

Abstract
Normal cells respond appropriately to various signals, while sustaining proper developmental programs and tissue homeostasis. Inappropriate signal reception, response or attenuation, can upset the normal balance of signaling within cells, leading to dysfunction or tissue malformation. To understand the molecular mechanisms that regulate protein-kinase-based signaling in the context of tissue morphogenesis, we analyzed the domain requirements of Drosophila Slpr, a mixed-lineage kinase (MLK), for Jun N-terminal kinase (JNK) signaling. The N-terminal half of Slpr is involved in regulated signaling whereas the C-terminal half promotes cortical protein localization. The SH3 domain negatively regulates Slpr activity consistent with autoinhibition via a conserved proline motif. Also, like many kinases, conserved residues in the activation segment of the catalytic domain regulate Slpr. Threonine 295, in particular, is essential for function. Slpr activation requires dual input from the MAP4K Misshapen (Msn), through its C-terminal regulatory domain, and the GTPase Rac, which both bind to the LZ–CRIB region of Slpr in vitro. Although Rac is sufficient to activate JNK signaling, our results indicate that there are Slpr-independent functions for Rac in dorsal closure. Finally, expression of various Slpr constructs alone or with upstream activators reveals a wide-ranging response at the cell and tissue level.
Keywords: Drosophila, JNK signaling, Dorsal closure, Kinase
Introduction
Protein kinases are a large and diverse superfamily that impacts virtually every fundamental process in cells (Manning et al., 2002). Mechanisms that regulate these enzymes are correspondingly diverse, including intra- and intermolecular interactions, posttranslational modification, subcellular compartmentalization, and feedback. Despite common themes in the regulation of protein kinase activity, the molecular details vary among kinase families and cell context. Consideration of these differences is imperative to fully understand the nature of substrate specificity, the consequences of kinase misregulation, and the potential for targeted therapeutic inhibitors and agonists. Various methodologies, including structural studies and in vitro manipulations, have increased our understanding of kinase biology significantly, however in vivo support for regulatory models derived from in vitro studies will help to consolidate physiologically relevant regulatory mechanisms.
In this report, we examine the molecular mechanisms regulating activation of the Drosophila mixed-lineage kinase (MLK), encoded by the slpr locus (Stronach and Perrimon, 2002), which is a member of the tyrosine-like kinase group (Manning et al., 2002). MLKs were named for their mixed homology kinase domains, with residues matching both tyrosine and serine/threonine kinases (Dorow et al., 1993); however, biochemical assays demonstrate specificity for serine and threonine residues (Gallo et al., 1994). MLKs are mitogen-activated protein kinase kinase kinases (MAP3Ks) that phosphorylate and activate MAP2K dual-specificity kinases, which in turn stimulate MAPKs of the Jun N-terminal kinase (JNK) and p38 families (Hirai et al., 1997; Kiefer et al., 1996; Rana et al., 1996; Teramoto et al., 1996; Tibbles et al., 1996). Seven mammalian MLKs have been identified, clustering into three subfamilies: the core MLKs (MLK1–4), the dual leucine zipper kinases (DLK and LZK), and the zipper sterile-α-motif kinase (ZAK) (for a review, see Gallo and Johnson, 2002). All family members activate the JNK pathway when overexpressed in cultured cells (Hirai et al., 1997; Liu et al., 2000; Merritt et al., 1999; Rana et al., 1996; Tibbles et al., 1996); however, their endogenous activities and regulation in response to distinct signals have been more difficult to discern (Craig et al., 2008). Genetic analyses using invertebrate models have shed light on functions for MLK and DLK family members in vivo. For instance, our previous studies implicated the Drosophila MLK, Slpr, in regulating JNK-dependent tissue morphogenesis (Polaski et al., 2006; Stronach and Perrimon, 2002), whereas the nematode mlk-1 gene is required for stress response to heavy metals (Mizuno et al., 2004). Both Drosophila and C. elegans DLK genes regulate neuronal synaptic structure and function via JNK or p38 MAPKs, respectively (Collins et al., 2006; Hammarlund et al., 2009; Nakata et al., 2005). The functional link between DLKs and nervous system development appears to be conserved in mammals as well (Hirai et al., 2006; Itoh et al., 2009). Targeted gene disruption of murine MLK core family members has been less revealing. Mlk1, Mlk2 double knockout mice appear normal, whereas Mlk3 mutant mice are viable but abnormal in some cytokine and metabolic stress signaling pathways (Bisson et al., 2008; Brancho et al., 2005; Jaeschke and Davis, 2007). Genetic analysis of ZAK has not been reported, although expression studies suggest a role in hypertrophic growth of cultured cardiomyoblasts, consistent with its expression in heart tissue (Huang et al., 2004; Liu et al., 2000).
Core MLKs have a Src-homology 3 (SH3) domain, a kinase domain, tandem leucine zippers (LZ) followed by a Cdc42-Rac interactive binding motif (CRIB) (Burbelo et al., 1995) and a long divergent C-terminus (Gallo and Johnson, 2002). Maximal activation of mammalian MLK3 protein in cultured cells is a multistep process, involving GTPase binding, relief of inhibition, dimerization and autophosphorylation (Bock et al., 2000; Leung and Lassam, 1998; Leung and Lassam, 2001; Vacratsis and Gallo, 2000; Zhang and Gallo, 2001). Complex multistep regulation of kinase activation has been studied extensively for members of the Raf and Src families, revealing that autoinhibition, membrane recruitment and regulatory phosphorylation are recurring themes (Boggon and Eck, 2004; Leicht et al., 2007).
To gain a better understanding of the mechanisms cells employ to engage kinases in signal transmission, we are examining the steps of Slpr activation during Drosophila embryonic dorsal closure. Among the MAP3Ks in Drosophila, Slpr is the only core MLK protein (Stronach, 2005), facilitating genetic analysis of its regulation in vivo. slpr was first identified as a locus required for JNK signaling during the process of dorsal closure (Stronach and Perrimon, 2002) wherein the ectoderm on each flank of the embryo is pulled toward the dorsal midline enclosing the embryo in a seamless epidermis (Harden, 2002; Jacinto et al., 2002b; Kiehart et al., 2000). Molecular markers reporting JNK signaling activity are present in cells leading the advancing epithelium, and mutations in JNK pathway components impair dorsal closure by disrupting both gene expression and organization of the cytoskeleton (Glise et al., 1995; Hou et al., 1997; Jasper et al., 2001; Kaltschmidt et al., 2002; Kockel et al., 1997; Riesgo-Escovar et al., 1996; Sluss et al., 1996; Stronach and Perrimon, 2002; Xia and Karin, 2004).
Although the core members of the Drosophila JNK pathway have been identified, the mechanism of pathway activation in the ectoderm during tissue closure is still poorly understood. Genetic evidence is consistent with Slpr functioning downstream of two inputs, the Rac GTPases and the MAP4K Misshapen (Stronach and Perrimon, 2002; Su et al., 1998), yet a mechanistic understanding of how they trigger Slpr activity has not been realized. In the work reported here, we examined the regulation of Slpr activation through a structure–function analysis of mutated and modified forms in vivo, in the context of tissue closure. Our data support a model for Slpr activation that includes multiple inputs, dual regulation by the SH3 domain, and modulation of subcellular distribution by the C-terminal domain. Moreover, we demonstrate a physical interaction between Slpr and Misshapen (Msn) that corroborates genetic data supporting their role in dorsal closure. Altogether, the modified forms of Slpr constitute a broad range of activities that define important functional domains and contribute to our understanding of how Slpr modulates JNK-dependent morphogenesis.
Results
Slpr structure–function analysis
To dissect the mechanisms that regulate Slpr activity in vivo, we have generated derivative constructs that harbor mutations or deletions of various domains and/or motifs conserved among MLK family members (Fig. 1). Their properties were assessed in transgenic animals in the following manner. First, to identify essential functions, transgenes were tested for the ability to rescue semi-viable and lethal slpr mutants (Fig. 2). Second, to indirectly assay catalytic activity in vivo, we asked what effect transgenic protein expression had on JNK signaling in the embryo during dorsal closure, using the puc-lacZ reporter, pucE69 (Fig. 3). Puc-lacZ is normally evident in the leading row of ectodermal cells undergoing closure and has been used extensively as a marker to evaluate JNK pathway activity. Third, to discern the impact of transgene expression on morphogenetic processes known to require endogenous Slpr function, dorsal and thorax closure were monitored for completion (Figs 2, 3). Finally, by correlating the activity of the transgenic Slpr derivatives with their subcellular protein localization in cells, we have identified protein domains and motifs that regulate Slpr distribution and function (Fig. 4).
Fig. 1.

Domain architecture and expression of Slpr and derived transgenic constructs. (A) Organization and position of Slpr domains (SH3, Src homology 3; LZ, leucine zipper; CRIB, Cdc42–Rac interactive binding) and mutant alleles. Constructs are shown as black lines with mutated amino acids noted. The kinase activation segment, flanked by conserved residues (underlined), shows putative Ser/Thr phosphorylation targets (bold). The sequence of the LZ–CRIB linker shows the proline residues (bold) that were targeted for mutagenesis. (B) Alignment of the activation segments of Drosophila Slpr (Dme) and its human (Hsa) homologs. Conserved phosphorylation sites are indicated by grey boxes. Black boxes identify experimentally determined primary phosphosites in the human MLKs. (C) Sequence alignment of the LZ–CRIB linkers of Slpr and its human homologs. Note the conserved proline residue(s) followed by a stretch of basic amino acids (grey boxes). (D) Western immunoblots of embryonic lysates expressing HA-epitope-tagged transgenic proteins. β-tubulin was used as a loading control. Transgenic proteins were stably expressed with the exception of the Cterm (asterisk), whose levels are reduced.
Fig. 2.

Rescue of slpr mutants by ubiquitous transgene expression. (A) Slpr transgenes (y-axis) were expressed with arm-G4 in each slpr mutant background (see Fig. 1 and its legend). The number of rescued slpr mutant males relative to FM7 male siblings was plotted as a percentage ‘rescue to adulthood’. The graph is based on data (supplementary material Table S1) from crosses with at least three transgenic lines per construct. Values >100% indicate that the recovery of rescued males exceeded sibling controls. (B) Rescue of the embryonic dorsal closure phenotype of slpr921 mutants as indicated by reduction in embryos with a severe phenotype. Representative cuticles are shown on the right. Raw data are given in supplementary material Tables S2, S3. Cuticles of slpr mutant progeny were unambiguously identified through genetic linkage with the gt mutant phenotype.
Fig. 3.

Effect of Slpr transgene expression on embryonic JNK target gene expression and tissue closure. (A) lacZ expression from the JNK-dependent pucE69 enhancer trap in embryos expressing the indicated Slpr transgenes with pnr-G4. Images are lateral views of three segments of stage 14 embryos focusing on the leading edge of the dorsal ectoderm. Dominant negative activity reduces the number of cells with active JNK signaling and puc-lacZ expression (c,d,e,h,j). Some transgenes have no effect (g,i), whereas others activate signaling, increasing the intensity and extent of lacZ expression (b,f,k,l). Scale bar: 10 μm. (B) Thr295 in the activation segment of Slpr kinase is essential for function. (a-e) Dorsolateral views of stage 16 embryos stained for Fasciclin III, revealing the ectodermal epithelium and extent of dorsal closure. Scale bar: 20 μm. (C) The effect of expression of Slpr transgenes, using pnr-G4, on the adult thorax morphology parallels the effect of transgene expression in the embryo. Dominant negative transgenes produce midline thorax clefts and widening of the space between adjacent dorsocentral bristles (a,d,g) compared with no transgene controls (b). Hyperactive Slpr variants promote loss of midline tissue, such as the scutellum, and narrowing of the inter-bristle space (c,f,i).
Fig. 4.

Cortical enrichment of Slpr is perturbed by loss of the C-terminal domain. (A) Ectopic Slpr-WT-HA expression in the pnr domain is detected with anti-HA immunostaining (green) compared with two polarized epithelial markers, Crumbs (crb; red a,a″, apical) and Discs large (dlg; red b,b″, lateral). Slpr is enriched cortically, and excluded from the nucleus, but not restricted to a particular apicobasal subdomain. (a,b) Dorsolateral view, apical section. Arrowhead in b indicates Slpr immunoreactivity in a cell protrusion emanating from the leading edge. (a′,b′) Ventrolateral view, apical section. (a″,b″) Cross section of dorsal ectoderm to visualize cell junctions; arrowheads point to Crb-positive apical junctions (a″) or Dlg-positive septate junctions (b″). (B) Ectopic expression of indicated transgenes in several segments of the lateral ectoderm within the pnr domain is detected with anti-SlprSH3 sera (a) or anti-HA immunostaining (b-l). Slpr-WT with or without the HA tag, the double A mutants, Cterm, ΔSH3, and Slpr AVA are cortically enriched (a-f,i,k,l), whereas SKLC-WT and SKLC-AAA are more diffusely distributed (d,h). SH3 alone is detected in the cytoplasm and nuclei of cells (j). Scale bar: 20 μm. (C) Slpr transgenic protein expression with arm-G4 is detected by anti-HA immunostaining (green) in slprBS06 mutant larval brains (a-c) and eye imaginal discs (d-f). In the absence of endogenous Slpr protein, Slpr-WT-HA is associated with the cell cortex in mushroom body (a,a′) and photoreceptor (d,d′) neurons. The Cterm has a similar cortical enrichment (c,c′,f,f′). By contrast, SKLC does not appear to localize at the cell membrane (b,b′,e,e′). Filamentous actin is highlighted with phalloidin as a counterstain (red, a-f). Higher magnifications of the boxed regions are shown on the right. Scale bars: 20 μm.
Mutations in activation segment residues disrupt Slpr function
The catalytic domain of many kinases includes a flexible activation segment, bounded by conserved DFG and APE motifs, which is often targeted for auto- and trans-phosphorylation (Nolen et al., 2004). Upon modification, the segment adopts a conformation that promotes substrate binding and catalysis (Nolen et al., 2004). The activation segment of Slpr contains conserved serine and threonine residues that are putative phosphorylation sites (Fig. 1B). If these residues are important for Slpr kinase structure or function, specific mutations are expected to decrease Slpr activity accordingly. Site-directed mutagenesis of all three Ser/Thr residues in the activation segment of Slpr to unphosphorylatable alanine (TST>AAA) renders the protein nonfunctional, based on several criteria. First, Slpr-AAA fails to rescue slpr mutant alleles when expressed ubiquitously with arm-G4 (Fig. 2A). Moreover, it behaves as a dominant negative protein with respect to stimulation of JNK pathway target gene expression and dorsal closure (Fig. 3Ac,Bb). Importantly, the transgenic protein is expressed at levels equivalent to the wild-type form (Fig. 1D) and localizes in a subcellular pattern indistinguishable from the wild-type form (Fig. 4Bc), demonstrating that mutating the activation segment does not result in a wholesale destabilization of the protein. Hence, these data show that Slpr-AAA is equivalent to a catalytically dead form (Slpr-KD), which suggests that the Ser/Thr residues are essential for function in vivo.
To pinpoint which residue is of primary importance, additional constructs were generated to produce proteins with two of the three residues changed to alanine (double A mutants, e.g. TST to TAA, ASA or AAT; Fig. 1A). Slpr-TAA and Slpr-ASA and the triple alanine mutant produced similar results in the rescue, JNK signaling and dorsal closure assays (Fig. 2A, Fig. 3Ad-e,Bc-d), suggesting that they are nonfunctional. Notably though, the Slpr-AAT mutant had a notable effect in JNK signaling and embryonic dorsal closure (Fig. 3Af,Be). Moreover, ubiquitous expression of Slpr-AAT resulted in recovery of a few rescued adults with the lethal slpr921 and slpr3P5 alleles (Fig. 2A). Indeed, many additional rescued males are observed as pharate adults (still in the pupal cases) among the progeny of these experimental crosses, implying that a significant fraction of embryos survive dorsal closure morphogenesis, whereas few if any rescued pharate males survived with expression of Slpr-AAA, -TAA, or -ASA. Together these data implicate threonine 295 as a crucial residue for Slpr-mediated JNK signaling.
The SH3 domain of Slpr negatively regulates JNK signaling
Many kinases that are stimulated by extracellular signals (e.g. Raf, Src) are inactive until a signal exceeds an activating threshold. Commonly, inducible kinases possess their own inhibitory domains to silence activity until the appropriate time. For mammalian MLK3, the SH3 domain is implicated in autoinhibition by binding to a proline-containing motif between the LZ and CRIB domains (Zhang and Gallo, 2001). However, the CRIB domain of yeast Ste20 kinase is implicated in negative autoregulation of its kinase domain, raising alternative possibilities (Lamson et al., 2002). To determine whether the SH3 domain of Slpr regulates its activity in vivo, three constructs were generated, SH3 only, ΔSH3 and Slpr-AVA (P472A, P474A; Fig. 1A,C). Expression of SH3 alone inhibited JNK signaling in the embryo (Fig. 3Aj). Nevertheless, it was possible to recover some adults that displayed a severe cleft thorax phenotype indicative of aberrant thorax closure and reduced JNK activity (Fig. 3Cg) (Agnes et al., 1999; Zeitlinger and Bohmann, 1999). Moreover, ubiquitous expression did not complement slpr mutants, but rather reduced recovery of slprBS06 mutants relative to the control (Fig. 2A) and failed to rescue the closure phenotype of the embryonic lethal slpr921 allele (Fig. 2B, supplementary material Tables S2, S3). Thus, like Slpr-KD and Slpr-AAA, the SH3 domain alone has dominant negative properties, disrupting endogenous Slpr-dependent JNK activation. By contrast, expression of a construct lacking the SH3 domain (ΔSH3) stimulated JNK signaling above that of Slpr-WT overexpression, with substantial upregulation of puc-lacZ away from the leading edge (Fig. 3Ak), epithelial puckering at the dorsal midline (not shown) and adult thorax phenotypes indicative of hyperactive JNK signaling (Fig. 3Cf) (Zeitlinger and Bohmann, 1999). Also, ΔSH3 weakly rescues slprBS06 mutants, resulting in an ~12% increase in recovery over animals without the transgene (Fig. 2A; supplementary material Table S1), and substantially rescues the embryonic dorsal open phenotype of slpr921 mutants (Fig. 2B; supplementary material Tables S2, S3), consistent with activation of JNK signaling. Altogether, these data support the hypothesis that the SH3 domain of Slpr is a negative regulatory domain; however, they do not reveal whether it inhibits Slpr activity by an intramolecular interaction.
To address this, a mutant was generated in which two proline residues, located between the LZ and CRIB domains of Slpr, were replaced by alanine (PVP>AVA). These residues are in a conserved region (Fig. 1C) important for SH3 binding in mammalian MLK3 (Zhang and Gallo, 2001). If the proline residues constitute a binding site for SH3 domain autoinhibition, then mutations that abrogate binding are expected to promote activation. Indeed, the Slpr-AVA protein is remarkably active. Embryonic expression of Slpr-AVA in the pnr-G4 domain resulted in a fivefold upregulation of puc-lacZ (Fig. 3Al, Fig. 5B) and a concomitant increase in the severity of the adult thorax phenotype relative to ΔSH3 expression (Fig. 3Cf,i), although very few of these adults were recovered. Together, these data corroborate results of in vitro assays that the SH3 domain functions in negative regulation, and the inhibitory site in the LZ–CRIB linker region is functionally conserved. In addition, our analysis reveals a potential positive contribution of the SH3 domain.
Fig. 5.

Slpr, Msn and Rac signaling modulate JNK pathway activity. (A) lacZ expression from the JNK-dependent pucE69 enhancer trap in embryos expressing the indicated transgenes with pnr-G4. (a-n) Representative lateral views of three segments of dorsal closure stage 14 embryos are focused on the leading edge of the dorsal ectoderm. Scale bar: 20 μm. (o-q) lacZ reporter expression in embryos at early (st 14, top row) and late (st 16, bottom row) stages of closure expressing the indicated transgenes. Lateral views are shown except in the bottom panel in o, which is a dorsal view. Arrowheads indicate the failure of dorsal closure. Scale bar: 50 μm. (B) Graph of puc-lacZ reporter expression in the indicated transgenic embryos (y-axis). Fluorescence extent and intensity from at least three independent samples for each genotype were quantified as described in the Materials and Methods, and plotted as mean area under the curve in arbitrary units (x-axis). Fold change relative to ‘no transgene’ control (black bar) is indicated on the right.
The Slpr C-terminus regulates protein distribution
The C-terminal half of MLK proteins is the least conserved among homologs although it tends to be rich in serine, threonine and proline residues (Phelan et al., 2001; Vacratsis et al., 2002). Yet, there are no recognizable protein domains to suggest a function. To investigate the function of the C-terminus of Slpr, two complementary constructs, SKLC-WT and Cterm, were analyzed (Fig. 1A). The constructs had neither dominant negative nor constitutive activity with respect to JNK target gene expression and tissue morphology (Fig. 3Ag,i,Ce,h); however, SKLC-WT expression supported rescue of slpr mutants, second only to the full length wild-type form (Fig. 2) indicating that this fragment, lacking the C-terminus, retains function. Incorporating the activation segment mutations to create SKLC-AAA abrogated function (Fig. 2, Fig. 3Ah,Cd), demonstrating the requirement for an intact kinase. Intriguingly, examination of SKLC transgenic protein distribution by immunofluorescence revealed a difference in subcellular localization compared with wild-type protein. Overexpressed full-length Slpr is cortically enriched, although not particularly biased in either apical or basolateral domains [see Polaski et al. (Polaski et al., 2006) and this work, Fig. 4A,Ba,b,Ca,d]. Conversely, the SKLC variants lacking the C-terminus were found to be diffusely distributed, with more extensive cytoplasmic and nuclear localization compared with the full-length protein (Fig. 4Bg,h,Cb,e). Cterm itself still appeared cortically enriched (Fig. 4Bi,Cc,f), localizing in a similar pattern to wild-type in epithelia and other cell types. Thus, Cterm promotes cortical enrichment, but otherwise has no rescue function (Fig. 2), and does not block endogenous signaling (Fig. 3Ai,Ch). Consistent with this interpretation, those Slpr variants that include the C-terminus, such as Slpr-WT and ΔSH3, were largely cortical and showed little cytoplasmic staining. By contrast, any variant that lacks the C-terminus, including SKLC, SK (not shown) and SH3, failed to be restricted to the cortex, localizing in the cytoplasm and nucleus to varying extents (Fig. 4B), illustrated most dramatically by the SH3 domain alone (Fig. 4Bj), which was found in the nucleus in most cells by the end of embryogenesis, although the significance of this localization is unclear.
Upstream input to Slpr activation: Rac1 and Misshapen
The role of Rac GTPase in JNK signaling and dorsal closure is separable
Biochemical and genetic evidence support a role for the GTPases, Rac and Cdc42, in stimulating JNK signaling via MLK proteins (Bock et al., 2000; Teramoto et al., 1996) by binding to the CRIB motif, typically in a nucleotide-dependent fashion (Burbelo et al., 1995). Indeed, full-length Slpr and SKLC bound preferentially to the GTP-loaded form of Rac and Cdc42 in in vitro pull down assays (Fig. 6) and although both are sufficient to upregulate JNK signaling when constitutive active forms are overexpressed during dorsal closure (Glise and Noselli, 1997), loss-of-function genetic data favor Rac over Cdc42 in the regulation of endogenous JNK signaling (Genova et al., 2000; Hakeda-Suzuki et al., 2002; Woolner et al., 2005). Thus, we chose to examine the impact of the association between Slpr and Rac1 for signaling and dorsal closure in vivo. To do so, the wild-type proteins were expressed individually, or together, using the pnr-G4, puc reporter system to monitor JNK signaling. Expression of Rac-WT or Slpr-WT alone in embryos resulted in nearly a twofold increase in intensity and extent of puc-lacZ expression compared with embryos lacking a transgene (Fig. 5Ad,h,i,B). Coexpression led to nearly a fourfold increase in β-gal staining (Fig. 5Am,B). If Rac activates Slpr, then dominant negative Slpr is expected to block Rac-dependent upregulation of JNK pathway activity. Curiously, Slpr-KD did not substantially block ectopic puc-lacZ expression induced by Rac-WT, despite the fact that Slpr-KD alone reduces expression almost completely (Fig. 5Aa,g,B). The same result was observed with other dominant negative Slpr derivatives, including SH3 alone (not shown). To examine the possibility that incomplete suppression of Rac-WT by Slpr-KD simply reflects inadequate levels of Slpr, we tested whether coexpression of dominant negative bsk (JNK) transgenes with Rac-WT would block reporter expression or not. Indeed they did, contrary to our result with Slpr-KD, arguing against inadequate levels of Slpr inhibition; however, morphological changes in these cells were not similarly suppressed (supplementary material Fig. S1). Thus, although JNK is necessary for transcription of puc, it appears not to be required for other Rac-dependent functions.
Fig. 6.

Slpr interacts preferentially with Rac-GTP and Cdc42-GTP. GST pull-down assay using the GTPases Rac and Cdc42, loaded with either GDP or GTP as indicated, and labeled Slpr WT or Slpr SKLC proteins from a rabbit reticulocyte lysate (35S-input). The Coomassie-Blue-stained gel (upper panels) and autoradiograph (lower panel) show expression of the fusion proteins and labeled input protein that is specifically bound, respectively.
Given that GTPase binding to the CRIB site of MLK3 is postulated to release SH3-mediated autoinhibition at the adjacent proline-dependent binding site (Zhang and Gallo, 2001), then expression of Slpr ΔSH3 is expected to bypass Rac-dependent activation. To test this, ΔSH3 was expressed alone or with a dominant negative form of Rac1 (Rac-DN) (Fig. 5Ao-q). As demonstrated previously, ΔSH3 stimulated JNK signaling (Fig. 3Ak, Fig. 5Aj). Expression of Rac-DN alone weakly diminished puc-lacZ expression (Fig. 5Ac,B), but strongly inhibited the progress of dorsal closure (Fig. 5Ap) as previously observed (Harden et al., 1995; Woolner et al., 2005). The Rac-DN phenotypes are clearly distinguishable from those of Slpr ΔSH3, allowing for epistasis analysis. If ΔSH3 activity is constitutive, downstream of Rac function, then coexpression with Rac-DN should be inconsequential, i.e. equivalent to ΔSH3 alone. As shown in Fig. 5Aq, with simultaneous expression of ΔSH3 and Rac-DN, a mixed phenotype was evident. JNK signaling was reduced relative to that when ΔSH3 was expressed alone (Fig. 5B), but remained hyperactivated compared with the level in control embryos (Fig. 5Ad,f,j,B). These data indicate that stimulation of JNK pathway activity downstream of Rac is insufficient to promote dorsal closure in the absence of functional Rac. In addition, maximal JNK reporter expression evoked by ΔSH3 also requires Rac function. Together these results implicate Rac in both JNK-dependent and -independent functions, reminiscent of our data with coexpression of Rac-WT and bsk-DN, in which JNK target gene expression and changes in cell morphology of the leading edge were distinct results of Rac function distinguished by their sensitivity to bsk-DN suppression.
Genetic analysis has implicated misshapen (msn) in JNK signaling during dorsal closure (Su et al., 1998). Msn is a MAP4K family kinase, and Slpr is predicted to be a substrate. To determine whether Msn promotes JNK signaling during dorsal closure in collaboration with Slpr, transgenes encoding the wild-type proteins were expressed individually or together using puc-lacZ as a readout for JNK signaling. Expression of Msn-WT or Slpr-WT resulted in a 1.4- to 2-fold upregulation of the reporter (Fig. 5Ae,i,B), whereas coexpression resulted in 2.6-fold upregulation, which is less than an additive effect (Fig. 5Ai,B); however, Slpr-KD did suppress Msn-dependent JNK signaling substantially (Fig. 5Ab), consistent with expected epistasis. These results suggest that although Msn might activate slightly more JNK signaling when Slpr is unlimited, Msn cannot maximally activate Slpr in the absence of additional input. Rac is the likely candidate for that input. Moreover, these results suggest a contingent order, where Rac activation of Slpr is prerequisite for further activation by Msn. To test this, Rac-WT and Msn-WT were coexpressed (Fig. 5Ak). Indeed, puc-lacZ staining was greater than for Rac alone, or Msn plus Slpr (3.4- vs 1.9- and 2.6-fold, respectively; Fig. 5B), but still less than for Rac and Slpr together (3.9-fold), suggesting a requirement for both Msn and Rac to activate Slpr. Maximal puc upregulation upon coexpression of Msn and Rac might not be possible owing to limited endogenous Slpr.
Msn CNH domain interacts with Slpr
If Msn activates JNK signaling via Slpr, then Msn is predicted to physically interact with Slpr, yet this has not been examined directly. Drosophila Msn contains an N-terminal kinase domain, a proline-rich central region, and a highly conserved regulatory citron homology (CNH) domain (Fig. 7A). One mammalian MAP4K family member, the hematopoietic progenitor kinase, interacts with MLK3 via the SH3 domain to activate JNK signaling (Kiefer et al., 1996; Leung and Lassam, 2001). Alternatively, another MAP4K, NCK-interacting kinase (NIK), mediates JNK pathway activation through direct binding of its C-terminus to the MAP3K MEKK1 (Su et al., 1997). Thus, although binding of the Slpr SH3 domain to centrally located proline motifs in Msn is a reasonable expectation, genetic rescue experiments with msn derivatives suggest that the central region is expendable for dorsal closure, whereas the kinase and C-terminus are essential (Su et al., 2000). To examine possible interactions, binding assays were conducted with GST-Msn fusion proteins and in-vitro-translated Slpr fragments. Tandem SH3 domains of the adapter protein Dock were included as a positive control for Msn binding. Fig. 7 shows that the central proline-rich region of Msn pulled down Dock, as expected; however, the Slpr SKLC and ΔSH3 fragments bound specifically to the GST–Msn–CNH derivative lacking the proline motifs but encompassing the C-terminal domain, whereas Dock did not (Fig. 7B). None of the fragments bound to GST alone. These data demonstrate that Msn and Slpr interact with each other, but not via a proline motif–SH3 interaction. To further narrow down the binding site for Msn–CNH on Slpr, we tested two shorter constructs, SK and LC, constituting two halves of the larger Slpr SKLC fragment. Specific binding was retained with the LC fragment, but not the SK (Fig. 7C), indicating that the conserved regulatory C-terminus of Msn interacts with a region of Slpr, encompassing the LZ, linker, and CRIB regions (aa 405–516).
Fig. 7.

The C-terminal CNH domain of Msn interacts in vitro with the LZ–CRIB region of Slpr. (A) Schematic domain organization of Msn showing the kinase domain, proline-rich region (PXXP), and regulatory C-terminal domain (also known as the CNH domain). Black lines represent GST fusion protein fragments with the amino acids indicated. Dock binds within the central region (dotted line). (B,C) GST pull-down assay with Msn fragments and labeled Dock, Slpr SKLC and Slpr ΔSH3 proteins from a wheatgerm lysate (35S-input) (B), or unlabeled Slpr SK and LC proteins from in vitro translation (C). The Coomassie-Blue-stained gels (upper panels) and autoradiograph or immunoblot (lower panels) show expression of the fusion proteins and input protein that is specifically bound. Molecular mass markers (kDa) are labeled.
Discussion
Negative regulation of Slpr function
Src family tyrosine kinases are autoinhibited through intramolecular interactions mediated by their SH2 and SH3 domains (Boggon and Eck, 2004). Raf Ser/Thr kinases are also subject to inhibition through intramolecular interactions between N- and C-terminal sequences (Wellbrock et al., 2004). Similarly, inhibition of MLK3 activity requires SH3 binding to a short peptide linker between the LZ and CRIB domains (Zhang and Gallo, 2001). Introduction of a Y52A substitution in the SH3 domain of MLK3 increases kinase activity by 2- to 2.5-fold, whereas a mutation in the putative binding site (P469A) abrogates SH3 binding and increases catalytic activity 4-fold (Zhang and Gallo, 2001). Our results with ΔSH3 and Slpr-AVA recapitulate these data in vivo using puc-lacZ as a surrogate for JNK signaling in the embryo (Fig. 3, Fig. 5B) consistent with the SH3 domain playing a negative role in Slpr activation by intramolecular inhibition (Fig. 8). If the SH3 domain bound elsewhere in Slpr or to a negative regulatory partner, mutating the prolines in the LZ–CRIB linker is not expected to relieve inhibition. This is not what we observed. Furthermore, elevation in signaling capacity of the proline substitution mutant relative to the SH3 deletion might indicate an additional positive role for the SH3 domain in MLK regulation (Zhang and Gallo, 2001). For example, once autoinhibition is relieved, the SH3 domain could recruit a positive regulator. Although our in vitro binding assays suggest that the SH3 domain is not responsible for binding the MAP4K Msn directly, relief of SH3-mediated inhibition could uncover the binding site for Msn in the vicinity of the PVP motif consistent with our mapping mapping of the interaction with Msn to the LC region of Slpr (Figs 7, 8).
Fig. 8.
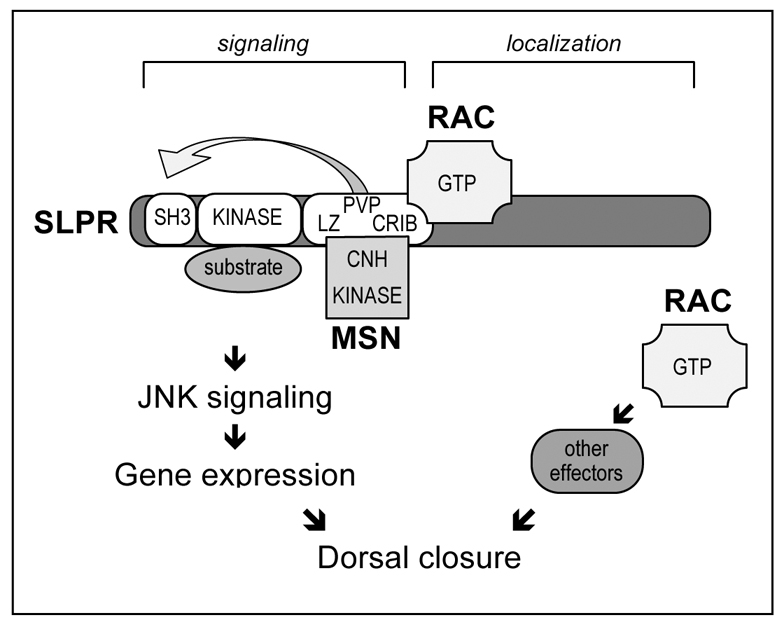
Model describing interactions between Slpr, Rac and Msn. The SKLC region of Slpr supports signaling functions whereas the C-terminal half is responsible for subcellular localization. The PVP motif in the LZ–CRIB linker region of Slpr is involved in SH3-mediated inhibition, which is relieved by interaction of the LZ–CRIB region with active Rac and the CNH domain of Msn to activate JNK signaling, gene expression and dorsal closure. Active Rac also participates in additional JNK-independent functions with other effectors to facilitate dorsal closure.
Complementing the observations that the SH3 domain of Slpr plays a negative regulatory role, we found that expression of the SH3 domain alone produced dominant negative phenotypes. Several mechanisms could account for this. The isolated SH3 domain could bind to the PVP site in endogenous Slpr preventing dimerization and activation; however, the SH3 is expected to be displaced upon Rac-GTP binding to the CRIB motif allowing further activation of endogenous Slpr. This scenario is inconsistent with our observations that JNK signaling is strongly downregulated by expression of the SH3 domain. The ectopic SH3 domain might simply be so abundant that it outcompetes endogenous activated Rac. Alternatively, the SH3 domain of Slpr might interfere with endogenous Slpr activity by titrating an input or effector molecule. Although this is consistent with our data that SH3 alone is epistatic to Msn (not shown), Msn binds in vitro to Slpr independently of the SH3 domain. Thus, an alternative SH3 binding partner remains a formal possibility. Finally, the SH3 domain might inhibit JNK signaling downstream of endogenous Slpr, but upstream of the transcriptional regulation of puc-lacZ, which might account for the curious result that over the course of dorsal closure progression, the SH3 transgenic protein became increasingly localized to the nucleus.
Regulation of Slpr subcellular distribution by the C-terminal region
Following the CRIB domain of Slpr is a long C-terminal region, constituting half of the total protein sequence. Extensive phosphorylation of the C-termini of MLK2 and MLK3 suggest that this region might serve a regulatory role as a substrate for proline-directed kinases engaged in feedback or crosstalk control (Phelan et al., 2001; Vacratsis et al., 2002). Thus, it was surprising to find that the SKLC-WT transgenic protein, lacking the C-terminus of Slpr, rescued slpr mutants to adulthood nearly as well as the epitope-tagged full-length version (Fig. 2A), indicating that the C-terminus is nonessential during development. Though SKLC overexpression in a wild-type background did not hyperactivate JNK signaling, it did not inhibit signaling either (Fig. 3), perhaps providing an appropriate intermediate level of signaling for rescue.
Detection of the tagged forms of Slpr using immunofluorescence did reveal a role for the C-terminus in proper protein localization. For example, those Slpr derivatives retaining the C-terminus were enriched at the cortex of epithelial cells, whereas those lacking it were predominantly cytoplasmic or nuclear, with variable localization at the cortex, perhaps reflecting dimerization with endogenous protein (Fig. 4). Supporting this, localization of transgenic protein in imaginal tissues dissected from slpr mutant larvae lacking endogenous protein revealed minimal cortical staining of the C-terminally truncated SKLC. Moreover, if the C-terminus is responsible for cortical enrichment, then it is predicted to localize at the cortex on its own. This prediction is borne out in several cell types, including embryonic ectoderm and larval brain and retina (Fig. 4). Thus, although the C-terminus is important for directing the correct subcellular localization of Slpr, it is not absolutely required for function, given that mutant animals expressing the SKLC form, lacking the localization region, survive nearly as well as those expressing the full-length form (Fig. 2). It remains to be determined how the C-terminal region of Slpr directs its localization.
Activating Slpr signaling
Many kinases require phosphorylation within their kinase domains to become maximally active. For example, mutations in the activation loop can render a kinase nonfunctional or hyperactive while leaving the active site intact (Nolen et al., 2004). The activation segment of Slpr is highly conserved, with three out of four putative phosphoacceptor residues being identical to human counterparts. Intriguingly, human MLK1 and MLK3 appear to rely on different threonine residues for primary activating autophosphorylation. Specifically, an MLK1 T312A mutant (Fig. 1B) is nearly inactive in vitro toward its substrate MKK4, with ~1% of the wild-type kinase efficiency, and also in vivo, as measured by JNK activation upon MLK1 transfection (Durkin et al., 2004). MLK3, by contrast, is nonfunctional when the first threonine (T277) is mutated to alanine, although S281 plays an auxiliary role; the equivalent residue T285 (to MLK1 T312) was not tested (Leung and Lassam, 2001). Finally, phosphorylation of the ultimate threonine residue (T290) in the activation segment of another MAP3K family member Cot (also known as Tpl2 and MAP3K8) is necessary but not sufficient for kinase activity in vitro (Luciano et al., 2004). To determine whether the three residues in the activation segment of Slpr are required for function, we analyzed alanine substitution mutants and implicate the terminal threonine residue (T295) as essential based on the ability to stimulate signaling in vivo (Fig. 3) and partially rescue slpr mutants (Fig. 2). Although these data clearly demonstrate a crucial role for T295, they do not indicate whether it is phosphorylated. Based on numerous kinase structures, T295 of Slpr is near the C-terminal anchoring motif of the activation segment, placing it in the P+1 loop, which contains residues contacting the peptide substrate (Nolen et al., 2004). Hence, this residue in Slpr might promote the integrity of the catalytic active site or stabilization of substrate binding, in addition to (or rather than) being a priming phosphorylation site. However, similar mutations in Cot and MLK1 do not interfere with the structural organization of the kinase domain in so far as protein binding interactions and limited protease digestion patterns are unaltered (Durkin et al., 2004; Luciano et al., 2004). Generation of a phosphomimetic form of Slpr will help to address the role of T295 phosphorylation.
Cumulative evidence supports a role for Rac and Cdc42 in the activation of mammalian MLK proteins, stimulating their kinase activity and downstream JNK signaling (Bock et al., 2000; Teramoto et al., 1996; Vacratsis and Gallo, 2000). The interaction is direct, via the CRIB motif in the MLKs, and requires the GTP-bound forms of Rac or Cdc42, not Rho (Bock et al., 2000; Burbelo et al., 1995; Nagata et al., 1998). Genetic analyses in Drosophila have implicated Rac family proteins (Rac1, Rac2, Mtl) and Cdc42 in the process of dorsal closure (Hakeda-Suzuki et al., 2002; Harden et al., 1995; Harden et al., 1999; Woolner et al., 2005), but proposed models suggest Rac GTPases are upstream of JNK signaling whereas Cdc42 acts downstream (Genova et al., 2000; Hakeda-Suzuki et al., 2002; Ricos et al., 1999; Woolner et al., 2005). Functionally, Rac proteins regulate actin cytoskeleton-based structures in the leading cells of the dorsal ectoderm, including a supracellular cable linking adhesive junctions between cells to maintain tension across the advancing epithelium (Jacinto et al., 2002a; Kiehart et al., 2000), and protrusions important for intercellular interactions across the midline during the zippering phase of closure (Hakeda-Suzuki et al., 2002; Jacinto et al., 2000; Woolner et al., 2005). The loss of these structures in compound Rac mutants resembles the phenotype of JNK pathway mutants; moreover, upregulation of JNK signaling partially rescues Rac mutant phenotypes suggesting that Rac proteins act upstream to direct JNK-dependent deployment of the actin cytoskeleton (Woolner et al., 2005).
Slpr is the best candidate linking Rac to the JNK pathway. slpr mutants dominantly suppress a Rac-induced phenotype in the developing Drosophila eye tissue (Stronach and Perrimon, 2002) and direct interactions between Slpr and active Rac are demonstrable (Fig. 6) (Neisch et al., 2010). Using the reagents developed in this study, we explored further whether Rac and Slpr cooperate during dorsal closure, as would be expected from existing models. Our coexpression studies demonstrate an additive effect on JNK target gene expression by Slpr and Rac-WT, but curiously, dominant negative forms of Slpr (SH3 and Slpr-KD) are ineffective at blocking ectopic signaling induced by Rac-WT (Fig. 5). The reason for this is unclear, but raises a few possibilities. For example, endogenous Rac might regulate JNK signaling primarily by an Slpr-dependent mechanism, but in the context of forced overexpression, might evoke Slpr-independent means to stimulate JNK. If so, convergence at the level of JNK is consistent with the evidence that dominant negative Bsk (JNK) is fully capable of blocking Rac-induced ectopic puc-lacZ expression (supplementary material Fig. S1). Alternatively, Rac might activate a pool of endogenous Slpr that is ‘resistant’ to inhibition by dominant negative Slpr. This could occur if Rac facilitates membrane association or dimerization of endogenous Slpr more effectively than dimerization with an inactivating kinase-dead form. Testing whether Rac induction of JNK signaling is sensitive to endogenous Slpr levels might address this possibility.
Although numerous studies support a positive role for Rac upstream of JNK signaling in dorsal closure, it is also curious that Rac-DN does not have a more dramatic negative effect on JNK signaling, given the expression results in a severe tissue-closure defect, confirming the inhibitory action of the dominant negative transgene (Fig. 5) (Raymond et al., 2001; Woolner et al., 2005). One explanation is that the two phenotypic outcomes, gene expression and tissue closure, require different Rac effectors (JNK-dependent and JNK-independent; Fig. 8) or unequal contributions of the Rac family members (Hakeda-Suzuki et al., 2002; Nolan et al., 1998). Alternatively, the puc-lacZ reporter might not be representative of all JNK pathway target genes, such as those required for cytoskeletal remodelling. In other words, incomplete loss of puc-lacZ expression as a result of Rac-DN does not preclude reduction in levels of expression of other genes below a point necessary to sustain dorsal closure. That said, ectopic JNK activation by Slpr ΔSH3 was insufficient to rescue the dorsal closure defects resulting from diminished Rac function (Fig. 5), further supporting a Slpr- and JNK-independent role for Rac in directing tissue closure. Protein kinase N is a good candidate to mediate JNK-independent signals from Rac-GTP (Lu and Settleman, 1999), although other effectors are possible. Altogether, our results are not entirely consistent with a simple epistatic relationship positioning Rac GTPase upstream of Slpr activation, especially in light of the results of similar experiments with Msn, which do support Msn as an upstream input. Cumulative data implicate Rac in multiple pathways.
The discovery of a role for Msn MAP4K in dorsal closure morphogenesis led to speculation that Msn and Rac might converge on a MAP3K to activate downstream JNK signaling in the Drosophila embryo (Su et al., 1998). Identification of Slpr as a MAP3K required for dorsal closure provided a candidate. Here we demonstrate binding between the C-terminal domain of Msn and the LZ–CRIB region of Slpr, independent of the SH3 domain of Slpr (Fig. 7). This result explains the genetic requirement for the kinase and C-terminal domains of Msn to rescue dorsal closure defects of msn mutants, while the proline-rich central domain is dispensible (Su et al., 2000). Moreover, as expected of a downstream transducer, dominant negative Slpr derivatives do block Msn-dependent upregulation of JNK signaling (Fig. 5). Although coexpression of Msn and Slpr does not activate JNK signaling to the extent seen with Rac and Slpr together, there is an enhancement relative to each alone. This result might reflect limiting levels of active Rac, consistent with a requirement for Rac as a primary positive input and Msn a secondary activating input. Indeed, previous studies using transfected mammalian cells demonstrated that Rac-DN inhibits JNK activation by Msn (Su et al., 1998). The binding site on Slpr for Msn is limited to the LZ, PVP-containing linker, and CRIB regions where Rac and the SH3 domain of Slpr also interact. Thus, it is possible that Rac binding (and relief of SH3 inhibition) subsequently exposes the interaction site for the Msn CNH domain. Interestingly, the C-terminal CNH domain of the mammalian Msn orthologue, NIK, interacts with another MAP3K, MEKK1, to elicit maximal activation of SAPK pathway activity (Su et al., 1997). Notably, the CNH domain of NIK, and MIG-15 in C. elegans, interacts with the cytoplasmic tail of β-integrin receptors (Poinat et al., 2002). Although this interaction has yet to be probed in Drosophila, the implication of JNK signaling being linked to integrin function in tissue closure is not unprecedented (Becker et al., 2000; Homsy et al., 2006; Kadrmas et al., 2004) and might provide additional insights into regulation of dorsal closure morphogenesis and JNK pathway activation in future studies.
Graded Slpr activity correlates with JNK reporter expression and tissue morphology
The structure–function analysis of Slpr benefits from the ability to detect different levels of JNK pathway activity via reporter gene expression in individual cells and within the whole tissue and organism. Our results in the embryo revealed that modified forms of Slpr have distinct and wide-ranging activities, from dominant negative to strongly hyperactive, implying that downstream signaling components are not limiting for signal transduction, at either the cell or tissue level. Also, the remarkably extensive range of transgene activities in the embryo, correlates precisely with the broad profile of thorax morphologies in the adult. These data demonstrate first, that the two processes, dorsal closure and thorax closure, are similarly sensitive across a wide range of Slpr activity; and second, that the plasticity in response at the cellular level is mirrored in the developmental flexibility at the organismal level, seen in the extreme range of thorax morphologies. These data might also indicate that graded changes in levels of Slpr-dependent JNK signaling in epithelial tissues results in concomitant changes in signaling output, rather than a binary response at a threshold level of pathway activity. Future work will investigate the underlying cellular mechanisms responsible for signaling output and altered tissue morphology.
Materials and Methods
Slpr transgenic constructs
All UAS–Slpr constructs described here were cloned into the pUASp vector (Rorth, 1998). UAS–Slpr-WT and UAS–Slpr-KD have been described previously (Polaski et al., 2006). UAS–Slpr-WT-HA (aa 1–1148) is equivalent to UAS–Slpr-WT except for the addition of two copies of the hemagglutinin epitope tag (HA) at the C-terminus. The HA tag is present in all subsequent constructs described here. UAS–SKLC-WT (aa 1–516) is a C-terminally truncated form of Slpr comprising only the SH3, kinase, LZ, and CRIB domains. UAS–SH3 (aa 1–114) is truncated after the SH3 domain. Both of these inserts were generated by standard PCR amplification, followed by restriction-enzyme digestion and ligation into the appropriate vector. All oligonucleotide primers used for cloning are listed in supplementary material Table S4.
Mutant (AAA, TAA, ASA, AAT, AVA, aa 1–1148) and N-terminally truncated forms of Slpr (ΔSH3 aa 110–1148, Cterm aa 537–1148) were generated using site-directed mutagenesis by overlap extension (SOEing) (Ho et al., 1989). For example, Slpr-AAA was made using overlapping mutagenic primers to change residues (T287, S291, and T295) in the kinase activation segment to alanine. Two regions were independently amplified and the products mixed, to generate, by overlap extension, the template for a second PCR amplification using the outermost original primers. The same strategy was employed for UAS–SKLC-AAA, replacing the wild-type fragment for the mutant fragment. Constructs were verified by dsDNA sequencing. P-element transformation was performed by Genetic Services, Inc (Sudbury, MA).
Western blot analysis
All UAS–Slpr-HA transgenic flies were crossed to arm-G4 flies and eggs were collected overnight at 25°C. After dechorionating and homogenizing in Laemmli sample buffer (SB), embryonic protein lysates were separated using SDS-PAGE, transferred to PVDF membrane, and probed with mouse anti-HA antibody (1:1000, Covance, 16B12). Mouse anti-β-tubulin (1:300, DSHB, E7) was used as a loading control. Low-molecular-mass transgenic proteins including SH3 were transferred in nonstandard buffer, lacking SDS (25 mM Tris, 190 mM glycine, 20% methanol).
Drosophila stocks
Chromosome location and Bloomington stock numbers are as follows: arm-G4 (II) BL#1560, pnrMD237/TM3Ser1{UAS–y+} (pnr-G4) BL#3039 (Calleja et al., 1996), pucE69 (puc-lacZ) (III) (Ring and Martinez Arias, 1993), UAS–Slpr-KD (II and III), UAS–Slpr-WT (II), slpr3P5, slpr921, slprBS06 (X) (Polaski et al., 2006), UAS–Rac1.L (Rac-WT) (II) BL#6293, UAS–Rac1.N17 (Rac-DN) (III) BL#6292 (Luo et al., 1994), UAS–msn.S (III) BL#5946 (Su et al., 1998), UAS–bskK53R (bsk-DN) (III) BL#9311.
Genetic rescue
Rescue of the embryonic dorsal closure phenotype of slpr921 mutants using a recombinant chromosome with the gtX11 allele was performed as described previously (Stronach and Perrimon, 2002).
For rescue of slpr mutants to adulthood, we crossed y93j w1118 slpr*/FM7;arm-G4 females with w1118/Y;UAS–slpr-Tg males for each slpr allele and raised the progeny at 21±0.5°C for moderate transgene expression. Mutant and FM7 males in the progeny were counted to quantify relative eclosion rate as an indication of transgene rescue. To avoid inadvertently including non-FM7 males that arise from non-disjunction of the maternal X chromosome, we counted only yellow−, non-FM7 males.
Immunofluorescence
Embryos were collected overnight, fixed with 4% formaldehyde in PEM buffer (100 mM Pipes, 2 mM EGTA, 1 mM MgSO4) and processed for indirect immunofluorescence (Patel, 1994). Larval tissues were dissected from third instar male animals that did not express FM7-linked GFP. Primary antibodies were rabbit anti-β-galactosidase at 1:1000 (Cappel), mouse anti-β-galactosidase at 1:1000 (Promega), mouse anti-phosphotyrosine 4G10 at 1:1000 (Upstate Biotech), mouse anti-HA 16B12 at 1:500 (Covance), affinity purified rabbit anti-Slpr at 1:400 (Polaski et al., 2006), mouse α-Crb Cq4 at 1:20, mouse anti-Dlg 4E3 at 1:800, mouse anti-FasIII 4G9 at 1:50 (DSHB). Texas-Red–phalloidin was used to visualize F-actin. All fluorophore-conjugated secondary antibodies were used at 1:200 dilution (Jackson Immunoresearch).
Image capture and processing
Adult flies were imaged using a Leica DFC300F camera mounted on a Leica MZ16 stereomicroscope. Images of stained embryos and larval tissues were obtained by laser confocal scanning microscopy (Bio-Rad Radiance 2000) using a Nikon E800 compound microscope. Figures were assembled with Adobe Photoshop.
Quantification of puc-lacZ expression
Cropped images of three segments of immunostained stage 14 embryos were imported into ImageJ software. Equivalent rectangular regions of interest were selected. A vertical profile plot of average grey values (y-axis) across the selection (x-axis) was generated for each region and the numerical values were imported into Microsoft Excel. The MODE (most common grey value) for each image was subtracted as background. The resulting values were used to calculate area under the curve (excluding negative values). The average of three to six areas per genotype were determined and plotted graphically. Error bars indicate the standard deviation (s.d.). Fold change was calculated relative to the ‘no transgene’ control.
In vitro binding assays
GST-Rac and GST-Cdc42 constructs were described previously (Lu and Settleman, 1999). To generate additional GST fusion protein constructs, Msn sequences were cloned into the pGEX-4T-1 vector (GE Healthcare). Msn-PRO encodes aa 321–768 of the 1102 aa isoform C. Msn-CNH encodes aa 767–1102 of isoform C. The Msn binding region of Dock, consisting of three SH3 domains (aa 141–398 of isoforms B and C), was cloned into the pET16b HIS-tag vector (Novagen) and used as a positive control in GST pull-down assays (Ruan et al., 1999). Slpr constructs used for in vitro synthesis were cloned into the pcDNA3.1 vector (Invitrogen). Dock and Slpr proteins were synthesized in rabbit reticulocyte or wheatgerm lysates using TnT® Coupled Transcription/Translation Systems (Promega). For expression of GST fusion proteins, freshly diluted overnight cultures of E. coli BL21 cells were grown for 1 hour and induced with 0.2 mM IPTG (isopropylthiogalactoside), then switched to growth at 21°C for 3 hours. After centrifugation, pellets were resuspended in phosphate–BME buffer [P-BME; 1× phosphate-buffered saline, 0.1 mM PMSF (phenylmethylsulphonyl fluoride), 0.001 mM EDTA, 0.013 mM β-mercaptoethanol] and lysed with 0.5 mg/ml lysozyme, incubated on ice for 30 minutes, repeatedly frozen and thawed, and sonicated. The homogenate was clarified by centrifugation and incubated with glutathione–agarose beads (50–100 μg/assay) overnight at 4°C. Beads were washed with P-BME buffer, blocked in 5% filtered BSA (in P-BME buffer) and washed again. At this point, GST-Rac and GST-Cdc42 were incubated with the appropriate nucleotide, either GDP or GTPγS. For the binding assay, 2–8 μl of in vitro translated protein lysates were incubated with the GST-beads in TIF buffer (150 mM NaCl, 20 mM Tris pH 8.0, 1 mM MgCl2, 0.1% NP40, 10% glycerol) overnight at 4°C. After washing six times with excess TIF buffer, beads were boiled in 2× SB for SDS-PAGE, western transfer, and autoradiography or immunoblotting.
Supplementary Material
Acknowledgments
We thank John Malta, James Matous and Stephanie Polaski for technical contributions to the rescue experiments, site-directed mutagenesis and DNA cloning, respectively. We acknowledge the Developmental Studies Hybridoma Bank (DSHB) developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242 for antibodies described in Materials and Methods. This work is supported by funding from the NIH (HD045836) to B.S. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/18/3177/DC1
References
- Agnes F., Suzanne M., Noselli S. (1999). The Drosophila JNK pathway controls the morphogenesis of imaginal discs during metamorphosis. Development 126, 5453-5462 [DOI] [PubMed] [Google Scholar]
- Becker E., Huynh-Do U., Holland S., Pawson T., Daniel T. O., Skolnik E. Y. (2000). Nck-interacting Ste20 kinase couples Eph receptors to c-Jun N-terminal kinase and integrin activation. Mol. Cell. Biol. 20, 1537-1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson N., Tremblay M., Robinson F., Kaplan D. R., Trusko S. P., Moss T. (2008). Mice lacking both mixed-lineage kinase genes Mlk1 and Mlk2 retain a wild type phenotype. Cell Cycle 7, 909-916 [DOI] [PubMed] [Google Scholar]
- Bock B. C., Vacratsis P. O., Qamirani E., Gallo K. A. (2000). Cdc42-induced activation of the mixed-lineage kinase SPRK in vivo. Requirement of the Cdc42/Rac interactive binding motif and changes in phosphorylation. J. Biol. Chem. 275, 14231-14241 [DOI] [PubMed] [Google Scholar]
- Boggon T. J., Eck M. J. (2004). Structure and regulation of Src family kinases. Oncogene 23, 7918-7927 [DOI] [PubMed] [Google Scholar]
- Brancho D., Ventura J. J., Jaeschke A., Doran B., Flavell R. A., Davis R. J. (2005). Role of MLK3 in the regulation of mitogen-activated protein kinase signaling cascades. Mol. Cell. Biol. 25, 3670-3681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo P. D., Drechsel D., Hall A. (1995). A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J. Biol. Chem. 270, 29071-29074 [DOI] [PubMed] [Google Scholar]
- Calleja M., Moreno E., Pelaz S., Morata G. (1996). Visualization of gene expression in living adult Drosophila. Science 274, 252-255 [DOI] [PubMed] [Google Scholar]
- Collins C. A., Wairkar Y. P., Johnson S. L., DiAntonio A. (2006). Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51, 57-69 [DOI] [PubMed] [Google Scholar]
- Craig E. A., Stevens M. V., Vaillancourt R. R., Camenisch T. D. (2008). MAP3Ks as central regulators of cell fate during development. Dev. Dyn. 237, 3102-3114 [DOI] [PubMed] [Google Scholar]
- Dorow D. S., Devereux L., Dietzsch E., De Kretser T. (1993). Identification of a new family of human epithelial protein kinases containing two leucine/isoleucine-zipper domains. Eur. J. Biochem. 213, 701-710 [DOI] [PubMed] [Google Scholar]
- Durkin J. T., Holskin B. P., Kopec K. K., Reed M. S., Spais C. M., Steffy B. M., Gessner G., Angeles T. S., Pohl J., Ator M. A., et al. (2004). Phosphoregulation of mixed-lineage kinase 1 activity by multiple phosphorylation in the activation loop. Biochemistry 43, 16348-16355 [DOI] [PubMed] [Google Scholar]
- Gallo K. A., Johnson G. L. (2002). Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell Biol. 3, 663-672 [DOI] [PubMed] [Google Scholar]
- Gallo K. A., Mark M. R., Scadden D. T., Wang Z., Gu Q., Godowski P. J. (1994). Identification and characterization of SPRK, a novel src-homology 3 domain-containing proline-rich kinase with serine/threonine kinase activity. J. Biol. Chem. 269, 15092-15100 [PubMed] [Google Scholar]
- Genova J. L., Jong S., Camp J. T., Fehon R. G. (2000). Functional analysis of Cdc42 in actin filament assembly, epithelial morphogenesis, and cell signaling during Drosophila development. Dev. Biol. 221, 181-194 [DOI] [PubMed] [Google Scholar]
- Glise B., Noselli S. (1997). Coupling of Jun amino-terminal kinase and Decapentaplegic signaling pathways in Drosophila morphogenesis. Genes Dev. 11, 1738-1747 [DOI] [PubMed] [Google Scholar]
- Glise B., Bourbon H., Noselli S. (1995). hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell 83, 451-461 [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S., Ng J., Tzu J., Dietzl G., Sun Y., Harms M., Nardine T., Luo L., Dickson B. J. (2002). Rac function and regulation during Drosophila development. Nature 416, 438-442 [DOI] [PubMed] [Google Scholar]
- Hammarlund M., Nix P., Hauth L., Jorgensen E. M., Bastiani M. (2009). Axon regeneration requires a conserved MAP kinase pathway. Science 323, 802-806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden N. (2002). Signaling pathways directing the movement and fusion of epithelial sheets: lessons from dorsal closure in Drosophila. Differentiation 70, 181-203 [DOI] [PubMed] [Google Scholar]
- Harden N., Loh H. Y., Chia W., Lim L. (1995). A dominant inhibitory version of the small GTP-binding protein Rac disrupts cytoskeletal structures and inhibits developmental cell shape changes in Drosophila. Development 121, 903-914 [DOI] [PubMed] [Google Scholar]
- Harden N., Ricos M., Ong Y. M., Chia W., Lim L. (1999). Participation of small GTPases in dorsal closure of the Drosophila embryo: distinct roles for Rho subfamily proteins in epithelial morphogenesis. J. Cell Sci. 112, 273-284 [DOI] [PubMed] [Google Scholar]
- Hirai S., Katoh M., Terada M., Kyriakis J. M., Zon L. I., Rana A., Avruch J., Ohno S. (1997). MST/MLK2, a member of the mixed lineage kinase family, directly phosphorylates and activates SEK1, an activator of c-Jun N-terminal kinase/stress-activated protein kinase. J. Biol. Chem. 272, 15167-15173 [DOI] [PubMed] [Google Scholar]
- Hirai S., Cui de F., Miyata T., Ogawa M., Kiyonari H., Suda Y., Aizawa S., Banba Y., Ohno S. (2006). The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J. Neurosci. 26, 11992-2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., Pease L. R. (1989). Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51-59 [DOI] [PubMed] [Google Scholar]
- Homsy J. G., Jasper H., Peralta X. G., Wu H., Kiehart D. P., Bohmann D. (2006). JNK signaling coordinates integrin and actin functions during Drosophila embryogenesis. Dev. Dyn. 235, 427-434 [DOI] [PubMed] [Google Scholar]
- Hou X. S., Goldstein E. S., Perrimon N. (1997). Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes Dev. 11, 1728-1737 [DOI] [PubMed] [Google Scholar]
- Huang C. Y., Chueh P. J., Tseng C. T., Liu K. Y., Tsai H. Y., Kuo W. W., Chou M. Y., Yang J. J. (2004). ZAK re-programs atrial natriuretic factor expression and induces hypertrophic growth in H9c2 cardiomyoblast cells. Biochem. Biophys. Res. Commun. 324, 973-980 [DOI] [PubMed] [Google Scholar]
- Itoh A., Horiuchi M., Bannerman P., Pleasure D., Itoh T. (2009). Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem. Biophys. Res. Commun. 383, 258-262 [DOI] [PubMed] [Google Scholar]
- Jacinto A., Wood W., Balayo T., Turmaine M., Martinez-Arias A., Martin P. (2000). Dynamic actin-based epithelial adhesion and cell matching during Drosophila dorsal closure. Curr. Biol. 10, 1420-1426 [DOI] [PubMed] [Google Scholar]
- Jacinto A., Wood W., Woolner S., Hiley C., Turner L., Wilson C., Martinez-Arias A., Martin P. (2002a). Dynamic analysis of actin cable function during Drosophila dorsal closure. Curr. Biol. 12, 1245-1250 [DOI] [PubMed] [Google Scholar]
- Jacinto A., Woolner S., Martin P. (2002b). Dynamic analysis of dorsal closure in Drosophila: from genetics to cell biology. Dev. Cell 3, 9-19 [DOI] [PubMed] [Google Scholar]
- Jaeschke A., Davis R. J. (2007). Metabolic stress signaling mediated by mixed-lineage kinases. Mol. Cell 27, 498-508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasper H., Benes V., Schwager C., Sauer S., Clauder-Munster S., Ansorge W., Bohmann D. (2001). The genomic response of the Drosophila embryo to JNK signaling. Dev. Cell 1, 579-586 [DOI] [PubMed] [Google Scholar]
- Kadrmas J. L., Smith M. A., Clark K. A., Pronovost S. M., Muster N., Yates J. R., 3rd, Beckerle M. C. (2004). The integrin effector PINCH regulates JNK activity and epithelial migration in concert with Ras suppressor 1. J. Cell Biol. 167, 1019-1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt J. A., Lawrence N., Morel V., Balayo T., Fernandez B. G., Pelissier A., Jacinto A., Martinez Arias A. (2002). Planar polarity and actin dynamics in the epidermis of Drosophila. Nat. Cell Biol. 4, 937-944 [DOI] [PubMed] [Google Scholar]
- Kiefer F., Tibbles L. A., Anafi M., Janssen A., Zanke B. W., Lassam N., Pawson T., Woodgett J. R., Iscove N. N. (1996). HPK1, a hematopoietic protein kinase activating the SAPK/JNK pathway. EMBO J. 15, 7013-7025 [PMC free article] [PubMed] [Google Scholar]
- Kiehart D. P., Galbraith C. G., Edwards K. A., Rickoll W. L., Montague R. A. (2000). Multiple forces contribute to cell sheet morphogenesis for dorsal closure in Drosophila. J. Cell Biol. 149, 471-490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kockel L., Zeitlinger J., Staszewski L. M., Mlodzik M., Bohmann D. (1997). Jun in Drosophila development: redundant and nonredundant functions and regulation by two MAPK signal transduction pathways. Genes Dev. 11, 1748-1758 [DOI] [PubMed] [Google Scholar]
- Lamson R. E., Winters M. J., Pryciak P. M. (2002). Cdc42 regulation of kinase activity and signaling by the yeast p21-activated kinase Ste20. Mol. Cell. Biol. 22, 2939-2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leicht D. T., Balan V., Kaplun A., Singh-Gupta V., Kaplun L., Dobson M., Tzivion G. (2007). Raf kinases: function, regulation and role in human cancer. Biochim. Biophys. Acta 1773, 1196-1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung I. W., Lassam N. (1998). Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J. Biol. Chem. 273, 32408-32415 [DOI] [PubMed] [Google Scholar]
- Leung I. W., Lassam N. (2001). The kinase activation loop is the key to mixed lineage kinase-3 activation via both autophosphorylation and hematopoietic progenitor kinase 1 phosphorylation. J. Biol. Chem. 276, 1961-1967 [DOI] [PubMed] [Google Scholar]
- Liu T. C., Huang C. J., Chu Y. C., Wei C. C., Chou C. C., Chou M. Y., Chou C. K., Yang J. J. (2000). Cloning and expression of ZAK, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-alpha motif. Biochem. Biophys. Res. Commun. 274, 811-816 [DOI] [PubMed] [Google Scholar]
- Lu Y., Settleman J. (1999). The Drosophila Pkn protein kinase is a Rho/Rac effector target required for dorsal closure during embryogenesis. Genes Dev. 13, 1168-1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano B. S., Hsu S., Channavajhala P. L., Lin L. L., Cuozzo J. W. (2004). Phosphorylation of threonine 290 in the activation loop of Tpl2/Cot is necessary but not sufficient for kinase activity. J. Biol. Chem. 279, 52117-52123 [DOI] [PubMed] [Google Scholar]
- Luo L., Liao Y. J., Jan L. Y., Jan Y. N. (1994). Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev. 8, 1787-1802 [DOI] [PubMed] [Google Scholar]
- Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002). The protein kinase complement of the human genome. Science 298, 1912-1934 [DOI] [PubMed] [Google Scholar]
- Merritt S. E., Mata M., Nihalani D., Zhu C., Hu X., Holzman L. B. (1999). The mixed lineage kinase DLK utilizes MKK7 and not MKK4 as substrate. J. Biol. Chem 274, 10195-10202 [DOI] [PubMed] [Google Scholar]
- Mizuno T., Hisamoto N., Terada T., Kondo T., Adachi M., Nishida E., Kim D. H., Ausubel F. M., Matsumoto K. (2004). The Caenorhabditis elegans MAPKphosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. EMBO J. 23, 2226-2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K., Puls A., Futter C., Aspenstrom P., Schaefer E., Nakata T., Hirokawa N., Hall A. (1998). The MAP kinase kinase kinase MLK2 co-localizes with activated JNK along microtubules and associates with kinesin superfamily motor KIF3. EMBO J. 17, 149-158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata K., Abrams B., Grill B., Goncharov A., Huang X., Chisholm A. D., Jin Y. (2005). Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 120, 407-420 [DOI] [PubMed] [Google Scholar]
- Neisch A. L., Speck O., Stronach B., Fehon R. G. (2010). Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J. Cell Biol. 189, 311-323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan K. M., Barrett K., Lu Y., Hu K. Q., Vincent S., Settleman J. (1998). Myoblast city, the Drosophila homolog of DOCK180/CED-5, is required in a Rac signaling pathway utilized for multiple developmental processes. Genes Dev. 12, 3337-3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B., Taylor S., Ghosh G. (2004). Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15, 661-675 [DOI] [PubMed] [Google Scholar]
- Patel N. H. (1994). Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. In Drosophila melanogaster: Practical Uses in Cell and Molecular Biology, Vol. 44 (ed. Goldstein L. S. B., Fyrberg E. A.), pp. 446-487 San Diego: Academic Press; [DOI] [PubMed] [Google Scholar]
- Phelan D. R., Price G., Liu Y. F., Dorow D. S. (2001). Activated JNK phosphorylates the c-terminal domain of MLK2 that is required for MLK2-induced apoptosis. J. Biol. Chem. 276, 10801-10810 [DOI] [PubMed] [Google Scholar]
- Poinat P., De Arcangelis A., Sookhareea S., Zhu X., Hedgecock E. M., Labouesse M., Georges-Labouesse E. (2002). A conserved interaction between beta1 integrin/PAT-3 and Nck-interacting kinase/MIG-15 that mediates commissural axon navigation in C. elegans. Curr. Biol. 12, 622-631 [DOI] [PubMed] [Google Scholar]
- Polaski S., Whitney L., Barker B. W., Stronach B. (2006). Genetic analysis of slipper/mixed lineage kinase reveals requirements in multiple Jun-N-terminal kinase-dependent morphogenetic events during Drosophila development. Genetics 174, 719-733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A., Gallo K., Godowski P., Hirai S., Ohno S., Zon L., Kyriakis J. M., Avruch J. (1996). The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J. Biol. Chem. 271, 19025-19028 [DOI] [PubMed] [Google Scholar]
- Raymond K., Bergeret E., Dagher M. C., Breton R., Griffin-Shea R., Fauvarque M. O. (2001). The Rac GTPase-activating protein RotundRacGAP interferes with Drac1 and Dcdc42 signalling in Drosophila melanogaster. J. Biol. Chem. 276, 35909-35916 [DOI] [PubMed] [Google Scholar]
- Ricos M. G., Harden N., Sem K. P., Lim L., Chia W. (1999). Dcdc42 acts in TGF-beta signaling during Drosophila morphogenesis: distinct roles for the Drac1/JNK and Dcdc42/TGF-beta cascades in cytoskeletal regulation. J. Cell Sci. 112, 1225-1235 [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar J. R., Jenni M., Fritz A., Hafen E. (1996). The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev. 10, 2759-2768 [DOI] [PubMed] [Google Scholar]
- Ring J. M., Martinez Arias A. (1993). puckered, a gene involved in position-specific cell differentiation in the dorsal epidermis of the Drosophila larva. Developmement Suppl, 251-259 [PubMed] [Google Scholar]
- Rorth P. (1998). Gal4 in the Drosophila female germline. Mech. Dev. 78, 113-118 [DOI] [PubMed] [Google Scholar]
- Ruan W., Pang P., Rao Y. (1999). The SH2/SH3 adaptor protein dock interacts with the Ste20-like kinase misshapen in controlling growth cone motility. Neuron 24, 595-605 [DOI] [PubMed] [Google Scholar]
- Sluss H. K., Han Z., Barrett T., Goberdhan D. C., Wilson C., Davis R. J., Ip Y. T. (1996). A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 10, 2745-2758 [DOI] [PubMed] [Google Scholar]
- Stronach B. (2005). Dissecting JNK signaling, one KKKinase at a time. Dev. Dyn. 232, 575-584 [DOI] [PubMed] [Google Scholar]
- Stronach B., Perrimon N. (2002). Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 16, 377-387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. C., Han J., Xu S., Cobb M., Skolnik E. Y. (1997). NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J. 16, 1279-1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. C., Treisman J. E., Skolnik E. Y. (1998). The Drosophila Ste20-related kinase misshapen is required for embryonic dorsal closure and acts through a JNK MAPK module on an evolutionarily conserved signaling pathway. Genes Dev. 12, 2371-2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. C., Maurel-Zaffran C., Treisman J. E., Skolnik E. Y. (2000). The Ste20 kinase misshapen regulates both photoreceptor axon targeting and dorsal closure, acting downstream of distinct signals. Mol. Cell. Biol. 20, 4736-4744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramoto H., Coso O. A., Miyata H., Igishi T., Miki T., Gutkind J. S. (1996). Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J. Biol. Chem. 271, 27225-27228 [DOI] [PubMed] [Google Scholar]
- Tibbles L. A., Ing Y. L., Kiefer F., Chan J., Iscove N., Woodgett J. R., Lassam N. J. (1996). MLK-3 activates the SAPK/JNK and p38/RK pathways via SEK1 and MKK3/6. EMBO J. 15, 7026-7035 [PMC free article] [PubMed] [Google Scholar]
- Vacratsis P. O., Gallo K. A. (2000). Zipper-mediated oligomerization of the mixed lineage kinase SPRK/MLK-3 is not required for its activation by the GTPase cdc 42 but Is necessary for its activation of the JNK pathway. Monomeric SPRK L410P does not catalyze the activating phosphorylation of Thr258 of murine mitogen-activated protein kinase kinase 4. J. Biol. Chem. 275, 27893-27900 [DOI] [PubMed] [Google Scholar]
- Vacratsis P. O., Phinney B. S., Gage D. A., Gallo K. A. (2002). Identification of in vivo phosphorylation sites of MLK3 by mass spectrometry and phosphopeptide mapping. Biochemistry 41, 5613-5624 [DOI] [PubMed] [Google Scholar]
- Wellbrock C., Karasarides M., Marais R. (2004). The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5, 875-885 [DOI] [PubMed] [Google Scholar]
- Woolner S., Jacinto A., Martin P. (2005). The small GTPase Rac plays multiple roles in epithelial sheet fusion-dynamic studies of Drosophila dorsal closure. Dev. Biol. 282, 163-173 [DOI] [PubMed] [Google Scholar]
- Xia Y., Karin M. (2004). The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell. Biol. 14, 94-101 [DOI] [PubMed] [Google Scholar]
- Zeitlinger J., Bohmann D. (1999). Thorax closure in Drosophila: involvement of Fos and the JNK pathway. Development 126, 3947-3956 [DOI] [PubMed] [Google Scholar]
- Zhang H., Gallo K. A. (2001). Autoinhibition of mixed lineage kinase 3 through its Src homology 3 domain. J. Biol. Chem. 276, 45598-45603 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.