

Abstract
Alpha-solenoid proteins are suggested to constitute highly flexible macromolecules, whose structural variability and large surface area is instrumental in many important protein-protein binding processes. By equilibrium and nonequilibrium molecular dynamics simulations, we show that importin-β, an archetypical α-solenoid, displays unprecedentedly large and fully reversible elasticity. Our stretching molecular dynamics simulations reveal full elasticity over up to twofold end-to-end extensions compared to its bound state. Despite the absence of any long-range intramolecular contacts, the protein can return to its equilibrium structure to within 3 Å backbone RMSD after the release of mechanical stress. We find that this extreme degree of flexibility is based on an unusually flexible hydrophobic core that differs substantially from that of structurally similar but more rigid globular proteins. In that respect, the core of importin-β resembles molten globules. The elastic behavior is dominated by nonpolar interactions between HEAT repeats, combined with conformational entropic effects. Our results suggest that α-solenoid structures such as importin-β may bridge the molecular gap between completely structured and intrinsically disordered proteins.
Introduction
Solenoid proteins, consisting of repeating arrays of simple basic structural motifs, account for >5% of the genome of multicellular organisms (1). Their fold often forms extended superhelical structures, which expose large surface areas to solvent. Because of this feature, solenoid proteins play central roles in the shaping of cellular scaffolds and many other cellular processes that require multiple protein-protein interactions. Examples include pathways of endocytosis, receptor-mediated nucleocytoplasmic transport, protein sorting, and the scaffold structure of the nuclear pore complex (2–5).
In structural studies, it was suggested that α-solenoids form especially flexible structures, which may fall between typical globular structured and intrinsically disordered proteins (6,7). Their tertiary structure usually displays high flexibility, whereas their secondary structure elements are thought to remain stable and well defined (6–8). This feature actually also bears close resemblance to the so-called molten globule state of proteins—a third protein structural state lying between the fully folded and completely unfolded states, in which stable secondary structure elements are believed to be arranged into a flexible, loosely packed tertiary fold (9–12). Despite their enormous biological importance, the principles governing flexibility and structural integrity of solenoid proteins have received limited attention so far.
Recently, single molecule spectroscopy methods, such as atomic force microscopy and optical or magnetic tweezers, have become useful tools to study the response of macromolecules to external forces (13). Whereas most studies on repeat proteins focus on the folding and unfolding mechanism (14–16), an atomic force microscopy study on different ankyrin repeat constructs showed that ankyrin repeats behave as Hookean springs with spring constants of 1.5–23 × 10−3 N/m (17). This finding suggests that such springlike features may be of general importance for the function of α-helical repeat proteins.
Importin-β is an archetypical α-solenoid and one of the best-studied repeat proteins. It is the main player in nucleocytoplasmic transport, mediating the principal pathway of protein import into the cell nucleus (3). Its extended superhelical structure, built from 19 so-called HEAT repeats, a structural motif composed of two α-helices linked by a short loop (18), is instrumental in binding and wrapping around cargo proteins destined to the nucleus. Simultaneously, importin-β interacts with nuclear pore complexes (19). To perform its function, importin-β must be able to structurally adapt to cargo proteins of various size and shape as well as to its effector protein RanGTP (20–22). Intriguingly, it has been suggested that importin-β complexes are capable of storing energy through a huge springlike deformation of this superhelix that is then released, upon dissociation, to drive disassembly and support high transport rates (20,22).
To test this hypothesis, we have performed extended equilibrium and nonequilibrium molecular dynamics (MD) simulations of the yeast importin-β solenoid (Kap95p, yImpβ). Our stretching simulations show that yImpβ exhibits an extraordinary degree of elasticity. Most unexpectedly, extensions by nearly a factor of two are found to be fully reversible on very short timescales. Our studies enabled us to determine the molecular spring constant of yImpβ and thus to calculate the amount of energy stored in molecular complexes of yImpβ. They also reveal the structural determinants underlying the unique level of elasticity in yImpβ, and highlight a marked difference to the usual construction principle of rigid proteins.
Methods
All simulations were based on an open unbound conformation of the importin-β homolog Kap95p (yImpβ). The relaxed structure with an elongation of ∼13.5 nm was simulated from PDB file 2BKU (22). The structure was obtained by free MD simulations of yImpβ without RanGTP as described in Zachariae and Grubmüller (8).
All subsequent MD simulations were performed using GROMACS 3.3 and 4.0 (23,24). The OPLS all-atom force field (25) and the TIP4P water model (26) were used. In all simulations, sodium and chloride ions corresponding to a salt concentration of 150 mM were added. All bond lengths were restrained using the LINCS algorithm (27). The temperature was kept constant at 300 K using the Berendsen thermostat (28) with a coupling time of 0.1 ps. The Berendsen barostat (28) was used with a coupling time of 1 ps and an isotropic compressibility of 4.5 × 10−5 bar−1 to maintain a constant pressure of 1 bar. Lennard-Jones interactions were cut off beyond 1 nm. Electrostatic interactions were calculated explicitly below 1 nm and via particle-mesh Ewald summation (29,30) beyond that distance. An integration time step of 2 fs was used. All simulations performed are summarized in Table S1 in the Supporting Material.
For unbiased simulations of yImpβ, the protein was placed in a dodecahedral box with box vectors of ∼18-nm length. Together with added water molecules and ions, these systems consisted of ∼550,000 particles. Preceding the production runs, 1000 steps of steepest-descent energy minimization, with a subsequent 1-ns simulation, and position restraints of 1.66 N/m on heavy protein atoms, were performed.
For stretching simulations, the protein was oriented along the box vectors, so that the main dimension of the protein pointed in z direction. The length of the box vectors was chosen to ensure that the minimal distance from protein atoms to the box boundaries does not fall below 1 nm. Subsequently, the box was enlarged by 15 nm along z, resulting in a box of 10 × 10 × 30 nm. Water and ions were added, resulting in ∼390,000 particles. One-thousand steps of steepest-descent energy minimization with a subsequent 1-ns simulation and position restraints of 1.66 N/m on heavy protein atoms were performed. All stretching simulations are based on the coordinates of this system. To stretch the protein, the Cα-atom of the N-terminus was fixed by position restraints with a force constant of 1.66 N/m, while a pulling potential with a force constant of 0.83 N/m was moved with constant velocity in z direction (see Fig. 1) (31). The pulling potential was acting exclusively in z direction. The simulations were stopped after 15 nm of spring movement. To determine the spring constant, a second set of simulations was performed with the setup as described above, but which were stopped after 5 nm of spring movement.
Figure 1.
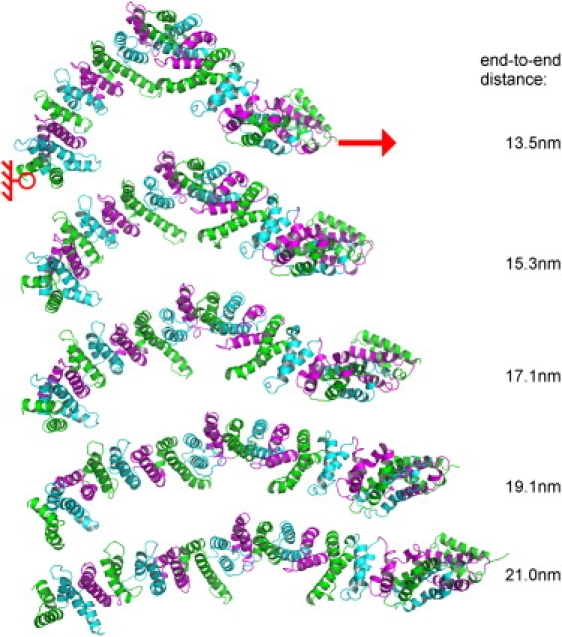
Snapshots during stretching of yImpβ. Different colors represent different HEAT repeats. The Cα atom of the N-terminus was kept fixed (red circle) while a moving harmonic potential was applied to the Cα atom of the C terminus (red arrow). (Right) Numbers denote the end-to-end distance of the protein.
Relaxation simulations formed a third group of simulations. Here, structures from stretching simulations were taken and simulated freely, i.e., without the stretching potential. Two approaches were used to generate the simulation systems. In the first case (elongations of 15.3, 17.1, and 19.1 nm), a snapshot of only the protein was used. Subsequently, a new box with a minimal distance to the protein of 1 nm was built and filled with water and ions. Before production runs, energy minimization and equilibration were performed as described before. In the second case (elongation of 21.0 nm), a snapshot of the whole simulation system including solvent atoms was taken and used as input for further unbiased simulations. In this instance, further energy minimizations or equilibration simulations were not needed.
To estimate entropy changes upon extension, in a fourth group of simulations, snapshots from elongated conformations of yImpβ at 13.4, 14.6, 15.3, 16.3, 17.1, 18.1, and 19.1 nm were taken and the Cα atoms of the termini were subjected to position restraints of 1.66 N/m. In this way, extended trajectories of the protein at these fixed elongations were obtained.
To quantify the equilibrium fluctuations of yImpβ, a sum of n weighted Gaussian functions with center μi, width σi, and weighting factor wi according to
| (1) |
was used to describe the histogram of elongations d of the protein. From the width of the Gaussians, force constants ki were derived via
where kB is the Boltzmann constant and T the temperature used in the simulations. The statistical error was estimated as follows: For all five subsets of four out of five trajectories, spring constants and center positions of the Gaussians were determined as above. The variance of the obtained values was used as an estimate of the error.
To quantify the flexibility of amino-acid side chains, we used dihedral order parameters for torsion angles from side chains (χ-angles), as described in van der Spoel and Berendsen (32). Briefly, from using the probability distribution p(θ) of a given dihedral angle θ, the autocorrelation function C(∞) of this dihedral angle at infinite times is calculated,
| (2) |
yielding an order parameter S2D = C(∞) (33) between zero (full flexibility) and one (no flexibility). For each residue, the lowest calculated order parameter was used. Calculations were performed using the GROMACS tool g_chi (32).
Entropies were calculated according to Schlitter (34), based on a principal component analysis of the backbone motions of yImpβ. To obtain a sufficiently equilibrated ensemble, the first 5 ns of simulated time were omitted from the entropy calculations. Entropies were calculated from sections of the trajectory of increasing length starting at a length of twindow = 20 ns. Each subsequent trajectory part twindow was chosen to be 5 ns longer than the previous one. A linear fit of the calculated entropies as a function of 1/twindow was used to extrapolate entropy estimates for infinite trajectory length. As an estimate for the accuracy of the determined entropies, the statistical uncertainty of the intersection of the line fit with the y axis was used.
Simulations of the four-helix bundle protein Rop were based on the PDB structure 1ROP (35). The protein was placed in a dodecahedral box with box vectors of length 6.8 nm. Water molecules and sodium and chloride ions according to a salt concentration of 150 mM were added. Energy minimization and equilibration were performed as described above. Subsequently, a simulation of 300-ns length was carried out. All other parameters were identical to the simulations described above.
Results and Discussion
Fully reversible elastic stretching
To characterize the flexibility of yImpβ, we carried out 42 independent force probe simulations. In all simulations, the protein was stretched by applying a moving harmonic potential to the C-terminus, while the position of the N-terminus was kept fixed. The elongation of yImpβ was measured as the distance between the Cα atoms of the N- and C-termini.
Fig. 1 shows snapshots from a representative slow (v = 1 m/s) stretching simulation. It is evident that, while yImpβ adopted a strikingly more elongated tertiary structure under mechanical stress, its helical secondary structure remained intact. No unfolding events were observed for elongations below ∼22 nm. Beyond that critical point, the structure showed local intermediate unfolding and separated into two segments of HEAT repeats, most frequently between HEAT repeats 4/5 and 14/15. These locations had also been suggested previously to form dynamic hot spots (8). Here, we focus at the elastic regime before unfolding.
To examine whether or not stretching of yImpβ is reversible, six simulations were carried out in which the stretching potential was turned off at different elongations of the protein. Four of these simulations were started at elongations of 15.3, 17.1, 19.1, and 21.0 nm from snapshots after stretching, carried out at 1 m/s (Fig. 2 A). To investigate a possible influence of stretching velocity, two more relaxation simulations were started from a stretching simulation with a 10-fold lower stretching velocity (0.1 m/s) at elongations of 15.1 and 17.3 nm (Fig. 2 B).
Figure 2.
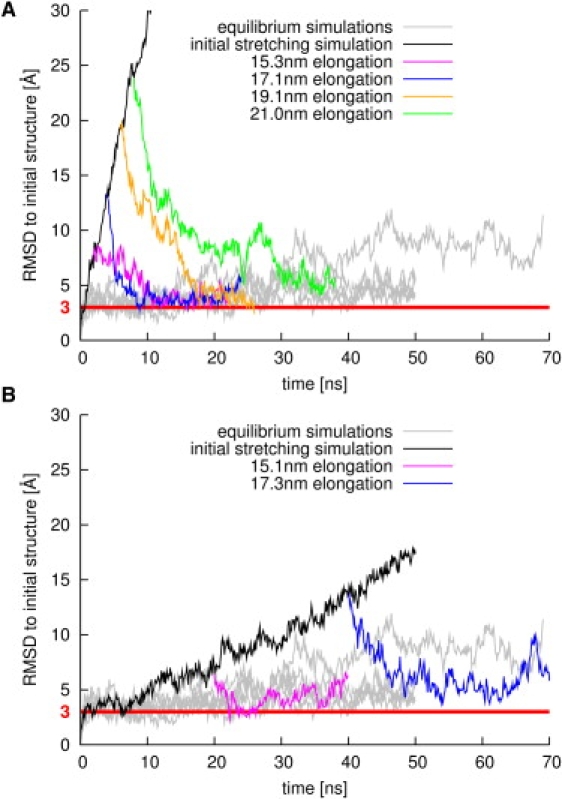
Backbone RMSD, with respect to the initial structure of yImpβ during stretching (black lines) and subsequent release (magenta, blue, orange, and green lines). For comparison, gray lines display data from equilibrium simulations. (Red horizontal line) RMSD value of 3 Å. (A) Stretching at 1 m/s and subsequent release at different elongations. (B) Stretching at 0.1 m/s and subsequent release at different elongations.
In each case, yImpβ was found to relax back to its original elongation. Strikingly, the initial structure was fully recovered, as testified by the low backbone root-mean-square deviation (RMSD) values with respect to the initial state (magenta, blue, and orange lines), which reached the equilibrium RMSD value of simulations of free yImpβ (gray lines). Relaxation simulations starting from initial RMSD values up to 20 Å returned to values at ∼3 Å. Even when stretched close to the limit of elastic elongation, the RMSD still dropped from 25 Å to 5 Å (green line).
These results demonstrate that yImpβ tolerates extreme extensions of more than twice the end-to-end distance of its RanGTP-bound structure (∼8.9 nm) (22), showing fully reversible recovery. Moreover, all relaxation processes occurred on timescales faster than ∼60 ns (Fig. S1 in the Supporting Material). This timescale is similar to that observed for the complete conformational transition between the nuclear and cytosolic states of the related nuclear transport receptor Cse1p (36), underscoring the functional relevance of these fast timescales for karyopherins. Furthermore, it can be seen from Fig. 2 B that fast relaxation occurs independent of the velocity of the stretching simulation carried out before. Such fully reversible elasticity (Fig. 2, A and B) is, to our knowledge, the largest seen for proteins to date, markedly exceeding that of ankyrin repeats (37) and that observed in the recent 2-ns simulations on the protein PR65, which also consists of HEAT repeats (38).
Spring constant of yImpβ
Stretching and subsequent fully reversible relaxation of yImpβ suggests that the protein indeed can act as a molecular spring. We have determined its molecular spring constant via two complementary and independent approaches.
First, yImpβ was subjected to weak mechanical stress, and its spring constant was obtained from the force-extension curve. The moving spring potential was chosen to advance slowly (v = 0.1 m/s) to minimize friction. The mean yImpβ spring constant kstr, determined from the slope of the force profiles of three independent stretching simulations (Fig. 3 A), was found to be (8 ± 1) × 10−3 N/m. The size of the observed force fluctuations is that expected for the probe spring constant of 0.83 N/m used in the stretching simulations (Fig. S2).
Figure 3.
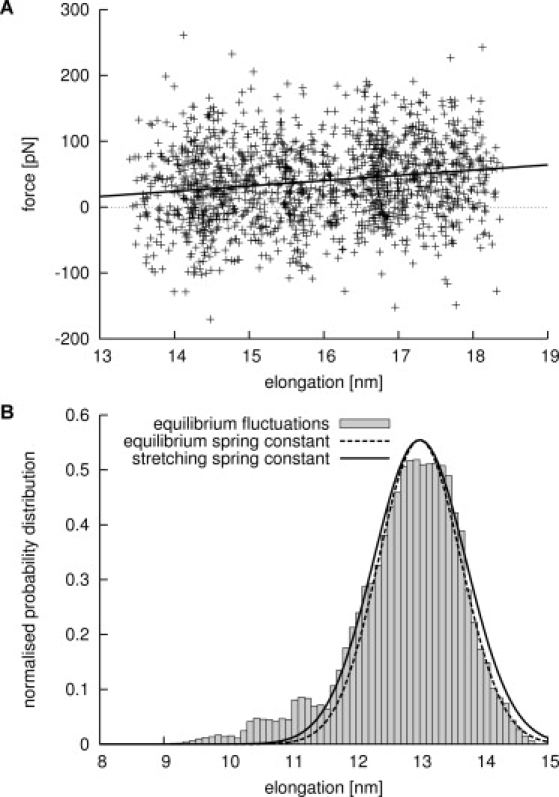
Determining the spring constant of yImpβ. (A) Typical force curve from a slow stretching simulation (crosses) and linear fit to the data (solid line). (B) Equilibrium fluctuation of elongations of yImpβ (shaded bars) and derivation of the spring constant. (Broken line) Boltzmann distribution of fluctuations according to the spring constant determined from equilibrium simulations (see text). For comparison, a Boltzmann distribution according to the spring constant determined in the stretching simulations is shown (solid line).
Secondly, extended equilibrium simulations were performed, from which the spring constant was obtained from the end-to-end distance distributions. Shaded bars in Fig. 3 B display equilibrium fluctuations of yImpβ elongations obtained from a 250-ns equilibrium simulation. As can be seen from the histogram, nearly all fluctuations of yImpβ can be described by a Gaussian function centered at 13 nm. The additional minor peaks at elongations of 11 nm and below belong to transient excursions to local energy minima (Fig. S1 and Fig. S3). The spring constant as well as its uncertainty was estimated from the width of the main peak by a statistical analysis as described in Methods. A spring constant of keq = (10 ± 4) × 10−3 N/m was found at an equilibrium elongation of (13.0 ± 0.2) nm (dashed line in Fig. 3 B). For comparison, the solid line in Fig. 3 B shows a Boltzmann distribution for a harmonic potential with spring constant kstr.
The values for the spring constant of yImpβ agree very well, characterizing yImpβ as a very soft spring, allowing large end-to-end equilibrium fluctuations of up to 2 nm. With this stiffness, yImpβ is slightly softer than the ankyrin repeats studied before (keq(experimental) = 1.5–23 × 10−3 N/m (17), keq(calculated) = 16.4 × 10−3 N/m (37)). It is far more rigid than DNA, which has a spring constant of 0.2–4.7 × 10−6 N/m upon elastic stretching (39). Viral shells, on the other hand, need to resist high pressures of up to 60 atm, and exhibit force constants ranging from ∼0.1 N/m to up to ∼4 N/m upon indention (40,41).
As calculated from its spring constant, yImpβ can store up to 40 kJ/mol in its compact RanGTP-bound state. This result suggests that this mechanical strain can indeed drastically reduce the binding free energy of the tight complex and thus enable disassembly by GTP hydrolysis which ultimately drives nucleocytoplasmic transport. Table 1 summarizes an extrapolation of these findings to complexes of human and mouse importin-β.
Table 1.
Estimation of stored energy in bound importin-β complexes according to an equilibrium elongation of 13 nm and a spring constant of 8 × 10−3 N/m, as derived from equilibrium and stretching simulations of yImpβ
Structural basis for the reversible elasticity of yImpβ
HEAT repeat proteins contain a well-described and extensive hydrophobic core, already detectable on a sequence level (18,21,42,43). Despite this pronounced hydrophobic core, α-solenoids appear to differ strongly from other α-helical proteins by their exceptionally high flexibility, characterized above. As a possible explanation, we hypothesized that the hydrophobic core connecting helices, despite its apparent regularity, shows differences compared to typical soluble proteins.
To test this idea, we compared successions of HEAT repeats (Fig. 4, B and D) with the repressor of primer (Rop) protein from Escherichia coli, a soluble protein that has a paradigmatic, well-studied regular hydrophobic core (Fig. 4, A and C). Indeed, Rop forms a dimer of two α-helices each, connected by a short loop, bearing close resemblance to a pair of HEAT repeats. The length of the α-helices is ∼45 amino acids in HEAT repeats and ∼60 in Rop, and the interhelical angle is ∼20° in both cases, demonstrating close structural similarity (35,43–45). Also, no marked differences in the packing efficiency of the hydrophobic cores are observed (Fig. 4, A and B). Therefore, a simple structural explanation is not sufficient to explain the enhanced flexibility of yImpβ. Moreover, during stretching, the contact pattern between the residues in the hydrophobic core of yImpβ remains unchanged (not shown).
Figure 4.
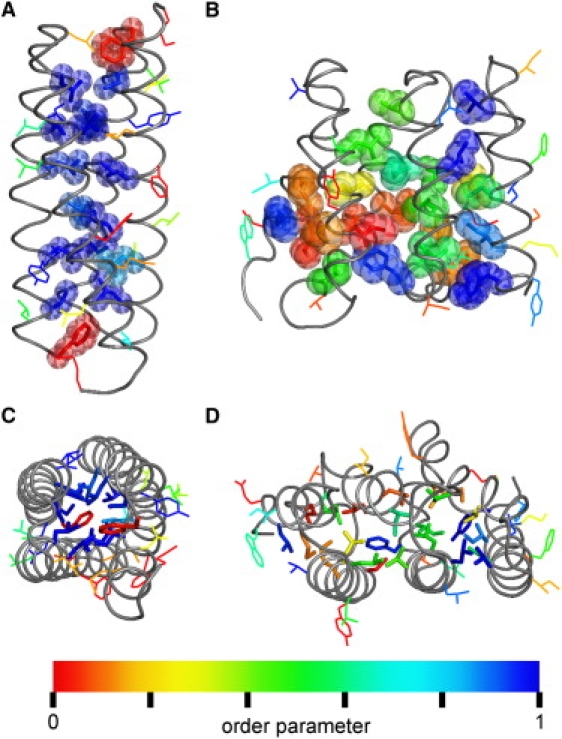
Comparison of the flexibility of hydrophobic side chains of Rop and yImpβ. (A and C) Side and top view of Rop. (B and D) Side and top view of HEAT repeats 4–6 from yImpβ. (Gray tubes) The protein backbone. Hydrophobic residues belonging to the respective hydrophobic core are shown as van der Waals spheres (A and B) or sticks (C and D), and as lines otherwise. The coloring reflects dihedral order parameters S2D (32). Values near 1 indicate rigid side chains (blue); low values reveal increased rotameric flexibility (red).
We therefore focused on the internal dynamics of the hydrophobic core as a second possible determinant for global yImpβ flexibility. The dihedral order parameter S2D (32) was used as a measure for the flexibility of the side chains (color-coded in Fig. 4). As can be seen, Rop core side chains are very rigid, i.e., they are confined to single conformers, whereas most yImpβ core side chains are rotationally nearly as flexible as surface-exposed side chains, despite their buried position.
This observation suggests that, by virtue of this molten globulelike conformational flexibility, the hydrophobic core of yImpβ can adapt to external forces, exerted by mechanical stress or binding interactions with other proteins. The mechanical properties of such an adaptable hydrophobic core are clearly seen in force-probe simulations, by comparing Rop and a succession of HEAT repeats from yImpβ. The Rop dimer did not display significant RMSD changes up to mechanical forces of 1200 pN, but then ruptured abruptly at a high force of 2900 pN. In contrast, yImpβ responded to mechanical stress gradually and in a controlled manner by small changes in the structure between HEAT repeats, as evidenced by stepwise small increases in backbone RMSD (Fig. 5).
Figure 5.
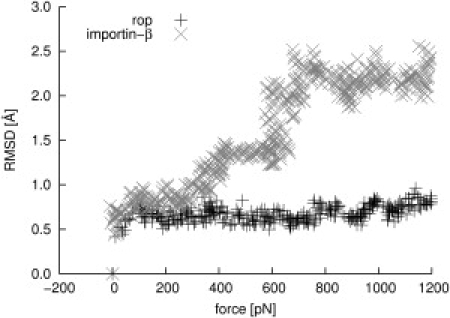
RMSD of Rop (shaded) and HEAT repeats 11 and 12 from yImpβ (solid) during stretching in force-probe MD simulations. For Rop, a moving harmonic potential was applied to the center-of-mass of both chains. For yImpβ, HEAT repeats 11–13 were simulated without connecting loops between HEAT repeats. A moving harmonic potential was applied to the centers-of-mass of HEAT repeats 11 and 13. In both cases, the harmonic potentials were moved perpendicular to the main longitudinal axis. The x axis shows the sum of the applied forces on both chains and HEAT repeats, respectively.
This behavior is also reflected in the energetics of the system. According to the modular composition of yImpβ, we first focused on energy contributions within and between adjacent HEAT repeats. Sequence-distal interactions were found not to contribute significantly to the overall energetics of the protein and were therefore not considered here.
Fig. 6 displays intrarepeat and interrepeat Coulombic and Lennard-Jones energies, respectively, as a function of the elongation of the protein, averaged over 0.1-nm intervals. For averaging, 250-ns equilibrium and ∼740-ns stretching simulations were merged, altogether totaling ∼990 ns of simulation. It is evident from Fig. 6 D that the largest contribution to the total increase in enthalpy upon elongation comes from Lennard-Jones interactions between neighboring HEAT repeats. By comparison, intrarepeat Lennard-Jones interactions (Fig. 6 B) as well as intra- and intermolecular Coulomb interactions (Fig. 6, A and C) only show relatively low increases with elongation.
Figure 6.
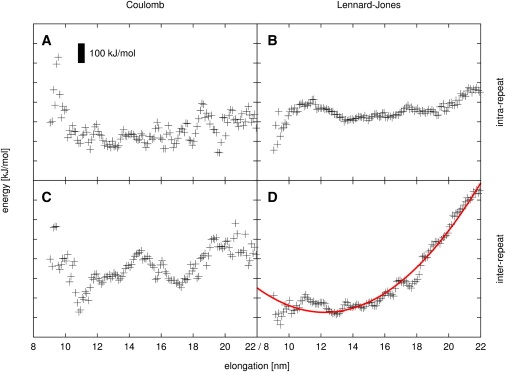
Dependence of enthalpic interactions in yImpβ on molecular elongation. (A) Intra-HEAT-repeat Coulombic interactions. (B) Intra-HEAT-repeat Lennard-Jones interactions. (C) Inter-HEAT-repeat Coulombic interactions. (D) Inter-HEAT-repeat Lennard-Jones interactions and harmonic fit (red line). (Black bar) Energy interval of 100 kJ/mol.
As the quadratic fit shows, the dominant interrepeat Lennard-Jones energy is well described by a harmonic potential with a spring constant of 23 × 10−3 N/m. When combined, all four energy contributions yield a spring constant of 47 × 10−3 N/m and an equilibrium elongation of 13.2 nm (blue line in Fig. 7 B, see below).
Figure 7.
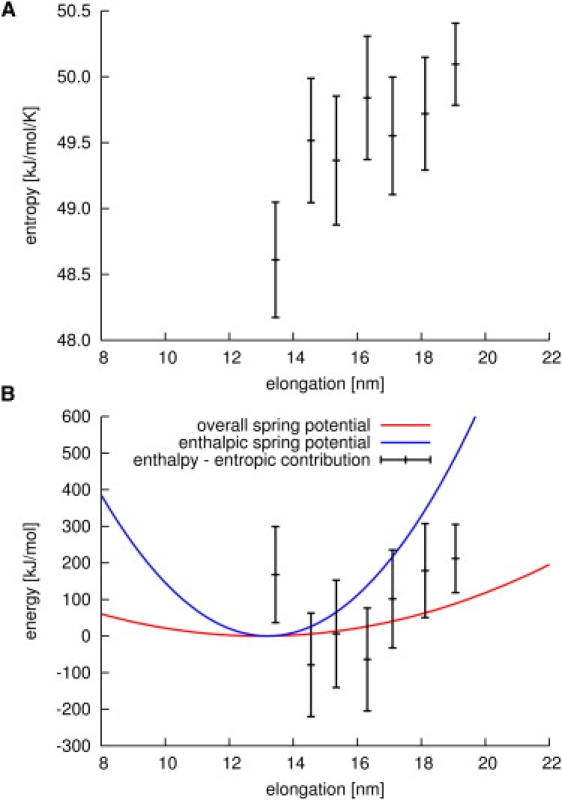
(A) Entropy estimation for different elongations of yImpβ. (B) Spring potential derived from purely enthalpic terms (blue line), and spring potential observed in equilibrium and stretching simulations (red line). The discrepancy between the two potentials is resolved when the entropic contribution to the free energy is subtracted from the purely enthalpic potential (black bars).
This equilibrium elongation agrees very well with the equilibrium end-to-end distance obtained from our free and stretching simulations. In contrast, the obtained spring constant is ∼4 times larger, which suggests that additional, compensating contributions must be relevant.
To identify these contributions, the change of entropy with elongation was estimated (Fig. 7 A), using the quasiharmonic approximation formulated by Schlitter (34). To this end, we carried out extended simulations, in which the end-to end-distance of yImpβ was kept fixed at different values, whereas motions on all other degrees of freedom of the protein were left unrestricted.
Fig. 7 A shows that, indeed, the entropy of yImpβ rises with increasing elongation of the protein. This finding is in contrast to the simple worm-like chain model of polymer chains, which would predict an entropy decrease. We attribute this rise in entropy to the unusual flexibility of the hydrophobic core discussed above, which, upon stretching, facilitates relaxation and leads to an enhanced flexibility of the entire protein.
Fig. 7 B shows that this rise in entropy indeed accounts for the discrepancy between the spring constant derived from purely enthalpic terms (blue line) and the overall spring constant observed for yImpβ in equilibrium and stretching simulations (red line). When the entropic contribution to the free energy (Fig. 7 A) is subtracted from the enthalpic part (Fig. 7 B, blue line), agreement within the error bars with the overall spring constant of yImpβ (Fig. 7 B, red line) is obtained (Fig. 7 B, black bars). In summary, the reversibly elastic, springlike behavior of yImpβ can be largely ascribed to contributions from Lennard-Jones interactions of the hydrophobic core between HEAT repeats and to substantial entropy changes associated with different degrees of elongation.
Conclusions
Our results characterize the α-solenoid yImpβ as a soft nanospring that tolerates up to twofold extensions without rupture of its hydrophobic core and is capable of fully reverting back to its original structure after stress release. Upon stretching, the hydrophobic core between HEAT repeats is able to adapt to the external changes by an internal rearrangement of hydrophobic residues, while the contact pattern between hydrophobic core residues is fully preserved. Our analysis of side-chain flexibilities suggests that yImpβ and, by similarity, also most other α-solenoid structures, exhibit a molten globulelike hydrophobic core and that this core structure underlies the flexibility of these proteins. Molten globule states are defined as possessing stable secondary structure elements and variable tertiary structure, as seen here, and they are assumed to differ from fully ordered states by relatively loose packing of the hydrophobic core, allowing side-chain rearrangements (11). Our simulations demonstrate that large conformational changes of the global structure of α-solenoids can be effected by the accumulation of many small changes between repeating secondary structure units. Alpha-solenoids thus bridge the structural gap between fully folded and intrinsically unfolded proteins both in terms of global structure and the mobility of core side chains (6–8,46). These unexpectedly highly flexible proteins also will deserve further studies addressing the question of whether conformational selection or induced fit is the predominant mechanism in their protein-protein binding pathways (47).
Acknowledgments
We thank Bostjan Kobe, Peter Hinterdorfer, and Ziv Reich for helpful discussions.
This work was partially funded by grant No. RGP53/2004 from the Human Frontier Science Program.
Supporting Material
References
- 1.Kajander T., Cortajarena A.L., Regan L. A new folding paradigm for repeat proteins. J. Am. Chem. Soc. 2005;127:10188–10190. doi: 10.1021/ja0524494. [DOI] [PubMed] [Google Scholar]
- 2.Hierro A., Rojas A.L., Hurley J.H. Functional architecture of the retromer cargo-recognition complex. Nature. 2007;449:1063–1067. doi: 10.1038/nature06216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 2007;8:195–208. doi: 10.1038/nrm2114. [DOI] [PubMed] [Google Scholar]
- 4.Fotin A., Cheng Y., Walz T. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature. 2004;432:573–579. doi: 10.1038/nature03079. [DOI] [PubMed] [Google Scholar]
- 5.Brohawn S.G., Partridge J.R., Schwartz T.U. The nuclear pore complex has entered the atomic age. Structure. 2009;17:1156–1168. doi: 10.1016/j.str.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kobe B., Kajava A.V. When protein folding is simplified to protein coiling: the continuum of solenoid protein structures. Trends Biochem. Sci. 2000;25:509–515. doi: 10.1016/s0968-0004(00)01667-4. [DOI] [PubMed] [Google Scholar]
- 7.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 8.Zachariae U., Grubmüller H. Importin-β: structural and dynamic determinants of a molecular spring. Structure. 2008;16:906–915. doi: 10.1016/j.str.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Ptitsyn O.B., Pain R.H., Razgulyaev O.I. Evidence for a molten globule state as a general intermediate in protein folding. FEBS Lett. 1990;262:20–24. doi: 10.1016/0014-5793(90)80143-7. [DOI] [PubMed] [Google Scholar]
- 10.Tsou C.L. Inactivation precedes overall molecular conformation changes during enzyme denaturation. Biochim. Biophys. Acta. 1995;1253:151–162. doi: 10.1016/0167-4838(95)00172-5. [DOI] [PubMed] [Google Scholar]
- 11.Ptitsyn O.B. How the molten globule became. Trends Biochem. Sci. 1995;20:376–379. doi: 10.1016/s0968-0004(00)89081-7. [DOI] [PubMed] [Google Scholar]
- 12.Vamvaca K., Vögeli B., Hilvert D. An enzymatic molten globule: efficient coupling of folding and catalysis. Proc. Natl. Acad. Sci. USA. 2004;101:12860–12864. doi: 10.1073/pnas.0404109101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neuman K.C., Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods. 2008;5:491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serquera D., Lee W., Itzhaki L.S. Mechanical unfolding of an ankyrin repeat protein. Biophys. J. 2010;98:1294–1301. doi: 10.1016/j.bpj.2009.12.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Werbeck N.D., Rowling P.J.E., Itzhaki L.S. Shifting transition states in the unfolding of a large ankyrin repeat protein. Proc. Natl. Acad. Sci. USA. 2008;105:9982–9987. doi: 10.1073/pnas.0705300105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rief M., Pascual J., Gaub H.E. Single molecule force spectroscopy of spectrin repeats: low unfolding forces in helix bundles. J. Mol. Biol. 1999;286:553–561. doi: 10.1006/jmbi.1998.2466. [DOI] [PubMed] [Google Scholar]
- 17.Lee G., Abdi K., Marszalek P.E. Nanospring behavior of ankyrin repeats. Nature. 2006;440:246–249. doi: 10.1038/nature04437. [DOI] [PubMed] [Google Scholar]
- 18.Andrade M.A., Bork P. HEAT repeats in the Huntington's disease protein. Nat. Genet. 1995;11:115–116. doi: 10.1038/ng1095-115. [DOI] [PubMed] [Google Scholar]
- 19.Liu S.M., Stewart M. Structural basis for the high-affinity binding of nucleoporin Nup1p to the Saccharomyces cerevisiae importin-β homologue, Kap95p. J. Mol. Biol. 2005;349:515–525. doi: 10.1016/j.jmb.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Conti E., Müller C.W., Stewart M. Karyopherin flexibility in nucleocytoplasmic transport. Curr. Opin. Struct. Biol. 2006;16:237–244. doi: 10.1016/j.sbi.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Cingolani G., Petosa C., Müller C.W. Structure of importin-β bound to the IBB domain of importin-α. Nature. 1999;399:221–229. doi: 10.1038/20367. [DOI] [PubMed] [Google Scholar]
- 22.Lee S.J., Matsuura Y., Stewart M. Structural basis for nuclear import complex dissociation by RanGTP. Nature. 2005;435:693–696. doi: 10.1038/nature03578. [DOI] [PubMed] [Google Scholar]
- 23.van der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 24.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 25.Jorgensen W.L., Tirado-Rives J. The OPLS potential functions for proteins. Energy minimizations for crystals of cyclic-peptides and crambin. J. Am. Chem. Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 26.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Comput. Phys. 1983;79:926–935. [Google Scholar]
- 27.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 28.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 29.Darden T., York D., Pedersen L. Particle mesh Ewald: an nlog(n) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 30.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald potential. J. Chem. Phys. 1995;103:8577–8592. [Google Scholar]
- 31.Grubmüller H., Heymann B., Tavan P. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 32.van der Spoel D., Berendsen H.J.C. Molecular dynamics simulations of Leu-encephalin in water and DMSO. Biophys. J. 1997;72:2032–2041. doi: 10.1016/S0006-3495(97)78847-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- 34.Schlitter J. Estimation of absolute and relative entropies of macromolecules using the covariance matrix. Chem. Phys. Lett. 1993;6:617–621. [Google Scholar]
- 35.Banner D.W., Kokkinidis M., Tsernoglou D. Structure of the ColE1 Rop protein at 1.7 Å resolution. J. Mol. Biol. 1987;196:657–675. doi: 10.1016/0022-2836(87)90039-8. [DOI] [PubMed] [Google Scholar]
- 36.Zachariae U., Grubmüller H. A highly strained nuclear conformation of the exportin Cse1p revealed by molecular dynamics simulations. Structure. 2006;14:1469–1478. doi: 10.1016/j.str.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 37.Sotomayor M., Corey D.P., Schulten K. In search of the hair-cell gating spring elastic properties of ankyrin and cadherin repeats. Structure. 2005;13:669–682. doi: 10.1016/j.str.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 38.Grinthal A., Adamovic I., Kleckner N. PR65, the HEAT-repeat scaffold of phosphatase PP2A, is an elastic connector that links force and catalysis. Proc. Natl. Acad. Sci. USA. 2010;107:2467–2472. doi: 10.1073/pnas.0914073107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bustamante C., Smith S.B., Smith D. Single-molecule studies of DNA mechanics. Curr. Opin. Struct. Biol. 2000;10:279–285. doi: 10.1016/s0959-440x(00)00085-3. [DOI] [PubMed] [Google Scholar]
- 40.Ivanovska I.L., de Pablo P.J., Wuite G.J. Bacteriophage capsids: tough nanoshells with complex elastic properties. Proc. Natl. Acad. Sci. USA. 2004;101:7600–7605. doi: 10.1073/pnas.0308198101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zink M., Grubmüller H. Mechanical properties of the icosahedral shell of southern bean mosaic virus: a molecular dynamics study. Biophys. J. 2009;96:1350–1363. doi: 10.1016/j.bpj.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andrade M.A., Petosa C., Bork P. Comparison of ARM and HEAT protein repeats. J. Mol. Biol. 2001;309:1–18. doi: 10.1006/jmbi.2001.4624. [DOI] [PubMed] [Google Scholar]
- 43.Kresse H.P., Czubayko M., Bloecker H. Four-helix bundle topology re-engineered: monomeric Rop protein variants with different loop arrangements. Protein Eng. 2001;14:897–901. doi: 10.1093/protein/14.11.897. [DOI] [PubMed] [Google Scholar]
- 44.Munson M., Balasubramanian S., Regan L. What makes a protein a protein? Hydrophobic core designs that specify stability and structural properties. Protein Sci. 1996;5:1584–1593. doi: 10.1002/pro.5560050813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Willis M.A., Bishop B., Brunger A.T. Dramatic structural and thermodynamic consequences of repacking a protein's hydrophobic core. Structure. 2000;8:1319–1328. doi: 10.1016/s0969-2126(00)00544-x. [DOI] [PubMed] [Google Scholar]
- 46.Gsponer J., Babu M.M. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009;99:94–103. doi: 10.1016/j.pbiomolbio.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 47.Lange O.F., Lakomek N.A., de Groot B.L. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 48.Mitrousis G., Olia A.S., Cingolani G. Molecular basis for the recognition of snurportin 1 by importin β. J. Biol. Chem. 2008;283:7877–7884. doi: 10.1074/jbc.M709093200. [DOI] [PubMed] [Google Scholar]
- 49.Lee S.J., Sekimoto T., Yoneda Y. Crystal structure of importin-β and SREBP-2 complex. Science. 2003;302:1513–1514. doi: 10.1126/science.1088372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.