

Abstract
Recent experiments pointed to the potential importance of ion correlation for multivalent ions such as Mg2+ ions in RNA folding. In this study, we develop an all-atom model to predict the ion electrostatics in RNA folding. The model can treat ion correlation effects explicitly by considering an ensemble of discrete ion distributions. In contrast to the previous coarse-grained models that can treat ion correlation, this new model is based on all-atom nucleic acid structures. Thus, unlike the previous coarse-grained models, this new model allows us to treat complex tertiary structures such as HIV-1 DIS type RNA kissing complexes. Theory-experiment comparisons for a variety of tertiary structures indicate that the model gives improved predictions over the Poisson-Boltzmann theory, which underestimates the Mg2+ binding in the competition with Na+. Further systematic theory-experiment comparisons for a series of tertiary structures lead to a set of analytical formulas for Mg2+/Na+ ion-binding to various RNA and DNA structures over a wide range of Mg2+ and Na+ concentrations.
Introduction
RNAs are highly charged polyanions. Metal ions, which can bind to RNAs to mediate the electrostatic interactions, are critical for RNA stability. Therefore, quantitative predictions for the ion-binding properties are essential for understanding the ion-dependent RNA folding thermodynamics and kinetics (1–17). RNA folding is often hierarchical (1,2): the secondary structural segments (helices, loops) are formed first, followed by the formation of tertiary contacts in the presence of Mg2+ or high monovalent salt (1,2). Most thermodynamic measurements for RNAs have been focused on the folding of secondary structural segments at a fixed standard salt condition, i.e., 1 M Na+ (18–23). These parameters have led to accurate predictions for RNA secondary structures and folding stabilities (18–23). More recent experimental and computational studies have extended the helix and loop thermodynamic parameters to different ionic conditions (19,24–27).
In contrast, our ability to predict the ion-dependent stability for tertiary structural folding is relatively limited (2–17). One of the difficulties comes from the fact that ion-RNA interactions are much more complicated for tertiary structural folding. The folding of a tertiary structure involves very high buildup of RNA backbone. The massive buildup of negative charges in the nucleotide backbone leads to significant binding of the counterions around RNA. The high concentration of the counterions around RNA causes strong coupling (correlation) between the ions, i.e., ions form a network due to the strong Coulomb interactions and, hence, the discrete distributions (instead of the mean-field distribution) of the ions become important. The effect can be quite significant for multivalent ions such as Mg2+ ions, which involve strong Coulomb interactions. The results from several experiments have suggested the potential significance of ion correlation for the binding of multivalent to nucleic acids (6,28).
The strong ion correlation prevents the use of any mean-field type of treatment for the multivalent ions. In general, there have been two major types of polyelectrolyte theories: the counterion condensation theory (29) and the Poisson-Boltzmann (PB) theory (30–34). Both theories have been quite successful in predicting a variety of electrostatic properties for nucleic acids and proteins (29–33). However, they both neglect the ion correlation effect, which can be important for multivalent ions such as Mg2+ (17,35–40). For example, the PB theory assumes the mean fluidlike ion distribution and thus ignores ion-ion correlation and ion fluctuation. Moreover, the counterion condensation theory is based on the simplified line-charge model and double-limit law, and thus is not applicable for complex nucleic acid structures with finite size in a solution of finite ion concentration.
Recently, motivated by the importance of ion correlation and fluctuation effects, we developed the tightly bound ion (TBI) model (37). The model takes into account the correlations between bound ions and the ion-binding ensembles (37). Experimental comparisons showed that the inclusion of ion correlations and binding ensembles in the TBI model leads to the improved predictions on salt-dependent thermodynamics of DNA/RNA helices/hairpins (25–27) and DNA helix assembly/bending (38–41). However, the model is based on simplified coarse-grained structures (37,42) originally developed for DNA helices. Because an RNA tertiary fold involves structural complexity far beyond helices, the coarse-grained model (for helices) cannot treat RNA tertiary folds. Therefore, we need a model that can handle the ion effects with ion correlations for RNA tertiary folds at the all-atom level.
The model developed here may lead to significant applications to the quantitative prediction of the ion effects in RNA tertiary structural folding, such as the folding of RNA kissing loop complex. Kissing loop complex is a tertiary structural motif (2,43) formed by basepairing between two hairpin loops. The motif is a highly frequently one occurring in RNA tertiary folds (2,44–46,48–50). The formation of a kissing complex involves significant buildup of the nucleotide backbone charges. As a result, the folding stability and kinetics of kissing loop complex critically depends on the ion-mediated electrostatic interactions. For example, Mg2+ ions can alter the unfolding and refolding rates by a factor of 102–103 and the saturating Mg2+ ions can cause a large change ΔG ∼10 kBT in the folding stability of a kissing loop (44,45), where kB is the Boltzmann constant and T is the temperature.
To date, no computational model has been developed to quantitatively predict the ion binding properties by considering the ion correlation effect for RNAs at the level of atomic structures. Here we develop an atomistic TBI model that can treat complex atomic nucleic acid structures, and employ the atomistic TBI model to predict the Na+/Mg2+-binding to RNA and DNA structures and the ion-dependent folding stability of HIV-1 DIS-type RNA kissing complex in Na+/Mg2+ solutions. Throughout the article, we emphasize the comparisons between the theoretical predictions and the experimental data. Furthermore, based on the theoretical predictions and the theory-experiment comparisons, we will derive analytical formulas for the Na+/Mg2+-binding. Such formulas may be practically useful for data analysis, experimental design, and quantitative predictions for the ion-dependence of RNA folding.
Methods
Structural model
The study covers a range of nucleic acid structures, including DNA and RNA duplexes, RNA pseudoknots, tRNAs, and the folded and unfolded forms of RNA kissing loop complexes. Below we describe how these structures are generated in the computational study.
For the canonical A-RNA and B-DNA duplexes, we use the software X3DNA (51) to construct the structure. For the native structure of the beet western yellow virus (BWYV) pseudoknot (Protein Data Bank (PDB) code: 437D) (52), the 58-nucleotide (nt) fragment of ribosomal RNA (rRNA, PDB code: 1HC8) (53), and the yeast tRNAPhe (PDB code: 1TRA) (54), we adopted the Protein Data Bank structure (see Fig. 1). For the HIV-1 dimerization initiation signal (DIS)-type kissing complexes (KC-1 and KC-2 in Fig. 1 G), because the wide-type HIV-1Lai sequence and the KC-1 and KC-2 kissing complexes show high similarities in basepairing patterns in the kissing interface and in the hairpins (49,50,55) and the kissing complexes generally have the similar coaxially stacked structures, we can use the wide-type HIV-1Lai (55,56) (PDB code: 2B8S) structure to model the three-dimensional all-atom structures of KC-1 and KC-2 kissing complexes (55,56).
Figure 1.
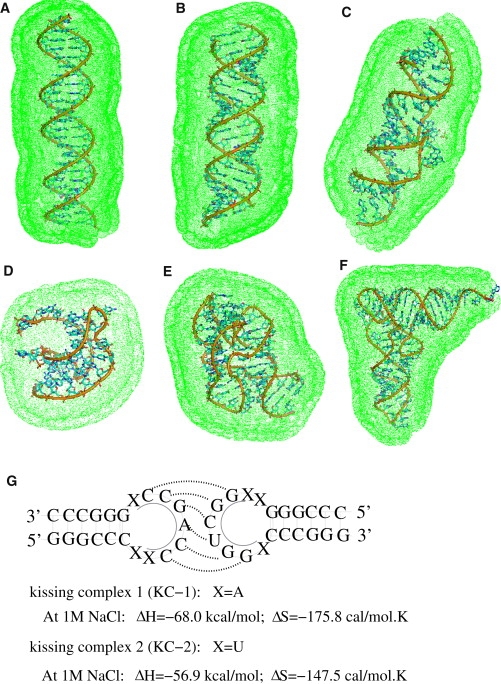
(A–F) The tightly bound regions in 0.1 M Mg2+ solution: (A) A 24-bp DNA duplex (structure produced from X3DNA (51)). (B) A 24-bp RNA duplex (structure produced from X3DNA (51)). (C) HIV-1 DIS kissing complex (PDB code: 2B8S) (55). (D) Beet Western yellow virus (BWYV) pseudoknot fragment (PDB code: 437D) (52). (E) A 58-nt ribosomal RNA (rRNA) fragment (PDB code: 1HC8) (66). (F) Yeast tRNAPhe (PDB code: 1TRA) (54). The figure for the three-dimensional structures is generated by the software PyMol (http://pymol.sourceforge.net/). (Green dots) Boundary of the tightly bound regions defined in the TBI model (37–40). (G) Illustration of the HIV-1 DIS type RNA loop-loop kissing complexes (KC-1 and KC-2) used in the calculations. The thermodynamic parameters for KC-1 and KC-2 at 1 M NaCl are taken from the experimental measurements (49,50). (Shaded lines) Basepairs at the kissing interface.
Tightly bound ion model at all-atom level
The original tightly bound ion (TBI) model was based on coarse-grained structures (37–40). Here we develop a new TBI model that can treat all-atom nucleic acid structures.
For multivalent ions such as Mg2+ ions, the higher charge and the resultant strong Coulomb interaction between the ions can cause strong coupling (correlation) between the ions. To treat such an effect, according to the correlation strength, we classify the counterions into two types (37–41): the strongly and weakly correlated ions, denoted as the tightly bound and diffusive ions, respectively. Correspondingly, the space occupied by these ions is denoted as the tightly bound region and diffusive region, respectively (see Fig. 1 for examples of the tightly bound regions for the different structures). For the diffusive ions, we use the mean-field (PB) approach, while for the tightly bound ions, we must consider discrete modes of ion distributions to account for the ion-ion correlation effect. For a mixed monovalent and multivalent ion solution, because the correlation effect for the monovalent ions is negligible, monovalent ions can be treated as diffusive ions that form a background for multivalent ion binding.
Evaluation of ion correlation requires enumeration of the discrete distributions of the tightly bound ions and calculation of the free energy of the system for each given ion distribution mode. To enumerate the discrete ion distributions, for an N-nucleotide (nt) RNA, we divide the tightly bound region into N cells, each around a phosphate. There exist a large number of modes (i.e., binding modes) of ion distributions represented as the different ways to partition the tightly bound ions into the different cells. A tightly bound cell contains the phosphate and the groove space that surrounds the respective phosphate. The tightly bound ions move inside the respective cells. The total partition function Z is given by the summation over all the possible binding modes M,
| (1) |
where ZM is the partition function for a given binding mode M (37–40),
| (2) |
Here Z(id) is the partition function for the uniform ion solution (without the polyelectrolyte). The value cz is the concentration of z-valent counterions. The value Nb is the number of tightly bound ions and
is the volume integral for the Nb tightly bound ions. The value ΔGb is the mean Coulombic interaction energy between the tightly bound charges (including the phosphate groups and the tightly bound ions). The value ΔGd is the sum of the free energy for the electrostatic interactions between the diffusive ions, and among the diffusive ions and the tightly bound charges and the entropic free energy of the diffusive ions. The expression ΔGbpol is the (Born) self-polarization energy for the tightly bound charges (40). The calculations for ΔGb, ΔGd, and ΔGbpol are described in the Supporting Material and also in Tan and Chen (40).
Based on the partition function Eq. 1, the electrostatic free energy for RNA in ion solutions is computed by
and other electrostatic properties can also be computed (17).
Computation procedure and atomistic parameter sets
In both the TBI model and the PB calculations, for an atomistic nucleic acid structure (e.g., from the Protein Data Bank or the Nucleic Acid Database), the different types of atoms are treated as spheres with the radii determined by the respective van der Waals radii which has been tabulated in Table S1 in the Supporting Material. Each phosphate (P) atom is considered to carry one unit negative charge at its center, while other types of atoms (C, O, N, etc.) are treated as neutral spheres. The molecular van der Waals boundary defines the dielectric boundary. The computational process of the TBI model is briefly described as follows:
During the first step, for the given all-atom structure, we solve the PB equation to obtain the counterion distributions, from which we determine the tightly bound region for the multivalent counterions (37–40).
During the second step, we compute the pairwise potentials of mean force Φ1(i) and Φ2(i, j) and Born energy Φ0(i); see the Supporting Material. To account for the dielectric discontinuity, we apply the generalized Born model for the calculations of the pairwise charge-charge interactions and the self-polarization energy of discrete charges (57–59). The Born radii for discrete charges are estimated from the pairwise method proposed by Hawkins et al. (58), and the scaling parameters for atoms are taken the values in Liu et al. (59); see Table S2. The volume exclusion between ions and nucleic acid atoms are accounted for by a truncated Lennard-Jones potential,
for r < 1 or U = 0 for r > 1, where r is the distance between an ion and an atom in the unit of the sum of their radii. The parameter u0 is taken as 0.35 because the exclusion between the hydrated ions and the molecular atoms are soft due to the soft H-atom exclusion (60). The calculated potentials of mean force Φ1(i) and Φ2(i, j) and the Born energy Φ0(i) are tabulated and stored for the following calculations of the partition function.
During the third step, we enumerate the possible binding modes. For each mode, we calculate ΔGb, ΔGd, and ΔGbpol (Eq. 2). The sum over the binding modes gives the total partition function Z (Eq. 1) (17,37,38), from which we can calculate the electrostatic free energy.
The ions are hydrated and the ionic radii are equal to 3.5 Å for Na+, 4.5 Å for Mg2+, and 4 Å for K+ (37–40), respectively. We also use a smaller divalent ion (with radius ∼3.5 Å) for the purpose of examining the ion size effect in the PB calculations. Here, the dielectric constant ɛ of nucleic acid interior is set to be 20, and ɛ of solvent is set as the value of water (37–40). In the PB calculation, a thin charge-free layer of thickness of a cation radius is added to the molecular surface to account for the excluded volume layer of the cations (37–40). The detailed parameter sets are given in the Supporting Material.
Results and Discussion
In the following sections, we apply the atomistic TBI model to investigate the properties of Mg2+ binding to the different nucleic acid structures, including duplexes and tertiary folds in mixed Na+/Mg2+ solutions. We will then compute the ion-dependent folding stability of HIV DIS type RNA kissing complex for a broad range of Na+/Mg2+ concentrations. All the theoretical predictions will be compared with the available experimental data. Furthermore, to examine the significance of ion correlation effect, the TBI-predictions will also be compared against the PB results. Based on the systematic quantitative predictions, we will derive a set of analytical formulas to for the estimation of the Na+/Mg2+ binding to nucleic acids.
Mg2+/Na+ binding to RNA/DNA
In the TBI model, the number of the so-called bound ions includes both the tightly bound Mg2+ ions and the diffusively bound ions (in the excess of bulk concentrations). Thus, the number of the bound Mg2+ and Na+ ions per nucleotide, fMg2+ and fNa+, can be calculated according to the Boltzmann distribution of the diffusive ions (26)
| (3) |
where is the mean fraction of the tightly bound Mg2+ per nucleotide, which is given by the average over all the possible tightly bound modes M:
Here, Nb is the number of the tightly bound ions for mode M, ZM is the partition function for mode M, Z is the total partition function given by Eq. 1, and N is the number of the phosphates (nucleotides) of the RNA. We note that the fractions of bound ions can be measured for the different nucleic acids. Therefore, in our theory-experiment comparisons, we adopt the data directly from the experimental literature (12,28,61–68).
We applied the TBI model to predict the properties of ion binding to different structures from short and long helices to complex RNA tertiary folds. Because the competition behaviors between Mg2+ and Na+ binding to different nucleic acids share some general features, in the following, we will first discuss the general behavior in Na+/Mg2+-binding and then analyze the specific features for different RNA/DNA structures.
General features in Mg2+/Na+-binding
We calculated the amount of Mg2+- and Na+-bound ions for a 24-bp DNA duplex, a 40-bp RNA and the DNA duplexes, a BWYV pseudoknot fragment, a 58-nt rRNA fragment, and the yeast tRNAPhe, respectively. From Figs. 2 and 3, we can see the following general features for various RNAs/DNAs.
Figure 2.
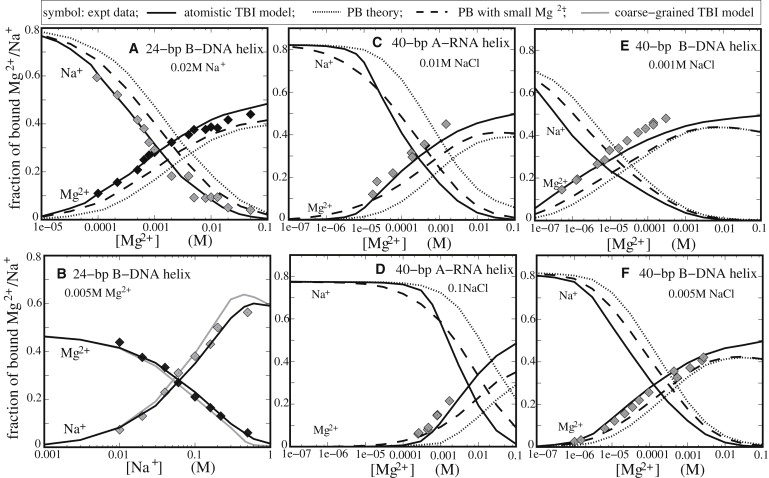
The Mg2+ and Na+ binding fractions per nucleotide for oligomeric DNA and RNA helices as functions of [Mg2+] or [Na+]. (Symbols) Experimental data. (Solid lines) Atomistic TBI predictions. (Dotted lines) PB predictions. (Dashed lines) PB predictions with a smaller Mg2+ radius (≃3.5 Å). (Shaded lines) TBI predictions on a coarse-grained DNA duplex. (A) A 24-bp B-DNA duplex in 20 mM Na+ (28,63). (B) A 24-bp B-DNA duplex in 5 mM Mg2+ (28,63). (C and D) A 40-bp RNA duplex in 10 mM [Na+] and 100 mM [Na+], respectively. The experimental data are for poly(A.U) (64). (E and F) A 40-bp DNA duplex in 1 mM [Na+] and 5 mM [Na+], respectively. The experimental data are for calf thymus DNA (65).
Figure 3.
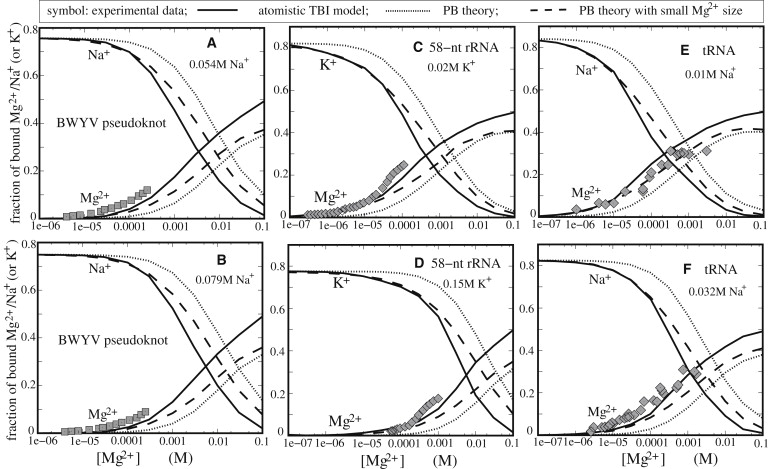
The Mg2+ and Na+ (or K+) binding fractions per nucleotide for various RNA tertiary structures. (Symbols) Experimental data. (Solid lines) Atomic TBI predictions. (Dotted lines) PB predictions. (Dashed lines) PB predictions with a smaller Mg2+ radius (≃3.5 Å). (A and B) BWYV pseudoknot fragment at different Na+ concentrations: 54 mM Na+ and 79 mM Na+, respectively. The experimental data are adopted from Soto et al. (12). (C and D) A 58-nt rRNA fragment at different K+ concentrations: 20 mM K+ and 150 mM K+, respectively. The experimental data are from Grilley et al. (66). (E and F) Yeast tRNAPhe at different Na+ concentrations: 10 mM Na+ and 32 mM Na+, respectively. The experimental data was taken from Rialdi et al. (67) and Römer and Hach (68) for panels A for panels B, respectively.
Mg2+-binding and Na+-binding are competitively coupled, i.e., the increase of [Mg2+] ([Na+]) enhances Mg2+(Na+)-binding and suppresses Na+(Mg2+)-binding. Mg2+ has higher valency than Na+. Therefore, the same level of charge neutralization for RNA backbone requires a smaller number of bound Mg2+ ions and hence a smaller cost of the translational entropy upon ion binding, causing a stronger preference of Mg2+ binding than Na+.
For both Mg2+ and Na+ binding, the TBI predictions agree well with the experimental data for a variety of nucleic acid structures over a wide range of [Mg2+] and [Na+]. The PB theory, which ignores ion correlation, underestimates the amount of the bound Mg2+, especially at high [Mg2+]. This is because the Coulomb correlation (coupling) between the bound ions causes the ions to self-organize and reach low-energy states. Such correlation-induced low-energy states cannot be accounted for by a mean-field theory such as PB.
For very low [Mg2+], however, the TBI model gives the same results as the PB. This is because the number of bound Mg2+ is small and the average distance between the bound ions is large at low [Mg2+]; therefore, the interion correlation is weak.
The above findings are also in accordance with the results from the Monte Carlo study (61).
The high level of ion binding for Mg2+ at lower (millimolar) ion concentration than Na+ goes beyond the effect of the ionic strength. For example, for a 24-bp DNA duplex, 0.4 mM and 5 mM [Mg2+] are equivalent to 20 mM and 160 mM [Na+] (shown in Fig. 2, A and B), respectively, i.e.,
The unusually efficient role of the Mg2+ may come from the higher ionic charge of Mg2+, which results in the stronger ability of the Mg2+ ions in RNA backbone neutralization and the stronger Coulomb correlation between the bound ions. The Coulombic correlation between the bound ions can significantly enhance the stability of the ion-RNA system (8).
In general, a smaller ionic radius can enhance the ion-RNA interaction and ion binding. Indeed, the use of a small Mg2+ size in the PB calculation (by decreasing the width of ion exclusion layer (30); see Methods) can improve the PB-based predictions on Mg2+-binding. However, such improvement is not sufficient to offset the difference between the PB prediction and the experimental data. Therefore, the theory-experiment difference for the PB may not just come from the effect of the ionic radius.
In addition to the above general features, the TBI model also predicts the differences in the ion binding properties for a great variety of different nucleic acid structures.
24-bp DNA duplex
Experiments based on the direct counting of the number of the bound ions (62) for a 24-bp DNA duplex (28,63) suggested that PB underestimates the Mg2+-binding and correspondingly overestimates the Na+-binding (28). The predictions from our TBI model, which explicitly accounts for the ion correlation and complete ensemble of ion distribution, show significant improvement for the predictions for both Na+ and Mg2+ binding over a wide ranges of ion concentrations (see Fig. 2, A and B). We note that Chu et al. (63) developed a size-modified PB theory to quantify the ion binding using adjustable effective ion sizes. Their method works well for mixed monovalent ion solution while the results are not completely satisfactory for Mg2+ solution (63). In contrast, the excellent agreement between the experimental data and the TBI predictions and the imperfect agreement between experiment and the PB/size-modified PB suggest that the interion Coulombic correlations and ion-binding ensembles may be important for the binding of multivalent ions such as Mg2+.
40-bp RNA and DNA duplexes
To examine the ability of the atomistic TBI model to treat long helices, we apply the model to treat the Mg2+- and Na+-binding to a 40-bp RNA duplex and a 40-bp DNA duplex. As shown by the theory-experiment comparisons in Fig. 2, C–F (64,65), TBI gives much more reliable predictions than PB, which tends to underestimate the binding of Mg2+. Similar to our conclusions for the ion-binding to the 24-bp DNA, we find that Mg2+ is much more efficient than Na+ in neutralizing the 40-bp duplexes. For example, for the 40-bp RNA duplex, 0.09 mM and 2.5 mM Mg2+ can approximately cause the same neutralization as 10 mM and 100 mM Na+, respectively. The overall agreements between the TBI predictions and the experimental data in Fig. 2, C–F, show that the helix-end effect for Mg2+-binding to the 40-bp duplexes is minor, in accordance with the previous analysis on Mg2+-binding to a DNA helix (40).
BWYV pseudoknot fragment
Fig. 3, A and B, shows the Mg2+ and Na+ binding fractions for a BWYV pseudoknot fragment predicted by the TBI model and the PB theory, along with the experimental data (12). The concentration dependence of Mg2+ and Na+ binding shows a trend similar to the one in Fig. 2. Theory-experiment comparison indicates that the TBI model gives reliable predictions for this 29-nt complex RNA tertiary structure (12). As shown in Fig. 3, A and B, 1.6 mM and 2.5 mM Mg2+ have about the same effect on RNA backbone neutralization as the 54 mM and 79 mM Na+, respectively. For the same RNA, a previous PB calculation yields ion binding slightly lower than the experimental results (12). We note that the PB calculation was based on an ion-exclusion layer of 2 Å, effectively corresponding to a small radius of 2 Å for both Mg2+ and Na+ ions. Small ion size results in enhancement of ion binding predicted by PB, and partially offsets the deficiency in ion binding caused by the neglect of ion correlation for the Mg2+ ions.
58-nt ribosomal RNA fragment
To investigate the ion-binding in a mixed Mg2+/K+ solution, we model K+ as a monovalent ion with a hydrated radius 4 Å, slightly larger than 3.5 Å for Na+. Fig. 3, C and D, shows that TBI gives reliable predictions for the Mg2+ and K+ binding to a 58-nt rRNA fragment (66). In contrast, the PB theory underestimates the Mg2+ binding and overestimates the K+ binding. Moreover, Mg2+ shows much higher efficiency in ion-binding and charge neutralization than K+. Specifically, 0.17 mM and 2.8 mM Mg2+ have the same effect in backbone charge neutralization as 20 mM and 150 mM K+, respectively. In contrast, we note that a previous PB calculation for the system involves much smaller radii for the (hydrated) ions (through the 2 Å ion-exclusion layer) for both K+ and Mg2+ (66).
Yeast tRNAPhe
To extend the TBI analysis to larger and more complex structures, we investigated the ion-binding properties for yeast tRNAPhe. We quantify the Na+-Mg2+ competition in the binding to yeast tRNAPhe (54). Fig. 3, E and F, shows that, as compared with the experimental data (67,68), the TBI model gives better predictions than the PB theory, which underestimates the Mg2+ binding. Decreasing the Mg2+ size in the PB calculations would improve the prediction. However, the improvement is not sufficient to make up the deficiency in the PB prediction presumably caused by the neglect of the correlation effect. For neutralizing the tRNAPhe, 0.07 mM and 0.4 mM Mg2+ have the same effect as 10 mM and 32 mM Na+, respectively.
Atomistic TBI versus coarse-grained TBI
By considering correlated ion distributions, the previous coarse-grained TBI model is able to give reliable predictions for the ion effects for simple nucleic acid structures such as DNA and RNA helices (25,26), DNA and RNA hairpin stability (27), and DNA helix assembly/bending (38–40). However, the coarse-grained model, originally derived for helices, cannot treat single-stranded regions of the structure and other, more-complex structures such as the tertiary packing of loops and helices. Therefore, a TBI model that can handle atomic structures of RNAs becomes indispensable for the study of ion effect in tertiary structural folding. As shown above by the theory-experiment comparisons, our atomistic TBI model can indeed give reliable predictions for Mg2+/Na+-binding properties for complex RNA tertiary folds such as BWYV pseudoknot, rRNA fragment, and yeast tRNA.
It is important to note that the previous coarse-grained model cannot represent the above tertiary folds in a reliable way, therefore, for the tertiary folds, a direct comparison between the present atomic TBI model and the previous coarse-grained TBI model is not possible. However, such a direct comparison is possible for simple nucleic acid helices (see Fig. 2 B).
Fig. 2 B shows the Mg2+/Na+-binding as a function of [Na+] for a 24-bp DNA helix immersed in a solution with 5 mM Mg2+. Detailed comparisons between the present atomic TBI and the previous coarse-grained TBI models and experimental data show the following results:
-
1.
Both models give overall reliable predictions.
-
2.
In the Mg2+-dominating region (low [Na+]), the two models give nearly identical predictions.
-
3.
In the Na+-dominating region (high [Na+]), the coarse-grained model slightly overestimates (underestimates) the Na+-binding (Mg2+-binding).
The very slight discrepancy between the two models might come from the molecular shape and volume which directly influence the accumulations of Na+ ion cloud near the DNA surface. In addition, for the 24-bp duplex, the atomic TBI model costs about twice the computational time as the coarse-grained model because the all-atomic structure considers more atoms (spheres) than the coarse-grained one.
In the TBI calculations, we also examined the sensitivity of the TBI predictions to the ion sizes. We have previously shown that smaller ions are more efficient in ionic neutralization and stabilization of the folded (or compact) structures (25,26,40). Here, for a 24-bp DNA helix in 20 mM Na+ and 10 mM Mg2+, the atomic TBI predicts that the decrease of Mg2+ size from 4.5 Å to 4 Å would cause the decrease in electrostatic free energy by ∼10% and the increase in tightly bound Mg2+ number by ∼4%. The decrease in Na+ size also enhances the Na+ binding, but the effect is not as strong as for Mg2+.
Na+-binding versus Mg2+-binding: analytical formulas
Charge neutralization
The TBI model allows us to systematically compute the mean number of bound ions per nucleotide and for the different RNA structures and different ion concentrations in a mixed Na+/Mg2+ solution. Based on the TBI calculations, equating and (i.e., equal charge neutralization) leads to the following empirical conversion formula between the ion concentrations,
| (4) |
where [Na+]Mg and [Mg2+] are the concentrations in millimolar (mM). Physically, for a nucleic acid in a mixed Na+/Mg2+ solution with a specific [Mg2+], such [Mg2+] can achieve the same ionic neutralization as the [Na+]Mg defined by Eq. 4. Our data for a variety of RNA structures discussed in the preceding sections suggest that, as an approximation, the chain length (N-nt) and the radius of gyration (Rg) of RNA backbone may be the two (low-resolution) determinants for parameters A and B,
| (5) |
where Rg0 is the radius of gyration for the corresponding A-form N-nt RNA helix. Equations 4 and 5 partially reflect the effects of the number N of the phosphate charges and the compactness Rg of RNA backbone. It is important to note that the Rg2-dependence reflects the effect of RNA surface charge density on ion-binding.
We calculated R0g for RNA duplexes with N nucleotides and found Rg0 = 0.406N + 130/(N + 11), where N ≳ 10-nt. As shown in Fig. S5 in the Supporting Material, Eq. 4 fits the results from the exact calculations quite well for both RNA duplexes and complex RNA tertiary structures.
Ion-binding profile in a mixed Mg2+/Na+ solution
Based on Eq. 4, we can derive an analytical formula for the [Na+]/[Mg2+]-dependent ion-binding profiles,
| (6) |
where [Na+]Mg is given by Eq. 4 and and are the mean numbers of bound ions per nucleotide for pure Mg2+ (i.e., [Na+] = 0) and pure Na+ (i.e., [Mg2+] = 0, [Na+]Mg = 0) solutions, respectively. Strictly, and depend on the charge density of nucleic acids and ion concentration. From the calculations over the extensive nucleic acid molecules, and only changes slightly for various molecules, as shown in Figs. 2 and 3. Here, for simplicity, we approximately use the constants of and for the extensive RNA and DNA molecules.
Extensive tests based on the experimental data for various RNA (and DNA) structures show that the analytical formulas above (Eqs. 4 and 6) are quite accurate over a wide range of ionic conditions (see Fig. S6 in the Supporting Material).
Ion-dependent stability of RNA loop-loop kissing complex
We model the folding stability of RNA kissing complex based on a two-state model: the kissing complex and the unkissed state (i.e., two dissociated hairpins) (see Fig. 1 G). The difference in the charge distributions of the kissing and the unkissed states leads to the ion-dependence of the folding stability of the kissing complex.
The stability of the kissing complex can be quantified by the free energy difference ΔGoT between the two states:
| (7) |
As an approximation, ΔGoT is decomposed into two parts: the electrostatic contribution and the nonelectrostatic (chemical) contribution (25,26),
| (8) |
| (9) |
where the electrostatic free energies ΔGelT (Na+/Mg2+) and ΔGelT (1 M Na+) are calculated from the polyelectrolyte theory (the TBI model) and ΔGoT (1 M Na+) is obtained from experimental measurement. From ΔGT, the melting temperature Tm can be computed for a given strand concentration CS (19,26).
We focus on two specific kissing complexes (KC-1 and KC-2 in Fig. 1). With the TBI model, we calculate the free energy change ΔG37 at 37°C and the melting temperature Tm as functions of Na+ and Mg2+ concentrations.
ΔG37 for KC-1 and KC-2 in Na+ solution
Fig. 4 A shows the folding free energy ΔG37 as a function of [Na+] for KC-1 and KC-2. The increase of [Na+] causes the decrease of ΔG37, indicating that Na+ favors the formation of the kissing complex over the unkissed state. This is because Na+-binding causes reduced Coulomb repulsion between the backbone charges and a lower free energy, and such Na+-induced stabilizing force is more significant for a more densely charged state—the kissing complex.
Figure 4.
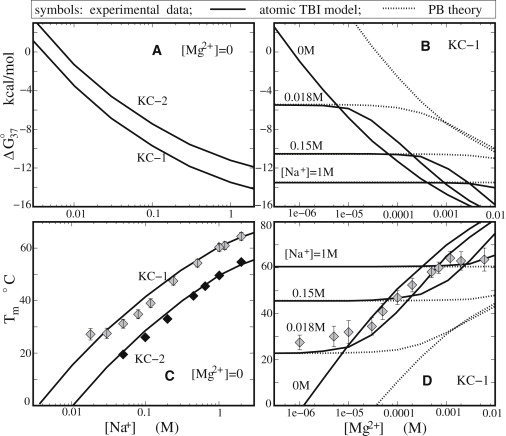
The free energies ΔG37 (A and B) and melting temperature Tm (C and D) as functions of the Na+ concentration (A and C) and the Mg2+ concentration (B and D). (C) The experimental data for KC-1 and KC-2 are from Weixlbaumer et al. (49) and Lorenz et al. (50), respectively. (D) The experimental data are from Weixlbaumer et al. (49). (Solid lines) TBI predictions. (Dotted lines) PB predictions.
Tm for KC-1 and KC-2 in Na+ solution
In accordance with the Na+-dependence of ΔG37, the melting temperatures Tm increases with increasing [Na+], as shown in Fig. 4 C, indicating the higher stability at high [Na+]. Theory-experiment comparison shows that the TBI predictions agree well with the experimental data for both complexes (49,50).
ΔG37 for KC-1 in mixed Na+/Mg2+ solution
In a pure Mg2+ solution ([Na+] = 0), the increase of [Mg2+] causes the decrease in ΔG37 and higher stability of the complex (see Fig. 4 B), which is similar to the trend for the Na+-dependence. However, Mg2+ is much more efficient than Na+ to achieve the same folding free energy. For example, 0.4 mM Mg2+ and 100 mM Na+ cause approximately the same free energy ΔG37 (∼−10 kcal/mol) for KC-1. The efficient role of Mg2+ is due to the higher ionic charge of Mg2+ and the possible ion correlation effect, as discussed above. For a pure Mg2+ solution, the PB theory predicts much higher ΔG37 than the TBI calculations, which is in accordance with the predictions on the Mg2+-binding for other structures discussed in the preceding sections.
In a mixed Na+/Mg2+ solution, there are apparently three regions in the ΔG37 curves as a result of the interplay between the Na+ and Mg2+ binding (26,27): Mg2+ dominant, Na+-Mg2+ competitive, and Na+ dominant regions. When [Mg2+] is high (relative to [Na+]), Mg2+ dominates the system. In this case, ΔG37 is close to the value for the corresponding pure Mg2+ solution and PB underestimates the Mg2+-induced stabilization. With the addition of Na+, Na+ begins to compete against Mg2+ in ion-binding and pushes Mg2+ away from the RNA surface, resulting in a weakened effect of Mg2+. Because the (divalent) Mg2+ is much more efficient than the (monovalent) Na+ in stabilizing the folded RNA, the weakened Mg2+-binding (and the enhanced Na+-binding) could lead to (slight) destabilization of the kissing complex. When [Na+] becomes high enough, Na+ would dominate the ion effect. In this case, ΔG37 becomes close to the value of the pure Na+ and the TBI model and the PB theory give the same results.
Tm for KC-1 in a mixed Na+/Mg2+ solution
The salt dependence of the melting temperature Tm in mixed Na+/Mg2+ is in accordance to ΔG37, as shown in Fig. 4 D. For the Na+- or Mg2+-dominated regime, Tm is close to the value of the corresponding pure Na+ or Mg2+. For the Na+-Mg2+ competing regime, the addition of Na+ causes slight increase in the Tm as compared with the pure Mg2+. As discussed above and previously (26,27), this is because the addition of Na+ counteracts the efficient role of Mg2+.
Fig. 4 D also shows that the TBI predictions are in good agreement with the available experimental data (49), while the PB theory underestimates Tm for the concentration range where Mg2+ ions play a role. The significant deviation between the PB prediction and the experimental data may be attributed to the neglect of ion-ion correlations and ion-binding ensemble, as discussed above.
Kissing complex versus duplex
The salt (Na+ and Mg2+)- dependence of Tm for the kissing complexes is stronger than that for the corresponding duplex with the same kissing and terminal mismatch sequences (26,49) (see Fig. 1). This is because the kissing complex involves much higher charge buildup, and hence, a much stronger ion dependence than the duplex.
At high salt (1M NaCl), the kissing complex has higher stability than the corresponding duplex. The higher stability may come from two contributions. First, because the initial unkissed state (dissociated hairpins) has a much smaller entropy than free single-strand chains (27), the formation of the kissing complex from the unkissed hairpins involves a smaller conformational entropy loss than the formation of the duplex from single-stranded chains. Second, the two hairpin stems are coaxially stacked in the kissing complex (49,50,55), and can further enhance the stability of the complex.
Kissing complexes KC-1 versus KC-2
Tm values of KC-1 and KC-2 have the similar [Na+]-dependence and very different [Mg2+]-dependence (49,50). The experimental Mg2+-dependence of Tm for KC-2 is weaker than that for KC-1 (49,50) (not shown here). This may be attributed to the sequence-dependent specific binding of Mg2+, while Na+ ions have negligible contribution to the specific binding. Experiments suggest that the uronic acid (U)-rich sequence of KC-2 would help the Mg2+ specific binding through the attraction between the negatively charged O atom (in U) and the positively charged Mg2+ (55), and the effect may be even more pronounced for the dangling U-sequence near the kissing interface (55); see KC-2 in Fig. 1. As a result, [Mg2+]-dependence of Tm for KC-2 is weaker due to the (weakly [Mg2+]-dependent) neutralization by the specific binding of Mg2+ (50).
Conclusions
This tightly bound ion model (TBI) enables all-atom treatment for the ion correlation and ion fluctuation effects which can be important for multivalent ions such as Mg2+ ions in RNA folding. The availability of the high-resolution all-atom RNA/DNA structures motivates the development of an all-atom model for the TBI theory. Based on the atomistic TBI model, we systematically quantify the Na+/Mg2+-binding to different nucleic acid structures as well as the salt-dependent RNA kissing complex stability. Our main conclusions are the following.
-
1.
Mg2+ is much more efficient in ionic neutralization than Na+, and such high efficiency goes beyond the mean-field electrostatic effect such as the ionic strength. The high efficiency of Mg2+ is stronger for compact tertiary folds than for extended states.
-
2.The Mg2+ binding and Na+ binding are competitively coupled in a mixed Na+/Mg2+ solution. There exist approximately three distinctive regimes for the Na+-Mg2+ competitive binding:
-
I.For [Mg2+] > > [Na+], Mg2+ binding is dominant and Na+ binding is suppressed. In this case, the PB underestimates the Mg2+-binding possibly due to the neglect of the ion correlation and fluctuation effects. In contrast, the TBI model gives much improved predictions.
-
II.As [Na+] increases, Na+-Mg2+ competition becomes stronger. Because of the high efficiency of Mg2+-binding, Mg2+-binding may still outcompete Na+-binding; thus, PB may still underestimate the Mg2+-binding.
-
III.[Na+] >> [Mg2+], Na+-binding becomes dominant. In this case, PB and TBI give the same predictions.
-
I.
-
3.
There exists a reliable analytical formula for the Mg2+/Na+-binding as a function of [Mg2+] and [Na+], the chain length, and the structure compactness of nucleic acids.
-
4.
The kissing complex formation is electrostatically highly unfavorable because of the strong Coulombic repulsion. Na+ and Mg2+ can effectively reduce such repulsion and make the complex formation less unfavorable. The Na+/Mg2+-dependence of kissing complex is very strong. The TBI model gives reliable predictions for the ion-dependent folding stability of the kissing loop complexes.
The excellent agreement between the TBI predictions and the experimental data for ion-binding properties and folding free energies indicates that the model is quite reliable. However, the model has several limitations.
In the TBI model and the PB calculations, we assume that each phosphate has one negative unit charge and treat all other types of atoms as neutral ones without considering the detailed partial charges.
The nonideality of the ionic solution is neglected and ions are assumed to be completely dissociated from each other. The nonideality effect of the solution, which can become important at high salt concentrations, may contribute to the overestimation of ion-binding (at high salt concentration) (47).
We ignore the effect of specific ion binding. The contribution of the specific binding to the global ion-dependence of the folding stability might not be significant. However, specific ion-binding may be important for the sequence-sensitive features of the ion effect for many tertiary folds such as the KC-2 kissing complex.
In the TBI model, the interactions in the tightly bound region are calculated as the sum of the pairwise potentials of mean forces where multibody effects are neglected. The experimental comparisons for the binding curves suggest that the TBI model generally tends to overestimate ion binding at high (divalent) ion concentrations.
All the above approximations (ideality of the ionic solution, pairwise potential of mean force) can contribute to such theory-experiment discrepancy. Further development of the model should include the improvement for the above approximations. A possible way for further development of the model may involve a combination of the present TBI theory and computer simulations. For ion-binding properties to RNAs, computer simulations (48,69) can be highly valuable in providing detailed ion distributions, sequence effects, and the ion-related dynamics. Detailed simulations can also provide effective tests and validations for many theoretical predictions such as the empirical formula that we derived in this study.
Acknowledgments
Most numerical calculations involved in this research were performed on the high-performance computer cluster at the University of Missouri Bioinformatics Consortium.
This research was supported by the National Science Foundation of the United States through grant No. MCB0920067 and by the National Science Foundation of China through grant Nos. 10844007 and 30670487, and by the Program for New Century Excellent Talents in University of China and the Chutian Scholar Program of Hubei Province.
Supporting Material
References
- 1.Brion P., Westhof E. Hierarchy and dynamics of RNA folding. Annu. Rev. Biophys. Biomol. Struct. 1997;26:113–137. doi: 10.1146/annurev.biophys.26.1.113. [DOI] [PubMed] [Google Scholar]
- 2.Tinoco I., Jr., Bustamante C. How RNA folds. J. Mol. Biol. 1999;293:271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 3.Sosnick T.R., Pan T. RNA folding: models and perspectives. Curr. Opin. Struct. Biol. 2003;13:309–316. doi: 10.1016/s0959-440x(03)00066-6. [DOI] [PubMed] [Google Scholar]
- 4.Woodson S.A. Metal ions and RNA folding: a highly charged topic with a dynamic future. Curr. Opin. Chem. Biol. 2005;9:104–109. doi: 10.1016/j.cbpa.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Thirumalai D., Hyeon C. RNA and protein folding: common themes and variations. Biochemistry. 2005;44:4957–4970. doi: 10.1021/bi047314+. [DOI] [PubMed] [Google Scholar]
- 6.Chu V.B., Herschlag D. Unwinding RNA's secrets: advances in the biology, physics, and modeling of complex RNAs. Curr. Opin. Struct. Biol. 2008;18:305–314. doi: 10.1016/j.sbi.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Draper D.E. RNA folding: thermodynamic and molecular descriptions of the roles of ions. Biophys. J. 2008;95:5489–5495. doi: 10.1529/biophysj.108.131813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S.J. RNA folding: conformational statistics, folding kinetics, and ion electrostatics. Annu. Rev. Biophys. 2008;37:197–214. doi: 10.1146/annurev.biophys.37.032807.125957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rook M.S., Treiber D.K., Williamson J.R. An optimal Mg2+ concentration for kinetic folding of the Tetrahymena ribozyme. Proc. Natl. Acad. Sci. USA. 1999;96:12471–12476. doi: 10.1073/pnas.96.22.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takamoto K., He Q., Brenowitz M. Monovalent cations mediate formation of native tertiary structure of the Tetrahymena thermophila ribozyme. Nat. Struct. Biol. 2002;9:928–933. doi: 10.1038/nsb871. [DOI] [PubMed] [Google Scholar]
- 11.Koculi E., Hyeon C., Woodson S.A. Charge density of divalent metal cations determines RNA stability. J. Am. Chem. Soc. 2007;129:2676–2682. doi: 10.1021/ja068027r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soto A.M., Misra V., Draper D.E. Tertiary structure of an RNA pseudoknot is stabilized by “diffuse” Mg2+ ions. Biochemistry. 2007;46:2973–2983. doi: 10.1021/bi0616753. [DOI] [PubMed] [Google Scholar]
- 13.Stellwagen E., Dong Q., Stellwagen N.C. Quantitative analysis of monovalent counterion binding to random-sequence, double-stranded DNA using the replacement ion method. Biochemistry. 2007;46:2050–2058. doi: 10.1021/bi062132w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu X., Andresen K., Pollack L. Abrupt transition from a free, repulsive to a condensed, attractive DNA phase, induced by multivalent polyamine cations. Phys. Rev. Lett. 2008;101:228101. doi: 10.1103/PhysRevLett.101.228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlatterer J.C., Kwok L.W., Pollack L. Hinge stiffness is a barrier to RNA folding. J. Mol. Biol. 2008;379:859–870. doi: 10.1016/j.jmb.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipfert J., Sim A.Y., Doniach S. Dissecting electrostatic screening, specific ion binding, and ligand binding in an energetic model for glycine riboswitch folding. RNA. 2010;16:708–719. doi: 10.1261/rna.1985110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan Z.J., Chen S.J. Predicting electrostatic forces in RNA folding. Methods Enzymol. 2009;469:465–487. doi: 10.1016/S0076-6879(09)69022-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Serra M.J., Turner D.H. Predicting thermodynamic properties of RNA. Methods Enzymol. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 19.SantaLucia J., Jr. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathews D.H., Sabina J., Turner D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 21.Chen S.J., Dill K.A. RNA folding energy landscapes. Proc. Natl. Acad. Sci. USA. 2000;97:646–651. doi: 10.1073/pnas.97.2.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang W.B., Chen S.J. RNA hairpin-folding kinetics. Proc. Natl. Acad. Sci. USA. 2002;99:1931–1936. doi: 10.1073/pnas.032443099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuker M. MFold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Owczarzy R., You Y., Walder J.A. Effects of sodium ions on DNA duplex oligomers: improved predictions of melting temperatures. Biochemistry. 2004;43:3537–3554. doi: 10.1021/bi034621r. [DOI] [PubMed] [Google Scholar]
- 25.Tan Z.J., Chen S.J. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys. J. 2006;90:1175–1190. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan Z.J., Chen S.J. RNA helix stability in mixed Na+/Mg2+ solution. Biophys. J. 2007;92:3615–3632. doi: 10.1529/biophysj.106.100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan Z.J., Chen S.J. Salt dependence of nucleic acid hairpin stability. Biophys. J. 2008;95:738–752. doi: 10.1529/biophysj.108.131524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai Y., Greenfeld M., Herschlag D. Quantitative and comprehensive decomposition of the ion atmosphere around nucleic acids. J. Am. Chem. Soc. 2007;129:14981–14988. doi: 10.1021/ja075020g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manning G.S. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. 1978;11:179–246. doi: 10.1017/s0033583500002031. [DOI] [PubMed] [Google Scholar]
- 30.Gilson M.K., Sharp K.A., Honig B. Calculating the electrostatic potential of molecules in solution: method and error assessment. J. Comput. Chem. 1987;9:327–335. [Google Scholar]
- 31.You T.J., Harvey S.C. Finite element approach to the electrostatics of macromolecules with arbitrary geometries. J. Comput. Chem. 1993;14:484–501. [Google Scholar]
- 32.Baker N.A., Sept D., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boschitsch A.H., Fenley M.O. A new outer boundary formulation and energy corrections for the nonlinear Poisson-Boltzmann equation. J. Comput. Chem. 2007;28:909–921. doi: 10.1002/jcc.20565. [DOI] [PubMed] [Google Scholar]
- 34.Zhou Y.C., Feig M., Wei G.W. Highly accurate biomolecular electrostatics in continuum dielectric environments. J. Comput. Chem. 2008;29:87–97. doi: 10.1002/jcc.20769. [DOI] [PubMed] [Google Scholar]
- 35.Grochowski P., Trylska J. Continuum molecular electrostatics, salt effects, and counterion binding—a review of the Poisson-Boltzmann theory and its modifications. Biopolymers. 2008;89:93–113. doi: 10.1002/bip.20877. [DOI] [PubMed] [Google Scholar]
- 36.Wang K., Yu Y.X., Gao G.H. Density functional study on the structural and thermodynamic properties of aqueous DNA-electrolyte solution in the framework of cell model. J. Chem. Phys. 2008;128:185101. doi: 10.1063/1.2918342. [DOI] [PubMed] [Google Scholar]
- 37.Tan Z.J., Chen S.J. Electrostatic correlations and fluctuations for ion binding to a finite length polyelectrolyte. J. Chem. Phys. 2005;122:44903. doi: 10.1063/1.1842059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan Z.J., Chen S.J. Ion-mediated nucleic acid helix-helix interactions. Biophys. J. 2006;91:518–536. doi: 10.1529/biophysj.106.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan Z.J., Chen S.J. Electrostatic free energy landscapes for nucleic acid helix assembly. Nucleic Acids Res. 2006;34:6629–6639. doi: 10.1093/nar/gkl810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan Z.J., Chen S.J. Electrostatic free energy landscapes for DNA helix bending. Biophys. J. 2008;94:3137–3149. doi: 10.1529/biophysj.107.122366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen G., Tan Z.J., Chen S.J. Salt-dependent folding energy landscape of RNA three-way junction. Biophys. J. 2010;98:111–120. doi: 10.1016/j.bpj.2009.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montoro J.C.G., Abascal J.L.F. Ionic distribution around simple DNA models. I. Cylindrically averaged properties. J. Chem. Phys. 1995;103:8273–8284. [Google Scholar]
- 43.Cruz J.A., Westhof E. The dynamic landscapes of RNA architecture. Cell. 2009;136:604–609. doi: 10.1016/j.cell.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Li P.T., Bustamante C., Tinoco I., Jr. Unusual mechanical stability of a minimal RNA kissing complex. Proc. Natl. Acad. Sci. USA. 2006;103:15847–15852. doi: 10.1073/pnas.0607202103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li P.T., Tinoco I., Jr. Mechanical unfolding of two DIS RNA kissing complexes from HIV-1. J. Mol. Biol. 2009;386:1343–1356. doi: 10.1016/j.jmb.2009.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lambert D., Leipply D., Draper D.E. The influence of monovalent cation size on the stability of RNA tertiary structures. J. Mol. Biol. 2009;390:791–804. doi: 10.1016/j.jmb.2009.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen A.A., Pappu R.V. Quantitative characterization of ion pairing and cluster formation in strong 1:1 electrolytes. J. Phys. Chem. B. 2007;111:6469–6478. doi: 10.1021/jp0708547. [DOI] [PubMed] [Google Scholar]
- 48.Chen A.A., Draper D.E., Pappu R.V. Molecular simulation studies of monovalent counterion-mediated interactions in a model RNA kissing loop. J. Mol. Biol. 2009;390:805–819. doi: 10.1016/j.jmb.2009.05.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weixlbaumer A., Werner A., Schroeder R. Determination of thermodynamic parameters for HIV DIS type loop-loop kissing complexes. Nucleic Acids Res. 2004;32:5126–5133. doi: 10.1093/nar/gkh841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lorenz C., Piganeau N., Schroeder R. Stabilities of HIV-1 DIS type RNA loop-loop interactions in vitro and in vivo. Nucleic Acids Res. 2006;34:334–342. doi: 10.1093/nar/gkj435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu X.J., Olson W.K. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su L., Chen L., Rich A. Minor groove RNA triplex in the crystal structure of a ribosomal frameshifting viral pseudoknot. Nat. Struct. Biol. 1999;6:285–292. doi: 10.1038/6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conn G.L., Gittis A.G., Draper D.E. A compact RNA tertiary structure contains a buried backbone-K+ complex. J. Mol. Biol. 2002;318:963–973. doi: 10.1016/S0022-2836(02)00147-X. [DOI] [PubMed] [Google Scholar]
- 54.Westhof E., Sundaralingam M. Restrained refinement of the monoclinic form of yeast phenylalanine transfer RNA. Temperature factors and dynamics, coordinated waters, and base-pair propeller twist angles. Biochemistry. 1986;25:4868–4878. doi: 10.1021/bi00365a022. [DOI] [PubMed] [Google Scholar]
- 55.Ennifar E., Walter P., Dumas P. Crystal structures of coaxially stacked kissing complexes of the HIV-1 RNA dimerization initiation site. Nat. Struct. Biol. 2001;8:1064–1068. doi: 10.1038/nsb727. [DOI] [PubMed] [Google Scholar]
- 56.Mujeeb A., Clever J.L., Parslow T.G. Structure of the dimer initiation complex of HIV-1 genomic RNA. Nat. Struct. Biol. 1998;5:432–436. doi: 10.1038/nsb0698-432. [DOI] [PubMed] [Google Scholar]
- 57.Still W.C., Tempczyk A., Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990;112:6127–6129. [Google Scholar]
- 58.Hawkins G.D., Cramer C.J., Truhlar D.G. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995;246:122–129. [Google Scholar]
- 59.Liu H.Y., Kuntz I.D., Zou X. Pairwise GB/SA scoring function for structure-based drug design. J. Phys. Chem. B. 2004;108:5453–5462. [Google Scholar]
- 60.Cheatham T.E., 3rd, Young M.A. Molecular dynamics simulation of nucleic acids: successes, limitations, and promise. Biopolymers. 2000;56:232–256. doi: 10.1002/1097-0282(2000)56:4<232::AID-BIP10037>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 61.Ni H.H., Anderson C.F., Record M.T. Quantifying the thermodynamic consequences of cation (M2+, M+) accumulation and anion (X−) exclusion in mixed salt solutions of polyanionic DNA using Monte Carlo and Poisson-Boltzmann calculations of ion-polyion preferential interaction coefficients. J. Phys. Chem. B. 1999;103:3489–3504. [Google Scholar]
- 62.Das R., Travers K.J., Herschlag D. Determining the Mg2+ stoichiometry for folding an RNA metal ion core. J. Am. Chem. Soc. 2005;127:8272–8273. doi: 10.1021/ja051422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chu V.B., Bai Y., Doniach S. Evaluation of ion binding to DNA duplexes using a size-modified Poisson-Boltzmann theory. Biophys. J. 2007;93:3202–3209. doi: 10.1529/biophysj.106.099168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krakauer H. The binding of Mg++ ions to polyadenylate, polyuridylate, and their complexes. Biopolymers. 1971;10:2459–2490. doi: 10.1002/bip.360101209. [DOI] [PubMed] [Google Scholar]
- 65.Clement R.M., Sturm J., Daune M.P. Interaction of metallic cations with DNA. VI. Specific binding of Mg2+ and Mn2+ Biopolymers. 1973;12:405–421. [Google Scholar]
- 66.Grilley D., Misra V., Draper D.E. Importance of partially unfolded conformations for Mg2+-induced folding of RNA tertiary structure: structural models and free energies of Mg2+ interactions. Biochemistry. 2007;46:10266–10278. doi: 10.1021/bi062284r. [DOI] [PubMed] [Google Scholar]
- 67.Rialdi G., Levy J., Biltonen R. Thermodynamic studies of transfer ribonucleic acids. I. Magnesium binding to yeast phenylalanine transfer ribonucleic acid. Biochemistry. 1972;11:2472–2479. doi: 10.1021/bi00763a014. [DOI] [PubMed] [Google Scholar]
- 68.Römer R., Hach R. tRNA conformation and magnesium binding. A study of a yeast phenylalanine-specific tRNA by a fluorescent indicator and differential melting curves. Eur. J. Biochem. 1975;55:271–284. doi: 10.1111/j.1432-1033.1975.tb02160.x. [DOI] [PubMed] [Google Scholar]
- 69.Chen A.A., Marucho M., Pappu R.V. Simulations of RNA interactions with monovalent ions. Methods Enzymol. 2009;469:411–432. doi: 10.1016/S0076-6879(09)69020-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.