

Abstract
Increasing evidence suggests that Alzheimer's disease (AD) is associated with oxidative damage that is caused in part by mitochondrial dysfunction. Here we investigated the feasibility of modifying Alzheimer pathology with the mitochondrial antioxidant coenzyme Q (CoQ). Exogenous CoQ protected MC65 neuroblastoma cells from amyloid precursor protein C-terminal fragment (APP CTF)-induced neurotoxicity in a concentration dependent manner, with concentrations of 6.25 µM and higher providing near complete protection. Dietary supplementation with CoQ at a dose of 10 g/kg diet to C65/Bl6 mice for one month significantly suppressed brain protein carbonyl levels, which are markers of oxidative damage. Treatment for one month with 2 g lovastatin/kg diet, which interferes with CoQ synthesis, resulted in a significant lowering of brain CoQ10 levels. Mitochondrial energetics (brain ATP levels and mitochondrial membrane potential) were unaffected by either CoQ or lovastatin treatment. Our results suggest that oral CoQ may be a viable antioxidant strategy for neurodegenerative disease. Our data supports a trial of CoQ in an animal model of AD in order to determine whether a clinical trial is warranted.
Keywords: Alzheimer’s disease, coenzyme Q, lovastatin, Tg2576, MC65
Introduction
Antioxidant treatment strategies have been proposed for Alzheimer’s disease (AD) because the neurodegenerative process in AD is marked by oxidative damage to the brain [1, 2]. Although the source of the oxidative damage remains uncertain, several lines of evidence have pointed to mitochondrial dysfunction as the cause [3] [4] [5]. “Mitochondrial antioxidants” are consequently candidate therapies for AD [6]. Coenzyme Q (CoQ) is especially attractive in light of favorable animal studies in a murine model of one neurodegenerative disease (Huntington’s disease) [7] and favorable clinical trials in another (Parkinson’s disease) [8]. In order to determine the feasibility of modifying AD pathology with CoQ, we examined 1) whether CoQ is capable of protecting neurons from amyloid precursor protein C-terminal fragment (APP CTF) - induced neurotoxicity, 2) whether CoQ enhances mitochondrial function in vitro, 3) whether chronic oral CoQ supplementation increases brain levels of CoQ, 4) whether chronic oral CoQ supplementation enhances mitochondrial function or attenuates oxidative damage in the brain, and 5) whether brain CoQ levels are depressed in the setting of AD pathology or in the setting of treatment with drugs which interfere with CoQ synthesis.
The hypothesis that brain CoQ levels may be lower in mice with AD pathology is made plausible by reports that brain CoQ levels are lower in murine models of Huntington’s disease, and other neurodegenerative disease characterized by oxidative damage [9]. The examination of the effects of drugs upon brain CoQ is motivated by the hypothesis that CoQ supplementation may be most appropriate for a sub-population of patients who have depressed tissue CoQ levels induced by common prescription drugs. The cholesterol-lowering statin drugs have been reported to depress tissue levels of CoQ [10] [11] [12] by inhibiting synthetic pathways which are shared by cholesterol and CoQ. Although the clinical significance of statin use upon CoQ synthesis remains uncertain, some investigators have even suggested that all statin users should be treated with supplemental CoQ [10]. Since statin drugs are currently under investigation for the treatment of AD, and since they are so widely prescribed already, it is plausible that AD patients taking statins might comprise a specific sub-population for whom CoQ therapy may be most appropriate. We consequently examined the effect of statin treatment upon brain levels of CoQ.
For CoQ supplementation, we used the highest dose reported in chronic feeding studies in mice [13]. Different forms of CoQ are designated by numerals that indicate the number of isoprenoid units incorporated into each molecule. The predominant form of CoQ in humans is CoQ10; the predominant form in mice is CoQ9. Mouse tissue levels of CoQ10 consequently have a greater capacity to increase with supplementation than do tissue levels of CoQ9, and most published reports have used CoQ10 supplementation in mice. Coenzyme Q10 was therefore selected for use in our studies. For depletion of CoQ, we used a clinically relevant, commonly used statin, lovastatin. The choice of a lipid soluble statin was intended to ensure brain penetration, and the dose was selected from a study in a murine model of AD which showed good tolerance and marked suppression of plasma cholesterol levels with chronic feeding [14].
In vitro studies were conducted in MC65 cells, an established human neuroblastoma line that conditionally expresses the first 17 amino-terminal residues and the 99 carboxy-terminal residues of amyloid precursor protein (APP CTF). Removal of tetracycline (tet) from these cells results in the initial expression of a species that migrates at approximately 10 kDa (the molecular weight of the APP CTF construct), with subsequent processing to a smaller band that co-migrates with Aβ1–40 but not Aβ1–42. This low molecular weight species undergoes progressive aggregation to high molecular weight bands. Both low and high molecular weight bands are recognized by antibodies to Aβ, including the 6E10 monoclonal antibody. Detectable APP CTF expression occurs within 3–4 h of tet withdrawal, appearance of aggregates and initial losses of culture viability occurs at about 2 days and near complete death occurs by approximately 3 days [15].
Materials and Methods
Cell culture, CoQ treatment, cell viability and in vitro measurements
MC65 cells were cultured and maintained in MEMα supplemented with 10% FBS (Gibco-BRL, Carlsbad, CA) and 1 µg/ml tet (Sigma-Aldrich, St. Louis, MO) as previously described [15]. For the experiments described here, confluent cells were trypsinized, washed with PBS, resuspended in OptiMEM without phenol red (Gibco/BRL, Carlsbad, CA), and plated in 48-well plates in fresh medium containing vehicle or desired concentrations of CoQ in the presence or absence of tet. Coenzyme Q (Sigma-Aldrich, St. Louis, MO) was prepared as a fresh ethanolic stock solution before each experiment. Final ethanol concentrations were 0.5%. All experiments were carried out in triplicate wells for each condition and repeated at least 3 times. Cell viability was measured using the CellTiter 96® Aqueous Non-Radioactive Cell Proliferation Assay (Promega Corporation, Madison, WI) according to manufacturer’s instructions. ATP levels were measured using the ATPlite 1step Luminescence ATP Detection Assay System (Perkin Elmer, Boston, MA) as per manufacturer’s instructions. Briefly, medium was removed from each treatment and replaced with 200 µl of ATP-lite substrate solution (diluted 50% with phosphate buffered saline). The plate was shaken for 2 min and luminescence read on a Victor3 Multilabel Reader (Perkin Elmer, Boston MA). ATP levels were quantified from an ATP standard curve. ATP levels were normalized to cell number by counting the average number of cells in replicate wells using a Bright Line Counting Chamber (Hausser Scientific, Horsham, PA). Mitochondrial membrane potential was determined by measuring the red fluorescence of the cationic dye JC-1. Briefly, cells were trypsinized, centrifuged at 800 × g for 4 min at 4°C, washed, then re-suspended at a concentration of 1 × 106 cells/ml in optimem. 5 × 105 cells were mixed with an equal volume of 5 µM JC-1 dissolved in staining buffer (20 mM MOPS, pH 7.5, 100 mM KCl, 10 mM ATP, 10 mM MgCl2, 10 mM sodium succinate and 1 mM EGTA, Sigma, St. Louis, MO) then incubated for 20 min at 37°C in a humidified atmosphere containing 5% CO2. Sodium azide (Sigma, St. Louis, Mo) was used as a control to inhibit mitochondrial membrane potential. The cell suspension was centrifuged at 800 × g for 4 min at 4°C, washed with ice cold staining buffer, re-suspended in 210 ml staining buffer and fluorescence read (excitaion 490 nm, emission 590 nm) on a Victor3 Multilabel Reader. Intracellular superoxide levels were measured 42 h following seeding using the nitroblue tetrazolium (NBT) assay as previously described [16]. Briefly, NBT (Sigma, St. Louis, MO) was added to the cell media at a final concentration of 1 mg/ml and incubated for 3 h at 37°C in a humidified atmosphere containing 5% CO2. The NBT-containing medium was removed, the cells washed twice with warm PBS followed by methanol, then air-dried. Intracellular blue formazan particles were dissolved by adding 120 µl/well 2M potassium hydroxide followed by 140 µl/well DMSO. The absorbance of dissolved NBT was measured at 620 nm using a Spectra Max Plus (Molecular Devices, Sunnyvale, CA). Hydrogen peroxide levels were determined in cell media using the Bioxytech® H2O2-560™ Quantitative Hydrogen Peroxide Assay (Oxis International, Foster City, CA) as per manufacturer’s instructions.
Mouse feeding protocol
Thirty-two 11-month old adult female C57BL/6 mice (22.8–29.2 g, Jackson Laboratories, Bar Harbor, ME) were used in the CoQ/lovasatin study. Mice were randomized to receive either Control diet (AIN-93M, Dyets Inc. Bethlehem, PA), AIN-93M supplemented with CoQ (Vitaline® Formulas, Green Bay, WI) at a dose of 10 g/kg diet or lovastatin (Axxora LLC, San Diego, CA) at a dose of 2 g/kg diet. Mice were allowed to acclimatize for at least 7 days before treatment and remained on their respective diets for one month. Throughout the study, each mouse was weighed and examined weekly to check for signs of dehydration, stress or toxicity. Eight month-old females (three C56Bl/6 and three transgenic mice bred from a breeding pair of Tg2576 mice generously provided by Dr. Karen Hsiao-Ashe, Mayo Clinic, MN) were used to compare CoQ levels between wild-type mice and Tg2576 mice. All mice were maintained in a climate-controlled environment with a 12-hr light/12-hr dark cycle, and diet and water supplied ad libitum. All procedures were conducted in accordance with the NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the institutional Animal Care and Use Committee of the Portland VA Medical Center.
Tissue Preparation
At the end of treatment, mice were sacrificed by CO2 inhalation and cervical dislocation. Brains were quickly removed and divided. Bilateral frontal cortex was dissected and frozen. The remaining forebrain from one hemisphere was committed for mitochondrial isolation (methods below). The contralateral hemisphere was frozen, as were the brainstem and cerebellum and heart.
CoQ determination
CoQ9 and CoQ10 from heart and brain tissues and mitochondria was extracted and measured by HPLC as previously described [17]. Briefly, tissue or mitochondria were homogenized using micro glass tissue grinder in 0.5ml potassium phosphate buffer pH 7.4 containing 1mM dithiothreitol (Invitrogen, Carlsbad, CA). The homogenate was treated with 2ml of ethanol containing 1µg/ml butylated hydroxytoluene (Sigma, St. Louis, MO) and ubiquinone was extracted with 5 ml hexane. After vigorous shaking, 4 ml of hexane layer was dried under N2. The residue was dissolved in 100 µl ethanol containing BHT and subjected to HPLC analysis. CoQ9 and CoQ10 were quantified using a Phenomenex Luna 3u (C18) column with a mobile phase methanol: hexane (90:10 v/v), detected at wavelength of 275 nm as previously described [18].
Brain mitochondria isolation and assessment of membrane potential
Mitochondria were isolated from mouse brain tissue using the MITOISO1 Mitochondrial Isolation Kit (Sigma, St. Louis, MO) according to manufacturer’s instructions. Briefly, 100mg of proximal left hemisphere was homogenized in 1ml of ice-cold Extraction Buffer containing 2mg/ml albumin. Samples were centrifuged at 1000 × g for 5 min at 4C. The supernatant was collected and centrifuged at 3500 × g for 10 min at 4C. The resulting pellet was re-suspended in Extraction Buffer and the differential centrifugation steps repeated to yield an enriched mitochondrial pellet. The mitochondrial-enriched pellet was suspended in 40µl 1x Storage Buffer and assayed immediately for protein concentration using the BCA protein assay. Mitochondrial membrane potential was immediately determined using the JC-1 membrane potential assay (Sigma-Aldrich, St. Louis, MO) according to manufacturer’s instructions. Briefly, the assays were conducted in a 2 ml reaction mixture containing JC-1 Assay Buffer and 25 µg mitochondrial protein. The reaction was initiated with the addition of JC-1 Stain and fluorescence was measured with a Victor3 Multilabel Reader with an excitation wavelength of 490nm and an emission wavelength of 590nm. Results were reported in fluorescence units/µg protein.
Brain ATP levels
ATP levels in right hemisphere homogenates were measured using the ATPlite 1step Luminescence ATP Detection Assay System. Briefly brain tissue was homogenized in buffer solution containing tris-buffered saline (TBS) and protease inhibitor cocktail VII (Calbiochem, San Diego, CA), and centrifuged at 33,000 RPM at 4C for 30 min. The resulting pellet was homogenized in buffer containing TBS, protease inhibitor cocktail VII and 1% Triton X100 (Roche, Indianapolis, IN), then centrifuged at 33,000 RPM at 4C for 30 min. The resulting supernatant (25 µl) was added to an equal volume of 20% trichloracetic acid (Sigma, St. Louis, MO), centrifuged at 10,000 × g 10 min, 4C, transferred to a fresh tube and diluted 50X with PBS containing calcium and magnesium (Invitrogen, Carlsbad, CA). The resulting supernatant (100 µl) was added to an equal volume of ATP light substrate solution and fluorescence measured on a Victor3 Mulitilabel Reader.
Brain oxidative damage markers
Frontal cortex samples were homogenized on ice in a buffer solution containing TBS, protease inhibitor cocktail VII and 1% Triton X100. Protein concentration was determined using the BCA Assay (Pierce Biotechnology, Rockford, IL). Protein carbonyls were determined by colorimetric ELISA using the Biocell Protein Carbonyl Enzyme Immuno-Assay Kit (Biocell Corp, Aukland, NZ) as per manufacturer’s instructions. Nitrotyrosine levels were measured by ELISA using methodology previously described [19] using the Bioxytech Nitrotyrosine Immunoassay (Oxis International, Inc., Foster City, CA). Malonaldehyde (MDA) levels were measured as an indicator of lipid peroxidation using the Bioxytevch LPO 586 Assay (Oxis International) as per manufacturer’s instructions, quantified using a standard curve and normalized to protein content.
Tocopherol Analysis
For the analysis of α- and γ-tocopherol, a modification of the method by Podda et al. [20] was used. Briefly, tissue was saponified with alcoholic KOH, extracted with hexane, dried under nitrogen, resuspended in 1:1 ethanol:methanol, then injected into an HPLC system. The HPLC system consisted of a Shimadzu LC-10ADvp controller, and a SIL-10ADvp auto injector with a 50µl sample loop. Tocopherols were detected using a LC-4B amperometric electrochemical detector (Bioanalytical Systems Inc., West Lafayette, IN, U.S.A.) with a glassy carbon working electrode, and a silver chloride reference electrode. The column used was a Waters Spherosorb ODS2 C-18 column, 100 × 4.6 mm, 3 µm particle size with a Waters Spherisorb ODS precolumn, 10 × 4.6 mm, 5 µm. An isocratic mobile phase delivery system was used, with a total run time of 6 minutes. The mobile phase used was 99:1 (v:v) methanol:water containing 0.1% (w:v) lithium perchlorate. The electrochemical detector was in the oxidizing mode, potential 500 mV, full recorder scale at 500 nA. Peak areas were integrated using Shimadzu Scientific 4.2 Class VP software package, and tocopherols were quantitated using authentic standards.
Statistical Analysis
Data were analyzed using SPSS statistical software version 13.0 (SPSS Inc, Chicago, IL). Statistical comparisons were performed with one-way analysis of variance followed by the Bonferroni post hoc test (comparisons between multiple groups).
Results
CoQ attenuates APP-CTF-induced neurotoxicity, depresses mitochondrial membrane potential, and suppresses oxidative stress in vitro
CoQ protected MC65 cells from APP CTF-associated toxicity in a concentration dependent manner, with an EC50 concentration of approximately 1.5 µM and concentrations of 6.25 µM and higher providing near complete protection from cytotoxicity (Fig. 1a). CoQ treatment had no effect on cellular ATP production (Fig. 1b), however CoQ (6.25 µM) significantly decreased mitochondrial membrane potential in MC65 cells as measured by the decrease in fluorescence of the fluorophore JC-1 (Fig. 1c). The cytochrome c oxidase inhibitor sodium azide was used as a control to prevent JC-1 fluorescence. Treatment with CoQ (6.25 µM) suppressed oxidative stress associated with APP CTF expression, as measured by intracellular superoxide levels (absorbance at 620 nm of nitroblue tetrazolium (NBT), Fig. 1d) and concentrations of hydrogen peroxide in the cell media (Fig. 1e).
Figure 1.
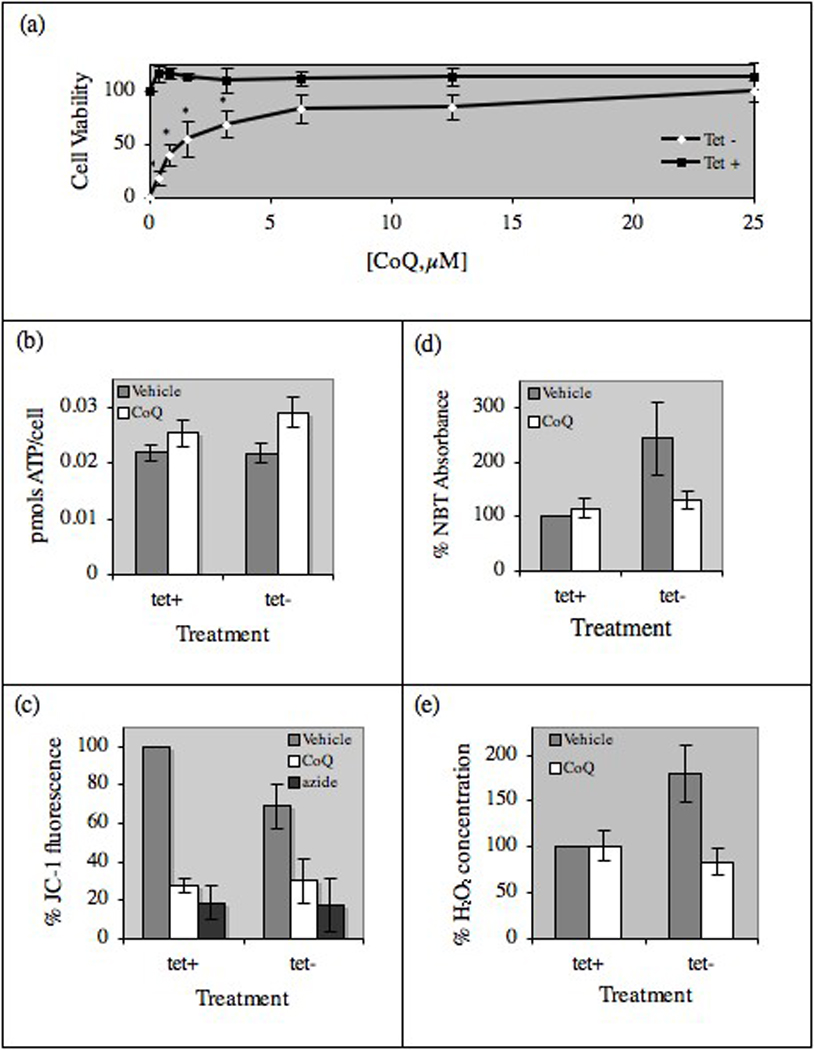
CoQ protects against APP CTF-associated toxicity in vitro. (a) MC65 cells were plated with (tet+) or without (tet−) tetracycline in the presence of increasing concentrations of CoQ for 2.75 days. Cell viability (determined by the MTS assay) is expressed as percent background-corrected absorbance of vehicle-treated tet+ cells. At CoQ doses of 6.25 µM and higher, there was no significant difference in cell viability between tet+ and tet− cells. (b) ATP concentration was determined at 2.0 days in tet+ and tet− cells treated with vehicle or 6.25 µM CoQ. (c) Mitochondrial membrane potential, expressed as percent JC-1 fluorescence compared to vehicle-treated tet+ cells, was determined at 2.0 days cells treated with vehicle or 6.25 µM CoQ. (d) Intracellular superoxide levels, measured at 2.0 days by the NBT assay in cells treated with vehicle or 6.25 µM CoQ, are expressed as % NBT absorbance compared to vehicle-treated tet+ cells. (e) Hydrogen peroxide levels were measured at 2.0 days in media from cells treated with vehicle or 6.25 µM CoQ using the Bioxytech Assay and expressed as % hydrogen peroxide concentration compared to vehicle-treated tet+ cells. All results are mean ± SE, n=3–5, bars with an * differ at p<0.05.
Effects of oral lovastatin and CoQ supplementation on tissue CoQ levels
Changes in body weight were used as criteria to evaluate potential toxicity. Weekly body weights were not changed by CoQ or lovastatin treatment (data not shown). Using values for weekly food consumption, animal weights and the concentration of CoQ and lovastatin in the formulated diets, we calculated that animals on the CoQ diet received an average daily dose of 1.0 g CoQ/kg body weight and 0.2 g lovastatin/kg body weight. Despite using a CoQ dose and delivery method previously reported to increase brain levels of CoQ, we did not see an increase in brain (Fig. 2a) or mitochondrial (Fig. 2b) levels of CoQ9 or CoQ10. As expected, lovastatin treatment reduced brain and mitochondrial levels of CoQ9 and CoQ10, although the effect on brain CoQ9 was not statistically significant.
Figure 2.
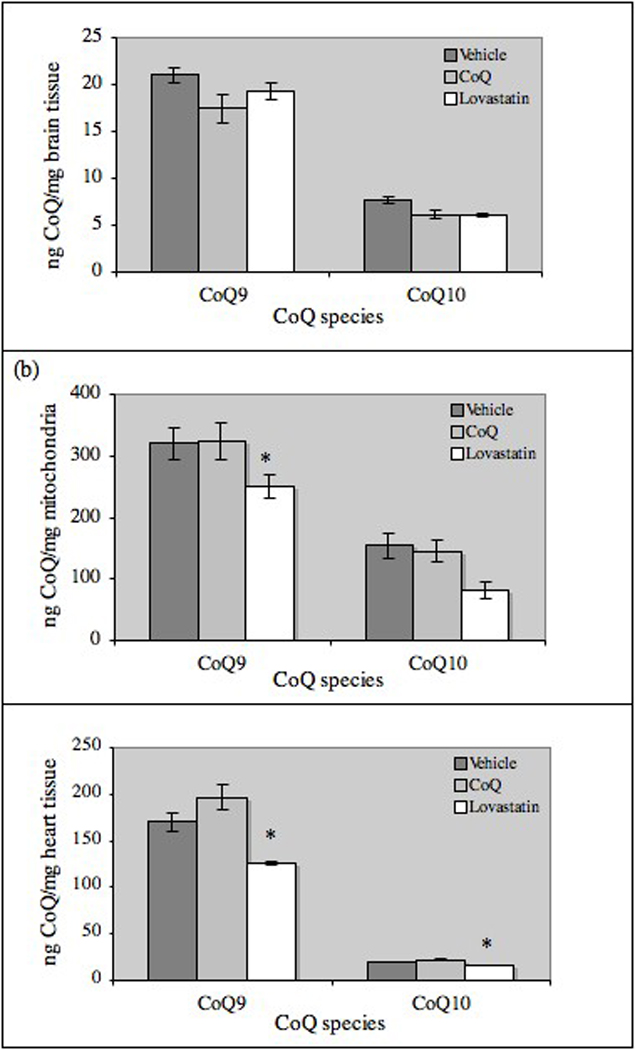
Effects of CoQ and lovastatin supplementation CoQ levels in wild-type mice. Homogenates of (a) left hemisphere brain tissue (Vehicle, n=10; CoQ, n=11; lovastatin, n=11), brain mitochondria (vehicle, n=4; CoQ, n=8; lovastatin, n=7) and (c) heart tissue (n=3/treatment) were extracted and analyzed for levels of CoQ9 and CoQ10 by HPLC. All results are mean ± SE, bars with an * differ at p<0.05.
In order to establish whether the doses used in our studies had effects on peripheral CoQ levels, CoQ9 and CoQ10 were measured in a subset of heart homogenates (n=3/treatment). Compared to vehicle treatment, CoQ supplementation increased heart levels of CoQ9 and CoQ10 by 15% and lovastatin significantly decreased levels of CoQ9 and CoQ10 by 25% (Fig. 2c). There was a non-significant trend for brain levels of CoQ9 and CoQ10 to be higher in young Tg2576 mice (which overexpress a mutant form of human APP) than in wild-type mice (data not shown).
Brain mitochondrial energetics are not improved by CoQ
Brain ATP levels (Fig. 3a) and mitochondrial membrane potential (JC-1 fluorescence, Fig. 3b) were unaffected by CoQ and lovastatin treatment.
Figure 3.
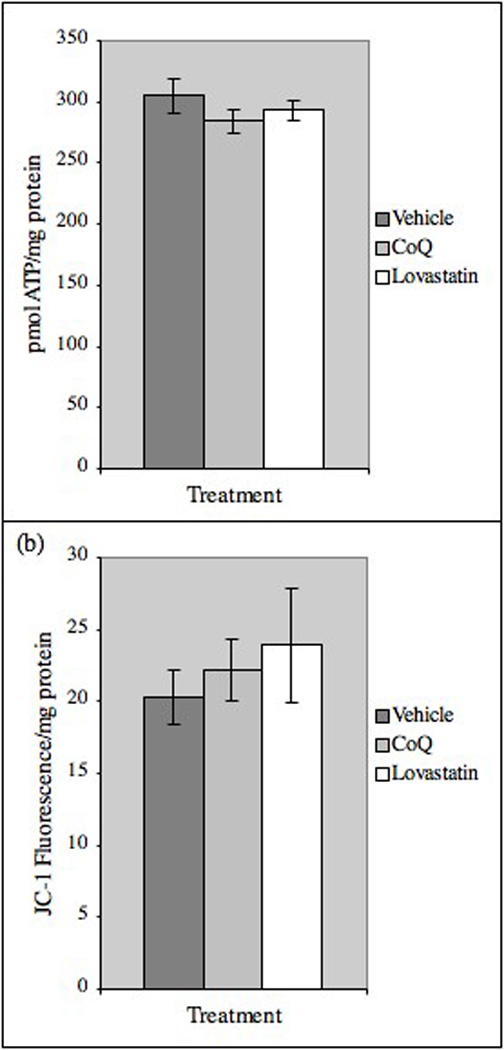
Tissue ATP levels and mitochondrial membrane potential are unaffected by lovastatin or CoQ treatment. (a) ATP levels were measured in right hemisphere homogenates. Results are mean ± SE (Vehicle, n=10; CoQ, n=11; lovastatin, n=10). (b) Mitochondria were isolated from brain tissue and membrane potential measured by JC-1 fluorescence/mg protein. Results are mean ± SE, (Vehicle, n=6; CoQ, n=7; lovastatin, n=8).
Brain protein oxidative damage is suppressed by CoQ
CoQ supplementation significantly suppressed brain levels of protein carbonyls, which are markers of oxidative damage (Fig. 4a). There was a non-significant trend to lower brain levels of nitrotyrosine (a marker of peroxynitrite-mediated damage to proteins, Fig. 4b) in CoQ-treated mice and no effect on lipid peroxidation (Fig. 4c) or levels of α-tocopherol (Fig. 4d) in brain homogenates from either CoQ or lovastatin-treated mice Levels of γ-tocopherol were below the level of detection.
Figure 4.
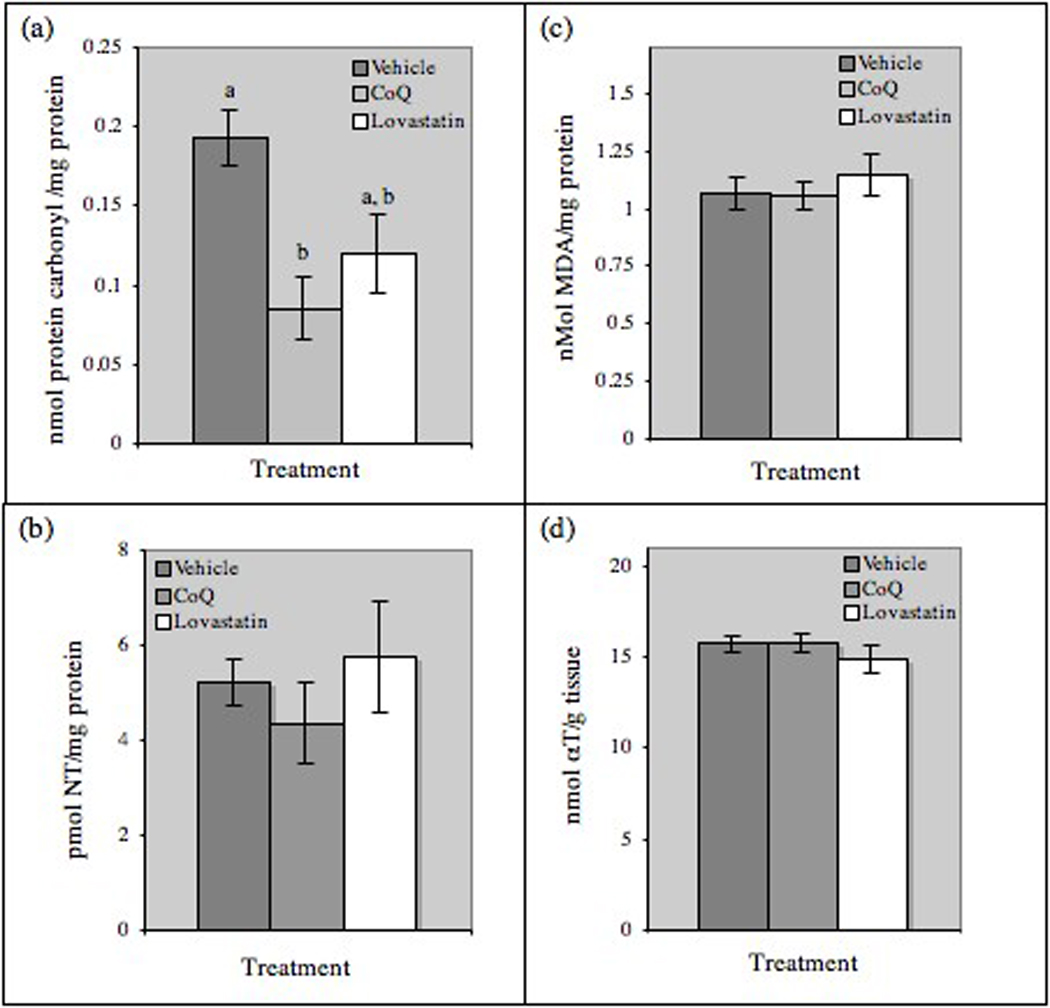
Effects if CoQ supplementation on brain oxidation. (a) Protein carbonyl content and (b) nitrotyrosine (NT) levels were measured in right hemisphere homogenates by commercial ELISA. (c) Malonaldehyde (MDA, an indicator of lipid peroxidation) and (d) α-tocopherol (αT) levels were measured in frontal cortex homogenates. Results are mean ± SE, (Vehicle, n=10; CoQ, n=11; lovastatin, n=11). Bars with different letters differ at P<0.05.
Discussion
These data support a trial of CoQ in an animal model of AD in order to determine whether a clinical trial is warranted. The in vitro experiments with MC65 neuroblastoma cells demonstrate that exogenous CoQ protects neuronal cells from APP CTF-mediated neurotoxocity, with an EC50 of approximately 1.5 µM. A dose of 6.25 µM provides complete protection from neurotoxicity and also suppresses hydrogen peroxide and superoxide production. These effects are not associated with any improvement in energy generation, but instead are associated with a suppression of mitochondrial membrane potential, consistent with previous reports that CoQ activates Uncoupling Protein 2 (UCP-2), a molecule which reduces oxidative stress by attenuating the membrane potential and the rate of energy production [21]. Although it was not possible to test the activation state of UCP-2 in these experiments, this hypothesis fits the in vitro data nicely.
The CoQ concentrations used in vitro are within the range seen in plasma from patients with AD (0.86 µM) [22] and Parkinson’s disease (0.5 µg/ml at baseline and 2 – 4 µg/ml with CoQ supplementation at 300–1200 mg/day) [8], and therefore appear to be feasible target levels in the brain. Although brain tissue and mitochondrial CoQ levels were not increased with oral CoQ supplementation, oxidative damage to brain proteins was attenuated by CoQ supplementation, so oral CoQ may be a viable antioxidant strategy for neurodegenerative disease, including AD.
The failure of CoQ supplementation to increase CoQ levels in brain homogenates or mitochondria was unexpected, but consistent with many other reports, which varied in dose and duration of CoQ supplementation, strain of rodent, and fractionation of brain tissue for analysis. We present a summary of published reports on brain levels of CoQ achieved by supplementation in Table I. Our finding that brain Coenzyme Q levels are not increased with oral administration is consistent with the findings in most [23] [24] [25], but not all [26] [27] published reports. The age of treated mice is not clearly explanatory, as brain CoQ is reported to be stable with normal mouse aging [28]. The discrepancy also does not appear to be explained by differences in the dose or duration of administration of CoQ when reviewed in the context of prior published studies (see Table I). Some investigators have noted that mitochondrial concentrations of CoQ, but not whole brain concentrations, may be affected by oral supplementation [24], while others [23] have also failed to find changes in brain mitochondria after CoQ supplementation. In summary, while mouse strain, mouse age, CoQ dose and CoQ duration of administration might have influenced the outcome of the experiments presented here, our observation that supplementation did not increase brain or mitochondrial CoQ is consistent with published reports.
Table I.
| Species | Strain | Sex | Age at Initiation |
Dose/Source | Duration | Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Rat | Sprague Dawley | male | 12 mo, 24 mo | 200 mg/kg/d In chow Vitaline formulas | 2 mo | Increased brain CoQ | Matthews 1998 |
| Rat | Fisher 344 | ? | 24 mo | Same | 1 mo | Increased brain CoQ | Matthews 1998 |
| Mouse | C57/Bl | male | 24 mo | CoQ10 123 mg/kg/d Sigma | 13 wk | No change in total brain, brain mitochondria, or synaptosomal CoQ (but increase in vit E) | Lass 1999 |
| Rat | Sprague dawley | male | 14 mo | 150 mg/kg/d calculated Tishcon | 4 wk, 13 wk | Statistically significant increase in total brain and brain mitochondria, magnitude small, in comparison to other organs. | Kwong 2002 |
| Mouse | C57Bl | male | 4 mo | 0.72 mg/g or 2.81 mg/g in chow Tishcon | 11 wk | No change in total brain; slight but statistically significant increase in brain mitochondria | Kamzolov 2003 |
| Mouse | C57Bl | male | 3 mo | (cont), 93 (low), or 371 mg (high) CoQ10/kg BW /d Tishcon | 1, 20, 32 mo | No increase in total brain or mitochondria, even after 20 mo (increases seen in all other organs) | Sohal 2006 |
Our experiments showed a statistically significant effect of lovastatin upon whole brain and brain mitochondrial CoQ10 levels, although the magnitude of reduction was quite modest in the brain. CoQ9 was significantly depressed in mitochondria, but not depressed in brain tissue. Since 2g/kg of lovastatin has previously been reported to markedly depress plasma cholesterol in Tg2576 mice [14], this dose appears to be clinically relevant and may even be at the limit of tolerability. Following one month of treatment, lovastatin-treated animals lost an average of 7% of their body weight (data not shown). Lovastatin treatment was not associated with increased indices of oxidative stress (protein carbonyls, nitrotyrosine and lipid peroxidation) despite the suppression of tissue CoQ. This discrepancy may be due to the cholesterol-lowering effects of lovastatin. Alternatively the brief duration of therapy may have limited the ability of lovastatin to affect markers of oxidative stress in this study. The next question to clarify before embarking on CoQ supplementation studies in AD is whether brains with AD pathology have a CoQ “deficiency” at baseline, which would greatly strengthen the rationale for CoQ therapy in AD. There is only one study of brain CoQ in human subjects with AD, and it showed that the expected age dependent decline in brain CoQ was not seen in AD patients, who paradoxically had higher brain CoQ levels than age matched control subjects [29]. Interestingly, we also found a trend toward increased brain CoQ in Tg2576 compared to wild type mice (although it is important to note that these mice were at an age before the appearance of overt AD pathology).
The suppression of oxidative damage in the brains of CoQ-fed mice, without an increase in brain levels of CoQ, may be explained in a number of possible ways. One possibility is that the ratio of reduced to oxidized CoQ might be favorably altered by CoQ supplementation, resulting in an antioxidant effect without a change in total brain CoQ. There may also be supplementation-induced changes in CoQ levels in specific cellular compartments that are not measurable in brain homogenates. It is also possible that systemic CoQ is able to achieve antioxidant effects by an indirect mechanism that may not require delivery of CoQ to the brain, by, for example, restoration of other brain antioxidants. This phenomenon has been demonstrated in vivo in an experiment showing that CoQ supplementation can increase brain levels of α-tocopherol without changing brain levels of CoQ [23]. We tested this possibility directly, however neither lovastatin or CoQ had an effect on brain levels of α-tocopherol, so this specific mechanism is not supported. This does not rule out the general possibility of systemic effects on oxidation status that do not require brain delivery of CoQ per se. For example, CoQ supplementation has also been shown to shift plasma aminothiol redox status toward a more antioxidative environment by increasing the reduced/oxidized glutathione ratio and decreasing cystienylglycine and homocysteine levels [24].
In conclusion, a large body of evidence implicates both mitochondrial dysfunction and oxidative damage in the pathogenesis of AD. Our results indicate that CoQ supplementation achieves antioxidant effects in vitro and in the brains of wild type mice. Future studies will investigate whether CoQ supplementation prevents oxidative stress and the impairment of mitochondrial function in Tg2576 mice. Additional research will also be necessary to determine the specific mechanism by which CoQ exerts antioxidant effects in vivo, and whether this antioxidant effect or associated mechanisms, such as increased UCP-2 activity, can be translated into meaningful neuroprotectant effects.
Acknowledgements
This work was supported by a Department of Veteran’s Affairs Merit Review Grant and R21 AG027445 to JFQ. The authors would like to thank Dr. Maret Traber and Scott Leonard for measuring tissue α- and γ-tocopherol levels and Vitaline® Formulas for their generous donation of CoQ.
References
- 1.Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, 2nd, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 2.Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. Lipid peroxidation in aging brain and Alzheimer's disease. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- 3.Smith MA, Drew KL, Nunomura A, Takeda A, Hirai K, Zhu X, Atwood CS, Raina AK, Rottkamp CA, Sayre LM, Friedland RP, Perry G. Amyloid-beta, tau alterations and mitochondrial dysfunction in Alzheimer disease: the chickens or the eggs? Neurochem Int. 2002;40:527–531. doi: 10.1016/s0197-0186(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 4.Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-Dependent Inhibition of Human Cytochrome c Oxidase by a Dimeric Conformer of Amyloid-{beta}1–42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 6.Beal MF. Coenzyme Q10 as a possible treatment for neurodegenerative diseases. Free Radic Res. 2002;36:455–460. doi: 10.1080/10715760290021315. [DOI] [PubMed] [Google Scholar]
- 7.Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, Juncos JL, Nutt J, Shoulson I, Carter J, Kompoliti K, Perlmutter JS, Reich S, Stern M, Watts RL, Kurlan R, Molho E, Harrison M, Lew M. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 9.Smith KM, Matson S, Matson WR, Cormier K, Del Signore SJ, Hagerty SW, Stack EC, Ryu H, Ferrante RJ. Dose ranging and efficacy study of high-dose coenzyme Q10 formulations in Huntington's disease mice. Biochim Biophys Acta. 2006;1762:616–626. doi: 10.1016/j.bbadis.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Langsjoen PH, Langsjoen AM. The clinical use of HMG CoA-reductase inhibitors and the associated depletion of coenzyme Q10. A review of animal and human publications. Biofactors. 2003;18:101–111. doi: 10.1002/biof.5520180212. [DOI] [PubMed] [Google Scholar]
- 11.Morisco C, Trimarco B, Condorelli M. Effect of coenzyme Q10 therapy in patients with congestive heart failure: a long-term multicenter randomized study. Clin Investig. 1993;71:S134–S136. doi: 10.1007/BF00226854. [DOI] [PubMed] [Google Scholar]
- 12.Yang HT, Lin SH, Huang SY, Chou HJ. Acute administration of red yeast rice (Monascus purpureus) depletes tissue coenzyme Q(10) levels in ICR mice. Br J Nutr. 2005;93:131–135. doi: 10.1079/bjn20041285. [DOI] [PubMed] [Google Scholar]
- 13.Witting PK, Pettersson K, Letters J, Stocker R. Anti-atherogenic effect of coenzyme Q10 in apolipoprotein E gene knockout mice. Free Radic Biol Med. 2000;29:295–305. doi: 10.1016/s0891-5849(00)00311-7. [DOI] [PubMed] [Google Scholar]
- 14.Park IH, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, Jung MW, Bang OY, Kim SU, Mook-Jung I. Lovastatin enhances Abeta production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging. 2003;24:637–643. doi: 10.1016/s0197-4580(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 15.Woltjer RL, Maezawa I, Ou JJ, Montine KS, Montine TJ. Advanced glycation endproduct precursor alters intracellular amyloid-beta/A beta PP carboxy-terminal fragment aggregation and cytotoxicity. J Alzheimers Dis. 2003;5:467–476. doi: 10.3233/jad-2003-5607. [DOI] [PubMed] [Google Scholar]
- 16.Choi HS, Kim JW, Cha YN, Kim C. A quantitative nitroblue tetrazolium assay for determining intracellular superoxide anion production in phagocytic cells. J Immunoassay Immunochem. 2006;27:31–44. doi: 10.1080/15321810500403722. [DOI] [PubMed] [Google Scholar]
- 17.Okamoto T, Fukui K, Nakamoto M, Kishi T, Okishio T, Yamagami T, Kanamori N, Kishi H, Hiraoka E. High-performance liquid chromatography of coenzyme Q-related compounds and its application to biological materials. J Chromatogr. 1985;342:35–46. doi: 10.1016/s0378-4347(00)84487-4. [DOI] [PubMed] [Google Scholar]
- 18.Pappu AS, Connor WE, Merkens LS, Jordan JM, Penfield JA, Illingworth DR, Steiner RD. Increased nonsterol isoprenoids, dolichol and ubiquinone, in the Smith-Lemli-Opitz syndrome: effects of dietary cholesterol. J Lipid Res. 2006;47:2789–2798. doi: 10.1194/jlr.M600295-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Quinn JF, Bussiere JR, Hammond RS, Montine TJ, Henson E, Jones RE, Stackman RW., Jr Chronic dietary alpha-lipoic acid reduces deficits in hippocampal memory of aged Tg2576 mice. Neurobiol Aging. 2007;28:213–225. doi: 10.1016/j.neurobiolaging.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 20.Podda M, Webber C, Traber MG, Packer L. Simultaneous determination of tissue tocopherols, tocotrienols, ubiquinols and ubiquinones. J Lipid Res. 1996;37:893–901. [PubMed] [Google Scholar]
- 21.Horvath TL, Diano S, Leranth C, Garcia-Segura LM, Cowley MA, Shanabrough M, Elsworth JD, Sotonyi P, Roth RH, Dietrich EH, Matthews RT, Barnstable CJ, Redmond DE., Jr Coenzyme Q induces nigral mitochondrial uncoupling and prevents dopamine cell loss in a primate model of Parkinson's disease. Endocrinology. 2003;144:2757–2760. doi: 10.1210/en.2003-0163. [DOI] [PubMed] [Google Scholar]
- 22.Quinn J, Suh J, Moore MM, Kaye J, Frei B. Antioxidants in Alzheimer's disease-vitamin C delivery to a demanding brain. J Alzheimers Dis. 2003;5:309–313. doi: 10.3233/jad-2003-5406. [DOI] [PubMed] [Google Scholar]
- 23.Lass A, Forster MJ, Sohal RS. Effects of coenzyme Q10 and alpha-tocopherol administration on their tissue levels in the mouse: elevation of mitochondrial alpha-tocopherol by coenzyme Q10. Free Radic Biol Med. 1999;26:1375–1382. doi: 10.1016/s0891-5849(98)00330-x. [DOI] [PubMed] [Google Scholar]
- 24.Kwong LK, Kamzalov S, Rebrin I, Bayne AC, Jana CK, Morris P, Forster MJ, Sohal RS. Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic Biol Med. 2002;33:627–638. doi: 10.1016/s0891-5849(02)00916-4. [DOI] [PubMed] [Google Scholar]
- 25.Sohal RS, Kamzalov S, Sumien N, Ferguson M, Rebrin I, Heinrich KR, Forster MJ. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic Biol Med. 2006;40:480–487. doi: 10.1016/j.freeradbiomed.2005.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamzalov S, Sumien N, Forster MJ, Sohal RS. Coenzyme Q intake elevates the mitochondrial and tissue levels of Coenzyme Q and alpha-tocopherol in young mice. J Nutr. 2003;133:3175–3180. doi: 10.1093/jn/133.10.3175. [DOI] [PubMed] [Google Scholar]
- 27.Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci USA. 1998;95:8892–8897. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lass A, Kwong L, Sohal RS. Mitochondrial coenzyme Q content and aging. Biofactors. 1999;9:199–205. doi: 10.1002/biof.5520090215. [DOI] [PubMed] [Google Scholar]
- 29.Edlund C, Soderberg M, Kristensson K, Dallner G. Ubiquinone, dolichol, and cholesterol metabolism in aging and Alzheimer's disease. Biochem Cell Biol. 1992;70:422–428. doi: 10.1139/o92-065. [DOI] [PubMed] [Google Scholar]