

Abstract
The transport cycle in the glutamate transporter (GlT) is catalyzed by the cotransport of three Na+ ions. However, the positions of only two of these ions (Na1 and Na2 sites) along with the substrate have been captured in the crystal structures reported for both the outward-facing and the inward-facing states of Gltph. Characterizing the third ion binding site (Na3) is necessary for structure-function studies attempting to investigate the mechanism of transport in GlTs at an atomic level, particularly for the determination of the sequence of the binding events during the transport cycle. In this study, we report a series of molecular dynamics simulations performed on various bound states of Gltph (the apo state, as well as in the presence of Na+, the substrate, or both), which have been used to identify a putative Na3 site. The calculated trajectories have been used to determine the water accessibility of potential ion-binding residues in the protein, as a prerequisite for their ion binding. Combined with conformational analysis of the key regions in the protein in different bound states and several additional independent simulations in which a Na+ ion was randomly introduced to the interior of the transporter, we have been able to characterize a putative Na3 site and propose a plausible binding sequence for the substrate and the three Na+ ions to the transporter during the extracellular half of the transport cycle. The proposed Na3 site is formed by a set of highly conserved residues, namely, Asp312, Thr92, and Asn310, along with a water molecule. Simulation of a fully bound state, including the substrate and the three Na+ ions, reveals a stable structure—showing closer agreement to the crystal structure when compared to previous models lacking an ion in the putative Na3 site. The proposed sequence of binding events is in agreement with recent experimental models suggesting that two Na+ ions bind before the substrate, and one after that. Our results, however, provide additional information about the sites involved in these binding events.
Introduction
Glutamate transporters (GlTs; also termed excitatory amino-acid transporters (EAATs)) belong to the family of secondary membrane transporters, which couple uphill translocation of the substrate across the membrane to the energetically favorable flow of ions down their concentration gradient. By coupling to the cotransport of three Na+ ions and one H+, and the countertransport of one K+, mammalian GlT transports one negatively charged glutamate across the membrane during each transport cycle (1–4). In contrast to the mammalian GlT, the transport mechanism in a bacterial homolog (Pyrococcus horikoshii (Gltph)) has been shown to be independent of H+ and K+ (5). Probably the most important aspect of stoichiometry in the function of GlTs is that of Na+:substrate. In mammalian GlT (EAAT3), the Na+:substrate stoichiometry has been well accepted to be 3:1. Based on the experimental measurement of the transport current reversal potential, the ratio of the coupling coefficient was given as 1 Glu−:3.17 Na+ in the mammalian GlT, although the dependence of the forward transport current on [Na+]out exhibited a Hill coefficient of only 2.25 (1). In contrast, the coupling stoichiometry of Na+:substrate in Gltph (the bacterial homolog for which high-resolution structures are available (6–8) and which has been used in this study) was less clear until recently. The results of functional studies of Gltph (5,7) are consistent with a mechanism in which substrate transport by Gltph is catalyzed by the cotransport of more than two Na+ ions. In particular, the study by Ryan et al. (5) reported the dependence of transport on [Na+]out with a Hill coefficient of 2.4 at 0.1 μM aspartate or 2.6 at 1 μM aspartate, indicating that very likely more than two Na+ ions are coupled to the transport of the substrate in each cycle. Very recently, the measurement of the uptake of 22Na+ and [14C] aspartate provided strong evidence that indeed three Na+ ions are cotransported along with the substrate during each transport cycle in Gltph (9).
The atomic-resolution crystal structures of Gltph (6–8) have provided a structural framework for understanding the transport mechanism of GlTs. Gltph shares ∼36% amino-acid identity with EAATs, with a large number of residues implicated in the binding and translocation of the substrate and cotransported ions highly conserved. Therefore, Gltph can serve as a structural model for understanding the mechanism of transport in EAATs (4,10). The structures of Gltph (6–8), solved in both the outward-facing and inward-facing states, reveal a trimeric architecture with each monomer composed of eight transmembrane helices (TM1–TM8) and two highly conserved helical hairpins (termed HP1 and HP2, respectively) forming the binding sites for the substrate and Na+ ions. The substrate-binding site is cradled by the two helical hairpins reaching from the opposite sides of the membrane. The structure of a single monomer in the outward-facing state (7) along with the binding sites are shown in Fig. 1.
Figure 1.
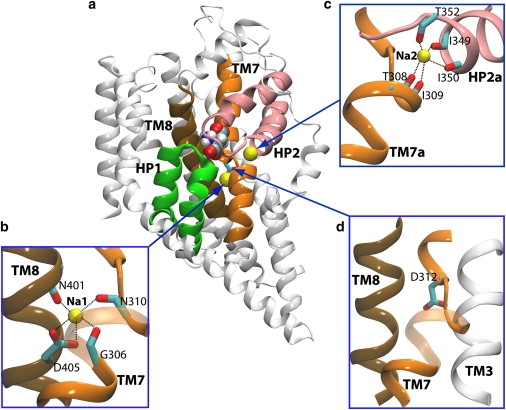
Structural features of Gltph. Note that the figures are made based on the crystal structure of outward-facing Gltph (PDB entry 2NWX). (a) The structure of Gltph monomer with bound substrate (shown in van der Waals representation) and the two structurally resolved Na+ ions at sites Na1 and Na2 (yellow spheres). Helical hairpins HP1 and HP2, and transmembrane helices TM7 and TM8, which together form the substrate and Na+ binding sites (green, pink, orange, and ochre, respectively). (b) The Na1 site composed of residues Gly306 and Asn310 on TM7, and Asn401 and Asp405 on TM8. (c) The Na2 site, located between two half-helices HP2a and TM7a, with the Na+ coordinated by three carbonyl oxygens from HP2 and two carbonyl oxygens from TM7a. (d) The Asp312 neighborhood (potential Na3 site). Asp312 is on the unwound part of TM7, with its side chain located behind TM7 pointing toward the interior of the transporter protein.
Crystallographic and thermodynamic studies of Gltph (7) have provided information on the binding sites of two Na+ ions (termed Na1 and Na2 in the crystal structures; see Fig. 1). The Na1 site, which is buried deeply within the protein, is formed by residues Gly306, Asn310, Asn401, and the negatively charged side chain of Asp405 (Fig. 1 b). The Na2 site is formed by five carbonyl oxygens located in two half helices from HP2 and TM7 (HP2a and TM7a), respectively, that stabilize the ion by their dipole moments (Fig. 1 c). However, the crystal structures of Gltph (6–8) do not provide any information on the third Na+ binding site (Na3). Note that the two Na+ ions are called here as Na1 and Na2, respectively, following the convention used in the crystal structures of Gltph (7,8). Also, note that we use Na3 to refer to the third Na+ binding site, which is not characterized in the crystal structures of Gltph (7,8). The numbering is used only to describe the binding sites (Na1, Na2, and Na3) and does not necessarily correspond to the sequence of binding of the three Na+ ions during the functional cycle of the transporter.
A number of experimental studies have investigated the sequence of binding of the substrate and the Na+ ions to mammalian GlTs during the transport cycle (11–17). Several studies indicated that at least one of the Na+ ions binds to the empty transporter before the substrate (glutamate) and at least one Na+ ion after the substrate (11–15). A more recent kinetic model for mammalian GlTs based on the measurement of transporter currents during glutamate uptake by EAAT2 (16) and voltage-clamp fluorimetry studies in EAAT3 (17) suggests that the empty transporter binds two Na+ ions before the substrate. Moreover, it has been suggested that one of the first two Na+ ions binds to GlT in a voltage-dependent manner (14,16–21), whereas the binding of the other two Na+ ions is voltage-independent (17).
Although these experiments have provided insightful information on the transport cycle in GlT, several key mechanistic details remain elusive. The binding sequence and coupling between the substrate and the three Na+ ions are not understood. Probably most importantly, the location of the third Na+ binding site (the Na3 site) has not been experimentally characterized. A recent theoretical study (22) employing electrostatic mapping calculations has examined potential Na+ binding sites in GlTs, but only near the substrate (within 10 Å of the α-carbon of the substrate) and by using static structures of Gltph and a homology model constructed for EAAT3. Here, taking Gltph as a model structure for understanding the transport mechanism of GlTs (4,5,10), and employing molecular dynamics simulations of a membrane-embedded model of Gltph, we focus on identifying the putative Na3 site and examine the effect and sequence of binding sequence of the substrate and the Na+ ions from the extracellular side. Upon analyzing the structure and dynamics of Gltph at different bound states, and by investigating the local conformational changes induced by each binding event, we identify a putative Na3 site in a highly conserved region. The results suggest that this binding site is only accessible from the extracellular solution through local conformational changes induced by Na+ binding to the Na1 site. Based on the results, we propose a sequence for the binding of the substrate and the three Na+ ions to Gltph from the extracellular side before the formation of the occluded state.
Methods
Here we briefly describe the simulation systems and protocols used in this study. A detailed description of this section is provided in the Supporting Material. The trimeric form of outward-facing Gltph (PDB code 2NWX (7)) embedded in a POPE lipid bilayer was used for all the simulations. A summary of the various bound states of Gltph investigated in this study is given in Table 1. Because each monomer functions independently (7,23,24), we have taken advantage of the trimeric configuration of the simulation systems, and in some of them we modeled individual monomers in different bound states. Some of the analyses are based on the notion that a Na+ binding site has to be accessible by water from outside before it can be reached by a Na+ ion. Each state has been simulated at least three times, e.g., systems S1-a, S1-b, and S1-c for the apo state. Note that the initial protein structure in all the simulations was the occluded state, i.e., the reported crystal structure of the outward-facing state of Gltph (7), unless specified otherwise. All the simulations were performed under periodic boundary conditions with a time step of 1 fs. A constant temperature (303 K) was maintained using Langevin dynamics with a damping coefficient of 0.5 ps−1. The Langevin piston method was employed to maintain a constant pressure of 1 atm with a piston period of 100 fs. Nonbonded interactions were calculated using a cutoff distance of 12 Å, and long-range electrostatic interactions were calculated using the particle-mesh Ewald method.
Table 1.
Summary of Gltph simulation systems reported in this study
| System | Monomer | Substrate | Na1 | Na2 | Na3 | Simulation time (ns) |
|---|---|---|---|---|---|---|
| S1-a | apo | – | – | – | – | 40 |
| S1-b | apo | – | – | – | – | 40 |
| S1-c | apo | – | – | – | – | 40 |
| S2-a | Na1-bound | – | + | – | – | 50 |
| S2-b | Na1-bound | – | + | – | – | 50 |
| S2-c | Na1-bound | – | + | – | – | 50 |
| S3-a | Substrate-bound | + | – | – | – | 30 |
| S3-b | Substrate-bound | + | – | – | – | 30 |
| S3-c | Substrate-bound | + | – | – | – | 30 |
| S4-a | Substrate/Na1-bound | + | + | – | – | 30 |
| S4-b | Substrate/Na1-bound | + | + | – | – | 30 |
| S4-c | Substrate/Na1-bound | + | + | – | – | 30 |
| Na3-a∗ | – | – | – | + | 20 | |
| Na3-b∗ | – | – | – | + | 20 | |
| Na3-c∗ | – | – | – | + | 20 | |
| Na3-d∗ | – | – | – | + | 20 | |
| Na3-e∗ | – | – | – | + | 20 | |
| Na3-f† | – | – | – | + | 20 | |
| Na3-g† | – | – | – | + | 20 | |
| Na3-h† | – | – | – | + | 20 | |
| Na3-i† | – | – | – | + | 20 | |
| Na3-j† | – | – | – | + | 20 | |
| Na1-boundD405N‡ | – | + | – | – | 30 | |
| Na1/Na3-bound§ | – | + | – | + | 20 | |
| Na1/Na3-bound¶ | – | + | – | + | 20 | |
| Fully bound | + | + | + | + | 20 |
Note that the systems were all simulated as trimers. However, individual monomers within the same trimer might have different bound states.
This simulation system is adopted from the equilibrated structure of the Na1-bound state at 50 ns (system S2-a).
The equilibrated structure of the Na1-bound state at 40 ns (system S2-a) is used as the starting point.
In this simulation system, Asp405 in TM8 was mutated to asparagine.
Na3 is in the putative binding site in this simulation system.
Na3 is in the intermediate binding site in this simulation system.
Results and Discussion
The transport cycle of Gltph involves cotransport of one substrate along with three Na+ ions (5,9). However, the crystal structure of outward-facing Gltph (7) provides information only on the binding sites for two of the Na+ ions (Na1 and Na2), and the position of the third Na+ binding site remains unknown. This study seeks to identify the unknown position of the third Na+ site (Na3) and to characterize the sequence and coupling between the binding of the substrate and the Na+ ions from the extracellular side to Gltph.
Water accessibility of potential ion-binding residues
A mutagenesis study (15) on EAAT3 (a mammalian GlT) has shown that Asp367 (corresponding to Asp312 in Gltph) is involved in the coordination of one of the Na+ ions during the transport cycle. In the crystal structure of Gltph (7), however, Asp312 is deeply buried in the protein with its side chain located behind TM7 (Fig. 1 d) and does not participate in any of the two identified Na+ binding sites; the distances between the carboxylate carbon of Asp312 and the Na+ ions in the Na1 and Na2 sites are 7.4 and 9.6 Å, respectively. Taken together, these results suggest that Asp312 is likely involved in the third Na+ binding site, namely the Na3 site. Asp312 might either directly bind the third Na+ ion during the substrate transport, or be involved in binding of the other Na+ ions in a later stage of the transport cycle.
Both the crystal structure of the TBOA-bound Gltph (7), and earlier simulation studies (25,26) indicated that in the apo state, the substrate binding site adopts an open conformation through an opening motion of the helical hairpin HP2, which completely exposes the substrate binding site to the extracellular solution. The opening of the substrate-binding site and its water accessibility do not necessarily result in the exposure of the Na+ binding sites. We will pay special attention here to the region around Asp312, which is potentially involved in the Na3 site (see above). In addition, we will also examine the exposure of Asp405, which is known to be involved in the Na1 binding site. The pKa values of Asp312 and Asp405 calculated by the program PROPKA 2.0 (27,28) which employs empirical relationships between protein pKa shifts and structures such as hydrogen bonding, desolvation effects, and charge-charge interactions) are ∼5.0 and 5.9, respectively, in the crystal structure of Gltph (7), suggesting that they preserve their deprotonated (charged) state in the protein, in agreement with experimental results (15). If these residues are inaccessible by extracellular water, it is safe to assume that they cannot be reached by extracellular Na+ ions either. In other words, water accessibility can be viewed as a prerequisite for ion binding to these residues.
The accessibility of extracellular water to Asp312 and Asp405 is monitored during the simulations of various bound states, namely, apo, Na1-bound, substrate-bound, and substrate/Na1-bound states. The results show that extracellular water can easily access Asp405 in all the states (Fig. 2), indicating the presence of a diffusion pathway for the extracellular Na+ ions to access the Na1 site in the apo state. However, neither in the apo state nor in the substrate-bound form is Asp312 accessible by extracellular water (Fig. 2). Somewhat unexpectedly, the extracellular solution was observed to gain access to, and eventually hydrate Asp312 significantly, only after the binding of a Na+ ion in the Na1 site in the empty transporter, that is, in the Na1-bound state (Fig. 2, b and f). Between three and six extracellular water molecules were found within 3.5 Å of the carboxylate group of Asp312 (Fig. 2 f) during the equilibration of the Na1-bound state. Therefore, it appears that the exposure of Asp312 to the extracellular solution can only be achieved after Na+ binding to the Na1 binding site, but lasts only until the binding of the substrate. (The data for the other two sets of simulations not shown in the main text are presented in Fig. S1 of the Supporting Material, clearly indicating consistent reproducibility of the results in all three independent sets of simulations.)
Figure 2.
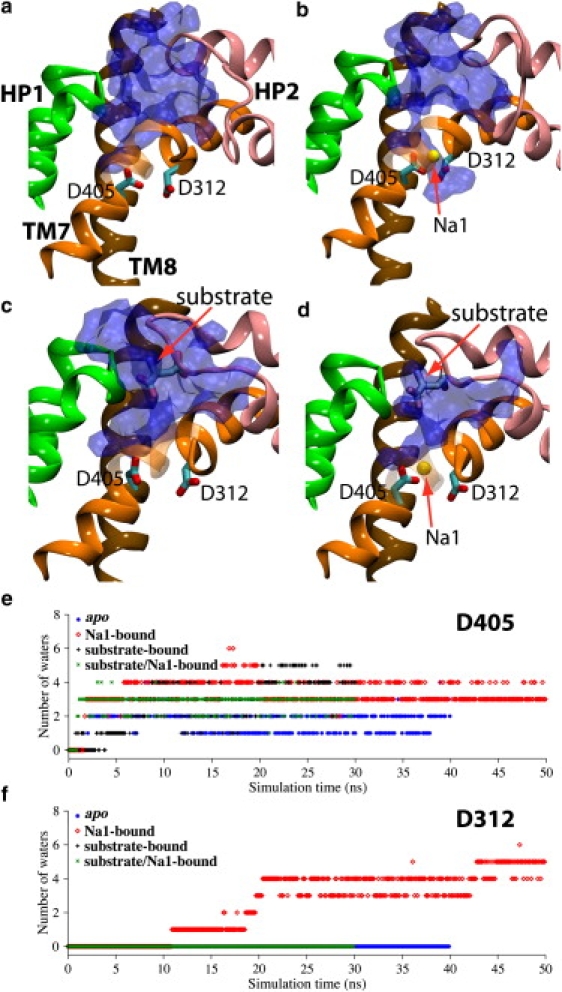
Water accessibility of Asp405 and Asp312 in the apo state (a), Na1-bound state (b), substrate-bound state (c), and the substrate/Na1-bound state (d). Water is represented by a blue molecular surface calculated using the MSMS algorithm in VMD. (e and f) Time series of the number of water molecules with 3.5 Å of the carboxylate groups of Asp405 (e) or Asp312 (f) in various bound states. Extracellular water can access Asp405 in all the simulated bound states, whereas the only system in which Asp312 can be accessed by extracellular water is the Na1-bound state. Note that the data is shown here for only one simulation set. See Fig. S1 for the other two sets.
Molecular mechanism of controlling access to Asp312
To elucidate the gating mechanism that controls the accessibility of Asp312 by the extracellular solution, we dissected the structural effects of the binding of the substrate and Na+ on the transporter protein. One of the most conserved regions of GlTs is the partially unwound portion of TM7 (6) (Fig. 3 f), namely residues 307–312 (TM7307–312), which is located between the two helical segments TM7a and TM7b. As will be discussed later, the conformation of the residues on this segment controls the access of the extracellular water to Asp312. We note that Asn310 and Asp312 on TM7307–312 have been shown to be essential for the function of GlTs (1,6,29–31).
Figure 3.
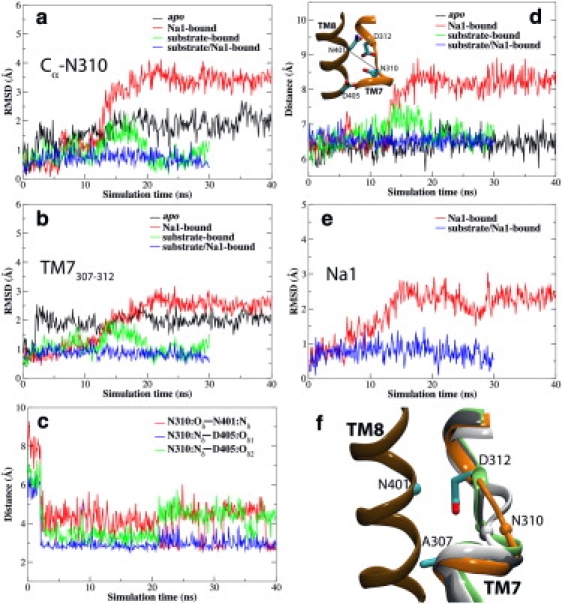
Na+ binding to the Na1 site induces the displacement of TM7307–312. Cα-RMSDs of Asn310 (a) and of TM7307–312 (residues 307–312) (b) in the simulated bound states. (c) Time evolution of the distances between Asn310:Oδ and Asn401:Nδ, between Asn310:Nδ and Asp405:Oδ1, and between Asn310:Nδ and Asp405:Oδ2 in the apo state. Note that the data shown in panels a–c are only for one set of the simulations. See Fig. S2 for the other two sets. (d) Time evolution of the distances between Asn401:Cα and Asn310:Cα in various bound states. (Inset) Structure of the region taken from the crystal structure showing the arrangement of the residues used for distance measurements. (e) Time evolution of the RMSDs of Na1 in the Na1-bound and substrate/Na1-bound states. (f) Comparison of the structure of TM7307–312 in the last frames of simulations of the apo state (white), Na1-bound state (orange), substrate-bound state (gray), and substrate/Na1-bound state (lime) after superposition using transmembrane segment TM8 (ochre). Cα of Asn401 on TM8 (cyan sphere). Cα of Asn310 in various bound states (shown in the same color representation as for TM7). In the Na1-bound state, TM7307–312 displays a large displacement from the crystal structure.
The time evolution of Cα root mean-square deviations (RMSDs) of TM7307–312 (Fig. 3 b) clearly shows that this region becomes more flexible upon the removal of the substrate (also see Fig. S2 for the data obtained from the other two sets of simulations not shown in Fig. 3). In the Na1-bound state, the Cα-RMSDs of TM7307–312 is the largest among the simulated systems, reaching ∼2.5 Å during the equilibration. The large flexibility of TM7307–312 in the absence of the substrate might offer an opportunity for the extracellular water to access Asp312 through the space between TM8 and TM7307–312. The time evolution of the distance between Asn401:Cα and Asn310:Cα (Fig. 3 d) displays a large increase by ∼2 Å during the simulation of the Na1-bound state, suggesting that Na+ binding to the Na1 site induces the displacement of TM7307–312 (Fig. 3 f). However, in the apo state, the distance between Asn401:Cα and Asn310:Cα remains almost unchanged, and a hydrogen bond between the side chains of Asn310 and Asp405 (Fig. 3 c, Fig. 4 a, and Fig. S3) prevents the displacement of TM7307–312 (Fig. 3 f). In addition, the change in the distance between Asn401:Nδ and Asn310:Oδ (Fig. 3 c) in the apo state shows that the side chain of Asn401 forms a hydrogen bond with Asn310. Inspecting the dynamics of apo Gltph, it becomes evident that, with reference to the crystal structure (7), the side chain of Asn310 moves toward Asp405 and that the side chain of Asn401 moves down toward Asn310—resulting in the formation of a hydrogen-bond network among Asn310, Asn401, and Asp405 (Fig. 4 a) that blocks the access of the extracellular water to Asp312. Disruption of this hydrogen-bond network is necessary to provide access to water, and as a result, Na+ ions to Asp312.
Figure 4.
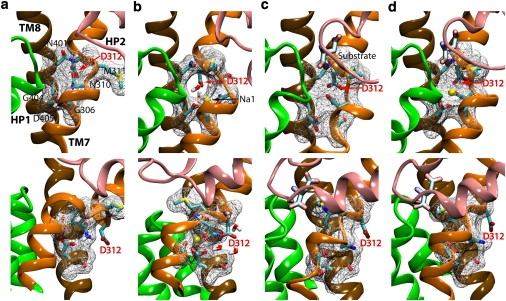
Mechanism of control of water accessibility of Asp312. Molecular surface representation of residues Gly306, Asn310, Met311, Asn401, Gly404, and Asp405 in the apo state (a), Na1-bound state (b), substrate-bound state (c), and substrate/Na1-bound state (d). (Upper and lower panels) Front and side (90°-rotated) views, respectively. Asp312 is shielded from the extracellular solution in the apo state and in the presence of the substrate. Na+ binding to the Na1 site (Na1-bound state) induces the exposure of Asp312 to the extracellular solution through the formation of a tunnel-like opening lined by residues Gly306, Asn310, Met311, Asn401, Gly404, and Asp405, but only in the absence of the substrate.
Na+ binding to the Na1 site (the Na1-bound state) results in the disruption of the aforementioned hydrogen-bond network, and the formation of a tunnel-like opening lined by six highly conserved residues (Gly306, Asn310, Met311, Asn401, Gly404, and Asp405), therefore allowing a deeper penetration of the extracellular water and hydration of Asp312 (Fig. 4 b; see also Fig. S4 for the other two simulation systems). In other words, Na+ binding to the Na1 site of the apo state provides access of the extracellular water to Asp312. In the Na1-bound state, the Na+ ion shows marked displacement from its original position (Fig. 3 e), an event which is lacking altogether in the presence of the substrate (Fig. 3 e). This displacement appears to be correlated with the large displacement of the TM7307–312 segment (Fig. 3 b and Fig. S2). An important mechanistic role might be attributed to these conformational changes, because Asp312 becomes accessible to the extracellular solution only upon Na+ binding to the Na1 site. Conformational changes induced by Na+ binding have been deduced from the studies characterizing the temperature dependence of the steady-state and pre-steady-state kinetics (32). Na+-induced conformational changes have also been indicated by the measurement of the transport current (14) and fluorescence signals (17) during the transport cycle, although these experiments measured structural changes in regions distant from the ion-binding sites.
In all simulations performed in the presence of the substrate, i.e., in the substrate-bound and substrate/Na1-bound systems, the Cα-RMSDs of TM7307–312 and the distance between Asn401:Cα and Asn310:Cα both deviate only marginally (Fig. 3, b and d) from the crystal structure (7), a behavior which is in sharp contrast to the Na1-bound systems described above. Our previous simulations (25) have shown that substrate binding to Gltph brings HP2 closer to HP1 and TM7a (Fig. 4, c and d). Coupling between HP2a and TM7a upon substrate binding confines the motion of TM7307–312 and prevents its displacement (Fig. 3 f), therefore impeding the access of extracellular water to Asp312. Taking a closer look at the structures in the substrate-bound and substrate/Na1-bound states (Fig. 4, c and d), it becomes evident that the access of extracellular water to Asp312 is completely blocked in these states by six residues (Gly306, Asn310, Met311, Asn401, Gly404, and Asp405). These results suggest that Na+ binding to the Na3 site has to take place before the substrate.
Unlike the Na1-bound state, Na+ binding to the Na1 site in the substrate-bound state (the substrate/Na1-bound state) does not induce an opening for the hydration of Asp312. The presence of the substrate in this state appears to reduce the fluctuation of Na1; the calculated RMSD of the Na+ ion in the Na1 site in the substrate/Na1-bound state is very small compared to those in the Na1-bound state (Fig. 3 e). In the substrate/Na1-bound state, Na1 is tightly bound by Asn310 and Gly306 on TM7 and by Asp405 on TM8. Mutagenesis experiments (15) have suggested that D367N mutation in EAAT3 (corresponding to Asp312 in Gltph) inhibits Na+ binding to the glutamate-free form of the transporter, but not to the glutamate-bound form. Our simulation results indicating that Na+ can reach Asp312 in the apo state, but not in the substrate-bound state, are in good agreement with these mutagenesis results (15). These experiments (15) have also suggested that D454N mutation in EAAT3 (corresponding to Asp405 in Gltph) does not affect Na+ binding to the glutamate-free form. Our simulation of the D405N mutant form of Gltph provides important information on the effect of this mutation. In the simulation of this mutant in the Na1-bound state, the Na+ ion in the Na1 site was found to move toward Asp312 and to become coordinated by residues Asn310, Asp312, and Asn405. This results in a configuration with no hydrogen bonds between Asn405 and Asn310, thus, allowing access of extracellular water to Asp312 (Fig. S5). These results suggest that the Na+ ion can still reach Asp312 in the D405N mutant, in keeping with the observed experimental results.
Putative Na3 binding site
Examination of the Na+-binding protein structures in the PDB indicates that a Na+ ion is usually coordinated on the average by five or six electron-donating atoms, most prominently oxygen atoms, which are provided by main-chain carbonyls, side-chain carbonyls, or by hydroxyl groups, as well as water molecules (33). The crystal structure of outward-facing Gltph (7) shows that the carboxylate group of Asp312 is deeply buried in the transporter protein between the transmembrane domains TM3, TM6, TM7, and TM8. Because TM3, TM6, and TM8 do not feature any broken helix structure near Asp312, main-chain carbonyls on these helices are unlikely to be involved in the coordination of Na+ ions around this region. The main-chain carbonyl of Asn310, which is located on the broken part of TM7, on the other hand, is involved in the Na1 site, suggesting that it likely does not participate in the binding of Na3, either. Inspecting the structure, the oxygen atoms of the side chains of Thr92 and Ser93 on TM3, Tyr247 on TM6, and Asn310 on TM7 are in close proximity to Asp312, suggesting that some of these residues might participate in the formation of the Na3 site.
To explore potential Na3 site in the vicinity of Asp312, we started from different snapshots taken from the simulation of the Na1-bound state, i.e., the only state in which water molecules were able to reach and hydrate Asp312, and after removal of the Na+ ion in the Na1 site replaced a water molecule randomly selected from the pool of water molecules in the vicinity of Asp312 by a Na+ at the beginning of 10 independent simulations (Na3-a–Na3-j; initial positions of the ions for systems Na3-a–Na3-e and systems Na3-f–Na3-j are shown in Fig. 5 a and in Fig. S6, respectively). In six simulations (Na3-c—Na3-e and Na3-h–Na3-j), the placed Na+ ion was observed to move into a common binding site formed by the carboxylate of Asp312, hydroxyl of Thr92, and the side-chain carbonyl of Asn310, as well as by one water molecule as depicted in Fig. 5 d. In this putative Na3 site, the ion is coordinated by five oxygen atoms, in agreement with existing structures of Na+-binding proteins (33). To further verify this putative Na3 binding site, we have also performed valence calculations for Na+ over the crystal structure of Gltph with substrate aspartate bound (PDB: 2NWX) using the program VALE (from Nayal and Di Cera (34)). A high probability of finding a Na+ ion in this region was found, as judged by valence values larger than 0.8 for the site formed by residues Asp312, Asn310, and Thr92 by these calculations (see Fig. S7). Moreover, the putative Na3 site suggested by our simulations is strongly supported by very recent mutagenesis experiments in EACC1 (35) showing that T101A mutation (corresponding to Thr92 in Gltph) dramatically decreases the apparent affinity of Na+ to the empty EAAC1, indicating the direct involvement of this conserved threonine in Na+ binding in GlTs (35).
Figure 5.
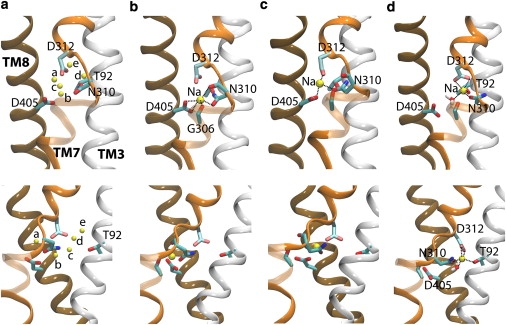
Putative Na3 site. (Upper and lower panels) Front and side (90°-rotated) views, respectively. (a) The initial five positions of Na3 in the Na3-probing simulations (systems Na3-a–Na3-e). (b) The last frame of the Na1-bound state showing the Na1 site. (c) The intermediate site in which Na+ is coordinated by Asn310, Asp312, and Asp405, identified in four out of 10 Na3-probing simulations (systems Na3-a, Na3-b, Na3-f, and Na3-g). (d) The putative Na3 site (identified in six out of 10 Na3-probing simulations (systems Na3-c, Na3-d, Na3-e, Na3-h, Na3-i, and Na3-j), which is composed of residues Asp312, Asn310, and Thr92, and one water molecule.
In the other four Na3 simulations (Na3-a, Na3-b, Na3-f, and Na3-g), in which the initial position of the placed Na+ ion is much closer to Asp405 than in the above six simulations (Na3-c–Na3-e and Na3-h–Na-j) (Fig. 5 a and Fig. S6), the placed Na+ ion moved toward Asp405 and was stabilized in a second site lined by the side chains of Asp405, Asp312, and Asn310 (Fig. 5 c). This site appears too close (within ∼3 Å) to the Na1 site, and, thus, might not be tolerated in the presence of an ion in the Na1 site (note that our exploratory Na3 simulations are done in the absence of an ion in the Na1 site). Moreover, the carboxylate group of Asp405 is completely involved in the coordination of Na1 according to the crystal structure (6,7), and, thus, cannot be involved in the Na3 binding site at the same time. To examine the stability of this site in the Na1/Na3-bound state, one Na+ ion was placed into the Na1 binding site in this Na3-bound state and the system was simulated for 20 ns (Table 1). The simulation results show that the Na+ ion in the second site displays a significant inward movement by ∼2 Å upon binding of a Na+ ion to the Na1 site, whereas a similar simulation performed on the system with ions in the Na1 site and in the putative Na3 site resulted in a stable configuration (see Fig. S8). Therefore, this second site formed by Asp405, Asp312, and Asn310, does not represent a stable Na+ site that can be occupied simultaneously with the Na1 site, i.e., this site cannot be the putative Na3 site. However, we can speculate, purely based on spatial arguments, that this second site (Fig. 5 c) might represent an intermediate state formed during the transition of the Na+ ion from the Na1 site (Fig. 5 b) to the putative Na3 site (Fig. 5 d) as it is located right in the middle of the Na1 and Na3 sites (see below), and because, in this site, the ion is jointly coordinated by both Asp405 and Asp312.
To further study the relevance of the identified Na3 site to the fully bound, occluded state of Gltph, the structural stability of this state (with the substrate and three Na+ ions) was examined in an independent 20-ns equilibrium simulation. The structure and dynamics of this fully bound state were compared with the crystal structure (7) and our previously reported substrate/Na1/Na2-bound state (25). In the fully bound state, the side-chain oxygen of Asn310 is involved in the coordination of the Na+ ion in the Na3 site (Fig. 6 c). The distance between Asn310:Oδ and Asp312:Cγ varies only slightly during the simulation of this state, thus, maintaining closely the configuration observed in the crystal structure (Fig. 6 f). In contrast, in the substrate/Na1/Na2-bound state, i.e., the state lacking a Na+ ion in the Na3 site, this distance increases significantly (Fig. 6 f), due to the large movement of the side chain of Asn310 away from the Na3 site and toward the Na1 site (Fig. 6 b). During this transition, the side-chain amino group of Asn310 forms hydrogen bonds with Asp312, and its side-chain oxygen establishes an interaction with the Na1 site. In summary, significant structural rearrangements within the region of the Na1 site and our putative Na3 site, although mostly localized, are observed in the simulation system without a Na+ ion in the Na3 site (i.e., the substrate/Na1/Na2-bound state). In contrast, the fully bound state appears to stay much closer to the crystal structure, supporting the notion that a Na+ ion is likely present in the crystal of Gltph that was used for structural determination (7), but could not be resolved, and, therefore, was not reported in the deposited structure.
Figure 6.
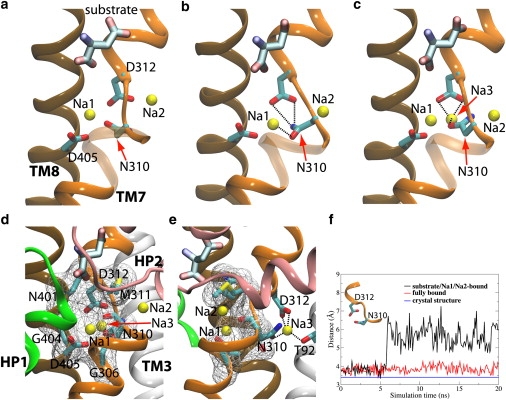
Structural comparison of the crystal structure, the substrate/Na1/Na2-bound state, and the fully bound state. (a) The crystal structure, in which the carbonyl of Asn310 points toward the carboxylate of Asp312 and stays in the putative Na3 site. (b) The last frame of the simulated substrate/Na1/Na2-bound state, in which the carbonyl of Asn310 shifts away from the Na3 site. Also note the backbone change in this region. (c) The last frame of the fully bound state (with the substrate, and three Na+ ions in Na1, Na2, and Na3 sites), in which the carbonyl of Asn310 is involved in the coordination of the Na+ ion in the Na3 site. Note that the conformations of residues Asp312 and Asn310 in this state are very similar to those in the crystal structure (a). (d and e) Front and side (90°-rotated) views showing a Na+ ion locked in the Na3 site inside the protein in the fully bound state. The access of the Na3 site to the extracellular solution is blocked by six residues: Gly306, Asn310, Met311, Asn401, Gly404, and Asp405. (f) Time evolution of the distance between Asp312:Cγ and N310:Oδ in the substrate/Na1/Na2-bound and fully bound states. For reference, the distance in the crystal structure is shown with a blue line.
In a recent report (22), other researchers employed electrostatic mapping with the program VALE (34) to the structure of Gltph and a homology model of EAAT3, in a search for the Na3 site. The authors, however, confined their search to a region within 10 Å radius of the substrate's α-carbon. The proposed Na3 site in that study, which is quite distinct from our putative Na3 site, is composed of several residues on HP2 as well as the γ-carboxylate group of the bound glutamate, i.e., the substrate (22). To examine the stability of a Na+ ion near the substrate, we set up a simulation in which one Na+ was placed into the proposed Na+ site in (22): the aspartate/Na1/Na2-bound state of Gltph. It was found that a Na+ ion in this site cannot be tolerated by the crystal structure, and the introduced Na+ ion resulted in very large deformation of the binding site, especially the substrate itself (see Fig. S9). The β-carboxylate group of the substrate flips away from the substrate-binding site toward this placed Na+ (Fig. S9). Therefore, this proposed site (22) might not represent a binding site for the cotransported Na+ ion. Note that the putative third Na+ binding site that we proposed is ∼12 Å away from the substrate's α-carbon.
The molecular surface representation of residues Gly306, Asn310, Met311, Asn401, Gly404, and Asp405 in the fully bound state shows that the entry pathway into the Na3 site from the extracellular side is blocked in this state (Fig. 6, d and e). Therefore, after the formation of the fully bound state, Na3 is locked into its binding site. This finding might explain why the replacement of Na+ by Tl+ in the binding assay of Gltph (7) has only revealed two Na+ ions (Na1 and Na2), because the entry pathway to the Na3 site is blocked in the presence of substrate.
Conclusions
Mechanistic implications
Based on the results of the simulations presented in this study, together with relevant experimental results, we can suggest a hypothetical sequence for binding events during the extracellular half-cycle of transport in Gltph (Fig. 7). Several relationships among the ion-binding sites and the substrate can be deduced from our simulations. We show that the access to the putative Na3 site, which is the deepest ion-binding site in Gltph, shows two major dependencies on the other ligands. First, the opening of the entry pathway to the Na3 site relies on the presence of a Na+ ion in the Na1 site, and secondly, substrate binding results in the occlusion of the entry pathway to the Na3 site, regardless of the Na1 occupancy. Furthermore, in our earlier study (25), we demonstrated that the binding of a Na+ ion to the Na2 site results in the complete occlusion of the binding sites for both the substrate and the other two ions, through conformational changes that lock HP2 in a fully closed state.
Figure 7.
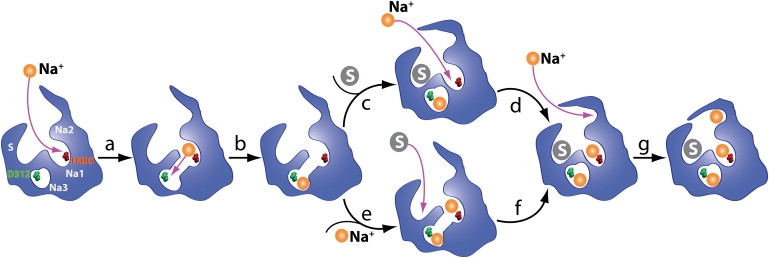
Schematic mechanism and sequence of binding of the substrate and three Na+ ions to Gltph. Na1, Na2, Na3, and S in the first figure mark the positions of the Na1, Na2, Na3, and substrate binding sites, respectively. (a) One Na+ binds to the Na1 site in the apo transporter resulting in the opening of a tunnel-like structure exposing D312. The same ion likely moves to the putative Na3 site located deep into the transporter (b). Next, either the second Na+ binds to the Na1 site followed by substrate binding (c and d), or the substrate binds to the Na+-bound transporter followed by the binding of the second Na+ ion to the Na1 site (e and f), resulting in the half-closure of the extracellular gate and the formation of the Na2 site. (g) One Na+ binds to the Na2 site, completing the binding process and resulting in the formation of the fully bound state.
Taking these results together, we propose the following model for the sequence of the binding events in Gltph. First, a Na+ ion binds to the Na1 site in the apo state, resulting in the opening of the entry pathway to the Na3 site. The next step is the binding of a Na+ ion to our putative Na3 site. This ion might be provided by one of the following two mechanisms:
-
1.
Diffusion of a second Na+ all the way from the extracellular space into the Na3 site; or
-
2.
Translocation of the ion already in the Na1 site into the Na3 site.
The latter mechanism (transition of the ion from the Na1 site to the Na3 site) will be followed by the entrance of a second Na+ ion into the Na1 binding site, or, more likely, is accompanied/induced by such an event (a knock-off mechanism). Although further investigation is required for establishing the mechanism, structural examination of the Na3 entry pathway and the close proximity of the Na+ ion bound in the Na1 site both appear to strongly favor the mechanism (2), i.e., the movement of the ion from the Na1 site to the Na3 site. The close proximity of the Na+ ion in the Na1 site to this pathway would make it difficult, due to repulsive forces, for a second ion to bypass and enter the pathway. The Na+ ion at the Na1 site itself, however, is in an optimal position to diffuse through this pathway. In fact, the ion in the Na1 site is already engaged in an indirect (water-bridged) interaction with the carboxylate group of Asp312, i.e., the Na3 site (Fig. 4 b) in the Na1-bound state. Therefore, Na1 appears very well posed to move deeper into the Na3 site, through an intermediate state in which the ion is coordinated by both Asp312 and Asp405 (Fig. 5 c). Interestingly, in four of the Na3-probing simulations (Na3-a, Na3-b, Na3-f, and Na3-g described above), such a configuration was indeed captured as a stable intermediate structure (see Fig. 5 d). Occupation of the Na3 site is followed by the binding of the substrate and a second Na+ ion to the Na1 site (Fig. 7), resulting in the partial closure of the extracellular gate (HP2). Finally, a Na+ ion binds in the Na2 site (25) and completes the formation of the fully bound, occluded state of the transporter (Fig. 7).
We note, however, that our current simulation results cannot fully resolve the sequence of binding the second Na+ ion and the substrate (Fig. 7). The Na1 site remains accessible from the extracellular solution, even after the binding of the substrate according to our simulations. A recent kinetic model (16,36) for mammalian GlTs suggests a binding sequence in which two Na+ ions bind before the substrate and the third ion binds after it. More-recent studies (13,17,35) on EAAT3 transporter also support the notion that two Na+ ions bind to GlT before the substrate, as evidenced by the observation that the binding of two Na+ ions to GlT forms a high-affinity substrate-binding site (35). Based on these results we prefer the scenario in which the second ion binds to the Na1 site before the substrate. However, in the provided schematic model we have opted to include the possibility of the substrate binding before the second ion. We also note that the experimental results used to infer information for the sequence of binding events often cannot identify which of the three sites is occupied first, and only suggest to us that two of them are occupied before the substrate and one after it. In this regard, simulation results complement the experimental results in providing a more-complete picture of the processes and events involved in the transport cycle of the transporter.
Acknowledgments
This study was supported by the National Institutes of Health (grant Nos. R01-GM086749, R01-GM067887, and P41-RR05969). The simulations were performed using the TeraGrid resources (grant No. MCA06N060).
Supporting Material
References
- 1.Zerangue N., Kavanaugh M.P. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 2.Levy L.M., Warr O., Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J. Neurosci. 1998;18:9620–9628. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pines G., Kanner B.I. Counterflow of L-glutamate in plasma membrane vesicles and reconstituted preparations from rat brain. Biochemistry. 1990;29:11209–11214. doi: 10.1021/bi00503a008. [DOI] [PubMed] [Google Scholar]
- 4.Kavanaugh M.P., Bendahan A., Kanner B.I. Mutation of an amino acid residue influencing potassium coupling in the glutamate transporter GLT-1 induces obligate exchange. J. Biol. Chem. 1997;272:1703–1708. doi: 10.1074/jbc.272.3.1703. [DOI] [PubMed] [Google Scholar]
- 5.Ryan R.M., Compton E.L., Mindell J.A. Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J. Biol. Chem. 2009;284:17540–17548. doi: 10.1074/jbc.M109.005926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yernool D., Boudker O., Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 7.Boudker O., Ryan R.M., Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 8.Reyes N., Ginter C., Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groeneveld M., Slotboom D.J. Na+:aspartate coupling stoichiometry in the glutamate transporter homologue Glt(Ph) Biochemistry. 2010;49:3511–3513. doi: 10.1021/bi100430s. [DOI] [PubMed] [Google Scholar]
- 10.Bendahan A., Armon A., Kanner B.I. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J. Biol. Chem. 2000;275:37436–37442. doi: 10.1074/jbc.M006536200. [DOI] [PubMed] [Google Scholar]
- 11.Wadiche J.I., Amara S.G., Kavanaugh M.P. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 12.Kanai Y., Nussberger S., Hediger M.A. Electrogenic properties of the epithelial and neuronal high affinity glutamate transporter. J. Biol. Chem. 1995;270:16561–16568. doi: 10.1074/jbc.270.28.16561. [DOI] [PubMed] [Google Scholar]
- 13.Watzke N., Bamberg E., Grewer C. Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J. Gen. Physiol. 2001;117:547–562. doi: 10.1085/jgp.117.6.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larsson H.P., Tzingounis A.V., Kavanaugh M.P. Fluorometric measurements of conformational changes in glutamate transporters. Proc. Natl. Acad. Sci. USA. 2004;101:3951–3956. doi: 10.1073/pnas.0306737101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tao Z., Zhang Z., Grewer C. Neutralization of the aspartic acid residue Asp-367, but not Asp-454, inhibits binding of Na+ to the glutamate-free form and cycling of the glutamate transporter EAAC1. J. Biol. Chem. 2006;281:10263–10272. doi: 10.1074/jbc.M510739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergles D.E., Tzingounis A.V., Jahr C.E. Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J. Neurosci. 2002;22:10153–10162. doi: 10.1523/JNEUROSCI.22-23-10153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koch H.P., Hubbard J.M., Larsson H.P. Voltage-independent sodium-binding events reported by the 4B-4C loop in the human glutamate transporter excitatory amino acid transporter 3. J. Biol. Chem. 2007;282:24547–24553. doi: 10.1074/jbc.M704087200. [DOI] [PubMed] [Google Scholar]
- 18.Auger C., Attwell D. Fast removal of synaptic glutamate by postsynaptic transporters. Neuron. 2000;28:547–558. doi: 10.1016/s0896-6273(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 19.Billups B., Rossi D., Attwell D. Anion conductance behavior of the glutamate uptake carrier in salamander retinal glial cells. J. Neurosci. 1996;16:6722–6731. doi: 10.1523/JNEUROSCI.16-21-06722.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otis T.S., Jahr C.E. Anion currents and predicted glutamate flux through a neuronal glutamate transporter. J. Neurosci. 1998;18:7099–7110. doi: 10.1523/JNEUROSCI.18-18-07099.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mim C., Balani P., Grewer C. The glutamate transporter subtypes EAAT4 and EAATs 1–3 transport glutamate with dramatically different kinetics and voltage dependence but share a common uptake mechanism. J. Gen. Physiol. 2005;126:571–589. doi: 10.1085/jgp.200509365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holley D.C., Kavanaugh M.P. Interactions of alkali cations with glutamate transporters. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009;364:155–161. doi: 10.1098/rstb.2008.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grewer C., Balani P., Rauen T. Individual subunits of the glutamate transporter EAAC1 homotrimer function independently of each other. Biochemistry. 2005;44:11913–11923. doi: 10.1021/bi050987n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koch H.P., Brown R.L., Larsson H.P. The glutamate-activated anion conductance in excitatory amino acid transporters is gated independently by the individual subunits. J. Neurosci. 2007;27:2943–2947. doi: 10.1523/JNEUROSCI.0118-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Z., Tajkhorshid E. Dynamics of the extracellular gate and ion-substrate coupling in the glutamate transporter. Biophys. J. 2008;95:2292–2300. doi: 10.1529/biophysj.108.133421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shrivastava I.H., Jiang J., Bahar I. Time-resolved mechanism of extracellular gate opening and substrate binding in a glutamate transporter. J. Biol. Chem. 2008;283:28680–28690. doi: 10.1074/jbc.M800889200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Robertson A.D., Jensen J.H. Very fast empirical prediction and interpretation of protein pKa values. Proteins Struct. Funct. Bioinf. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 28.Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins Struct. Funct. Bioinf. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 29.Borre L., Kanner B.I. Coupled, but not uncoupled, fluxes in a neuronal glutamate transporter can be activated by lithium ions. J. Biol. Chem. 2001;276:40396–40401. doi: 10.1074/jbc.M104926200. [DOI] [PubMed] [Google Scholar]
- 30.Zarbiv R., Grunewald M., Kanner B.I. Cysteine scanning of the surroundings of an alkali-ion binding site of the glutamate transporter GLT-1 reveals a conformationally sensitive residue. J. Biol. Chem. 1998;273:14231–14237. doi: 10.1074/jbc.273.23.14231. [DOI] [PubMed] [Google Scholar]
- 31.Rosental N., Bendahan A., Kanner B.I. Multiple consequences of mutating two conserved β-bridge forming residues in the translocation cycle of a neuronal glutamate transporter. J. Biol. Chem. 2006;281:27905–27915. doi: 10.1074/jbc.M600331200. [DOI] [PubMed] [Google Scholar]
- 32.Mim C., Tao Z., Grewer C. Two conformational changes are associated with glutamate translocation by the glutamate transporter EAAC1. Biochemistry. 2007;46:9007–9018. doi: 10.1021/bi7005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glusker J.P. Structural aspects of metal liganding to functional groups in proteins. Adv. Protein Chem. 1991;42:1–76. doi: 10.1016/s0065-3233(08)60534-3. [DOI] [PubMed] [Google Scholar]
- 34.Nayal M., Di Cera E. Predicting Ca2+-binding sites in proteins. Proc. Natl. Acad. Sci. USA. 1994;91:817–821. doi: 10.1073/pnas.91.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao Z., Rosental N., Grewer C. Mechanism of cation binding to the glutamate transporter EAAC1 probed with mutation of the conserved amino acid residue Thr101. J. Biol. Chem. 2010;285:17725–17733. doi: 10.1074/jbc.M110.121798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z., Tao Z., Grewer C. Transport direction determines the kinetics of substrate transport by the glutamate transporter EAAC1. Proc. Natl. Acad. Sci. USA. 2007;104:18025–18030. doi: 10.1073/pnas.0704570104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.