

Abstract
Four single-cysteine variants of the intrinsically disordered C-terminal domain of the measles virus nucleoprotein (NTAIL) were cyanylated at cysteine and their infrared spectra in the C≡N stretching region were recorded both in the absence and in the presence of one of the physiological partners of NTAIL, namely the C-terminal X domain (XD) of the viral phosphoprotein. Consistent with previous studies showing that XD triggers a disorder-to-order transition within NTAIL, the C≡N stretching bands of the infrared probe were found to be significantly affected by XD, with this effect being position-dependent. When the cyanylated cysteine side chain is solvent-exposed throughout the structural transition, its changing linewidth reflects a local gain of structure. When the probe becomes partially buried due to binding, its frequency reports on the mean hydrophobicity of the microenvironment surrounding the labeled side chain of the bound form. The probe moiety is small compared to other common covalently attached spectroscopic probes, thereby minimizing possible steric hindrance/perturbation at the binding interface. These results show for the first time to our knowledge the suitability of site-specific cysteine mutagenesis followed by cyanylation and infrared spectroscopy to document structural transitions occurring within intrinsically disordered regions, with regions involved in binding and folding being identifiable at the residue level.
Introduction
During the last decade, an increasing amount of experimental evidence has pointed out that as much as one-third of eukaryotic proteins contain long intrinsically disordered regions. Bioinformatics analysis indicates that 12% of these proteins are fully disordered (1). Intrinsically disordered proteins (IDPs) are ubiquitary proteins that fulfill essential biological functions while being devoid of highly populated secondary and tertiary structure under physiological conditions and in the absence of a partner (2). IDPs consist of dynamic ensembles of interconverting conformers that exert their biological function(s) by recognizing their binding partners through their disordered regions. In many cases, the lack of structure is required for biological function. The protein flexibility that is inherent to disorder confers functional advantages. An increased plasticity would 1), enable the binding of numerous structurally distinct targets; 2), provide the ability to overcome steric restrictions by enabling larger surfaces of interaction in protein complexes than those obtained with rigid partners; and 3), allow protein interactions to occur with both high specificity and low affinity (2–10).
These peculiar binding properties enable IDPs to exert their function within a complicated network of cellular and extracellular regulation mechanisms.
Although there are IDPs that carry out their function while remaining permanently disordered (e.g., entropic chains) (5), many of them undergo induced folding, i.e., a disorder-to-order transition upon binding to their physiological partners (11–13). Because of their inherent flexibility, IDPs escape atomistic description using x-ray crystallography: due to the considerable flexibility of the chain, IDPs generally fail to be crystallized and crystals can only be obtained for IDPs bound to a partner, provided that the disorder-to-order transition warrants a sufficient gain of rigidity compatible with the crystal nucleation process.
NMR spectroscopy, being able to provide information at the residue level, is in principle quite well suited to provide a meaningful description of IDPs and of their dynamics. It is, however, very demanding in terms of both protein amounts and costs related to uniform or selected isotopic enrichment strategies. Paramagnetic relaxation enhancement NMR, which relies on the covalent insertion of paramagnetic spin labels as relaxation agents and leads to intramolecular distance constraints, can be even more demanding in these aspects.
Site-directed spin labeling coupled to electron paramagnetic resonance (EPR) spectroscopy has been successfully used to map regions within IDPs involved in binding and folding (14,15). A novel approach relying on a combination of site-directed spin-labeling EPR spectroscopy and modeling of local rotation conformational spaces has been recently developed to model partly disordered protein complexes (16).
In continuous-wave EPR experiments, the linewidth of the probe's spin transitions can be used as a proxy for the nanosecond-timescale dynamics of the local structure (17), and thus EPR spin labels can be used to map structural transitions in proteins which undergo localized disorder-to-order transitions on binding to substrates (proteins, membranes, nucleic acids, or small-molecule targets) or other induced folding events. Multiple spin labels can be combined with pulsed EPR relaxation experiments to infer distances between the labels and thus to build up a picture of a structure from pairs of labels (18). Spin labels are usually introduced via disulfide bond formation at cysteine sites introduced by site-directed mutagenesis. The relative bulkiness of the resulting labeled side chain may, however, profoundly affect the folding or binding interface of a protein (19), thus leading to reduced complex formation (14). Therefore, useful EPR labeling sites are usually restricted to residues that either are solvent-exposed in the bound form or not otherwise involved in close protein-substrate interactions.
Optical techniques commonly applied to IDPs have their own inherent drawbacks. Covalently attached fluorescent probes have been inserted within disordered regions in attempts to document solution exposure (via quenching) and distances between sites (via specific contact quenching or fluorescent resonance energy transfer); these approaches usually suffer from the above-mentioned drawback, that polyaromatic fluorescent probes are usually much bulkier than the native side chain. Noncovalent fluorescent binding dyes can only provide information about the global structure of a protein with no site-specificity. Far-ultraviolet (UV) circular dichroism can report on secondary structure transitions accompanying binding, but this technique also only provides global information.
Despite the fact that infrared spectroscopy is inherently sensitive to picosecond-timescale dynamics of both the protein structure and the solvent, it is most often applied as a global probe of secondary structure, due to overlapping spectral bands in the amide region. One way to recover some site-specific information in infrared spectroscopy is to introduce artificial amino acids containing one or more functional groups that absorb in the clear region of the aqueous biomolecular infrared spectrum between 1900 and 2700 cm−1 (20–24). One artificial amino acid that can be introduced in proteins of any size without having to resort to peptide synthesis, semisynthesis, or amber stop codon suppression, is β-thiocyanatoalanine or cyanylated cysteine (20,25–27). The thiocyanate moiety is produced by posttranslational modification of the free thiol of cysteine, and can thus be inserted at any cysteine site resulting from mutagenesis or onto any natively solvent-exposed cysteine.
The side chain of this cyanylated cysteine contains a C≡N group whose stretching vibration absorbs at 2155–2164 cm−1 in typical protein environments (28). The frequency of the SC≡N stretching vibration depends on the hydrogen bonding character of its local environment; as with other nitriles, the frequency undergoes a blue-shift upon accepting a hydrogen bond at nitrogen (28,29). Water-exposed SC≡N bands are centered at ∼2163 cm−1, while SC≡N bands in hydrophobic solvents appear at lower frequencies. Thus, the frequency can be used to discriminate between water-exposed and water-excluded environments, i.e., a binding or folding interface or a lipid membrane (30). The CN stretching band of cyanylated cysteine is stronger (ɛ = 100–250 M−1 cm−1 depending on solvent (22,28,31)) than that of β-cyano-alanine (ɛ = ∼30 M−1 cm−1 (22)) or p-cyano-phenylalanine (ɛ = ∼40–60 M−1 cm−1 based on p-tolunitrile (21)), and weaker than the asymmetric N=N=N stretching vibration of β-azido-alanine (ɛ = ∼450 M−1 cm−1 (22)) and presumably also p-azido-phenylalanine (24). What is unique about the aliphatic SCN group is that it can be introduced posttranslationally, as opposed to all of the other artificial side chains just mentioned.
The SC≡N linewidth depends on the picosecond dynamics of its local environment (28,31), which for solvent-exposed probe groups comes almost exclusively from the solvent dipolar fluctuations. It has recently been shown that local helical secondary structure leads to a broadening of the C≡N stretching band due to a reversal of motional narrowing from the faster water motions around a more disordered secondary structure (32). Taken together, the structural dependence of the linewidth and the sensitivity to water exposure of the frequency suggest that cyanylated cysteine could be an ideal probe of structural transitions in IDPs. Its small size compared to any other covalently attached probe may mean that it could find even wider use than spin or fluorescent labels in characterizing the site-specific structure of disordered or extremely dynamic proteins.
To assess the ability of cyanylated cysteine's unique infrared (IR) band to probe structural transitions in IDPs, we have introduced a single cyanylated cysteine at selected sites of the well-characterized intrinsically unstructured C-terminal domain of the measles virus nucleoprotein (NTAIL) (33) (see Fig. 1). We then have monitored spectral changes upon addition of one of the physiological partners of NTAIL, namely the C-terminal X domain (XD) of the phosphoprotein that is known to trigger α-helical folding of NTAIL (for reviews see (34–36)).
Figure 1.
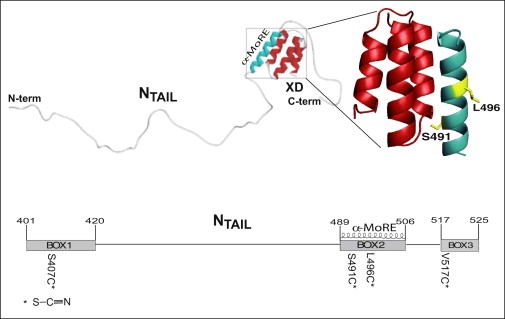
(Top) Model of the NTAIL-XD complex as derived by small-angle x-ray scattering studies highlighting the role of the α-MoRE in the interaction with XD (40). The structure of the chimera between XD and the NTAIL region encompassing residues 486–504 (PDB code 1T60) (39) is enlarged and the side chains of residues targeted for cysteine substitution and ensuing cyanylation are shown. The picture was drawn using PyMol (DeLano Scientific, San Carlos, CA). (Bottom) Schematic representation of NTAIL positions targeted for cysteine substitution and cyanylation (stars). The three conserved regions, namely Box1, Box2, and Box3, are also shown, as is the α-MoRE.
Within a conserved region of NTAIL (aa 489–506, Box2), an α-helical molecular recognition element (α-MoRE) involved in binding to XD and in induced folding was identified (37) and modeled in the crystal structure of XD (38,39) (see Fig. 1). Using small angle x-ray scattering, a low-resolution structural model of the complex between XD and the entire NTAIL domain has been obtained (40). This model revealed that most of NTAIL (aa 401–488) remains disordered in the complex with XD and does not establish contacts with XD. In addition, this model showed that beyond the α-MoRE, the C-terminal region is also likely involved in binding to XD. Involvement of the C-terminal Box3 (aa 517–525) in binding to XD was also supported by fluorescence spectroscopy studies which pointed out a dose-dependent modification in the environment of a Trp residue introduced at NTAIL position 518 upon addition of XD (40).
Two site-directed spin label (SDSL) EPR spectroscopy studies of the NTAIL-XD interaction have been recently reported (14,15). In these studies, 14 sites scattered within NTAIL were targeted for spin-labeling, and EPR spectroscopy of spin-labeled variants provided information on the mobility of the spin label and hence on the involvement of specific residues in complex formation. These spin-labeled variants enabled detailed mapping of the gain of rigidity of NTAIL in the presence of either the secondary structure stabilizer trifluoroethanol or XD. The mobility of the spin labels grafted within the 488–502 region was shown to be dramatically reduced upon binding to XD, with the spin-label grafted onto position 491 experiencing a highly restricted mobility due to its location at the XD interface (see Fig. 1). In addition, XD was shown to trigger α-helical folding of the 488–502 region only, while the 505–522 region, which is only moderately affected by XD, does not gain α-helicity. Strikingly, for this latter region, trifluoroethanol was shown to trigger reductions in the spin label mobility similar to those observed in the presence of XD, thus indicating a lack of direct contacts between Box3 and XD. The latter has been also recently confirmed by heteronuclear single quantum coherence experiments using 15N-labeled XD and a Box3 peptide (41). Recent in-depth spectroscopic studies of the NTAIL-XD interaction, which resulted in the assignment of the NMR spectrum of NTAIL in both the free and XD-bound form, showed that residues 483–506 of NTAIL are in intermediate exchange with XD, while the 475–482 and 507–525 regions are in fast exchange. Binding of XD triggers α-helical folding of Box2 and results in a gain of rigidity by Box3. The bound form consists of an ensemble of conformers in which Box3 has a reduced conformational freedom that may favor the establishment of weak, nonspecific contacts with XD (42). Using an original modeling approach to describe partly disordered protein complexes, which relies on a combination of SDSL-EPR spectroscopy and modeling of local rotation conformational spaces, the 490–500 region of NTAIL has been recently shown to be effectively prestructured in the absence of the partner. This approach also allowed quantitative estimation of the extent of α-helical sampling of the free form and confirmed that the 505–525 region of NTAIL does conserve a significant degree of conformational freedom even in the bound form (16).
In this work we have targeted for cysteine cyanylation four different sites within NTAIL, one falling in the N-terminal nonbinding region (Box1, position 407), two located within the α-MoRE (positions 491 and 496, with the side chain of the former pointing toward the XD binding interface), and the fourth in the C-terminus (Box3, position 517) (Fig. 1). The goals of this study are twofold: first, to use a relatively well-characterized IDP system to examine the sensitivity of cyanylated cysteine to local structural transitions; and second, to reexamine the structure of the XD-bound form of NTAIL via a new site-specific probe methodology.
Experimental Methods
Mutagenesis and expression
The four single-cysteine NTAIL variants, S407C, S491C, L496C, and V517C, were expressed and purified as reported previously (14,15), as was XD (38). The expression and solubility levels of the cysteine NTAIL variants were comparable to those obtained with wt NTAIL. Likewise, the purification yields were similar to those observed with the native form of the protein and were ∼10 mg of purified protein per liter of induced Escherichia coli culture.
Cyanylation of cysteine-containing variants
Before the cyanylation reaction, ∼2 mg of each single-cysteine variant was dissolved to roughly 500 μM concentration. A one-hundred molar excess of dithiothreitol (Sigma-Aldrich, St. Louis, MO) was added to produce the free thiol at cysteine. Dithiothreitol was removed via size exclusion chromatography on a Superdex 75 HR 10/300 column (GE Healthcare, Piscataway, NJ) with an elution buffer of degassed 70 mM phosphate buffer at pH 7.0. The fractions containing NTAIL were concentrated to 500 μL via ultrafiltration using centrifugal concentrators with a 5 kDa polyethersulfone membrane (Millipore, Billerica, MA). An eight-fold molar excess 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) (Pierce Chemical, Rockford, IL) was added from a 25 mM DTNB stock solution in 200 mM phosphate buffer. After 30 min, the reaction yield for the mixed disulfide was measured based on the release of TNB, using an extinction coefficient of 13600 cm−1 M−1 at 412 nm and a model No. V-570 UV/visible/near-infrared spectrophotometer (Jasco, Easton, MD). Sodium cyanide (NaCN) was then added at a 55× molar excess, leading to cyanylation of both the protein mixed disulfide and the remaining unreacted DTNB. After 30 min, excess reagents were removed via size exclusion chromatography on a PD-10 column packed with Sephadex G-25 (GE Healthcare) using 70 mM phosphate buffer at pH 7.0 as the elution buffer. Fractions containing NTAIL were collected and concentrated to ∼20 μL as described above. Cyanylated proteins are referred to as S407C∗, S491C∗, L496C∗, and V517C∗, where C∗ denotes cyanylated cysteine.
Preparation of XD-containing samples
Before the addition of XD, cyanylated NTAIL samples were transferred into a 20 mM phosphate buffer using a PD-10 size exclusion column and concentrated to 20 μL as described above. XD was added in a twofold molar excess to 20 μL (∼2 mM concentration) cyanylated NTAIL samples. The mixture was thereafter reconcentrated to 20 μL using a SVC 100 SpeedVac (Savant, Thermo Scientific, Asheville, SC). For all samples, a twofold molar excess of XD was expected to saturate NTAIL based on the previously determined dissociation constant (40). In the case of S491C∗, no spectral changes were observed at higher concentrations of XD, suggesting that this sample also was also fully bound at twofold molar excess.
Infrared spectroscopy
All infrared spectra were collected at 25°C in a 22-μm pathlength BioCell (BioTools, Jupiter, FL) with a Vertex 70 FTIR spectrometer (Bruker Optics, Billerica, MA), using 1024 scans at 2 cm−1 resolution. A quantity of 2 cm−1 resolution was used to speed data acquisition, because no significant differences were observed compared to samples collected at 1 cm−1 resolution. All samples exhibited an optical density of ∼500 μO.D., which is consistent with a concentration of 2.0–2.5 mM of cyanylated protein if the extinction coefficient is assumed to be ∼100 M−1 cm−1, from studies of methyl thiocyanate (MeSCN) in water. This concentration would correspond to a nearly quantitative yield of cyanylated protein as compared to the total concentration of protein as determined by UV absorbance at 280 nm. A background spectrum of buffer was subtracted from each sample infrared spectrum, and the baseline for the C≡N stretching region in all NTAIL samples was additionally corrected by fitting the baseline (excluding the region between 2140 and 2190 cm−1) to a polynomial and subtracting the fit. All NTAIL infrared spectra were reproduced using at least two different protein samples (from separate purifications) and two different authors. All NTAIL C≡N stretching bands were fit to a pseudo-Voigt profile that yields a single width parameter (full width at half-maximum, FWHM) used for analysis:
| (1) |
The fitted parameters reported vary by <0.2 cm−1 between samples in all cases.
Results and Discussion
We targeted for cyanylation four positions within NTAIL: one located within Box1 (position 407), two located within Box2 (e.g., 491 and 496) and one within Box3 (position 517) (Fig. 1). The rationale for these choices was that position 407 was known not to be involved in binding to XD (14,15,40) and could hence serve as a negative control, whereas the other three positions were known to be either severely (positions 491 and 496) or moderately (position 517) affected by XD (14). Previous site-directed spin-labeling EPR studies also showed that position 491 experiences a very restricted mobility upon complex formation, with the spin label exerting a steric hindrance leading to only 75% of complex formation even in the presence of saturating amounts of XD (14). As such, this position is particularly interesting and worth investigating by using the much smaller cyanylated cysteine probe group.
Cyanylated yields of all cysteine variants were near-quantitative according to UV absorbance at 412 nm, with no substantial variation between samples. For all samples, the maximum optical density of the C≡N stretching band was between 200 and 500 μO.D. Far-UV circular dichroism of cyanylated variants indicated negligible changes in the secondary structure when compared to the wild-type protein (see Supporting Material).
The infrared spectra for all cyanylated variants in the presence or absence of a twofold molar excess of XD are shown in Fig. 2, with maximum absorption frequencies and linewidths presented in Table 1. The two spectra for the S407C∗ protein are nearly superimposable, with no significant change in the frequency or linewidth of the C≡N stretching band probe in the presence of XD (Fig. 2 a), in agreement with previous studies pointing out lack of any involvement of Box1 in binding to XD. The infrared spectra of the S407C∗ protein therefore serve as a control, exhibiting the probe signal's near-zero natural variability in the absence of any local binding and folding events.
Figure 2.
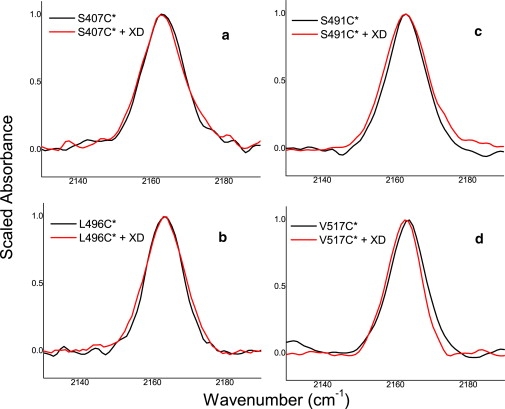
Infrared spectra in the C≡N stretching region for cyanylated NTAIL variants in the absence (black) and presence (red) of XD. (a) S407C∗, (b) L496C∗, (c) S491C∗, and (d) V517C∗.
Table 1.
Central frequencies and fitted linewidths (full width at half-maximum) for the C≡N stretching bands shown in Fig. 2
| Probe site | Unbound frequency/cm−1 | Unbound line width/cm−1 | Frequency with XD/cm−1 | Line width with XD/cm−1 |
|---|---|---|---|---|
| S407C∗ | 2163.4 | 13.0 | 2163.2 | 13.5 |
| L496C∗ | 2163.3 | 11.7 | 2163.2 | 13.6 |
| S491C∗ | 2163.1 | 12.4 | 2163.0 | 14.0 |
| V517C∗ | 2163.4 | 11.3 | 2162.4 | 11.0 |
All reported values are ±0.2 cm−1.
Conversely, the probe infrared spectrum for the L496C∗ protein does exhibit a change upon addition of XD (Fig. 2 b). The central frequency does not change, remaining 2163 cm−1 in both bound and unbound forms, indicating that the probe is always solvent-exposed. This is as expected according to the crystal structure of a chimeric construct encompassing XD and residues 486–504 of NTAIL (39), in which Box2 assumes a helical conformation with L496 pointing toward the solvent and away from the Box2/XD helical bundle interface (see Fig. 1). The linewidth, however, increases significantly upon binding to XD. This increase in linewidth reflects the structural transition of Box2 from a random coil to a helical structure upon binding, where this structural transition had also been documented by previous EPR spin labeling studies (14,15).
An increase in the L496C∗ probe's linewidth concomitant with formation of helical secondary structure agrees with recent observations made in water-soluble helical peptides (32), which indicate that an increase in the helical structure content near the cyanylated cysteine probe leads to an increase in the linewidth of the C≡N stretching vibration. Because the probe is solvent-exposed throughout the structural transition, a change in the dynamics of the water near the secondary structure (or possibly a change in the dipolar inhomogeneous distribution) is the likely source of the change in linewidth. Empirically, this observation in L496C∗ agrees with previous structural observations of the Box2 structural transition. This spectral change is the first indication in a true IDP that the linewidth of the C≡N stretching band is indeed sensitive to formation of secondary structure, despite the fact that the probe vibration is water-exposed throughout the structural transition.
As shown in Fig. 2 c, the C≡N stretching band of the S491C∗ protein does not red-shift as might be expected were it buried in the binding interface and shielded from the solvent. However, the linewidth does increase, indicating that the probe experiences a local structural transition from random coil to helix (as in the case of the L496C∗ protein): the increase in linewidth argues for the formation of helical structure in the vicinity of the 491 position. Upon comparison with the spectrum of the L496C∗ protein, and noting that all band frequencies in each case are 2163 cm−1, it also appears that the probe in the S491C∗ protein is also solvent-exposed regardless of whether XD is added.
Previous EPR studies revealed that grafting a nitroxide radical at position 491 of NTAIL impairs the ability of the protein to yield 100% complex formation with XD. Indeed, the experimentally observed EPR spectrum of this spin-labeled variant in the presence of a molar excess of XD was found to be composed of two signals: one arising from the unbound spin-labeled protein (25%) and one resulting from the spin-labeled protein bound to XD (75%) (14). It was concluded that the 491 side chain is able to adopt a number of different orientations, suggesting a high extent of possible conformational diversity at this particular residue. Occurrence of conformational diversity at this site in both the free and bound forms was also confirmed recently by Kavalenka et al. (16). Taking into account the ability of the spin-labeled S491C NTAIL protein to interact with XD despite the presence of the much bulkier nitroxide label, the present IR results do not argue for lack of complex formation between the cyanylated S491C∗ NTAIL variant and XD. Instead, they suggest that the cyanylated side chain of Cys491 is solvent-exposed and not buried at the binding interface as suggested by the crystal structure. This could be due to the length of the cyanylated cysteine side chain as compared to the serine residue it replaces here: the longer artificial side chain is apparently able to reach outside the binding interface so that the C≡N group is solvated. That the solvent exposure of the probe does not reflect lack of complex formation is further supported by the increase in linewidth in the spectrum of the variant in the presence of XD—a change reflecting formation of a helical structure in the vicinity of the 491 position. Discrepancy between the results of IR and EPR studies with the S491C-labeled variant illustrates the utility of the much smaller cyanylated cysteine side chain, which apparently can be grafted to a binding interface without substantially altering the bound conformation.
While the probe vibration of S491C∗ does not exhibit a red shift indicative of solvent exclusion upon binding to XD, the probe of the V517C∗ protein does red-shift by a small yet significant and reproducible amount (∼1 cm−1, Fig. 2 d). This shift away from 2163 cm−1 (which is the frequency for all other probe bands in this study) indicates the formation of an environment around position 517 that is no longer purely aqueous, and which has a substantially increased hydrophobic character.
To quantify the hydrophobicity of the environment around the cyanylated probe at position 517, we recorded the IR spectra of MeSCN in mixed solutions of tetrahydrofuran (THF) and water (Fig. 3). Due to its low dielectric constant and reduced polarity as compared to water, THF can be regarded as a reasonable bulk model for the interior of a protein or an environment of similar hydrophobicity, such as a lipid bilayer (21). As shown in Fig. 3, as the volume percentage of THF increases the central frequency decreases from 2163 cm−1 (in 100% water) to 2155 cm−1 (in 100% THF), with smaller frequency changes near the endpoints and larger changes with broad bands and apparently multiple spectral populations at intermediate solvent mixtures (40–60% THF). The spectra of the V517C∗ variant in the absence and presence of XD are comparable to the spectra of MeSC≡N in 100% water and 20% THF, respectively (for further analysis of these data, see Supporting Material). Although the behavior of MeSCN in mixed solvents is clearly nonmonotonic and likely influenced by specific solvation effects, Fig. 3 represents a starting point for interpreting the small yet significant shift observed for V517C∗ upon XD binding. Cyanylated cysteine frequencies of 2157–2158 cm−1 were recently observed for peptide side chains known to be buried in lipid micelles (30); compared to the frequency of 2162 cm−1 observed for V517C∗ in the presence of XD, this further indicates that the environment around the V517C∗ probe vibration is mostly, but not completely, aqueous.
Figure 3.
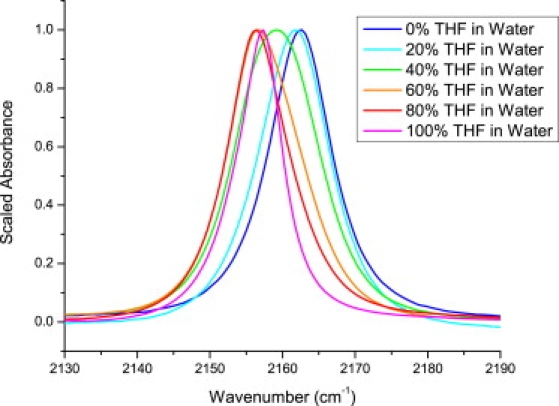
Infrared absorption bands in the C≡N stretching region for methyl thiocyanate in varying mixtures of THF with water.
The extent of water exclusion from the probe group in V517C∗ is small and the side chain is still largely water-exposed even in the XD-bound form. The structural role of Box3 in binding has not been completely elucidated yet. EPR data indicate that although the orientations of spin labels within Box3 are partially restricted in the bound form, they nevertheless retain a considerable mobility as compared to those within Box2 (16). Recent NMR data indicate that upon addition of XD and after α-helical folding of Box2, Box3 exhibits a reduced flexibility that could favor the establishment of numerous, transient, and aspecific contacts with XD (42). The conclusion of these prior EPR and NMR results is that Box3 remains unstructured when NTAIL binds to XD. However, if Box3 is removed, the binding between NTAIL and XD is weakened (40), indicating that Box3 provides substantial thermodynamic stability to the bound complex without structure formation. This means that Box3 residues may participate transiently in binding XD while retaining the ability to adopt a wide range of different conformations. This appears to be the case with the V517C∗ protein, where the C≡N band has revealed a transient hydrophobic contact at position 517 which could contribute to binding with XD.
Infrared absorption transitions in the 4–5 μm wavelength region have an intrinsic reporting timescale of 13–16 fs. If there is a substantial distribution of dipolar environments around the cyanylated cysteine chain of the V517C∗ variant, then the distribution should be represented at least partially in the lineshape of the probe vibration. There is not a substantial linewidth increase in the spectrum of the variant in the presence of XD as compared to the free form. This suggests that the vibrational chromophore of the bound form experiences an environment which is, on mean, more hydrophobic than pure water but does not sample a large diversity of hydrophobic or hydrophilic environments. The simplest explanation for the small frequency shift with little lineshape variation is that when NTAIL is bound to XD, residue 517 participates in a weak, nonspecific hydrophobic interaction with the Box2-XD moiety. The experimentally observed IR spectral behavior is thus in agreement with previous data designating Box3 as an unstructured and nonspecific contributor to NTAIL-XD complex formation. Using cyanylated cysteine to further map local hydrophobicity for Box3 in this complex is an appealing future possibility.
The dual sensitivity of the C≡N stretching band of cyanylated cysteine to local hydration and structure formation, as exemplified by its use in the NTAIL single-cysteine variants presented here, makes it a particularly attractive probe for characterization of site-specific structural changes in IDPs. A general approach to using cyanylated cysteine to map site-specific structural transitions in IDPs is suggested in Fig. 4. If a residue-specific response of the probe to a binding event is discovered, then the participation of neighboring residues could be mapped by moving the same probe to nearby sites. This study, when coupled with previous studies in model peptides involving both structure formation and folding, indicates that the probe vibration should be able to report solvent exposure, including hydrophobic contacts of varying degrees, as well as formation of structure even at solvent-exposed sites. Cysteine is rarely present in intrinsically disordered sequences, so the strategy of creating single-cysteine mutants of IDP sequences already in use for site-directed spin labeling studies should also be widely useful for incorporation of this new vibrational probe into IDPs, with more possible incorporation sites than for covalently attached spin labels or fluorophores.
Figure 4.
Flow chart suggesting an experimental strategy for mapping structural transitions in uncharacterized IDPs using cyanylated single-cysteine variants.
This study shows that vibrational spectroscopy of cyanylated cysteine is a method well suited to monitoring the induced folding events that NTAIL undergoes in the presence of XD, while providing information at the residue level. Therefore, it can be added to the panel of widely used experimental approaches to study induced folding events within IDPs (43–45).
Conclusions
Any new probe able to provide site-specific information about binding and possible associated secondary structure transitions and/or changes in solvent exposure in IDPs could see wide use. The behavior of cyanylated cysteine was studied in the context of four previously characterized single-cysteine NTAIL variants. The probe was shown to be sensitive to changes in both mean hydrophobicity of the probe vibration's environment, as well as to the electric dipolar dynamics of the same environment, which are each affected by changes in the local protein structure during binding events. Moreover, the covalently attached C≡N probe moiety has the advantage of being smaller than conventional fluorophores or spin labels and can be introduced into IDPs at nearly any site via cysteine site-directed mutagenesis. While applied to a well-characterized system here, this probe is expected to be very useful for characterizing other IDP systems for which structural data are scarce. The spectral signatures that have been obtained in this study should have a high predictive value: indeed, the spectral shape and location reflect the specific environment in the proximity of the label and the extent of its involvement in the interaction with a partner, while also providing information on possible transitions in local secondary structure. As such, one should be able to infer information on the chemical environment specific residues within any given IDP in the presence of partner(s) without any prior structural information on the bound form of the IDP.
Acknowledgments
C.H.L. acknowledges support from the Dreyfus Foundation (New Faculty Start-Up Award), Research Corporation (Cottrell College Science Award), and the National Institute of General Medical Sciences (grant No. R15-GM088749). D.M.S. acknowledges a summer fellowship enabled by a science education grant to Haverford College by the Howard Hughes Medical Institute. S.L. acknowledges support from the Agence Nationale de la Recherche, under the specific program “Microbiologie et Immunologie” (grant No. ANR-05-MIIM-035-02). The content is solely the responsibility of the authors and does not necessarily represent the views of any of the agencies above.
Supporting Material
References
- 1.Bogatyreva N.S., Finkelstein A.V., Galzitskaya O.V. Trend of amino acid composition of proteins of different taxa. J. Bioinform. Comput. Biol. 2006;4:597–608. doi: 10.1142/s0219720006002016. [DOI] [PubMed] [Google Scholar]
- 2.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 3.Dunker A.K., Brown C.J., Obradovic Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem. 2002;62:25–49. doi: 10.1016/s0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- 4.Dunker A.K., Garner E., Villafranca J.E. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac. Symp. Biocomput. 1998;3:473–484. [PubMed] [Google Scholar]
- 5.Dunker A.K., Lawson J.D., Obradovic Z. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 6.Dunker A.K., Obradovic Z. The protein trinity—linking function and disorder. Nat. Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 7.Fink A.L. Natively unfolded proteins. Curr. Opin. Struct. Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Gunasekaran K., Tsai C.J., Nussinov R. Extended disordered proteins: targeting function with less scaffold. Trends Biochem. Sci. 2003;28:81–85. doi: 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 9.Uversky V.N., Li J., Fink A.L. Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of α-synuclein assembly by β- and γ-synucleins. J. Biol. Chem. 2002;277:11970–11978. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 10.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 11.Dyson H.J., Wright P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 12.Uversky V.N., Oldfield C.J., Dunker A.K. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005;18:343–384. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- 13.Wright P.E., Dyson H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belle V., Rouger S., Longhi S. Mapping α-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins Struct. Funct. Bioinf. 2008;73:973–988. doi: 10.1002/prot.22125. [DOI] [PubMed] [Google Scholar]
- 15.Morin B., Bourhis J.M., Longhi S. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling electron paramagnetic resonance spectroscopy. J. Phys. Chem. B. 2006;110:20596–20608. doi: 10.1021/jp063708u. [DOI] [PubMed] [Google Scholar]
- 16.Kavalenka A., Urbančič I., Strancar J. Conformational analysis of the partially disordered measles virus N(TAIL)-XD complex by SDSL EPR spectroscopy. Biophys. J. 2010;98:1055–1064. doi: 10.1016/j.bpj.2009.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Columbus L., Hubbell W.L. A new spin on protein dynamics. Trends Biochem. Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 18.Schiemann O., Prisner T.F. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q. Rev. Biophys. 2007;40:1–53. doi: 10.1017/S003358350700460X. [DOI] [PubMed] [Google Scholar]
- 19.Langen R., Oh K.J., Hubbell W.L. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 20.Fafarman A.T., Webb L.J., Boxer S.G. Site-specific conversion of cysteine thiols into thiocyanate creates an IR probe for electric fields in proteins. J. Am. Chem. Soc. 2006;128:13356–13357. doi: 10.1021/ja0650403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Getahun Z., Huang C.Y., Gai F. Using nitrile-derivatized amino acids as infrared probes of local environment. J. Am. Chem. Soc. 2003;125:405–411. doi: 10.1021/ja0285262. [DOI] [PubMed] [Google Scholar]
- 22.Oh K.I., Lee J.H., Cho M. Beta-azidoalanine as an IR probe: application to amyloid Aβ(16-22) aggregation. J. Phys. Chem. B. 2008;112:10352–10357. doi: 10.1021/jp801558k. [DOI] [PubMed] [Google Scholar]
- 23.Waegele M.M., Tucker M.J., Gai F. 5-Cyanotryptophan as an infrared probe of local hydration status of proteins. Chem. Phys. Lett. 2009;478:249–253. doi: 10.1016/j.cplett.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye S.X., Huber T., Sakmar T.P. FTIR analysis of GPCR activation using azido probes. Nat. Chem. Biol. 2009;5:397–399. doi: 10.1038/nchembio.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Degani Y., Neumann H., Patchornik A. Selective cyanylation of sulfhydryl groups. J. Am. Chem. Soc. 1970;92:6969–6971. doi: 10.1021/ja00726a043. [DOI] [PubMed] [Google Scholar]
- 26.Degani Y., Patchornik A. Cyanylation of sulfhydryl groups by 2-nitro-5-thiocyanobenzoic acid. High-yield modification and cleavage of peptides at cysteine residues. Biochemistry. 1974;13:1–11. doi: 10.1021/bi00698a001. [DOI] [PubMed] [Google Scholar]
- 27.Doherty G.M., Motherway R., Malthouse J.P. 13C NMR of cyanylated flavodoxin from Megasphaera elsdenii and of thiocyanate model compounds. Biochemistry. 1992;31:7922–7930. doi: 10.1021/bi00149a025. [DOI] [PubMed] [Google Scholar]
- 28.Maienschein-Cline M.G., Londergan C.H. The CN stretching band of aliphatic thiocyanate is sensitive to solvent dynamics and specific solvation. J. Phys. Chem. A. 2007;111:10020–10025. doi: 10.1021/jp0761158. [DOI] [PubMed] [Google Scholar]
- 29.Choi J.H., Oh K.I., Cho M. Nitrile and thiocyanate IR probes: quantum chemistry calculation studies and multivariate least-square fitting analysis. J. Chem. Phys. 2008;128:134506. doi: 10.1063/1.2844787. [DOI] [PubMed] [Google Scholar]
- 30.McMahon H.A., Alfieri K.N., Londergan C.H. Cyanylated cysteine: a covalently attached vibrational probe of protein-lipid contacts. J. Phys. Chem. Lett. 2010;1:850–855. doi: 10.1021/jz1000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh K.I., Choi J.H., Cho M. Nitrile and thiocyanate IR probes: molecular dynamics simulation studies. J. Chem. Phys. 2008;128:154504. doi: 10.1063/1.2904558. [DOI] [PubMed] [Google Scholar]
- 32.Edelstein L., Stetz M.A., Londergan C.H. The effects of α-helical structure and cyanylated cysteine on each other. J. Phys. Chem. B. 2010;114:4931–4936. doi: 10.1021/jp101447r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longhi S., Receveur-Bréchot V., Canard B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003;278:18638–18648. doi: 10.1074/jbc.M300518200. [DOI] [PubMed] [Google Scholar]
- 34.Bourhis J.M., Canard B., Longhi S. Structural disorder within the replicative complex of measles virus: functional implications. Virology. 2006;344:94–110. doi: 10.1016/j.virol.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 35.Bourhis J.M., Longhi S. Measles virus nucleoprotein: structural organization and functional role of the intrinsically disordered C-terminal domain. In: Longhi S., editor. Measles Virus Nucleoprotein. Nova Science Publishers; Hauppauge, NY: 2007. [Google Scholar]
- 36.Longhi S., Oglesbee M. Structural disorder within the measles virus nucleoprotein and phosphoprotein. Protein Pept. Lett. 2010;17:961–978. doi: 10.2174/092986610791498894. [DOI] [PubMed] [Google Scholar]
- 37.Bourhis J.M., Johansson K., Longhi S. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 2004;99:157–167. doi: 10.1016/j.virusres.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Johansson K., Bourhis J.M., Longhi S. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 2003;278:44567–44573. doi: 10.1074/jbc.M308745200. [DOI] [PubMed] [Google Scholar]
- 39.Kingston R.L., Baase W.A., Gay L.S. Characterization of nucleocapsid binding by the measles virus and mumps virus phosphoproteins. J. Virol. 2004;78:8630–8640. doi: 10.1128/JVI.78.16.8630-8640.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bourhis J.M., Receveur-Bréchot V., Longhi S. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005;14:1975–1992. doi: 10.1110/ps.051411805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernard C., Gely S., Darbon H. Interaction between the C-terminal domains of N and P proteins of measles virus investigated by NMR. FEBS Lett. 2009;583:1084–1089. doi: 10.1016/j.febslet.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 42.Gely S., Lowry D.F., Longhi S. Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J. Mol. Recognit. 2010 doi: 10.1002/jmr.1010. In press. [DOI] [PubMed] [Google Scholar]
- 43.Eliezer D. Biophysical characterization of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2009;19:23–30. doi: 10.1016/j.sbi.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Receveur-Brechot V., Bourhis J.M., Longhi S. Assessing protein disorder and induced folding. Proteins Struct. Funct. Bioinf. 2006;62:24–45. doi: 10.1002/prot.20750. [DOI] [PubMed] [Google Scholar]
- 45.Uversky V.N., Longhi S., editors. Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation. John Wiley and Sons; Hoboken, NJ: 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.