

Abstract
Store-operated Ca2+ entry (SOCE) contributes to Ca2+ handling in normal skeletal muscle function, as well as the progression of muscular dystrophy and sarcopenia, yet the mechanisms underlying the change in SOCE in these states remain unclear. Previously we showed that calsequestrin-1 (CSQ1) participated in retrograde regulation of SOCE in cultured skeletal myotubes. In this study, we used small-hairpin RNA to determine whether knockdown of CSQ1 in adult mouse skeletal muscle can influence SOCE activity and muscle function. Small-hairpin RNA against CSQ1 was introduced into flexor digitorum brevis muscles using electroporation. Transfected fibers were isolated for SOCE measurements using the Mn2+ fluorescence-quenching method. At room temperature, the SOCE induced by submaximal depletion of the SR Ca2+ store was significantly enhanced in CSQ1-knockdown muscle fibers. When temperature of the bathing solution was increased to 39°C, CSQ1-knockdown muscle fibers displayed a significant increase in Ca2+ permeability across the surface membrane likely via the SOCE pathway, and a corresponding elevation in cytosolic Ca2+ as compared to control fibers. Preincubation with azumolene, an analog of dantrolene used for the treatment of malignant hyperthermia (MH), suppressed the elevated SOCE in CSQ1-knockdown fibers. Because the CSQ1-knockout mice develop similar MH phenotypes, this inhibitory effect of azumolene on SOCE suggests that elevated extracellular Ca2+ entry in skeletal muscle may be a key factor for the pathophysiological changes in intracellular Ca2+ signaling in MH.
Introduction
Store-operated Ca2+ entry (SOCE) represents an important pathway for controlling communications between the extracellular Ca2+ reservoir and the intracellular Ca2+ store to maintain Ca2+ homeostasis (1). In skeletal muscle, SOCE participates in Ca2+ handling during physiological conditions such as exercise and fatigue (2–5). Whereas elevated SOCE has been linked to the progression of muscular dystrophy in skeletal muscle (6,7), as well as the functional state of smooth muscle (8), compromised SOCE has recently been identified in aged skeletal muscle (9). It is well known that tight coupling between the sarcolemmal/transverse-tubular (T-tubule) membrane and sarcoplasmic reticulum (SR) is required for effective SOCE function (10). For example, altering the coupling between SR and T-tubule membranes through manipulation of junctophilin expression has been shown to influence SOCE activity and Ca2+ handling in skeletal muscle (11). Although this study reveals that a close physical juxtaposition of these membrane surfaces is essential for operation of SOCE in skeletal muscle, the molecular mechanisms controlling this signaling, particularly within the SR itself, have not been extensively studied.
We have previously shown that calsequestrin-1 (CSQ1), a SR-resident Ca2+-binding protein, participated in the generation of retrograde signals to the sarcolemmal/T-tubular membrane through the action of a Ca2+-binding motif located on the carboxyl-terminal region of CSQ1 (12). In particular, we found that overexpression of the full-length CSQ1 in cultured skeletal myotubes led to inhibition of SOCE, an effect that was absent with overexpression of CSQ1 lacking the Ca2+-binding motif. Additional evidence for the role of retrograde signaling from SR to sarcolemmal/T-tubular membrane was provided by our recent demonstration that azumolene, a compound binding to the ryanodine receptor (RyR) Ca2+ release machinery, could inhibit SOCE in skeletal muscle in a RyR conformation-dependent manner (13). Because dantrolene, an analog of azumolene, has been used for the clinical treatment of malignant hyperthermia (MH), these results suggest that extracellular Ca2+ entry may also participate in the pathophysiology of MH. Thus, SOCE may be a promising clinical target in the treatment of MH.
Paolini et al. (14) found that genetic ablation of CSQ1 altered ultrastructure of the SR and Ca2+ signaling of the skeletal muscle. The CSQ1 knockout mice develop a hypercontractile state at elevated ambient temperature with high mortality in male mice (15), a phenomenon identical to MH-susceptible, RyR1 mutation knock-in mice (16) and similar to the pathophysiology of MH observed in human patients (17). Currently, it is not clear whether ablation of CSQ1 alters Ca2+ permeability across the sarcolemmal/T-tubular membrane, nor do we know whether this could contribute to the phenotype of high ambient temperature-induced intracellular Ca2+ elevation observed in CSQ1 knockout animals. Examination of the physiological function of CSQ1 would benefit from a study where targeted ablation of CSQ1 in skeletal muscle can be achieved to minimize the potential adaptive developmental responses in the whole animal with systemic ablation of the gene.
Here we used small hairpin RNA (shRNA) to knock down CSQ1 expression in adult skeletal muscle fibers of living mice to test the hypothesis that reduced CSQ1 expression may cause elevation of extracellular Ca2+ entry into the myoplasm via SOCE. We found that a significant elevation in SOCE occurred in skeletal muscle with CSQ1 knockdown. This is accompanied by alteration of Ca2+ release from SR and sensitivity of SOCE to elevated temperature. Our data suggests that CSQ1 likely participates in maintaining the retrograde regulation of SOCE in skeletal muscle either in parallel with or as part of its function in Ca2+ buffering within the SR. Additionally, our results suggest that one potential mechanism for the MH-like symptoms observed in the CSQ1 knockout mice may be via Ca2+ entry through the SOCE pathway.
Materials and Methods
Plasmids
The shRNA specifically targeting mouse CSQ subtype 1 (shCSQ1) and scrambled shRNA (shCON) sequences were designed according to previous report (18) and the sequences were, respectively,
shCSQ1 sense 5′-GTACCTCCTGAAGAAGACAGCGTTTATTCAAGAGATAAACGCTGTCTTCTTCAGTTTTTGGAAA-3′,
antisense 5′-AGCTTTTCCAAAAACTGAAGAAGACAGCGTTTATCTCTTGAATAAACGCTGTCTTCTTCAGGAG-3′,
shCON sense 5′-GTACCTCTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTGGAAA-3′,
antisense 5′-AGCTTTTCCAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAAGAG 3′.
Sense and antisense oligonucleotides of shCSQ1 and shCON were annealed and cloned into psiRNA-hH1 G2 vector (InvivoGen, San Diego, CA). The two shRNA expression cassettes driven by a H1 promoter were then excised from the above vector and subcloned into a custom-made pCMS-mRFP vector with a red fluorescence protein (RFP) marker (19) driven under a separate promoter. Full-length CSQ1 cDNA (NM_009813) in pGEX4T-1 vector was subcloned into pMH6 vector (Roche, Branchburg, NJ).
Cell culture and gene transfection
HEK 293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. shCSQ1 or shCON pCMS-mRFP plasmids were cotransfected with CSQ1 cDNA plasmid at 9:1 molar ratio into HEK293 cells using transfection reagent GeneJammer (Stratagene, Cedar Creek, TX) according to the manufacturer's instructions (2 μg DNA: 4 μL reagent). Forty-eight hours later, transfected HEK 293 cells were lysed in RIPA buffer (150 mM NaCl, 20 mM NaPO4, 50 mM NaF, 2 mM EDTA, 0.1% SDS, 1% deoxycholic acid, 1% Triton X-100, 14 mM β-mercaptoethanol, 30 mM Na pyrophosphate, and 0.2 mM Na vanadate at pH 7.2), plus protease inhibitors consisting of 1 mM phenylmethanesulphonyl fluoride and 1% p8340 protease inhibitor cocktail from Sigma (St. Louis, MO). Twenty micrograms of protein from each sample was separated by 10% SDS electrophoresis and transferred to polyvinylidene difluoride membranes. The membrane was probed with rabbit anti-mouse CSQ1 antibody and mouse anti-mouse β-actin antibody (Sigma). Target proteins were detected using the ECL-Plus Luminescent Kit (GE Healthcare, Piscataway, NJ).
Flexor digitorum brevis muscle electroporation
The shRNA probes were introduced into flexor digitorum brevis (FDB) muscles of 2–3 month-old wild-type mice using an electroporation method. Briefly, mice were anesthetized with 100 mg/kg ketamine plus 12 mg/kg xylazine. Ten microliters of hyaluronidase (2 mg/mL in sterile saline) was injected subcutaneously into the footpad for digestion of collagen and extracellular matrix surrounding FDB muscle. Delivery of shCSQ1 or shCON plasmids into the FDB muscle was achieved by following the protocol of Cai et al. (20). To confirm effective knockdown of CSQ1 in FDB fibers, we developed a technique of Western blotting with manually selected individual FDB fibers with the help of Dr. Jingsong Zhou (Rush University, Chicago, IL). Briefly, 12–14 days after electroporation, mice were sacrificed by cervical dislocation and FDB fibers were enzymatically dissociated (21). Dissociated muscle fibers were plated onto a ΔTC3 culture dish and RFP-positive fibers were hand-picked under a stereo-fluorescence microscope using a 0.8–1.1 mm × 100 mm capillary tube (PYREX, Lowell, MA). These handpicked FDB fibers were lysed in RIPA buffer containing 1% protease inhibitor cocktail by vortexing and proteins were separated by 10% SDS electrophoresis in sample buffer containing 100 mM dithreitol. Western blotting for CSQ1, CSQ2, sarco/endoplasmic-reticulum calcium ATPase (SERCA1), Orai1, and stromal interaction molecule 1 (STIM1) were performed using rabbit polyclonal anti-CSQ1 and anti-CSQ2 antibodies (provided by Dr. Eun Kyung Kim, Department of Life Science, Gwangju Institute of Science and Technology, Gwangju, South Korea), mouse monoclonal anti-SERCA1 antibody (Affinity BioReagents, Rockford, IL), rabbit polyclonal anti-Orai1 antibody (Millipore, Billerica, MA), and mouse monoclonal anti-STIM1 (BD Bioscience, Franklin Lakes, NJ). Western blotting for α-sarcomeric actin (Sigma) was used as a loading control.
Measurement of intracellular Ca2+ and SOCE activity
Individual muscle fibers were plated on ΔTC3 dishes for experiments performed at room temperature (18 ± 2°C) and metal-coated ΔT4 dishes (Bioptechs, Butler, PA) for experiments under controlled bathing temperature. FDB fibers were loaded with 10 μM FURA-2 AM at room temperature for 1 h and treated with 40 μM n-benzyl-p-toluene sulfonamide (Sigma) for 15 min to prevent muscle contraction associated with intracellular Ca2+ release. Changes in FURA-2 fluorescence were measured with a spectrofluorometer (12) (Photon Technology International, Birmingham, NJ). For assessment of SOCE in adult skeletal muscle, Mn2+ quenching of FURA-2 fluorescence was used by following the protocol of Collet and Ma (22). For measurement of heat stress-induced changes in [Ca2+]i, FDB muscle fibers were bathed in 2 mM Ca2+ Tyrode solution and temperature of the culture medium was raised from 18°C to 39°C during 2–3 min under the control of a thermal controller (Bioptechs, Butler, PA). For assessment of changes in Ca2+ permeability across the sarcolemmal membrane under heat stress, muscle fibers were incubated in 0 mM Ca2+ Tyrode solution at 39°C, and 0.5 mM Mn2+ solution was perfused for 8 min. Mn2+ entry rate was calculated as the rate of decrease in FURA-2 fluorescence measured at excitation wavelength of 360 nm (ΔF360/s).
SR Ca2+ content was assessed by incubation of muscle fibers with 30 mM caffeine for 5 min and area under the curve (AUC; from fluorescence rise to first baseline reached) was calculated using the Integrate function of the FELIX software (Photon Technology International).
Statistics
Values were calculated as mean ± SE. Significance was determined by paired t-test for the densitometric assay and by Student's t-test for all other comparisons. A value of P < 0.05 was used as criteria for statistical significance. When other P-values indicate a greater level of statistical significance, it is noted in the individual figure legends.
Results
Effective and specific CSQ1 knockdown in adult muscle fiber using shRNA probe
Previous studies by Wang et al. (18) showed that a shRNA probe targeting nucleotide sequence 532–551 of the murine CSQ1 mRNA could effectively and specifically knock-down the expression of CSQ1, but not CSQ2, in C2C12 myotubes. We employed this probe in our study to knock-down CSQ1 expression in adult skeletal muscle. To identify transfected muscle fibers, we cloned the shCSQ1 sequence into a modified shRNA expression plasmid containing a RFP reporter gene (19) under the control of a separate promoter. Efficacy of the shCSQ1 for suppression of CSQ1 expression was first tested in HEK293 cells cotransfected with full-length murine CSQ1 cDNA. As shown in Fig. 1 a, HEK293 cells cotransfected with shCSQ1 and full-length CSQ1 displayed greatly reduced expression of exogenous CSQ1 protein compared with those transfected with full-length CSQ1 and a scrambled shRNA probe as a control (shCON).
Figure 1.
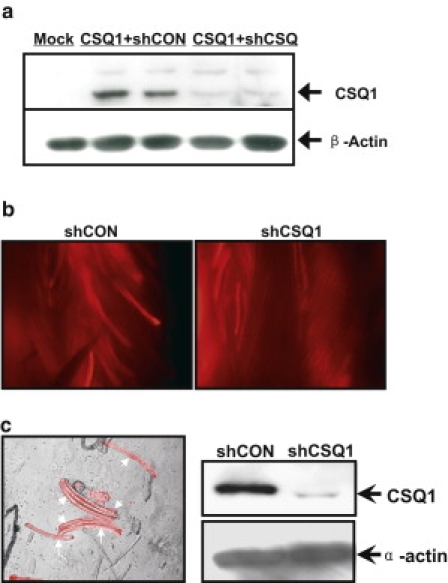
shRNA-mediated knockdown of CSQ1 from mammalian skeletal muscle. (a) Western blotting was performed for CSQ 1 and β-actin using HEK 293 cells ectopically cotransfected with full-length CSQ1 in pHM6 vector and shCSQ1 (CSQ1+shCON) or shCON (CSQ1+shCSQ) in pCMS-mRFP vector to confirm the effectiveness of shRNAs. Cells treated with transfection reagent only were included as a control (Mock). Duplicate wells for CSQ1+shCON and CSQ1+shCSQ were included and experiments were repeated twice. (b) Representative images for in vivo electroporation of shCSQ1 and shCON pCMS-mRFP plasmids into FDB muscles. High transfection rate (40∼70%) was achieved at 12∼14 days after electroporation for both constructs, as marked by the RFP expressions. (c) Representative images of enzymatically dissociated FDB fibers (arrows). Healthy shCSQ1-transfected fibers labeled with RFP fluorescence (left panel). A quantity of 15∼20 individual FDB fibers labeled with RFP were hand-picked from the shCON and shCSQ1-transfected muscles for Western blotting against mouse CSQ1 (right). Alpha-sarcomeric actin was included as a loading control (right panel). Experiments were repeated in 10 mice (20 FDB muscles).
To achieve efficient transfection of muscle fibers in living mice, we used electroporation-mediated delivery of plasmid DNA into FDB muscle bundles, following a published procedure (20). As shown in Fig. 1 b, 12∼14 days after electroporation, >40% of muscle fibers remain transfected, as indicated by the appearance of red fluorescence in the dissected FDB muscle bundles. The efficiency of transfection was similar between the shCSQ1 and shCON groups. Unless otherwise noted, all controls used in this study were performed with FDB fibers transfected with the shCON plasmid. An equal number of (15∼20) transfected fibers from both groups were hand-picked for Western blot analysis and experiments were repeated in 10 mice. As shown in Fig. 1 c, shCSQ1-treated FDB fibers displayed >80% knockdown of CSQ1, confirming the efficacy of this specific shRNA probe to suppress the expression of endogenous CSQ1 in adult skeletal muscle. Individual shCSQ1 or shCON-transfected FDB muscle fibers positive for red fluorescence were thus used for subsequent functional studies.
Increased SOCE activity in skeletal muscle after CSQ1 knockdown and inhibitory effect of azumolene
We have previously established that quenching of FURA-2 fluorescence by extracellular Mn2+ entry could be used to evaluate SOCE activation after depletion of SR Ca2+ store in skeletal muscle (13). Specifically, the rate of Mn2+-quenching of FURA-2 fluorescence at excitation wavelength of 360 nm, the isosbestic point that is independent of changes in intracellular Ca2+, provides a quantitative assessment of SOCE. Our first set of experiments was performed with 10 mM caffeine plus 5 μM ryanodine (C/R) to induce SR Ca2+ release via the RyR channel. As shown in Fig. 2 a, 5 min after bathing muscle fibers with C/R in an extracellular solution containing 0 mM Ca2+, addition of 0.5 mM Mn2+ produced quenching of the FURA-2 fluorescence in FDB fibers at ambient room temperature (18 ± 2°C) (Fig. 2 a, first trace). To normalize this data, FURA-2 fluorescence was completely quenched by permeabilization of the muscle fiber using 1% Triton at the end of the experiment. The rate of Mn2+-quenching is thus defined as the normalized F360 signal. Notice that FDB fibers treated with shCSQ1 displayed a significantly higher rate of Mn2+ quenching of FURA-2 (Fig. 2 a, third trace), suggesting that knockdown of CSQ1 leads to elevation of SOCE in skeletal muscle.
Figure 2.
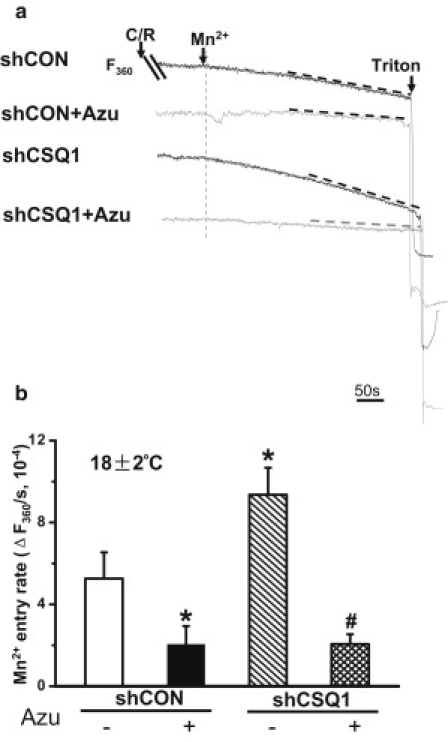
Elevation of RyR1-coupled SOCE in skeletal muscle with knockdown of CSQ1. (a) Representative traces of SOCE entry induced by submaximal concentration of caffeine plus ryanodine (C/R) were accessed by Mn2+ entry of Fura-2 fluorescence at excitation wavelength of 360 nm (F360). Individual traces were: shCON, shCON plasmid-transfected single FDB fiber pretreated by 0.1% DMSO; shCON+Azu, shCON plasmid-transfected FDB fiber pretreated by 20 μM azumolene; shCSQ1, shCSQ1 plasmid-transfected FDB fiber pretreated by 0.1% DMSO; and shCSQ1+Azu, shCSQ1 plasmid-transfected FDB fiber pretreated by 20 μM azumolene. (b) Quantification of SOCE activity in the above four groups by decrease of F360. (∗P < 0.05 compared to shCON group treated by DMSO; #P < 0.01 compared to shCSQ1 group treated by DMSO; n = 5–7 for each experiment condition. Experiments were performed at room temperature (18 ± 2°C).)
Based on our previous results showing that azumolene can inhibit RyR1-coupled extracellular Ca2+ entry (13), we tested whether azumolene could inhibit the elevated SOCE in shCSQ1 transfected muscle fibers. As shown in Fig. 2 a (second trace), inclusion of 20 μM azumolene substantially inhibited SOCE induced by C/R in the shCON group. Moreover, the elevated SOCE in shCSQ1-treated FDB fibers was inhibited by azumolene to a level similar to that of the shCON-transfected fiber (Fig. 2 a, fourth trace). Because azumolene was dissolved in dimethyl sulfoxide (DMSO, 0.1% final concentration), we treated the muscle fibers with 0.1% DMSO as a control. Data from multiple experiments (summarized in Fig. 2 b) show that the elevated Mn2+ entry associated with CSQ1-knockdown is suppressed by azumolene, supporting our hypothesis that CSQ1 plays an important role in RyR1-coupled activation of SOCE in skeletal muscle (12).
Assessment of SR Ca2+ handling in CSQ1 knockdown muscle fibers
To understand the mechanism behind the enhanced SOCE activity in skeletal muscle with knockdown of CSQ1, we first measured the resting cytosolic [Ca2+]i in shCON- and shCSQ1-treated muscle fibers. As shown in Fig. 3 b, when measurements were conducted at ambient room temperature, no significant differences were observed in [Ca2+]i between the shCON and shCSQ1 groups, suggesting that knockdown of CSQ1 did not affect Ca2+ homeostasis in muscle fiber in the resting state. To test whether knockdown of CSQ1 alters the movement of Ca2+ across the SR membrane as a result of RyR1 stimulation, we perfused individual FDB fibers with 30 mM caffeine for 5 min to completely mobilize SR Ca2+. As shown in Fig. 3 a and c, and consistent with the results of Paolini et al. (14), the SR Ca2+ content in shRNA-treated FDB fibers was significantly smaller than that of the control fiber, as determined by the calculated AUC. However, despite that, the Ca2+ efflux out of SR in the shRNA-treated FDB fibers displayed significantly faster kinetics as compared with shCON-treated fibers (Fig. 3, a and d), consistent with published data from Sztretye et al. (23). Both reduced SR Ca2+ content and accelerated Ca2+ release kinetics in the CSQ1 knockdown muscle fibers may contribute to the elevated SOCE activity.
Figure 3.
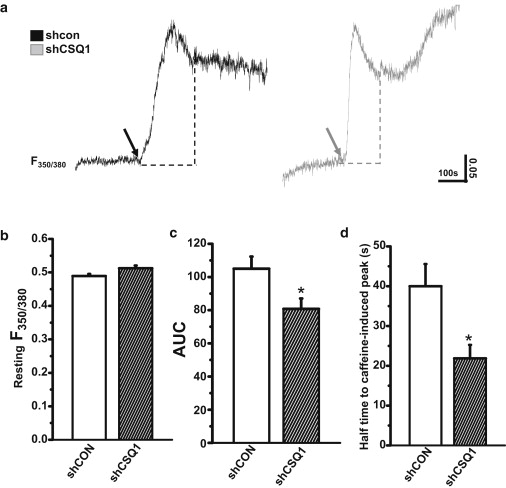
Altered SR Ca2+ release property after CSQ1 knockdown. (a) Representative traces of SR Ca2+ release induced by 30 mM caffeine (arrows) in shCON (solid trace) and shCSQ1-treated FDB fibers (shaded trace). Changes in FURA-2 fluorescence ratio at excitation wavelengths of 350:380 nm (F350/380) were measured and (dotted lines) area under the curve (AUC) was highlighted. Statistical results of (b) resting Ca2+ level and (c) AUC induced by caffeine and (d) half-time to peak in shCON (solid bar) and shCSQ1-treated FDB fibers (hatched bar). (∗P < 0.05; n = 8∼12. Experiments were performed at room temperature (18 ± 2°C).)
Increase in [Ca2+]i under heat stress and contribution of Ca2+ entry
Considering the intriguing observation that CSQ1-knockout mice display MH-like symptoms upon exposure to elevated ambient temperature (15), we tested whether knockdown of CSQ1 can alter the resting Ca2+ levels in isolated FDB muscle fibers at high temperatures. We found that increasing the temperature to >40°C frequently resulted in contracture of the shCSQ1-treated muscle fibers, which prevented our systematic evaluation of the effect of high-temperature on Ca2+ signaling. We therefore conducted our experiments within the temperature window of 18–39°C. As shown in Fig. 4, when the temperature surrounding the muscle fibers was elevated from 18 to 39°C, significantly higher elevation of intracellular Ca2+ was observed in shCSQ1-transfected fibers, compared with shCON-treated muscle fibers. This elevated intracellular Ca2+ could be reduced by application of 2 mM NiCl2 (Fig. 4, a and b). Because Ni2+ is a competitive blocker of Ca2+ channels on the sarcolemmal membrane, this result suggests that a component of temperature-dependent elevation of Ca2+ in shCSQ1-transfected muscle fibers may originate from the extracellular source.
Figure 4.
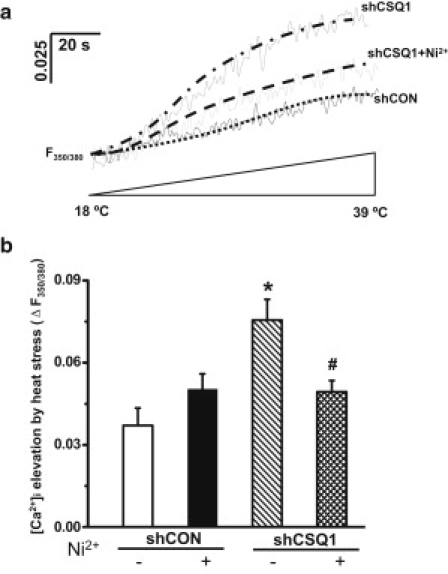
Exaggerated temperature-dependent intracellular Ca2+ ([Ca2+]i) elevation after knockdown of CSQ1. (a) Representative traces of [Ca2+]i elevation induced by moderate heat stress were shown by changes in F350/380 of FURA-2 fluorescence. Individual traces were: shCON, shCON plasmid-transfected single FDB fiber; shCSQ1, shCSQ1 plasmid-transfected FDB fiber; and shCSQ1+Ni2+, shCSQ1 plasmid-transfected FDB fiber treated by 2 mM NiCl2. (To avoid overlapping, trace for shCON-treated FDB fiber with preincubation of Ni2+ was not shown.) (b) Statistical results of [Ca2+]i elevation at 39°C compared to 18°C in shCON, shCON+Ni2+, shCSQ1, and shCSQ1+Ni2+ groups. (∗P < 0.05 compared to shCON group without Ni2+ treatment; #P < 0.05 compared to shCSQ1 group without Ni2+ treatment; n = 7–10.)
Sustained incubation of muscle fibers at >39°C with 2.5 mM Ca2+ present in the extracellular solution resulted in contracture of the fiber (data not shown), possibly due to additional activation of a heat-sensitive component of Ca2+ entry from the extracellular solution (24). This parallels the stiffening seen in the CSQ1 knockout mouse as MH is triggered (15).
To probe whether there were changes in Ca2+ permeability across the surface membrane with knockdown of CSQ1 in skeletal muscle, we performed experiments at 39°C with no Ca2+ present in the extracellular solution. Mn2+ quenching of FURA-2 fluorescence was used to monitor the changes in permeability of divalent cations across the surface membrane. As shown in Fig. 5 a (first trace), a significant component of Mn2+ influx was observed in shCON muscle fibers during heat stress. This entry rate was elevated with knockdown of CSQ1 (Fig. 5 a, third trace). Interestingly, the elevated Mn2+ entry in both the shCON- and shCSQ1-transfected fibers could be prevented by the addition of 20 μM azumolene (Fig. 5 a, second and fourth traces). Data from multiple experiments are summarized in Fig. 5 b. These results suggest that knockdown of CSQ1 expression could activate the RyR1-coupled SOCE in skeletal muscle under heat stress, which may contribute to the temperature-dependent elevation of cytosolic Ca2+ (Fig. 3).
Figure 5.
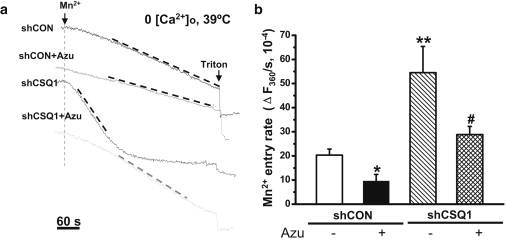
Enhanced Mn2+ entry activated by heat stress in CSQ1 knockdown muscle. (a) Representative traces of Mn2+ entry at F360 induced by heat stress in 0 [Ca2+]o bathing solution. Individual traces were: shCON, shCON plasmid-transfected single FDB fiber pretreated by 0.1% DMSO; shCON+Azu, shCON plasmid-transfected FDB fiber pretreated by 20 μM azumolene; shCSQ1, shCSQ1 plasmid-transfected FDB fiber pretreated by 0.1% DMSO; and shCSQ1+Azu, shCSQ1 plasmid-transfected FDB fiber treated by 20 μM azumolene. (b) Statistical comparison of SOCE activity (ΔF360/s, 10−4) in the above four groups. (∗∗P < 0.01 or ∗P < 0.05 compared to shCON group pretreated by 0.1% DMSO; #P < 0.05 compared to shCSQ1 group pretreated by 0.1% DMSO, n = 6–13.)
Assessment of changes in Ca2+ regulatory proteins with CSQ1 knockdown in skeletal muscle
Recent studies (25) have identified two molecular components of the SOCE machinery, with STIM1 functioning as the endoplasmic reticulum (ER)/SR Ca2+ sensor that undergoes conformational changes in response to depletion of the Ca2+ store inside the ER/SR. Clustering of STIM1 can then couple to activation of the Orai1 channel located on the plasma membrane to mediate SOCE. To test whether the elevated SOCE we observed with knockdown of CSQ1 in skeletal muscle was due to changes in expression of STIM1 and/or Orai1, we performed Western blot analysis with anti-STIM1 and -Orai1, as well as other Ca2+ regulatory proteins (e.g., SERCA1 and CSQ2) in skeletal muscle fibers derived from mice electroporated with shCON and shCSQ1 plasmids. For the purpose of quantification, 15∼20 FDB fibers (labeled with RFP fluorescence as a marker of transfection) were handpicked for the immunoblot analyses. As shown in Fig. 6, the expression levels of SERCA1, STIM1, and CSQ2 were not very different between the shRNA and shCON-treated muscle fibers, confirming the specificity of the shRNA probe to knock-down the expression of CSQ1 in skeletal muscle. Interestingly, expression of Orai1 was significantly lower in shCSQ1-treated muscle fibers compared with shCON-treated muscle fibers (P < 0.05, n = 8) (Fig. 6 b). Thus, changes in the expression of SOCE machinery cannot account for the elevated SOCE observed in skeletal muscle with knockdown of CSQ1. It is possible that compensatory reduction of Orai1 expression may be necessary for the survival of skeletal muscle fibers with reduced expression of CSQ1 in adaptation to the increased SOCE activity.
Figure 6.
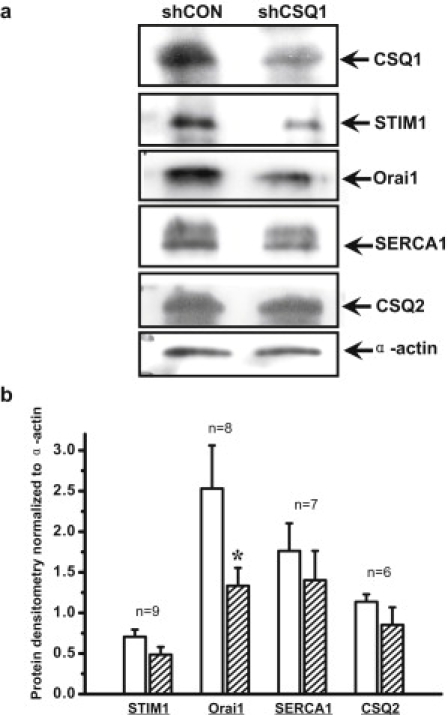
Reduced protein expression of SOCE Ca2+ conducting pore after CSQ1 knockdown. (a) Representative single-fiber Western blots for shCON and shCSQ1-treated FDB fibers, including STIM1, Orai1, SERCA1, CSQ2, and α-actin. (b) Densitometry results of protein expression levels after CSQ1 knockdown (hatched bar). (∗P < 0.05 compared to shCON-treated FDB fiber (open bar) by paired t-test. The n number for individual sets of proteins was labeled on the histogram.)
Discussion
In this study, we show that knockdown of CSQ1 influences Ca2+ signaling in adult skeletal muscle by perturbing Ca2+ entry across the sarcolemmal/T-tubular membrane, which increases the vulnerability of the skeletal muscle to heat stress. The elevated rate of Mn2+-entry observed in the shCSQ1-treated FDB fibers can be blocked by azumolene to a level comparable to that of the shCON-treated fibers, indicating that reduced CSQ1 expression may affect RyR1-coupled SOCE activation in skeletal muscle. Taking into account that systemic knockout of CSQ1 in mice leads to hypersensitivity of the animal to heat and volatile anesthetics with a phenotype resembling that of the MH (15), our data provide further evidence that increased activity of RyR1-coupled SOCE could be associated with the etiology of MH. The therapeutic effect of dantrolene, an azumolene analog used for clinical treatment of MH, seems to act through a mechanism that involves the suppression of RyR1-activated SOCE.
Operation of SOCE requires retrograde signaling from the ER/SR membrane to the sarcolemmal membrane, across a distance of ∼100 Å in the triad junction of skeletal muscle (26). Because CSQ1 is a Ca2+-binding protein located inside the SR lumen, changes in SOCE function associated with CSQ1 knockdown could reflect the following possibilities: 1), expression of the molecular components for SOCE function could be altered with reduced expression of CSQ1; 2), reduced CSQ1 expression likely alters the SR Ca2+ release process and affects the retrograde steps of SOCE activation; and 3), knockdown CSQ1 expression could alter the conformational coupling between the SR and sarcolemmal membrane components and thus indirectly influence permeability of Ca2+ across the sarcolemmal membrane. Our data provides some insights into these possibilities.
First, it is well known that STIM1 located on the ER membrane functions as the luminal Ca2+ sensor that controls opening of the Orai Ca2+ channel located on the plasma membrane for operation of SOCE (25). The changes in SOCE function could be a result of altered expression of either STIM1 or Orai1. Our Western blot data showing that knockdown of CSQ1 expression leads to reduced expression of Orai1 is counterintuitive to the elevated SOCE activity in skeletal muscle. We speculate that this may represent a compensatory mechanism for the survival of skeletal muscle in adaptation to the reduced CSQ1 expression and elevated SOCE activity. At present, we do not know if the increased SOCE activity associated with CSQ1 knockdown is due to an increase in preformed STIM1-Orai1 complexes in the sarcolemma or T-tubule membrane that can increase the efficiency of signal transduction and Ca2+ entry, or whether the expression of other Orai homologs, such as Orai2 and Orai3, is altered in skeletal muscle.
Second, as CSQ1 is a SR-resident Ca2+ buffering protein, altering expression level of CSQ1 would change the SR Ca2+ buffering capacity and accessibility of the free Ca2+ inside the SR. Previous studies showed that saponin-permeabilized CSQ1-null muscle fibers seem to have both a reduced response to electric stimulation and a smaller SR Ca2+ content (14). Recent studies by Sztretye et al. (23) also found that the kinetics of voltage-induced Ca2+ release and the evacuability of Ca2+ from the SR store in response to sustained voltage stimulation are modified with reduced CSQ1 expression. Our data demonstrate that the caffeine-releasable SR Ca2+ content was smaller and the Ca2+ release kinetics faster with reduced CSQ1 expression, consistent with these studies. The tight link between SR Ca2+ storage and SOCE activity has long been established (13), and it is conceivable that a reduced SR Ca2+ store—possibly due to compromised Ca2+ buffering capacity secondary to a reduced CSQ1 protein content in SR—leads to higher SOCE activity. In addition, the faster kinetics of SR Ca2+ release in the shCSQ1-treated muscle fiber could contribute to the elevated SOCE, possibly through enhanced clustering of STIM1 and Orai1, to excessively activate SOCE at the plasma membrane.
Third, it is well known that close conformational coupling between plasma membrane and ER membrane constituents is essential for efficient operation of SOCE (10,22) and graded activation of SOCE has been shown to couple to graded reduction of the SR Ca2+ store (10,13). Indeed, ultrastructural adaptations have been shown to occur in CSQ1 null muscle fibers (14), providing indirect evidence for the possibility of change in conformational coupling between the two membrane compartments in the absence of CSQ1 protein. In addition, a component of SOCE in skeletal muscle has been shown to involve conformational changes in the RyR1 (13). In this study, several lines of evidence imply that alteration in the conformational coupling might act synergistically with the altered SR Ca2+ release kinetics to activate SOCE in CSQ1-knockdown skeletal muscle. Here, we show that azumolene suppresses the elevated SOCE activity in CSQ1 knockdown muscle fibers. This drug has been shown to uncouple the tight link between SR Ca2+ depletion via RyR1 and the activation of SOCE (13). We also show that muscle fibers with reduced CSQ1 expression display increased vulnerability to heat-induced changes in Ca2+ permeability across the sarcolemmal membrane without substantial depletion of the SR Ca2+ store, pointing to the possibility of conformational changes in the SOCE machinery.
The steady-state resting Ca2+ levels do not show significant differences between the shCSQ1 and shCON muscle fibers at ambient room temperature, suggesting that integrity of the control mechanism for movement of extracellular Ca2+ across the sarcolemmal membrane is not likely to be altered with reduced CSQ1 expression in skeletal muscle under nonstressed condition. A sharp difference between the shCSQ1 and shCON muscle fibers is revealed in divalent cation permeability across the sarcolemmal membrane and cytosolic Ca2+ at elevated temperatures. Previous work by van der Poel and Stephenson (27) showed that increased temperature to 40°C led to increased SR Ca2+ leakage in mechanically skinned muscle fibers, presumably through temperature-induced superoxide production. Our data show that in addition to its effect on the SR Ca2+ transport process, increase in temperature likely has a separate effect on SOCE, because the inhibition of Ca2+ entry from the extracellular space by Ni2+ eliminates the difference in [Ca2+]i transients in shCON and shCSQ1-fibers (Fig. 4). Furthermore, although exposure of muscle fibers to higher temperatures may activate heat-sensitive Ca2+ entry into the cytosol, coordinated action of SERCA-mediated Ca2+ uptake into the SR (due to its high Q10 value) and Na+/Ca2+ exchanger- or plasma membrane Ca2+-ATPase-mediated Ca2+ extrusion mechanisms allows for tight control of Ca2+ homeostasis in the native skeletal muscle. This may explain the minimal effect of Ni2+ on the resting [Ca2+]i in shCON-treated muscle fibers at 39°C. Nevertheless, the clear effect of Ni2+ on the elevated [Ca2+]i and inhibition of azumolene on Mn2+-quenching of FURA-2 fluorescence at 39°C in shCSQ1-tranfected muscle fibers suggest that a significant component of the defective [Ca2+]i homeostasis under high temperature may be due to the elevated RyR1-coupled SOCE seen after knockdown of CSQ1.
Malignant hyperthermia is a skeletal muscle syndrome associated with anesthetic-triggered increases in [Ca2+]i and excessive production of body heat (17). Although many studies show that a disturbance of Ca2+ release from SR is associated with triggering an MH crisis, several recent studies have begun to reveal the contribution of SOCE to muscle physiology and disease. With regard to MH, in particular, elevated SOCE could be another pathway for the uncontrolled rise in cytoplasmic [Ca2+] associated with MH. Our data presented here add further insights into the physiological roles of CSQ1 in normal muscle function and its potential for dysfunction during disease states. The parallel changes of heat-sensitivity in CSQ1 knockdown fibers and susceptibility of CSQ1-null mice to MH-like episodes further suggest that CSQ1 could potentially participate in the etiology of MH. In addition, because many genetic mutations in CSQ1 are linked to skeletal muscle or cardiac dysfunction in human diseases, our data could have broad implications for clinical medicine. Targeting SOCE or CSQ1 could be a potential treatment of MH and/or the Ca2+-dependent pathology of the dystrophinopathies and sarcoglycanopathies (28).
Acknowledgments
We thank Dr. Jingsong Zhou for help with the single-muscle-fiber Western-blot technique.
This work was supported by grants from the National Institutes of Health (Bethesda, MD) to J.M. and N.W., a grant from the Korean Science and Engineering Foundation to D.H.K., a Scientist Development Award from the American Heart Association to X.Z., and a grant from the National Science Foundation of China to J.M. J.P. was supported by clinical funds of the Department of Anesthesiology, University of Pittsburgh School of Medicine.
References
- 1.Parekh A.B., Putney J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 2.Kurebayashi N., Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibers. J. Physiol. 2001;533:185–199. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao X., Yoshida M., Brotto M. Enhanced resistance to fatigue and altered calcium handling properties of sarcalumenin knockout mice. Physiol. Genomics. 2005;23:72–78. doi: 10.1152/physiolgenomics.00020.2005. [DOI] [PubMed] [Google Scholar]
- 4.Pan Z., Yang D., Ma J. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat. Cell Biol. 2002;4:379–383. doi: 10.1038/ncb788. [DOI] [PubMed] [Google Scholar]
- 5.Stiber J., Hawkins A., Rosenberg P. STIM1 signaling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008;10:688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mallouk N., Allard B. Ca2+ influx and opening of Ca2+-activated K+ channels in muscle fibers from control and MDX mice. Biophys. J. 2002;82:3012–3021. doi: 10.1016/S0006-3495(02)75642-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alderton J.M., Steinhardt R.A. Calcium influx through calcium leak channels is responsible for the elevated levels of calcium-dependent proteolysis in dystrophic myotubes. J. Biol. Chem. 2000;275:9452–9460. doi: 10.1074/jbc.275.13.9452. [DOI] [PubMed] [Google Scholar]
- 8.Leung F.P., Yung L.M., Huang Y. Store-operated calcium entry in vascular smooth muscle. Br. J. Pharmacol. 2008;153:846–857. doi: 10.1038/sj.bjp.0707455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao X., Weisleder N., Brotto M. Compromised store-operated Ca2+ entry in aged skeletal muscle. Aging Cell. 2008;7:561–568. doi: 10.1111/j.1474-9726.2008.00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Launikonis B.S., Ríos E. Store-operated Ca2+ entry during intracellular Ca2+ release in mammalian skeletal muscle. J. Physiol. 2007;583:81–97. doi: 10.1113/jphysiol.2007.135046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirata Y., Brotto M., Pan Z. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys. J. 2006;90:4418–4427. doi: 10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin D.W., Pan Z., Ma J. A retrograde signal from calsequestrin for the regulation of store-operated Ca2+ entry in skeletal muscle. J. Biol. Chem. 2003;278:3286–3292. doi: 10.1074/jbc.M209045200. [DOI] [PubMed] [Google Scholar]
- 13.Zhao X., Weisleder N., Ma J. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J. Biol. Chem. 2006;281:33477–33486. doi: 10.1074/jbc.M602306200. [DOI] [PubMed] [Google Scholar]
- 14.Paolini C., Quarta M., Protasi F. Reorganized stores and impaired calcium handling in skeletal muscle of mice lacking calsequestrin-1. J. Physiol. 2007;583:767–784. doi: 10.1113/jphysiol.2007.138024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dainese M., Quarta M., Protasi F. Anesthetic- and heat-induced sudden death in calsequestrin-1 knockout mice. FASEB J. 2009;23:1710–1720. doi: 10.1096/fj.08-121335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chelu M.G., Goonasekera S.A., Hamilton S.L. Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J. 2006;20:329–330. doi: 10.1096/fj.05-4497fje. [DOI] [PubMed] [Google Scholar]
- 17.Nelson T.E. Malignant hyperthermia: a pharmacogenetic disease of Ca2+ regulating proteins. Curr. Mol. Med. 2002;2:347–369. doi: 10.2174/1566524023362429. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y., Xu L., Meissner G. Knocking down type 2 but not type 1 calsequestrin reduces calcium sequestration and release in C2C12 skeletal muscle myotubes. J. Biol. Chem. 2006;281:15572–15581. doi: 10.1074/jbc.M600090200. [DOI] [PubMed] [Google Scholar]
- 19.Campbell R.E., Tour O., Tsien R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai C., Masumiya H., Ma J. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X., Weisleder N., Ma J. Uncontrolled calcium sparks act as a dystrophic signal for mammalian skeletal muscle. Nat. Cell Biol. 2005;7:525–530. doi: 10.1038/ncb1254. [DOI] [PubMed] [Google Scholar]
- 22.Collet C., Ma J. Calcium-dependent facilitation and graded deactivation of store-operated calcium entry in fetal skeletal muscle. Biophys. J. 2004;87:268–275. doi: 10.1529/biophysj.103.039305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sztretye M., Royer L., Rios E. Ca depletion and ablation of calsequestrin similarly increase the evacuability of the Ca store of skeletal muscle. Biophys. J. 2010;98(Suppl):295a. [Google Scholar]
- 24.Talavera K., Yasumatsu K., Nilius B. Heat activation of TRPM5 underlies thermal sensitivity of sweet taste. Nature. 2005;438:1022–1025. doi: 10.1038/nature04248. [DOI] [PubMed] [Google Scholar]
- 25.Prakriya M. The molecular physiology of CRAC channels. Immunol. Rev. 2009;231:88–98. doi: 10.1111/j.1600-065X.2009.00820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma J., Pan Z. Junctional membrane structure and store operated calcium entry in muscle cells. Front. Biosci. 2003;8:d242–d255. doi: 10.2741/977. [DOI] [PubMed] [Google Scholar]
- 27.van der Poel C., Stephenson D.G. Effects of elevated physiological temperatures on sarcoplasmic reticulum function in mechanically skinned muscle fibers of the rat. Am. J. Physiol. Cell Physiol. 2007;293:C133–C141. doi: 10.1152/ajpcell.00052.2007. [DOI] [PubMed] [Google Scholar]
- 28.Whitehead N.P., Yeung E.W., Allen D.G. Muscle damage in MDX (dystrophic) mice: role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006;33:657–662. doi: 10.1111/j.1440-1681.2006.04394.x. [DOI] [PubMed] [Google Scholar]