit in dopamine neurotransmission. The reversal of the D1 receptor agonist effect by SCH 23390, a D1 receptor antagonist, confirms the actions are due to stimulation of the D1 receptor rather than non-specific effects (Izco et al., 2007).
Endocannabinoid signal transduction mediated by CB1 receptors also plays a key role in the modulation of the rewarding effects of EtOH. Thus, CB1 knock-out mice show reduced EtOH consumption (Hungund et al., 2003; Thanos et al., 2005), and do not release dopamine in the nucleus accumbens in response to the administration of EtOH. The density of CB1 receptors and agonist-stimulated [35S]-GTPγS binding was reduced in the brain of DBA/2 (alcohol-avoiding) mice compared with C57BL/6 (alcohol-preferring) mice (Hungund and Basavarajappa, 2000; Basavarajappa and Hungund, 2001). In line with this, the CB1 antagonist, SR141716A, reduces the voluntary consumption of EtOH in C57BL/6 mice (Arnone et al., 1997), in Sardinian alcohol-preferring rats (Colombo et al., 1998) and in Long Evans rats (Freedland et al., 2001), suggesting that inhibiting the functionality of CB1 receptors reduces the motivation to consume EtOH. On the other hand, the acute administration of CB1 receptor agonists such as CP55940 or WIN 55212-2 increases the motivation to consume EtOH in Wistar rats and in EtOH-preferring rats, the effect being completely prevented by treatment with the CB1 antagonist, SR141617A (Gallate et al., 1999; Colombo et al., 2002). All these data suggest a specific role for CB1 receptors in EtOH-seeking behaviours.
We have now examined EtOH consumption and preference in METH-lesioned mice, and determined whether changes in these parameters are related to alterations in the cannabinoid system mediated by CB1 receptors. To this end, we studied the changes induced by METH on the density and function of CB1 receptors and on the levels of the 2-arachidonoyl glycerol (2-AG) and anandamide (AEA) in the limbic forebrain of mice with an established dopamine lesion induced by the drug. These endocannabinoids have been repeatedly involved in the motivating properties of EtOH and in the development of tolerance, dependence and relapse to alcohol (González et al., 2004;Basavarajappa and Hungund, 2005, Cailléet al., 2007). The activities of MAGL and fatty acid amide hydrolase (FAAH), the main enzymes involved in endocannabinoid metabolism, were also quantified. The ability of the CB1 antagonist AM251 [N-(piperidin-1-yl)-5-(4-iodophonyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] and of the inhibitor of monoacylglycerol lipase, N-arachidonoyl maleimide (NAM) to modulate EtOH intake and preference in METH-lesioned mice was also evaluated.
Methods
Animals, drug administration and experimental protocol
Adult male C57BL/6J mice (Harlan Iberica, Barcelona, Spain) initially weighing 25–30 g were housed in groups of 10, in standard cages (38 cm long × 22 cm wide × 15 cm high), in conditions of constant temperature (21 ± 2°C) and a 12 h light/dark cycle (lights on: 08 h 00 min) and given free access to food and water. The mice were randomly assigned to two treatment groups. Group I was injected with saline, while group II received METH (8 mg·kg−1, i.p., three times with a 3 h interval). Seven days later, the mice were given access to voluntary consume EtOH. Independent groups were killed 1 and 4 weeks after METH administration for determination of neurotoxicity and cannabinoid parameters. The protocol of METH administration used in this study induces neurotoxicity in dopamine nerve terminals 7 days later (Sanchez et al., 2003; Granado et al., 2009).
METH was dissolved in 0.9% w/v NaCl (saline) and injected at a volume of 10 mL·kg−1. Doses were quoted in terms of the base. EtOH drinking solutions were made up from absolute EtOH, and diluted with tap water to different concentrations. For injections, absolute EtOH was diluted with 0.9% saline to a 20% w/v solution. Quinine and sucrose were dissolved in tap water.
All experimental procedures were performed in accordance with the guidelines of the Animal Welfare Committee of the Universidad Complutense de Madrid (following European Council Directives 86/609/CEE and 2003/65/CE). Drug/molecular target nomenclature conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2008).
EtOH intake test
Voluntary EtOH consumption and preference were examined using a two-bottle free-choice paradigm (Naassila et al., 2002). Fluid was supplied by means of plastic serological pipettes of 25 mL graduated at 0.2 mL intervals (construction of the drinking tubes followed the method by Tordoff and Bachmanov (2005), and is given in detail on the Website http://www.monell.org/MMTPP.
Three days after METH or saline administration, the mice were individually housed in polycarbonate mouse cages (type 2, 22.0 cm long × 16.5 cm wide × 14 cm high, floor space 363 cm2, EH-II-1264, EHRET Gmbh & Co. KG Labor- und Pharmatechnik, Emmendingen, Germany) with standard rodent chow available ad libitum and habituated to drinking for 4 days from two pipettes of 25 mL, containing plain tap water, placed on the stainless steel cage top. Then, the mice were given access to two pipettes, one pipette containing plain water and the other EtOH in water. The EtOH concentration (v/v) was increased every 7 days, and the mice received solutions containing 3, 6, 10 and finally 20% EtOH over the course of the experiment. All drinking solutions were renewed every 2 days, at which time the position of the bottles was changed to avoid side preferences. Fluid intake was determined daily. Water consumption was calculated throughout the experiment. To obtain a measure of EtOH consumption that corrected for individual differences in mouse size, grams of EtOH consumed per kilogram of body weight per day were calculated for each mouse every day. Average EtOH consumption per day was calculated for each EtOH concentration. The EtOH preference ratio was calculated for each EtOH concentration as volume of EtOH solution consumed per total volume of fluid (water plus EtOH solution) consumed. At days 4 and 7 of each 7 day interval, the body weight was assessed. The cages were cleaned at 7 day intervals in the middle of each EtOH block to avoid interaction with changes in EtOH concentrations.
Pharmacological experiments with the highly selective CB1 receptor antagonist, AM251 (Lan et al., 1999) were performed in order to address the hypothesis that CB1 activation is involved in the observed changes in EtOH consumption in METH-lesioned mice. In these experiments, the mice were exposed to solutions containing 10% EtOH for 3 days; AM251 (5 mg·kg−1, i.p.) was given each of the 3 days before the onset of the dark–light cycle at a volume of 10 mL·kg−1. The lyophilized AM251 was first dissolved in dimethylsulphoxide (DMSO), and then a solution containing 0.9% (w/v) NaCl and Tween 80 (9:1) was slowly added to yield a final concentration of 0.125% DMSO for the vehicle. The control mice received vehicle at the same volume. The dose of AM251 used has been shown previously to be effective at inhibiting the actions of CB1 agonists (Landa et al., 2006).
NAM, a monoacylglycerol lipase inhibitor, was used to produce an increase in 2-AG content. In these experiments, the mice were exposed to solutions containing 6% EtOH for 3 days; NAM (3 mg·kg−1, i.p.) was given on each of the 3 days before the onset of the dark–light cycle at a volume of 10 mL·kg−1. NAM was first dissolved in EtOH, and then a solution 0.9% (w/v) NaCl was added to yield a final concentration of 1% EtOH for the vehicle. The control mice received vehicle at the same volume. The dose of NAM used has been shown previously to be effective at increasing 2-AG levels in mouse brain (Burston et al., 2008).
Sucrose and quinine consumption test
Three days after METH or saline administration, the mice were habituated in their home cage to drinking from two pipettes containing water for 4 days, and were then given plain tap water in one pipette and sucrose or quinine in the other pipette, using a protocol similar to that described for EtOH consumption. Two sucrose concentrations (0.17 and 1.7%) and two quinine concentrations (0.03 and 0.1 mM) were tested in the two-bottle preference test according to previously described protocols (Naassila et al., 2002). The mice had 5 days of access to each solution. The preference for each solution was calculated by dividing the volume of the taste solution consumed by the total volume of fluid (taste solution plus water) consumed.
Plasma EtOH concentration
To determine whether METH-pretreated mice metabolized EtOH differently from the controls, 7 days after METH and saline administration, the mice were administered an EtOH (3 g·kg−1, i.p., 20% w/v in saline). Samples of 20 µL of blood were collected from the tail in heparin-treated capillary tubes between 30 and 270 min after injection. At the end of collection, the samples were centrifuged for 6 min (Microcentrifuge MK5, model 01400-00, Analox, London, UK) and injected into an analyser (AM1, Analox). The rationale of the method consists of EtOH being oxidized by the enzyme alcohol oxidase in the presence of molecular oxygen; therefore, the rate of oxygen consumption is directly proportional to alcohol concentration. Plasma EtOH levels were calculated as mg·dL−1 using EtOH 300 mg·dL−1 as standard.
Dissection of limbic forebrain
The mice were killed by cervical dislocation and decapitation, and the brains were rapidly removed. After removal of the cerebellum and olfactory bulbs, a coronal cut across the optic chiasma using a mouse brain matrix (WPI, Aston, Stevenage, UK) was made. Forebrain tissue containing the nucleus accumbens and the anterior cingulate cortex was referred to as limbic forebrain (Wang et al., 2003).
Measurement of dopamine and metabolites
Catechol concentration was evaluated 1 and 4 weeks after METH exposure in order to determine the long-lasting depletion of dopamine concentration exerted by METH in the limbic forebrain. In addition, dopamine concentration was always evaluated at the end of each experimental protocol to confirm neurotoxicity (data not shown).
Dopamine and the metabolites, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), were measured by HPLC and electrochemical detection. The mobile phase consisted of KH2PO4 (0.05 M), octanesulphonic acid (0.4 mM), EDTA (0.1 mM) and methanol (16%), and was adjusted to pH 3 with phosphoric acid, filtered and degassed. The flow rate was 1 mL·min−1. The HPLC system consisted of a pump (Waters 510, Waters, Barcelona, Spain) linked to an automatic sample injector (Loop 200 µL, Waters 717 plus Autosampler), a stainless steel reversed-phase column (Spherisorb ODS2, 5 µm, 150 × 4.6 mm; Waters) with a precolumn and a coulometric detector (Coulochem II, Esa, Chelmsford, MA, USA). The working electrode potential was set at 400 mV with a gain of 1 µA (for dopamine) and 500 nA (for the remaining compounds). The current produced was monitored by using integration software (Clarity, DataApex, Prague, Czech Republic).
[3H]-WIN 35428 binding
[3H]-WIN 35428 binding was measured by modification of the method described in detail by Segal et al. (2003), and used as an index of dopamine transporter density. One and four weeks after METH administration, the animals were killed, and their brains were rapidly removed and dissected on ice within 2 min. Limbic forebrain was sonicated in ice-cold sodium phosphate buffer (20 mM; pH 7.4) containing sucrose (0.32 M). The homogenate was centrifuged at 30 000×g for 15 min at 4°C. The supernatant was discarded and the wash procedure was repeated twice more. The pellet was finally resuspended in 60 volumes of homogenization buffer. The assay solution (500 µL) contained [3H]-WIN 35428 (5 nM), desipramine (300 nM) and 100 µL tissue preparation (approx. 80 µg protein). Non-specific binding was carried out in the presence of cocaine (30 µM). The reaction mixture was incubated for 90 min at 4°C. The assay was terminated by rapid filtration, and radioactivity was counted by scintillation spectrometry.
Quantitative analysis of endocannabinoid levels
The LC–ESI–MS/MS method used for endocannabinoid analysis in rat brain tissue was based on the method of Richardson et al. (2007) with a minor change in sample extraction procedure. The samples were dissected on ice, stored at −80°C and thawed on ice immediately before analysis. Internal standards [10 µL of 2-AG-d8 (100 µM) and 15 µL of arachidonoyl ethanolamine (AEA)-d8 (28 µM)] were added to each sample, blank sample (0.4 mL water) and QC sample (0.4 mL human plasma). Ethyl acetate/hexane (9:1 v/v, 1.2 mL) was added to each sample followed by sonication on ice, then 0.4 mL water was added to each tube, and samples were vortexed and sonicated for 30 min and then centrifuged (7000×g for 10 min at 4°C). The extraction was repeated twice, and the supernatant was pooled and evaporated to dryness in a vacuum evaporator at room temperature (20°C). Prior to analysis, each sample extract was reconstituted in 50 µL of acetonitrile, of which 10 µL was used for analysis by HPLC (Agilent 1100; Agilent Technologies, Waldbron, Germany), in line with a mass spectrometry system consisting of a Micromass Quattro Ultima triple quadrupole mass spectrometer (Waters, Manchester, UK) equipped with an electrospray ionization (ESI) interface. Liquid chromatographic separation of endocannabinoids was achieved by using a Thermo Hypersil-Keystone Hypurity Advance column (100 × 2.1 mm, 3 µm) with guard column (C8 Hypurity Advance cartridges, Thermo Fisher Scientific, Runcorn, UK). Mobile phase A consisted of an aqueous solution of ammonium acetate (1 g·L−1) and formic acid (0.1%); mobile phase B was composed of ammonium acetate (1 g·L−1) and formic acid (0.1%) dissolved in acetonitrile. The flow rate was 300 µL·min−1, and column pressure was 80–90 bars. AEA, 2-AG, AEA-d8 (internal standard) and 2-AG-d8 (internal standard) were supplied by Axxora (Bingham, Nottingham, UK). Two batches of quality control human plasma standard samples were used to confirm the day-to-day accuracy of the method, one batch containing an endocannabinoid concentration in the low range and the other with concentrations of endocannabinoids in the high range in human plasma. Quantification of the endocannabinoids was by the internal standard method, using fully extracted calibration standards for each of the endocannabinoids.
CB1 binding
The density of CB1 receptors was assessed base on the method previously described by Wang et al. (2003). Briefly, brain tissue samples from individual mice were homogenized in 3 mL of ice-cold TME (50 mM Tris, 1 mM EDTA, 3 mM MgCl2, pH 7.4) buffer containing 320 mM sucrose. The homogenate was centrifuged at 40 000×g for 15 min at 4°C. The pellet was then resuspended in the same buffer and washed twice more with subsequent centrifugations. The pellet was finally resuspended in TME buffer to yield brain homogenates with a protein concentration of 0.5 mg·mL−1 after protein determination by the Bradford method. Binding was carried out with 50 µg of membrane protein in TME buffer supplemented with 5 mg·mL−1 of fatty acid-free BSA in the presence of a saturating concentration (5 nM) of [3H]-N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride ([3H]-SR141716A) in a total volume of 500 µL at 30°C for 1 h, and the reaction was stopped by the addition of 2 mL of ice-cold buffer (50 mM Tris–HCl, 1 mg·mL−1 BSA, pH 7.4) followed by vacuum filtration through fibreglass filters (Filter Mat 11731, Skatron Instruments A.S, Lierbyen, Norway) pre-soaked in 0.1% polyethylenimine. Non-specific binding was measured in the presence of 10 µM unlabelled AM251. Retained radioactivity was quantified by liquid scintillation spectrometry after extraction overnight in 2 mL of scintillation liquid. All binding assays were performed in triplicate. Specific binding was defined as the difference between total binding and non-specific binding, and was expressed as fmol [3H]-SR141617A mg−1 protein. The 5 nM saturating concentration of the radioligand was determined from saturation curves constructed using different concentrations (0.2–10 nM) of the CB1 receptor agonist [3H]-SR141716A using untreated mouse brain membranes prepared in a similar way. Data were analysed by computer-assisted non-linear regression analysis using GraphPad Prism software (San Diego, CA, USA), and receptor density (Bmax) and affinity constant (KD) data were found to be 1725 ± 76 fmol·mg−1 protein and 1.10 ± 0.10 nM respectively.
[35S]-GTPγS binding
Agonist-stimulated [35S]-GTPγS binding was measured by the method adapted from that of Sim et al. (1995). Briefly, an aliquot of 50 µg of the same membrane protein used for CB1 binding for each sample was incubated in 500 µL of assay buffer (50 mM Tris–HCl, 0.2 mM EGTA, 3 mM MgCl2, 100 mM NaCl, pH 7.4) containing 1% fatty acid-free BSA and 30 µM GDP. After 15 min of incubation at room temperature, 0.64 nM [35S]-GTPγS was added to the reaction tubes in the presence or absence of 100 nM (6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol (HU210), and 10 µM unlabelled GTPγS was added for the determination of non-specific binding. Triplicate aliquots were incubated at 30°C for 1 h, and the reaction was terminated by the addition of 400 µL of ice-cold 50 mM Tris–HCl buffer, pH 7.4, followed by vacuum filtration through fibreglass filters (Filter Mat 11731, Skatron Instruments A.S) pre-soaked in 0.1% polyethylenimine. Bound radioactivity was quantified by liquid scintillation spectrometry after extraction overnight in 2 mL of scintillation liquid as before. As for the above assay, 30 µM GDP concentration was defined from previous experiments using a range of GDP concentrations of 0.1–100 µM. HU210-stimulated [35S]-GTPγS binding was defined as a percentage increase above basal levels.
FAAH activity
FAAH activity was measured by determining the conversion of AEA to arachidonic acid and ethanolamine by brain homogenates (30 µg of protein), which were incubated in a 200 µL reaction volume during 30 min at 37°C and pH 7.6 in the presence of a final concentration of 16 µM AEA mixture (non-radioactive AEA containing trace amounts of [3H]-AEA). The reaction was stopped by placing the tubes in ice, and after the addition of two volumes of chloroform/methanol (1:1), vortexing and centrifugation, 200 µL of the methanol/buffer phase containing the [3H]-ethanolamine released in the reaction was analysed by liquid scintillation counting. FAAH activity was expressed as nanomoles of product formed per minute and per milligram protein (Rubio et al., 2009).
MAGL activity
MAGL activity was analysed as described by Ghafouri et al. (2004) with minor modifications. The technique is based on the measurement of the [3H]-glycerol formed from hydrolysis of 2-oleoyl-[3H]-glycerol ([3H]-2-OG). Limbic forebrain was homogenized in 50 mM Tris pH 8 containing 320 mM sucrose and protein inhibitors. After centrifugation at 100 000×g for 60 min at 4°C, 16 µg of the cytosolic protein fraction was added to a 16 µM solution containing a mixture of unlabelled 2-OG and [3H]-2OG, and the enzymatic reaction was performed in a 10 mM Tris–HCl buffer, pH 7.2, containing 1 mM EDTA, 0.1% fatty acid-free BSA and 5% DMSO for 10 min at 37°C. After that, the reaction was stopped by adding 400 µL of chloroform : methanol (1:1). The samples were centrifuged (1000×g for 10 min), and a 200 µL aliquot of the upper aqueous phase was counted. MAGL activity was expressed as nmol·mg−1 protein·min−1.
Statistics
Statistical analyses of EtOH consumption, preference measurements and EtOH plasma levels were performed using the statistical computer package BMDP/386 Dynamic (BMDP Statistical Solutions, Cork, Ireland). Data were analysed by anova with repeated measures (program 5V). Both used treatment as the between-subjects factor, and EtOH or time concentration as the repeated measure. Data from sucrose and quinine preference, CB1 density and activity and MAGL and FAAH activities were analysed using two-way anova followed by Bonferroni test. The same method was used to compare data from dopamine transporter density, dopamine and metabolite levels. The effects of the CB1 antagonist AM251 and the MAGL inhibitor NAM on EtOH consumption and preference were analysed using one-way anova followed by Newman–Keuls multiple comparison test where significant differences occurred. The same method was used to compare endocannabinoid levels. Student's t-test for unpaired observations was used to compare data from food consumption and weight gain. Differences were considered significant at P < 0.05.
Materials
(+)-METH.HCl, quinine and sucrose were obtained from Sigma-Aldrich (Madrid, Spain), and absolute EtOH (Absolute EtOH aditio) was from Panreac (Barcelona, Spain). HU210, AM251 and NAM were obtained from Tocris Bioscience (Bristol, UK). 2-OG was obtained from Sigma-Aldrich. [3H]-2-OG (2-mono-oleoyl glycerol rac [glycerol-1,2,3-3H]; specific activity = 40 Ci·mmol−1) and [3H]-AEA (ethanolamine-1-3H; specific activity = 60 Ci·mol−1) were obtained from American Radiolabelled Chemicals, Inc (St. Louis, MO, USA). [3H]-WIN 35428 (specific activity = 85.9 Ci·mmol−1) and [35S]-GTPγS (specific activity = 1250 Ci·mmol−1) were obtained from PerkinElmer (Madrid, Spain). [3H]-SR14167A (specific activity = 42 Ci·mmol−1) was obtained from GE Healthcare (Madrid, Spain). Desipramine and HPLC standards were obtained from Sigma-Aldrich. Methanol and HClO4 were obtained from Merck Chemicals (Madrid, Spain). All other chemical reagents were obtained from Sigma-Aldrich.
Results
Alcohol consumption test
Figure 1 shows the intake of and preference for EtOH of control and METH-exposed mice in the two-bottle free-choice procedure with increasing concentrations of EtOH. METH-pretreated mice showed increased consumption of EtOH solutions (Figure 1A) and greater preference ratios (Figure 1B) compared with controls. Both saline- and METH-pretreated mice showed increasing preference with increasing EtOH concentration up to 10%, with the 20% solution presenting a lower preference, which is consistent with an aversive component of taste at higher EtOH concentrations. Relative fluid consumption (mL·g−1·day−1, Figure 1C) was similar in the two groups, indicating that the increased EtOH consumption by METH-pretreated mice was not caused by an overall increase in total amount of fluid consumed. No significant change in body weight was observed during EtOH exposure. At the end of EtOH exposure (4 weeks), the gain in body weight (g) for the different groups was as follows: saline-pretreated mice exposed to EtOH: 1.02 ± 0.25 (n= 10); METH-pretreated mice exposed to EtOH: 0.98 ± 0.25 (n= 10) (F1,18= 0.10, P= 0.92). There was no difference in food consumption (g·kg−1·day−1) between groups: saline-pretreated mice exposed to EtOH: 154 ± 4.5 (n= 10); METH-pretreated mice exposed to EtOH: 151 ± 3.4 (n= 10) (F1,18= 0.58, P= 0.57).
Figure 1.
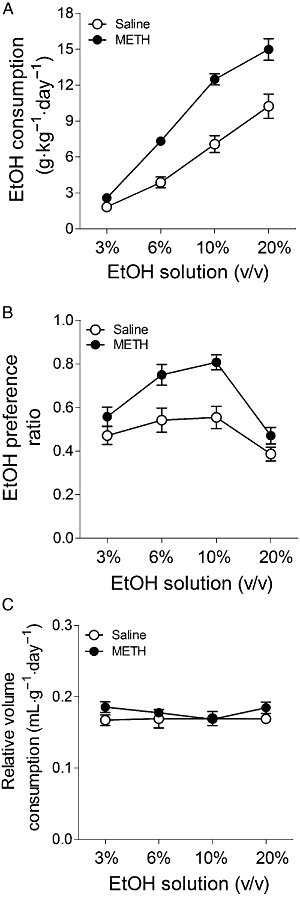
(A) Consumption of and (B) preference for EtOH, and (C) relative intake of fluid in mice exposed to METH (8 mg·kg−1, i.p., three times every 3 h) or saline 7 days before starting the two-bottle free-choice paradigm. The mice had 7 days of access to each EtOH solution (3, 6, 10 and 20% EtOH). (A) Consumption (g·kg−1·day−1) of each EtOH solution (average of 7 days). There was a significant effect of pretreatment (F1,30= 21.23, P < 0.0001), EtOH concentration (F3,30= 113.43, P < 0.0001) and interaction F3,30= 6.86, P= 0.0001. (B) Preference ratio (volume of EtOH consumed/total volume of fluid consumed). There was a significant effect of pretreatment (F1,30= 9.37, P= 0.0016), EtOH concentration (F3,30= 28.69, P < 0.0001) and interaction (F3,30= 4.13, P= 0.0042). (C) Relative fluid intake (mL·g−1·day−1) at each EtOH concentration (average of 7 days). Results shown as mean ± SEM, n= 10–20.
These differences in EtOH consumption and preference do not appear to be secondary to differences in acute EtOH clearance, because plasma EtOH concentration during 4.5 h after 3 g·kg−1 EtOH administration did not differ between saline- and METH-treated animals (Figure 2).
Figure 2.
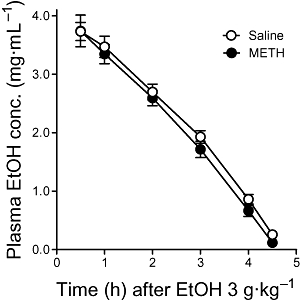
Plasma EtOH concentration after EtOH injection (3 g·kg−1, i.p.) in mice exposed to METH or saline 7 days before. The samples were collected from the tail up to 4.5 h after EtOH administration. There was a significant effect of time (F5,85= 156.97, P < 0.0001), but pretreatment (F1,17= 0.91, P= 0.31) and interaction (F5,85= 0.61, P= 0.64) were not significant. Results shown as mean ± SEM, n= 7–11.
Sucrose and quinine voluntary consumption tests
To determine whether differences in EtOH consumption and preferences might reflect changes in taste preferences or calorie needs caused by METH treatment, drinking studies with sucrose and quinine were performed. With regard to the sucrose data (Figure 3A), two-way anova revealed that there was a significant difference in sucrose preference across the two concentrations in control mice, the greater preference appearing with the highest sucrose concentrations (F1,39= 65.11, P < 0.0001). This pattern was not altered by METH pretreatment (F1,39= 1.46, P= 0.24, n.s.). Interaction was not significant (F1,39= 2.38, P= 0.13, n.s.). As expected, preference for the highest concentration of quinine (0.1 mM) was significantly lower than that observed for 0.03 mM (F1,34= 22.08, P < 0.0001, Figure 3B). This effect was not modified by METH pretreatment (F1,34= 1.02, P= 0.32, n.s.). Interaction was not significant (F1,34= 1.11, P= 0.30, n.s.).
Figure 3.
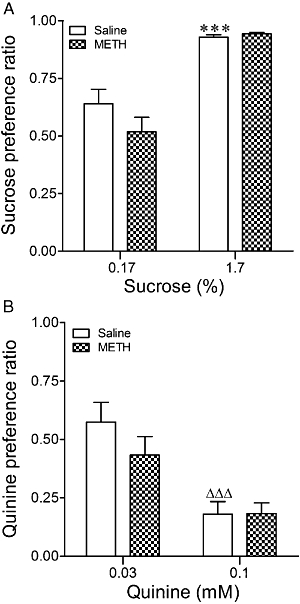
Preference ratios for sucrose (A) and quinine (B) in mice exposed to METH (8 mg·kg−1, three times every 3 h, i.p.) or saline 7 days before starting the two-bottle free-choice paradigm. The mice had 5 days of access to each sucrose or quinine solution with an interval of 4 days between the highest sucrose concentration and the lowest quinine concentration. Preference ratio (volume of sucrose or quinine/total volume of fluid consumed). Results shown as mean ± SEM, n= 8–12. ***P < 0.001, different from 0.17% sucrose preference in saline-pretreated mice. ΔΔΔP < 0.001, different from 0.03 mM quinine preference in saline-pretreated mice.
Endocannabinoid levels in METH-lesioned mice
While there was an increase in 2-AG levels in the limbic forebrain 7 days after METH administration (F2,22= 7.16, P= 0.0045), the levels of AEA were lower than those observed in the saline-treated group (F2,20= 4.56, P= 0.025). No differences were found in the levels of these compounds 4 weeks after METH injection (Figure 4).
Figure 4.
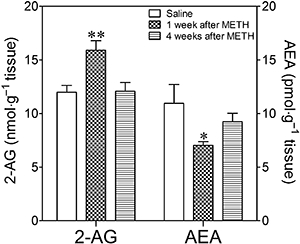
Endocannabinoid levels in the limbic forebrain of mice injected with METH (8 mg·kg−1, i.p., three times every 3 h) or saline. 2-AG and AEA levels were quantified 1 and 4 weeks after treatment. Results shown as mean ± SEM, n= 5–9. *P < 0.05; **P < 0.01, different from saline-treated mice.
MAGL and FAAH activities in METH-lesioned mice
Two-way anova showed a significant effect of METH treatment on MAGL activity (F1,23= 5.77, P= 0.02), no effect of time (F1,23= 2.40, P= 0.13), but a significant interaction (F1,23= 7.73, P= 0.01). In agreement with the 2-AG levels, MAGL activity was 25% lower in the limbic forebrain 7 days after METH injection. This effect was not evident at the 4 week time-point (Figure 5A). By contrast, analysis of FAAH hydrolytic activity by two-way anova revealed that there was no effect of METH (F1,30= 0.032, P= 0.860), time (F1,30= 0.573, P= 0.455) or interaction (F1,30= 3.07, P= 0.090) (Figure 5B).
Figure 5.
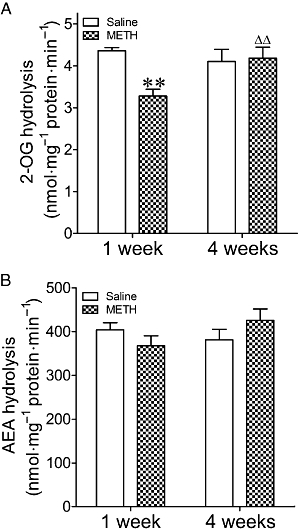
(A) 2-OG and (B) AEA hydrolysis in the limbic forebrain of mice exposed to METH (8 mg·kg−1, i.p., three times every 3 h) or saline 1 or 4 weeks before. Results shown as mean ± SEM, n= 6–10. **P < 0.01, different from saline-treated mice. ΔΔP < 0.01, different from METH-treated mice 1 week before.
CB1 receptor density and agonist-stimulated [35S]-GTPγS binding assay
Two-way anova revealed that at a saturating concentration of [3H]-SR141716A (5 nM), there was no difference in CB1 receptor densities and affinities between membrane preparations of limbic forebrain of METH-pretreated mice and those found in the saline-pretreated group (F1,30= 0.00, P= 0.99), indicating that the binding capacity is similar in the two groups. Time elapsed after treatment did not alter [3H]-SR141716A binding (F1,30= 0.24, P= 0.62) (Table 1).
Table 1.
Density and coupling of CB1 receptors in the limbic forebrain of mice exposed to METH or saline 1 or 4 weeks before
| CB1 receptor density | HU210-stimulated CB1 receptor–G protein coupling | |||
|---|---|---|---|---|
| Time after treatment (week) | Saline | METH | Saline | METH |
| 1 | 1057 ± 62 | 1049 ± 14 | 137 ± 8 | 130 ± 4 |
| 4 | 1028 ± 26 | 1037 ± 18 | 145 ± 6 | 134 ± 3 |
METH was administered at a dose of 8 mg·kg−1, i.p., three times every 3 h. Cannabinoid CB1 receptor-binding sites were measured using [3H]-SR141716A. Data are expressed in fmol·mg−1 protein (mean ± SEM, n= 6–10). HU210-stimulated [35S]-GTPγS binding shown as percentage of basal stimulated binding (mean % ± SEM., n= 6–10).
There was no change in the HU210-stimulated incorporation of [35S]-GTPγS in the limbic forebrain of METH-pretreated mice compared to the saline group (F1,30= 2.06, P= 0.16). The value was similar 1 and 4 weeks after treatment (F1,30= 0.83, P= 0.37) (Table 1).
Effect of AM251 and NAM on the METH-induced increase in intake of and preference for EtOH
One-way anova revealed that there was a significant effect of AM251 administration on intake of (F3,52= 8.27, P= 0.0001) and preference for EtOH (F3,54= 5.11, P= 0.0036), but not on the relative fluid consumption (F3,55= 0.52, P= 0.67). METH-pretreated mice showed increased consumption of EtOH (Figure 6A) and a greater preference ratio (Figure 6B) compared with the saline group. The relative volume of fluid consumption was similar in the two groups (Figure 6C). Administration of the CB1 antagonist, AM251 (5 mg·kg−1) to METH-pretreated mice on each of the 3 days of 10% EtOH exposure significantly reduced the consumption of and preference for EtOH (Figure 6A,B). AM251 did not modify the relative liquid intake (Figure 6C). Although the effect was not significant, AM251 reduced consumption (29%) and preference (35%) for EtOH in saline-pretreated mice (Figure 6A,B).
Figure 6.
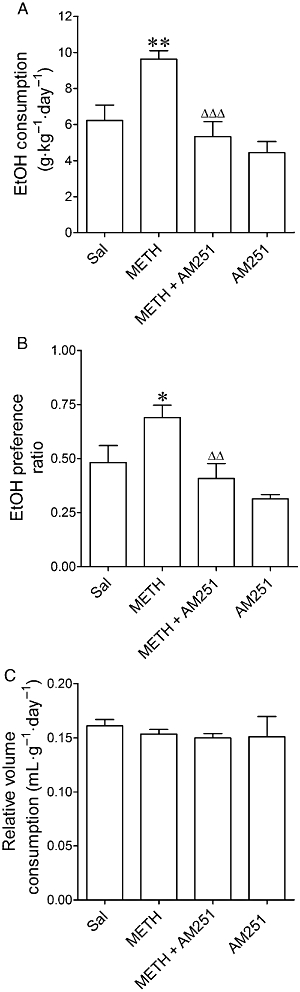
Effects of AM251 on (A) the consumption of and (B) preference for EtOH and on (C) the relative intake of fluid in mice exposed to METH (8 mg·kg−1, i.p., three times every 3 h) or saline 7 days before starting the two-bottle free-choice paradigm. The mice were treated with AM251 (5 mg·kg−1, i.p.) on each of the 3 days of exposure to 10% EtOH. Preference ratio for EtOH was calculated as the volume of EtOH consumed/total volume of fluid consumed. Relative fluid intake is shown as mL·g−1·day−1. Results shown as mean ± SEM, n= 7–17. *P < 0.05, **P < 0.01, different from saline-pretreated mice. ΔΔP < 0.01, ΔΔΔP < 0.001, different from the METH-pretreated group.
To mimic the effects of reduced MAGL activity in limbic brain of METH-treated mice and thus validate its functional consequences for EtOH consumption, the MAGL inhibitor NAM was injected on each of the 3 days of 6% EtOH exposure. NAM increased EtOH consumption (F3,36= 20.00, P= 0.0001) and preference (F3,36= 10.72, P= 0.0006) in METH- and saline-pretreated mice, but did not alter the relative volume consumed (F3,36= 2.45, P= 0.08) (Figure 7).
Figure 7.
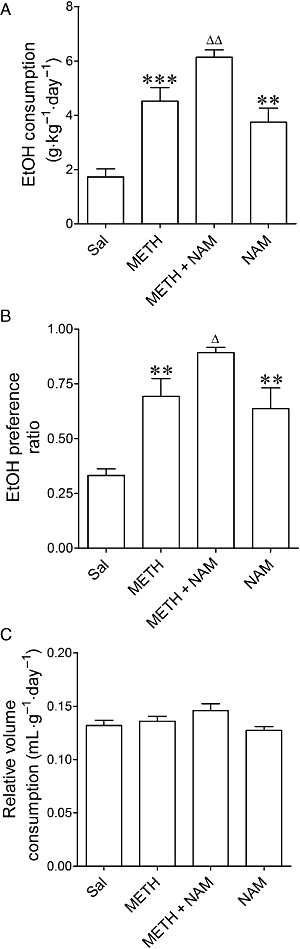
Effects of NAM on (A) the consumption of and (B) preference for EtOH, and on (C) the relative intake of fluid in mice exposed to METH (8 mg·kg−1, i.p., three times every 3 h) or saline 7 days before starting the two-bottle free-choice paradigm. The mice were treated with NAM (3 mg·kg−1, i.p.) on each of the 3 days of exposure to 6% EtOH. Preference ratio for EtOH was calculated as the volume of EtOH consumed/total volume of fluid consumed. Relative fluid intake is shown as mL·g−1·day−1. Results shown as mean ± SEM, n= 6–11. **P < 0.01, ***P < 0.001, different from saline-pretreated mice. ΔP < 0.05, ΔΔP < 0.01, different from the METH-pretreated group.
Catechol concentration and dopamine transporter density in METH-lesioned mice
Analysis of dopamine and HVA data by two-way anova revealed that there was a significant effect of treatment (F1,27= 60.84, P < 0.0001 for dopamine; F1,25= 38.68, P < 0.0001 for HVA), time (F1,27= 5.03, P= 0.03 for dopamine; F1,25= 4.95, P= 0.04 for HVA) and interaction (F1,27= 7.38, P= 0.011 for dopamine; F1,25= 5.67, P= 0.025 for HVA). Regarding DOPAC data, two-way anova indicated that there was a significant effect of treatment (F1,23= 29.11, P < 0.0001) and time (F1,23= 7.65, P= 0.011, but not interaction (F1,23= 3.011, P= 0.096). Analysis of dopamine transporter data indicated that there was a significant effect of treatment (F1,17= 77.68, P < 0.0001), but not time (F1,17= 2.69, P= 0.16) nor interaction (F1,17= 0.17, P= 0.72).
Administration of METH (8 mg·kg−1, three times at three hourly intervals) decreased the concentration of dopamine, DOPAC and HVA, and the density of dopamine transporters in the limbic forebrain 1 and 4 weeks after injection. Except for dopamine transporters, there was some degree of recovery, the depletion being less pronounced 4 weeks after treatment (Figure 8).
Figure 8.
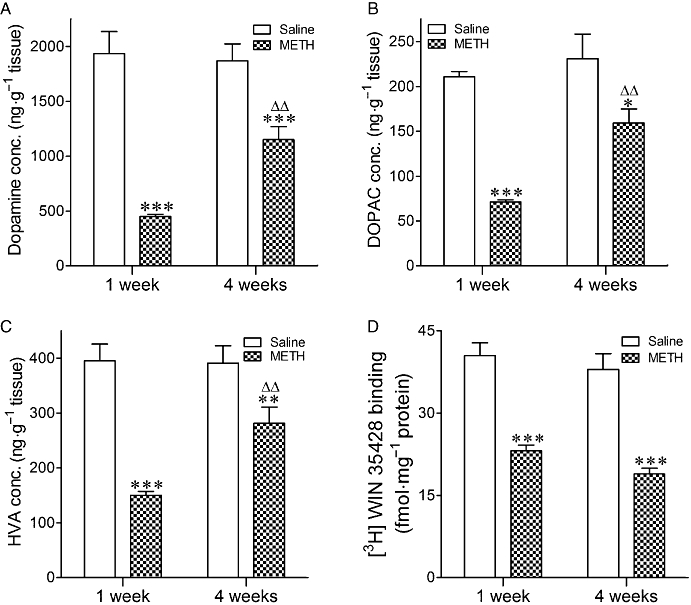
Concentration of (A) dopamine, (B) DOPAC, (C) HVA and (D) [3H]-WIN 34428 binding in the limbic forebrain of mice exposed to METH (8 mg·kg−1, i.p., three times every 3 h) or saline 1 and 4 weeks before. Results shown as mean ± SEM, n= 4–10. *P < 0.05, **P < 0.01, ***P < 0.001, different from the corresponding saline-treated mice. ΔΔP < 0.01, different from mice injected with METH 1 week before being killed.
Discussion
Using a two-bottle free-choice paradigm, this study shows that mice exposed to a neurotoxic dose of METH and consequently showing a long-term loss of dopamine concentration and dopamine transporters in the limbic forebrain, exhibit higher consumption of and preference for EtOH over water. These differences between the two groups of mice do not reflect a more global change in taste preferences or calorie needs because consumption of sucrose- or quinine-containing solutions was similar in METH-lesioned and saline-treated mice. In addition, preference ratios for EtOH, sucrose and quinine are in accordance with those previously described (Naassila et al., 2004; Lewis et al., 2005; Izco et al., 2007). In the current study, METH- and saline-injected animals did not differ in terms of volume of fluid consumed, indicating that the increased EtOH intake by METH-injected mice was not caused by an overall increase in the total amount of fluid consumed. Food consumption was also similar in both groups, indicating that the altered EtOH intake does not result from changes in appetite.
Previous studies have reported that 7 days after repeated administration of METH, time at which EtOH exposure was started, there is a decrease in striatal dopamine concentration (Sanchez et al., 2003), and in the immunoreactivity for dopamine transporters and tyrosine hydroxylase (Granado et al., 2009). The current study extends these findings by demonstrating that the reduction of dopamine concentration and dopamine transporters induced by METH is also evident in the limbic forebrain and that it persists for at least 4 weeks after injection. Nevertheless, some degree of recovery occurs because the loss of dopamine content observed 7 days after METH is more pronounced than that detected 4 weeks after METH.
Differences in alcohol consumption between saline- and METH-treated mice are not likely to be related to changes in EtOH metabolism because the blood EtOH concentration in METH-treated mice was not different from that observed in saline-treated mice during the 4.5 h after the injection of a single dose of EtOH.
Because endocannabinoid signalling through CB1 receptors has been shown to positively modulate various aspects of alcohol-seeking behaviour, we tested the effect of a CB1 antagonist on the elevated EtOH intake observed in METH-treated mice. Administration of AM251 reduced the consumption of and preference for EtOH in METH-treated mice without modifying the relative volume intake. In addition, as expected, AM251 reduced consumption (29%) and preference (35%) for EtOH in saline-treated mice, although this was not significant. These findings are in line with previous studies showing reduced preference for EtOH in B6 (EtOH preferring) and D2 (EtOH non-preferring) strains of mice following pharmacological blockade with SR141716A given at a similar dose to that used in the current study (Vinod et al., 2008b). Chronic CB1 receptor blockade with SR141716A also significantly decreased EtOH intake in C57BL/6J mice in a two-bottle-choice paradigm (Thanos et al., 2005; Kelaïet al., 2006). SR141716A and AM251 have similar behavioural activity in C57BL/6J mice (McMahon and Koek, 2007), and both are equipotent at blocking the hypothermic effects induced by Δ9-tetrahydrocannabinol and WIN 55212-2, and at attenuating the hypoactivity induced by the two agonists (McMahon and Koek, 2007). Consistent with these observations, CB1 knock-out mice show greatly reduced EtOH consumption and preference (Hungund et al., 2003; Wang et al., 2003; Naassila et al., 2004; Vinod et al., 2008b).
Given the evidence of a role for the CB1 receptor in mediating EtOH-seeking behaviour, we sought to determine whether METH-treated animals might show changes in CB1 density and activity, which might explain the difference in drinking behaviour displayed by the lesioned mice. However, radioligand binding studies showed no differences between the groups in either of these parameters in limbic forebrain.
Endocannabinoids are synthesized and released from depolarized neurones and act retrogradely to stimulate CB1 receptors located on pre-synaptic terminals (for review, see Alger, 2002). Thus, the next aim of our study was to determine whether the neuronal damage induced by METH was associated with changes in the endocannabinoid system, and whether these changes might be of functional importance in the differential EtOH intake observed in the METH-treated group. Indeed, 7 days after METH administration, 2-AG levels were increased in limbic forebrain and at the same time there was a decrease in the activity of MAGL, suggesting that 2-AG metabolism is impaired in the limbic brain of METH-treated mice, thereby contributing to the accumulation of 2-AG.
Conversely, a reduction in AEA was found in the limbic brain of METH-treated mice compared with saline, while FAAH activity did not differ between the two groups. Although the precise molecular mechanism is not known, there is abundant evidence indicating that the two endocannabinoids are differentially regulated in the limbic brain of mice showing dopaminergic neuronal damage. In this line, it has been shown that lesioning dopaminergic neurones with 6-hydroxy dopamine produces a threefold increase in AEA, but not 2-AG, content within the striatum (Gubellini et al., 2002). This increase was reversed when dopamine was restored by chronic l-DOPA treatment (Maccarrone et al., 2003). It has also been reported that monoamine depletion by reserpine results in a 2.5-fold increase in both AEA and 2-AG content within the striatum (Di Marzo et al., 2000). These findings are consistent with the hypothesis that decreased dopaminergic transmission regulates endocannabinoid content within forebrain regions that receive dopaminergic innervation.
Evidence in the literature indicates that endogenous dopamine modulates endocannabinoid content within the limbic forebrain via activation of D1 or D2 receptors, and that dopamine through D1 activation reduces AEA content in limbic forebrain (Patel et al., 2003). Although we have no evidence of changes in D1 receptor density following METH administration, previous studies in our laboratory have shown that MDMA produces a similar dopaminergic lesion and also increases D1 receptor density in the nucleus accumbens. Therefore, this could be an explanation for the reduced AEA levels following METH administration. On the other hand, there is evidence that METH increases COX-2 protein levels in areas showing dopamine loss 3 days after injection (Kita et al., 2000). This enzyme is also involved in AEA metabolism (Guindon and Hohmann, 2008), so we cannot rule out its involvement in the changes observed.
NAM is a recently developed, highly selective and potent MAGL inhibitor that increases the endogenous levels of 2-AG in the brain and raises the potency of 2-AG, but not AEA in agonist-stimulated [35S]-GTPγS binding assay, a measure of G-protein activation (Burston et al., 2008). In vitro, NAM prevents cerebellar membrane-mediated degradation of 2-AG at a relatively low concentration (Saario et al., 2005; Blankman et al., 2007). Here, we report that administration of NAM enhances the increase in the consumption of and preference for EtOH observed in both saline- and METH-treated mice. The fact that MAGL inhibition in control mice increases EtOH consumption to a similar extent to that produced by METH lesion indicates that 2-AG-elevated levels in key brain regions of the reward system are involved in mediating the EtOH-seeking behaviour of lesioned mice. The effect of NAM on EtOH consumption and preference in saline-treated mice is presumably due to a regulatory role of basal 2-AG and CB1 activation, which is consistent with the tendency of AM251 to reduce EtOH consumption and preference in these animals.
It is of interest that boosting 2-AG levels (by NAM; this study) or AEA levels (by inhibitors of FAAH pharmacologically or genetically; Vinod et al., 2008a) can both increase EtOH consumption and preference in mice. Although we did not observe an increase in AEA content after METH administration, the rise in 2-AG seems to be sufficient to cause CB1 receptor activation and the consequent increase in EtOH drinking behaviour.
In summary, this study indicates for the first time that the long-lasting dopamine neurotoxicity induced by METH is associated with changes in the endocannabinoid system that predispose mice to high voluntary consumption of and increase preference for EtOH. In addition, it suggests that the widely described impairment of dopamine function produced by METH in human beings (Volkow et al., 2001) may increase the risk for EtOH abuse in consumers.
Acknowledgments
The authors thank Plan Nacional sobre Drogas (Ministerio de Sanidad, grant nos. PR75/06-15077; 2008/074), Ministerio de Ciencia y Tecnologia (grant no. SAF2007-65175), Ministerio de Sanidad (RTA-RD06/0001/006; FIS PI07/0892), UCM-CAM (grant nos. CCG07-UCM/SAL-2588, CCG08-UCM/SAL-3935) and Fundación Mutua Madrileña for financial support.
Glossary
Abbreviations:
- AEA
arachidonoyl ethanolamine
- 2-AG
2-arachidonoyl glycerol
- DMSO
dimethylsulphoxide
- DOPAC
dihydroxyphenyl acetic acid
- EtOH
ethanol
- FAAH
fatty acid amide hydrolase
- HU210
(6aR)-trans-3-(1,1-dimethylheptyl)-6a,7,10,10a-tetrahydro-1-hydroxy-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol
- HVA
homovanillic acid
- MAGL
monoacylglycerol lipase
- METH
methamphetamine
- NAM
N-arachidonoyl maleimide; N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride
- R141716A
N-(piperidin-1-yl)-5-(4-iodophonyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- AM251; SKF81297
3,4-6-chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Arnone M, Maruani J, Chaperon F, Thiébot MH, Poncelet M, Soubrié P, et al. Selective inhibition of sucrose and ethanol intake by SR 141716, an antagonist of central cannabinoid (CB1) receptors. Psychopharmacology. 1997;132:104–106. doi: 10.1007/s002130050326. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS, Hungund BL. Cannabinoid receptor agonist-stimulated [35S]guanosine triphosphate gammaS binding in the brain of C57BL/6 and DBA/2 mice. J Neurosci Res. 2001;64:429–436. doi: 10.1002/jnr.1094. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS, Hungund BL. Role of the endocannabinoid system in the development of tolerance to alcohol. Alcohol Alcohol. 2005;40:15–24. doi: 10.1093/alcalc/agh111. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burston JJ, Sim-Selley LJ, Harloe JP, Mahadevan A, Razdan RK, Selley DE, et al. N-arachidonoyl maleimide potentiates the pharmacological and biochemical effects of the endocannabinoid 2-arachidonoylglycerol through inhibition of monoacylglycerol lipase. J Pharmacol Exp Ther. 2008;327:546–553. doi: 10.1124/jpet.108.141382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillé S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH. Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci. 2007;27:3695–3702. doi: 10.1523/JNEUROSCI.4403-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo G, Agabio R, Lobina C, Reali R, Vacca G, Gessa GL. Stimulation of locomotor activity by voluntarily consumed ethanol in Sardinian alcohol-preferring rats. Eur J Pharmacol. 1998;357:109–113. doi: 10.1016/s0014-2999(98)00560-3. [DOI] [PubMed] [Google Scholar]
- Colombo G, Serra S, Brunetti G, Gomez R, Melis S, Vacca G, et al. Stimulation of voluntary ethanol intake by cannabinoid receptor agonists in ethanol-preferring sP rats. Psychopharmacology. 2002;159:181–187. doi: 10.1007/s002130100887. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Hill MP, Bisogno T, Crossman AR, Brotchie JM. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson's disease. FASEB J. 2000;14:1432–1438. doi: 10.1096/fj.14.10.1432. [DOI] [PubMed] [Google Scholar]
- Freedland CS, Sharpe AL, Samson HH, Porrino LJ. Effects of SR141716A on ethanol and sucrose self-administration. Alcohol Clin Exp Res. 2001;25:277–282. [PubMed] [Google Scholar]
- Gallate JE, Saharov T, Mallet PE, McGregor IS. Increased motivation for beer in rats following administration of a cannabinoid CB1 receptor agonist. Eur J Pharmacol. 1999;370:233–240. doi: 10.1016/s0014-2999(99)00170-3. [DOI] [PubMed] [Google Scholar]
- Ghafouri N, Tiger G, Razdan RK, Mahadevan A, Pertwee RG, Martin BR, et al. Inhibition of monoacylglycerol lipase and fatty acid amide hydrolase by analogues of 2-arachidonoylglycerol. Br J Pharmacol. 2004;143:774–784. doi: 10.1038/sj.bjp.0705948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González S, Valenti M, de Miguel R, Fezza F, Fernández-Ruiz J, Di Marzo V, et al. Changes in endocannabinoid contents in reward-related brain regions of alcohol-exposed rats, and their possible relevance to alcohol relapse. Br J Pharmacol. 2004;143:455–464. doi: 10.1038/sj.bjp.0705963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granado N, Ares-Santos S, O'Shea E, Vicario-Abejón C, Colado MI, Moratalla R. Selective vulnerability in striosomes and in the nigrostriatal dopaminergic pathway after methamphetamine administration: early loss of TH in striosomes after methamphetamine. Neurotox Res. 2009 doi: 10.1007/s12640-009-9106-1. Sep 4 [Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubellini P, Picconi B, Bari M, Battista N, Calabresi P, Centonze D, et al. Experimental parkinsonism alters endocannabinoid degradation: implications for striatal glutamatergic transmission. J Neurosci. 2002;22:6900–6907. doi: 10.1523/JNEUROSCI.22-16-06900.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. A physiological role for endocannabinoid-derived products of cyclooxygenase-2-mediated oxidative metabolism. Br J Pharmacol. 2008;153:1341–1343. doi: 10.1038/bjp.2008.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H, Cadet JL. p53-Knockout mice are protected against the long-term effects of methamphetamine on dopaminergic terminals and cell bodies. J Neurochem. 1997;69:780–790. doi: 10.1046/j.1471-4159.1997.69020780.x. [DOI] [PubMed] [Google Scholar]
- Hogan KE, Staal RGW, Sonsalla PK. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: disparity between homogenates and vesicle preparations. J Neurochem. 2000;74:2217–2220. doi: 10.1046/j.1471-4159.2000.0742217.x. [DOI] [PubMed] [Google Scholar]
- Hungund BL, Basavarajappa BS. Distinct differences in the cannabinoid receptor binding in the brain of C57BL/6 and DBA/2 mice, selected for their differences in voluntary ethanol consumption. J Neurosci Res. 2000;60:122–128. doi: 10.1002/(SICI)1097-4547(20000401)60:1<122::AID-JNR13>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C. Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J Neurochem. 2003;84:698–704. doi: 10.1046/j.1471-4159.2003.01576.x. [DOI] [PubMed] [Google Scholar]
- Itzhak Y, Martin JL, Ail SF. nNOS inhibitors attenuate methamphetamine-induced dopaminergic neurotoxicity but not hyperthermia in mice. Neuroreport. 2000;11:2943–2946. doi: 10.1097/00001756-200009110-00022. [DOI] [PubMed] [Google Scholar]
- Izco M, Marchant I, Escobedo I, Peraile I, Delgado M, Higuera-Matas A, et al. Mice with decreased cerebral dopamine function following a neurotoxic dose of MDMA (3,4-methylenedioxymethamphetamine, ‘ecstasy’) exhibit increased ethanol consumption and preference. J Pharmacol Exp Ther. 2007;322:1003–1012. doi: 10.1124/jpet.107.120600. [DOI] [PubMed] [Google Scholar]
- Kelaï S, Hanoun N, Aufrère G, Beaugé F, Hamon M, Lanfumey L. Cannabinoid–serotonin interactions in alcohol-preferring vs. alcohol-avoiding mice. J Neurochem. 2006;99:308–320. doi: 10.1111/j.1471-4159.2006.04054.x. [DOI] [PubMed] [Google Scholar]
- Kita T, Shimada K, Mastunari Y, Wagner GC, Kubo K, Nakashima T. Methamphetamine-induced striatal dopamine neurotoxicity and cyclooxygenase-2 protein expression in BALB/c mice. Neuropharmacology. 2000;39:399–406. doi: 10.1016/s0028-3908(99)00175-6. [DOI] [PubMed] [Google Scholar]
- Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, et al. Structure–activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. Med Chem. 1999;42:769–776. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- Landa L, Slais K, Sulcova A. Impact of cannabinoid receptor ligands on behavioural sensitization to antiaggressive methamphetamine effects in the model of mouse agonistic behaviour. Neuro Endocrinol Lett. 2006;27:703–710. [PubMed] [Google Scholar]
- Lewis SR, Ahmed S, Dym C, Khaimova E, Kest B, Bodnar RJ. Inbred mouse strain survey of sucrose intake. Physiol Behav. 2005;85:546–556. doi: 10.1016/j.physbeh.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Gubellini P, Bari M, Picconi B, Battista N, Centonze D, et al. Levodopa treatment reverses endocannabinoid system abnormalities in experimental parkinsonism. J Neurochem. 2003;85:1018–1025. doi: 10.1046/j.1471-4159.2003.01759.x. [DOI] [PubMed] [Google Scholar]
- McMahon LR, Koek W. Differences in the relative potency of SR 141716A and AM 251 as antagonists of various in vivo effects of cannabinoid agonists in C57BL/6J mice. Eur J Pharmacol. 2007;569:70–76. doi: 10.1016/j.ejphar.2007.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann H, Ladenheim B, Hirata H, Moran TH, Cadet JL. Differential toxic effects of methamphetamine (METH) and methylenedioxymethamphetamine (MDMA) in multidrug-resistant (mdr1a) knockout mice. Brain Res. 1997;769:340–346. doi: 10.1016/s0006-8993(97)00754-3. [DOI] [PubMed] [Google Scholar]
- Naassila M, Ledent C, Daoust M. Low ethanol sensitivity and increased ethanol consumption in mice lacking adenosine A2A receptors. J Neurosci. 2002;22:10487–10493. doi: 10.1523/JNEUROSCI.22-23-10487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naassila M, Pierrefiche O, Ledent C, Daoust M. Decreased alcohol self-administration and increased alcohol sensitivity and withdrawal in CB1 receptor knockout mice. Neuropharmacology. 2004;46:243–253. doi: 10.1016/j.neuropharm.2003.09.002. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- Patel S, Rademacher DJ, Hillard CJ. Differential regulation of the endocannabinoids anandamide and 2-arachidonoylglycerol within the limbic forebrain by dopamine receptor activity. J Pharmacol Exp Ther. 2003;306:880–888. doi: 10.1124/jpet.103.054270. [DOI] [PubMed] [Google Scholar]
- Richardson D, Ortori CA, Chapman V, Kendall DA, Barrett DA. Quantitative profiling of endocannabinoids and related compounds in rat brain using liquid chromatography–tandem electrospray ionization mass spectrometry. Anal Biochem. 2007;360:216–226. doi: 10.1016/j.ab.2006.10.039. [DOI] [PubMed] [Google Scholar]
- Rubio M, de Miguel R, Fernández-Ruiz J, Gutiérrez-López D, Carai MA, Ramos JA. Effects of a short-term exposure to alcohol in rats on FAAH enzyme and CB1 receptor in different brain areas. Drug Alcohol Depend. 2009;99:354–358. doi: 10.1016/j.drugalcdep.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Saario SM, Salo OM, Nevalainen T, Poso A, Laitinen JT, Järvinen T, et al. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoyl-glycerol in rat cerebellar membranes. Chem Biol. 2005;12:649–656. doi: 10.1016/j.chembiol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Sanchez V, Zeini M, Camarero J, O'Shea E, Bosca L, Green AR, et al. The nNOS inhibitor, AR-R17477AR, prevents the loss of NF68 immunoreactivity induced by methamphetamine in the mouse striatum. J Neurochem. 2003;85:515–524. doi: 10.1046/j.1471-4159.2003.01714.x. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R, O'Neil ML, Melega WP, Cho AK. Escalating dose methamphetamine pretreatment alters the behavioral and neurochemical profiles associated with exposure to a high-dose methamphetamine binge. Neuropsychopharmacology. 2003;28:1730–1740. doi: 10.1038/sj.npp.1300247. [DOI] [PubMed] [Google Scholar]
- Sim LJ, Selley DE, Childers SR. In vitro autoradiography of receptor-activated G proteins in rat brain by agonist-stimulated guanylyl 5′-[gamma-[35S]thio]-triphosphate binding. Proc Natl Acad Sci USA. 1995;92:7242–7246. doi: 10.1073/pnas.92.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Jochnowitz ND, Zeevalk GD, Oostveen JA, Hall ED. Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res. 1996;738:172–175. doi: 10.1016/0006-8993(96)00995-x. [DOI] [PubMed] [Google Scholar]
- Staal RG, Hogan KA, Liang CL, German DC, Sonsalla PK. In vitro studies of striatal vesicles containing the vesicular monoamine transporter (VMAT2): rat versus mouse differences in sequestration of 1-methyl-4-phenylpyridinium. J Pharmacol Exp Ther. 2000;293:329–335. [PubMed] [Google Scholar]
- Thanos PK, Dimitrakakis ES, Rice O, Gifford A, Volkow ND. Ethanol self-administration and ethanol conditioned place preference are reduced in mice lacking cannabinoid CB1 receptors. Behav Brain Res. 2005;164:206–213. doi: 10.1016/j.bbr.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Tordoff MG, Bachmanov AA. Monell Mouse Taste Phenotyping Project. 2005. Construction of drinking tubes. Available at http://www.monell.org/MMTPP/ (accessed 15 January 2009.
- United Nations Office on Drugs and Crime (UNODC) Annual report 2008. 2008. Available at http://www.unodc.org/documents/about-unodc/AR08_WEB.pdf (accessed 1 June 2009.
- Vinod KY, Sanguino E, Yalamanchili R, Manzanares J, Hungund BL. Manipulation of fatty acid amide hydrolase functional activity alters sensitivity and dependence to ethanol. J Neurochem. 2008a;104:233–243. doi: 10.1111/j.1471-4159.2007.04956.x. [DOI] [PubMed] [Google Scholar]
- Vinod KY, Yalamanchili R, Thanos PK, Vadasz C, Cooper TB, Volkow ND, et al. Genetic and pharmacological manipulations of the CB(1) receptor alter ethanol preference and dependence in ethanol preferring and nonpreferring mice. Synapse. 2008b;62:574–581. doi: 10.1002/syn.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu J, Harvey-White J, Zimmer A, Kunos G. Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc Natl Acad Sci USA. 2003;100:1393–1398. doi: 10.1073/pnas.0336351100. [DOI] [PMC free article] [PubMed] [Google Scholar]