

Abstract
Charged amino acids such as Arginine play important roles in many membrane-mediated biological processes such as voltage gating of ion channels and membrane translocation of cell penetration peptides. It is well established that local membrane deformation and formation of water defects are crucial to the stabilization of charged species in contact with the membrane, which suggests that mechanical properties of the membrane are relevant although a clear connection has not been established. As a quantitative measure, we study how changes in the composition and therefore mechanical properties of a lipid bilayer influence the pKa of Arg in the membrane center using free energy simulations. Compared to previous studies in a single-component lipid bilayer containing saturated lipids or lipids with a modest degree of unsaturation, substantially larger pKa shifts are observed in the presence of highly unsaturated lipid tails and cholesterol. Moreover, the underlying molecular mechanisms for the pKa perturbation are distinct in different systems, with the unsaturated lipid tails mainly destabilizing the charged state of Arg and the cholesterol stabilizing the neutral state of Arg. The observed behaviors in both cases are at odds with predictions based on mechanical considerations at a mesoscopic level—highlighting that, while mechanical considerations are useful for stimulating hypothesis, their applicability to dissecting phenomena at the molecular-length scale is rather limited.
Introduction
Although continuum mechanics and continuum electrostatic models have played important roles in understanding many biological processes that involve large-scale deformation of lipid membranes (1–5), studies that are more recent have highlighted the importance of local adjustments in lipid membrane and lipid/protein interface. One of the much-discussed subjects in this regard in recent literature concerns the mechanism and energetics of moving a charged group through a lipid membrane. A pioneering continuum electrostatics model (6) that treated the lipid membrane as a low-dielectric slab found an immense energy barrier of ∼40 kcal/mol. However, Hessa et al. (7,8) recently proposed a new biological hydrophobicity scale based on a translocon assay, in which the transfer free energy of charged residues into the lipid membrane is much smaller than the prediction of the dielectric model, although uncertainties remain regarding the proper interpretation of the results due to the complex nature of the translocon assay (9–11). Atomistic simulations using the voltage-sensing domain of a potassium channel (12) have highlighted that the membrane can locally deform and create water defects, stabilizing charged residues that are in close contact with the hydrophobic region of the lipid. Understanding of this local deformation has been further refined recently via combined molecular dynamics (MD) simulations, solid-state NMR, and neutron diffraction experiments (13). An interesting continuum model (14), featuring a continuum membrane that can locally deform, was shown to be able to reproduce the free energy profile for the translocation of a charged amino acid through lipid membrane (15).
These recent discussions suggest that a change in the mechanical properties of the lipid membrane can potentially have a major impact on the energetics of processes that implicate charged amino acids in a membrane environment. However, because the deformation associated with the water defects occurs on a molecular scale (as opposed to large-scale membrane deformation implicated in, for example, mechanosensation (2) or membrane remodeling (16)), the magnitude of the mechanical effect is not clear. To shed light into this subject, which has potentially broad relevance to biomembrane-mediated processes, we have chosen to investigate a specific problem—i.e., whether changes in the composition, and therefore mechanical properties of a lipid bilayer, can significantly shift the microscopic pKa of a charged residue, such as arginine (Arg).
Because Arg residues play an important role in the voltage-sensing domain of potassium channels (17), cell penetration peptides (18–20) (and in an innovative approach for probing structural changes in membrane proteins (21), the pKa of Arg in different regions of a lipid membrane) have been studied by several recent MD simulations (22–26). These employed different computational protocols (e.g., alchemy thermodynamic integration (26) versus potential of mean force simulations (22–25)) and different force fields. It is encouraging that results of these studies are in semiquantitative agreement, showing that although the pKa of an Arg is substantially lowered in the membrane, the magnitude of the shift is fairly modest such that even in the middle of the bilayer, the pKa is (slightly) above 7.0. In other words, local membrane deformation (i.e., formation of water defects) provides sufficient stabilization that an Arg likely remains protonated even in the middle of the bilayer.
All these previous simulations, however, studied an Arg (or its analog) in a single-component zwitterionic lipid bilayer with either fully saturated lipid tails (DPPC (22,23,26)) or a modest degree of unsaturation (DOPC (24,25)). Realistic cellular membranes, however, are much more complex in composition (27,28) and therefore differ in mechanical properties (i.e., the energy cost for deformation). For example, a common component in many cellular membranes is cholesterol, which is known to increase the bending moduli of lipid bilayers (29); mixing lipids with saturated and unsaturated tails also modulates the structural (30) and mechanical properties (31) of membranes. Therefore, studying the dependence of Arg's pKa on the membrane composition provides a well-defined and realistic example of how membrane heterogeneity modulates the energetics of membrane-mediated processes that involve charged species. Moreover, the results can be used to quantitatively test or improve sophisticated continuum models (14,32) or coarse-grained particle models of membranes (33,34), thereby facilitating the development of computational models that can potentially bridge a broad range of length scales involving biomembranes (2,4,35).
In the following, we present our Methods, concentrating primarily on the systems setup for monitoring the convergence of thermodynamic integration simulations. We then present Results and Discussions. Finally, we offer our Conclusions. The main finding of the work is that the pKa of Arg in the membrane can be substantially lowered by highly unsaturated lipid tails and cholesterol—a prediction that we hope will stimulate experimental investigations. Interestingly, the underlying molecular mechanisms for the pKa perturbation are distinct in different systems, with the unsaturated lipid tails mainly destabilizing the charged state of Arg, while cholesterol stabilizes the neutral state of Arg. The observed behaviors in both cases are also at odds with predictions based on mechanical considerations at a mesoscopic level, which highlights that while mechanical considerations are useful for stimulating hypothesis, their applicability to dissecting phenomena at the molecular length-scale is rather limited.
Methods
System setup and molecular dynamics protocols
To study effects of lipid types and cholesterol on the protonation state of an Arg analog in the center of membrane, pKa shift of the Arg analog is calculated in five bilayer systems: pure 1,2-di-palmitoyl-sn-glycero-3-phosphatidylcholine (DPPC) and 1-stearoyl-2-docosahexaenoyl-sn-glycero-3-phosphatidylcholine (SDPC), and SDPC/1-stearoyl-2-docosahexaenoyl-sn-glycero-3-phosphatidylethanolamine (SDPE) mixtures with 0, 20, and 40% cholesterol (see Fig. 1 for structures).
Figure 1.
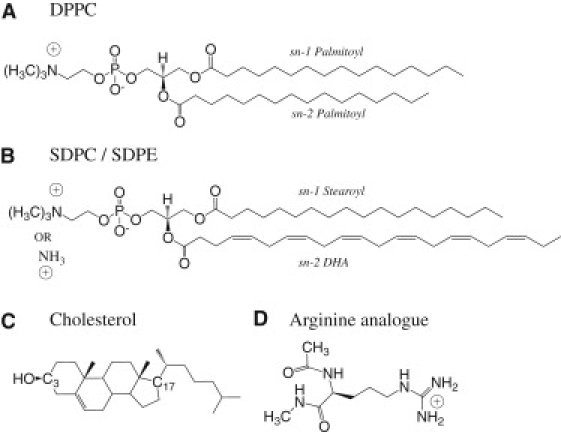
Chemical structures of lipids and the Arginine analog considered in this work. (A) 1,2-di-palmitoyl-sn-glycero-3-phosphatidylcholine (DPPC). (B) 1-stearoyl-2-docosahexaenoyl-sn-glycero-3-phosphatidylcholine (SDPC) and 1-stearoyl-2-docosahexaenoyl-sn-glycero-3-phosphatidylethanolamine (SDPE). (C) Cholesterol. (D) Arginine analog.
The CHARMM force field (36,37) is used to describe all components of the system, including the Arg analog (22) and docosahexaenoic acid (DHA) (38). All the simulations are carried out using the modified GROMACS 3.3.3 package (39). An Arg analog is restrained to the center-of-membrane using a harmonic force constant of 1000 kJ/mol/nm2 with the PULL module of GROMACS. The DPPC and SDPC systems are simulated in the NPAT ensemble (40) under two conditions of constant area-per-lipid and temperature: 64 Å2, 323 K, and 70 Å2 (41), 311 K, respectively. The mixed SDPC/SDPE lipid bilayer systems with 0, 20, and 40% cholesterol are simulated under a constant surface-tension, ∼25 dyne/cm, and temperature, 311 K (NPγT); the area-per-lipid for the cholesterol-free system is 69.2 ± 0.2 Å2, which is close to available experimental value for a pure SDPC bilayer (41). In addition, SDPC and SDPC/SDPE systems are simulated with a higher surface-tension, ∼38 dyne/cm, which is a value commonly used with the CHARMM lipid force field (37,42) and which corresponds to a higher area-per-lipid, ∼77 Å2. See the Supporting Material for details.
Thermodynamic integration calculations and convergence tests
We use thermodynamic integration to compute the pKa shift of an Arg in the membrane center relative to bulk solution, where the charged guanidinium group is gradually switched to a neutral one using a coupling parameter λ; i.e., the guanidinium group is fully charged at λ = 0 and neutral at λ = 1. The charging free energy is calculated by integrating the corresponding free energy derivative with respective to λ,
| (1) |
where 〈…〉λ indicates an ensemble average at a given λ – window and ΔU = U0 – U+ is the instantaneous energy gap between the charged and neutral forms of Arg in a given environment. For the system size used in this study, corrections for finite-size and Ewald summation for systems with a net charge were found to be small (26,43). The pKa shift relative to the bulk clearly is related to the difference in the charging free energy in the membrane center and in bulk solution,
| (2) |
where we use the double-approximate sign (≈) to highlight the key assumption that the pKa shift is dictated by the difference in charging free energy in different environments, which is likely valid (44,45). In Results and Discussions, the pKa results are presented in terms of absolute pKa values, which are obtained by adding the calculated pKa shifts to the experimental pKa of Arg in solution (12.5) (46).
As highlighted in many recent microscopic pKa calculations (26,45,47,48), it is important to carefully monitor the convergence of thermodynamic integration calculations. This is particularly important for the current problem because the titration process potentially implicate several slow processes such as cholesterol flip-flop, water penetration into the membrane, and lateral diffusion of lipids. Accordingly, most simulation windows are in the range of 100–200 ns (see the Supporting Material and Table 1 for details).
Table 1.
Results of thermodynamic integration for the calculation of pKa of an Arg analog in the membrane center
| Δ∂G/∂λ (kcal/mol)/sampling time (ns)∗ | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Lipid | Chol. † | Temp.‡ |
|
λ = 0.0 | 0.5 | 1.0 | −ΔΔGchg¶ | pKa¶ | |
| DPPC‖ | 0 | 323 | 64 | −9.5 (0.7)/18 | −13.9 (1.0)/29 | 14.3 (1.3)/42 | −6.9 (1.0) | 7.7 (0.7) | |
| DPPC | 0 | 323 | 64 | −7.8 (0.8)/65 | −12.5 (1.7)/111 | 10.4 (3.2)/108 | −5.6 (1.4) | 8.6 (0.9) | |
| SDPC | 0 | 311 | 70 | −12.1 (1.7)/118 | −20.0 (1.6)/138 | 11.8 (2.0)/154 | −10.1 (1.2) | 5.3 (0.9) | |
| SDPC/E | 0 | 311 | 70 | −12.4 (1.2)/114 | −22.4 (2.5)/145 | 9.2 (3.4)/282 | −12.0 (1.8) | 3.9 (1.2) | |
| SDPC/E | 20 | 311 | — | −11.6 (1.6)/188 | −17.3 (1.3)/128 | −2.6 (2.8)/140 | −12.2 (1.2) | 3.8 (0.9) | |
| SDPC/E | 40 | 311 | — | −11.9 (0.9)/95 | −23.3 (2.4)/148 | −5.4 (2.1)/110 | −16.0 (1.4) | 1.2 (1.0) | |
| SDPC | 0 | 311 | 77 | −10.2 (1.5)/92 | −20.8 (1.6)/113 | 10.4 (2.7)/137 | −10.4 (1.3) | 5.1 (0.9) | |
| SDPC/E | 0 | 311 | 77 | −9.8 (3.1)/143 | −18.4 (1.4)/171 | 11.1 (2.0)/213 | −8.9 (1.4) | 6.1 (1.0) | |
| SDPC/E | 20 | 311 | — | −15.2 (1.4)/135 | −19.3 (1.5)/133 | 8.5∗∗ (3.4)/186 | −11.3 (1.4) | 4.5 (1.0) | |
| SDPC/E | 40 | 311 | — | −14.2 (1.8)/126 | −20.5 (1.4)/111 | 0.9 (5.0)/183 | −13.6 (1.8) | 2.9 (1.3) | |
Differences in the free-energy derivatives (Eq. 1) in the membrane center relative to bulk solvent, Δ∂G/∂λ = ∂G/∂λcenter – ∂G/∂λsolvent, are shown. Numbers in parentheses are statistical errors determined by block-averaging (57).
Cholesterol concentration (%).
Temperature (K).
Area-per-lipid (Å2).
ΔΔGchg is computed by integrating Δ∂G/∂λ over λ; pKa is computed based on Eq. 2 and the solution reference value of 12.5 (46).
Thermodynamic integration results from our previous work (26), with a different set of force-field (OPLS) parameters for the neutral state of Arg.
See Effects of Cholesterol.
Results and Discussions
In the following, we systematically explore the effects of several factors on the computed pKa of an Arg in the center of the membrane bilayer. Due to the complexity of the problem, sampling remains a challenging issue (especially in the presence of cholesterol), despite the fact that most windows have been sampled with >100 ns of molecular dynamics (MD) simulations. Therefore, considerable uncertainty remains regarding the computed pKa values. Nevertheless, by comparing different systems and investigating trends in the free energy derivatives, meaningful conclusions can be drawn regarding factors that contribute most significantly to the pKa of Arg in the membrane.
Effects of force-field parameters for Arg
First, we briefly comment on the effect of using a slightly different set of force-field parameters for the neutral state of the Arg analog in this study. In our recent study (26), we have adopted partial charges from the OPLS (49) force field for the neutral state; this was motivated by the observation that partial charges for the charged guanidinium group in CHARMM and OPLS force fields are identical. In this work, we have used the set of parameters newly developed by Li et al. (22) for the neutral Arg side chain; this set was developed in the framework of the CHARMM 22 force field (36) and therefore was expected to be more compatible with the lipid force fields used here. As shown in Table 1, the effect of using different force fields for the neutral state of Arg has a relatively minor effect on the computed pKa. The free energy derivatives become slightly lower by 1–2 kcal/mol at all λ-values and the computed pKa at the membrane center is 8.6, as compared to the value of 7.7 in our recent work (26) and the value of ∼8.0 in the work of Li et al. (22). This result again supports that Arg remains protonated even in the membrane center, due to stabilization from lipid headgroups and water molecules in a locally deformed DPPC bilayer.
Effects of tail unsaturation: DPPC versus SDPC
As shown by their chemical structures in Fig. 1, SDPC and DPPC have identical headgroups but different tails; while DPPC has fully saturated tails (C16:0), SDPC tails are longer and have a high degree of unsaturation (C22:6 for DHA; C18:0 for stearoyl acid). Accordingly, an SDPC bilayer and a DPPC bilayer have two major differences. On the one hand, a SDPC bilayer is slightly thicker by ∼2–3 Å (e.g., see Fig. S4 in the Supporting Material), which makes the membrane center less accessible to solvent. On the other hand, a SDPC bilayer is likely more deformable than a DPPC bilayer; for example, polyunsaturation of lipid tails is known to decrease the bending modulus of a membrane by more than a factor of two (31). Therefore, depending on which factor dominates, one may expect either a larger or a smaller energetic penalty associated with the formation of water defects for stabilizing a net charge in the membrane, which would lead to a higher or lower pKa for an Arg in an SDPC bilayer than in a DPPC bilayer.
As shown in Table 1, regardless of the surface-tension (and therefore area-per-lipid), the calculated pKa of Arg in the membrane center in an SDPC bilayer is substantially reduced compared to that in a DPPC bilayer; the shift is >3 pKa units and suggests that Arg is much more likely deprotonated in a SDPC bilayer. The free energy derivatives in Table 1 from different SDPC/Arg simulations further suggest that the major contributions to the larger pKa shift come from λ-windows with the charged Arg (λ = 0, 0.5). In Fig. 2, we compare the cumulative number of phosphate oxygen and water oxygen near Cζ in Arg from charged(λ = 0)/neutral(λ = 1) state simulations with DPPC and SDPC bilayers. In the charged (λ = 0) window, the Arg+ is stabilized by water oxygen and phosphate oxygen in the water defect in both DPPC and SDPC bilayers. However, the size and composition of the water defect are different in the two bilayers. The water defect in a DPPC bilayer has more water but less phosphate groups compared to the SDPC case (Fig. 2, A and B); in total, the water defect in a DPPC bilayer has ∼3–5 more oxygen atoms within 8 Å compared to that in a SDPC bilayer, regardless of surface-tension (Fig. 2 C and Fig. S3 c). As expected, the number of polar species in the water defect is highly correlated to ∂G/∂λ; as shown in Fig. 3, A and B, the larger the water defect, the smaller drop in ∂G/∂λ compared to the bulk solution value.
Figure 2.
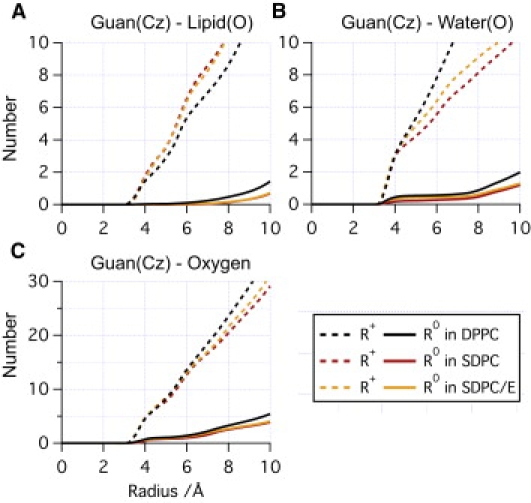
Cumulative radial distribution functions for (A) lipid oxygen, (B) water oxygen, and (C) total oxygen with respect to the guanidinium Cζ in three systems without cholesterol. Note that more water molecules are in contact with Arg+ in DPPC, while a smaller number of lipid headgroups are in contact with Arg+ in the bilayer with saturated hydrocarbon tails (DPPC) than in bilayers with polyunsaturated tails (SDPC and SDPC/E). In all figures in the main text, for the SDPC-containing systems, the results shown are from low surface-tension simulations; the corresponding data at high surface-tension are included in the Supporting Material.
Figure 3.
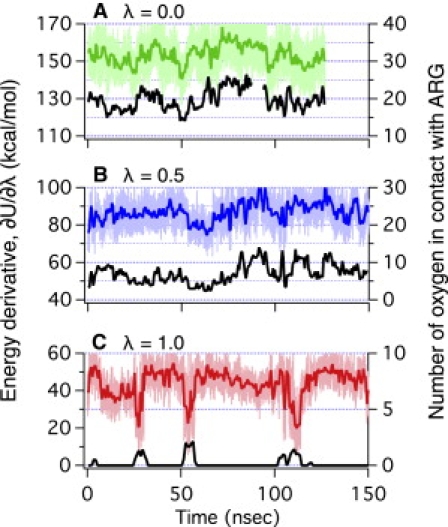
(A–C) Energy gap (in color, ΔU = U0 – U+ in Eq. 1) and the number of oxygen (in black) in the vicinity (≤8 Å) of Arg Cζ at three charge-states as a function of time in the SDPC system under low surface-tension. Note that the energy gap is positively correlated to the number of oxygen at charged states (λ = 0.0, 0.5), while it is negatively correlated at the neutral state (λ = 1.0).
The key question is then what causes the smaller size of the water defect in a SDPC bilayer compared to a DPPC bilayer, which seems contradictory to the higher degree of deformability of a SDPC bilayer due to the unsaturated lipid tails. Although an SDPC bilayer is slightly thicker than a DPPC bilayer, this is not an important factor because the water defect is even smaller in a SDPC bilayer at high surface-tension (not shown). Important clues come from the cylindrically averaged number density plot of lipid phosphorus, tail carbon, and water oxygen at ∼Cζ (Fig. 4), which indicates that not only lipid headgroup density but also tailgroup density is higher near Arg+ in a SDPC bilayer than in a DPPC bilayer. Examination of snapshots from the simulations (e.g., Fig. 4 D) suggests that it is mainly the DHA of SDPC that wraps around Arg+. Although calculation of binding affinity of lipids to an Arg analog embedded in the membrane as a function of the degree of tail-unsaturation is beyond the scope of this study, one can reasonably expect that poly-unsaturation increases the binding affinity of lipids because it enhances the space-filling ability of lipids. More importantly, the higher binding affinity of poly-unsaturated lipid tails to the Arg analog increases the density of headgroups near the Arg analog, and replacement of water with headgroups decreases the total oxygen number near the Arg analog, as shown in the radial distribution functions in Fig. 2. As a result, ∂G/∂λ values at λ = 0 or 0.5 are smaller in an SDPC bilayer than in a DPPC bilayer, which leads to drops in Arg pKa by as much as ∼3–4 units.
Figure 4.
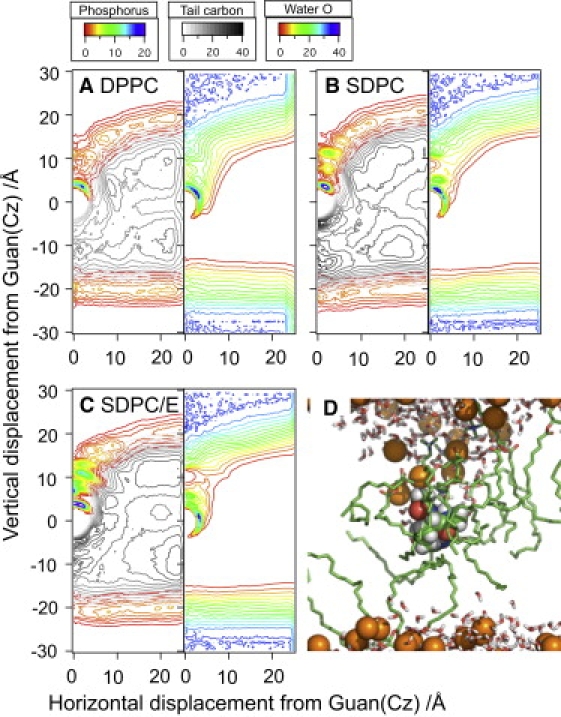
(Color online) Number density maps (in the unit of nm−3) of phosphorus/hydrocarbon (left) and water oxygen (right) in three cholesterol-free bilayers (A) DPPC, (B) SDPC, and (C) SDPC/E with a fully charged Arg at the origin. Note that both head- and tailgroups of unsaturated lipids have higher affinity to Arg+ than those of DPPC. Consequently, there are smaller numbers of water molecules in the water defect in SDPC and SDPC/E bilayers than in a DPPC bilayer, which is consistent with the radial distribution plots in Fig. 2. (D) Typical configuration of SDPC with its DHA wrapping around Arg+. (Green) Lipids in contact with Arg+ (hydrogen not included for clarity). (Orange spheres) Phosphorus in the headgroup.
Overall, these results support the argument that the larger pKa shift for an Arg in an SDPC bilayer than in a DPPC bilayer is due mainly to the mechanical flexibility of the poly-unsaturated tails in SDPC. However, the way that mechanical flexibility works on a molecular level is completely different from the expectation based on a mesoscopic consideration. The flexible DHA tails enhance lipid packing around Arg+ and this effect has little to do with mesoscopic mechanical properties such as bending modulus, which would lead to the incorrect prediction that Arg+ is better stabilized in a more deformable SDPC bilayer. For example, Choe et al. (14) predicted pKa ∼7 in the center of membrane with mechanical parameters from a typical saturated bilayer using their novel model that combines continuum mechanics and continuum electrostatics; the membrane deformation energy with a charged Arg in the center was estimated to be ∼5 kcal/mol. Because mechanical components and nonmechanical components are decoupled in their model, one can reasonably assume that changes in mechanical parameters (e.g., bending moduli) affect only the mechanical deformation energy. In addition, one expects that the mechanical deformation energy is largely negligible with a neutral Arg in the membrane center, given that the degree of membrane deformation is small. Therefore, the transfer free-energy of Arg+ can decrease, at most, 5 kcal/mol, while that of a neutral Arg is almost constant, with a greater number of flexible membranes. This change corresponds to an increase in pKa by <4 units, which is of the opposite sign to the current result from microscopic simulations.
Effects of headgroup type: SDPC versus SDPC/E
As shown in Fig. 1, the difference between SDPC and SDPE is relatively minor, with SDPE having a smaller headgroup (-NH3+ rather than -NMe3+). Therefore, mixing SDPE with SDPC is not expected to lead to any large change in the membrane mechanical properties or thickness (and therefore, in the pKa of an Arg in the membrane center). This is indeed observed in the free energy simulations. As shown in Table 1, the computed pKa values for an Arg in SDPC and SDPC/E bilayers differ by ∼1 pKa unit, which is not significant considering the statistical errors associated with the current simulations. For example, the difference in pKa at low surface-tension results from the free energy derivatives at neutral (λ = 1) states; although ∂G/∂λ in SDPC and SDPC/E shows similar trends before 100 ns, only that in SDPC/E has a period when low ∂U/∂λ appears frequently for ∼70 ns (compare panels b and c of Fig. S1). This occurs because one or two water molecules penetrate into the membrane and remain near Arg0 for a long time (>5 ns), whereas most water molecules have a resident time of <200 ps. Those localized water molecules stabilize a neutral Arg more than they do a charged Arg, which leads to a lower pKa, as will be discussed further in the next section.
Effects of cholesterol
With cholesterol molecules in the membrane, it is well established that the lipid tails tend to be more ordered, that the bending modulus of the membrane becomes enhanced, and that the membrane thickens (50,51). Therefore, it is expected that adding cholesterol can further increase the pKa shift of Arg in the membrane center, due presumably to additional energy penalty for the local membrane deformation and water defect formation in the charged state of Arg.
The current free energy simulations, with up to 40% cholesterol in a SDPC/E bilayer, have shown that the pKa of Arg in the membrane center indeed tends to further decrease compared to cholesterol-free cases—although the magnitude is somewhat modest, especially considering the statistical uncertainty that remains in our 100–200 ns per λ-state simulations. Nevertheless, a striking observation is that the free energy derivatives for the charged windows (λ = 0, 0.5) do not tend to change significantly in the presence of cholesterol, while the free energy derivatives for the neutral state (λ = 1.0) consistently (except for 20% cholesterol with a high surface-tension, see Conclusions below) show dramatic decreases (>10 kcal/mol) relative to cholesterol-free cases. This strongly suggests that the effect of cholesterol on Arg pKa is not mechanical in nature and warrants further analysis.
First, we examine the time-dependence of the free energy derivative contribution, which is the energy gap between the neutral and charged state of Arg in the presence of interactions with the environment (i.e., ΔU = U0 – U+ in Eq.1). As shown in Fig. S1, c–e, the energy gap is rather well behaved for λ = 0, 0.5 for both cholesterol-free and cholesterol-rich membrane simulations. By contrast, the energy gap for the neutral-Arg (λ = 1) exhibits considerably larger fluctuations and occasional jumps, especially with cholesterol present; along this line, requiring the energy gap data to satisfy all four statistical tests (see the Supporting Material) is crucial for ensuring that simulations are sufficiently long to average over a significant number of jumps. Further analysis of the simulation results suggests that these large variations in the energy gap are correlated with changes in the number of polar groups (e.g., oxygen in water and hydroxyl in cholesterol) in contact with the neutral Arg. With a neutral Arg in the membrane, the degree of local membrane deformation is substantially smaller than with a charged Arg; as a result, the number of polar groups in contact with the Arg changes much more rarely for λ = 1 compared to the number of polar groups for the charged states (λ = 0, 0.5), which is why the λ = 1 window takes substantially longer to converge than other windows. In the presence of cholesterol, the number of water molecules in contact with the neutral Arg tends to be larger (see below), which leads to more dramatic variations in the energy gap.
The importance of polar groups, such as water, in contact with the neutral Arg on the free energy derivative can be further illustrated by the distribution of the energy gap. As shown in Fig. 5, the energy gap distribution is approximately Gaussian (45,52) for the λ = 0 window regardless of cholesterol concentration; moreover, the center of the distribution is nearly independent of the cholesterol concentration. These observations suggest that the charged state of Arg is well stabilized by the local deformation of the membrane, with or without cholesterol, and that the environment of the charged Arg equilibrates rather quickly on the simulation timescale. By contrast, for the neutral state (λ = 1), the energy gap distributions are highly non-Gaussian, especially for high cholesterol concentrations, which indicate the presence of multiple environments of the neutral Arg. A simple decomposition of the distributions confirms that, as the cholesterol concentration increases, the number of environmental states (which correspond to different numbers of polar groups in contact with the neutral Arg) also increases.
Figure 5.
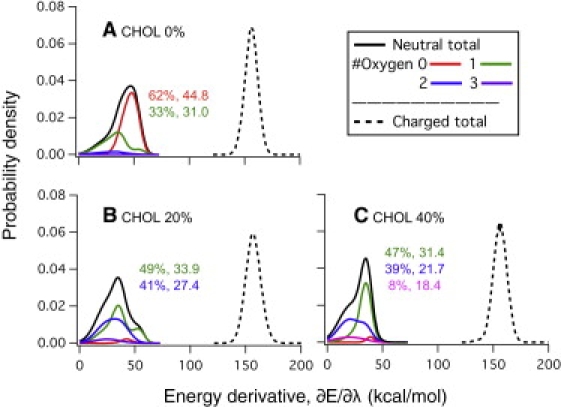
(A–C) Histograms of the energy gap, ∂U/∂λ = U0 – U+, at the charged and neutral Arg states in SDPC/E systems (dotted and solid lines, respectively). Histograms at the neutral state, which deviate significantly from the Gaussian distribution, are further decomposed into contributions from substates by the number of oxygen atoms in contact with the guanidinium group; the population and the average ∂U/∂λ of each substate are given in the corresponding color. Note that the population for the substate in which the guanidinium group has no contact with oxygen decreases significantly as the cholesterol concentration increases while the average ∂U/∂λ for the substates is largely independent of cholesterol concentration.
To help provide further insights into the structural characterization of the neutral Arg's environment, we examine both cumulative radial distribution functions (Fig. 6) and number density plots (Fig. 7) for key groups around the guanidium of Arg. As shown in Fig. 6 A, the distribution of phosphate groups near the guanidium (<10 Å) is largely independent from cholesterol, regardless of the charge state of Arg. The numbers of water molecules differ a bit in different simulations (Fig. 6 B), although the total number of oxygen atoms (including water, cholesterol hydroxyl, and lipid phosphate and ester groups) is remarkably insensitive to the cholesterol concentration when Arg is charged. This once again argues that the local membrane deformation in the presence of a charged Arg is fairly insensitive to the presence of cholesterol; this is because cholesterol molecules can easily disperse locally to avoid the local deformation of the membrane (see Fig. S7). With a neutral Arg, by contrast, the number of oxygen atoms is rather sensitive to the presence of cholesterol (Fig. 6 D). With 40% cholesterol, the current simulations find, consistently, a larger number of water molecules in contact with the neutral Arg than for other cases (Fig. 6 B, for a snapshot, see Fig. 7 B); with 20% cholesterol, two >100-ns simulations with different surface-tensions find that different numbers of cholesterols also get involved in stabilizing the neutral Arg (Fig. 6 C, for a snapshot, see Fig. 7 A). These trends are further elaborated with the number density plots in Fig. 7, which reveal that both water and cholesterol hydroxyl oxygen can be highly localized in the vicinity (<5 Å) of a neutral Arg. Number densities also indicate that neutral Arg can still maintain minor water defects at 20% cholesterol, but that such defects disappear completely at 40% cholesterol. Moreover, a strong water band within 5 Å of the neutral Arg at 40% cholesterol indicates that the complete removal of water defects by high cholesterol concentration creates a relatively stable local free-energy minimum for water near the neutral guanidinium.
Figure 6.
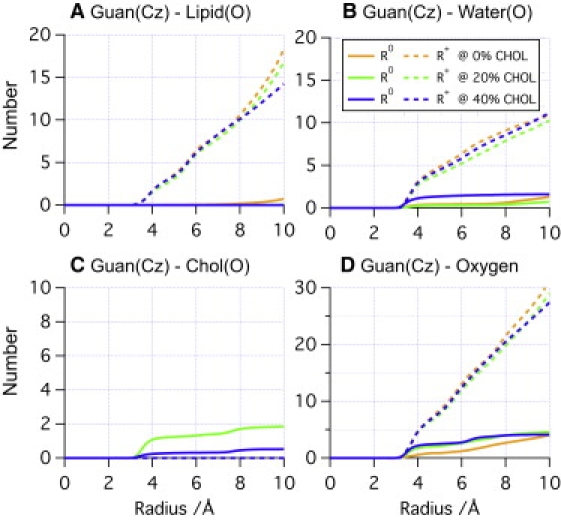
Cumulative radial distribution functions of (A) phosphate oxygen, (B) water oxygen, (C) cholesterol oxygen, and (D) total oxygen near Arg in SDPC/E simulations with different cholesterol concentrations. Note that the total number of oxygen in contact with the neutral guanidinium group is highly correlated with the significant drop of Δ < ∂U/∂λ(λ = 1.0) > in Table 1.
Figure 7.
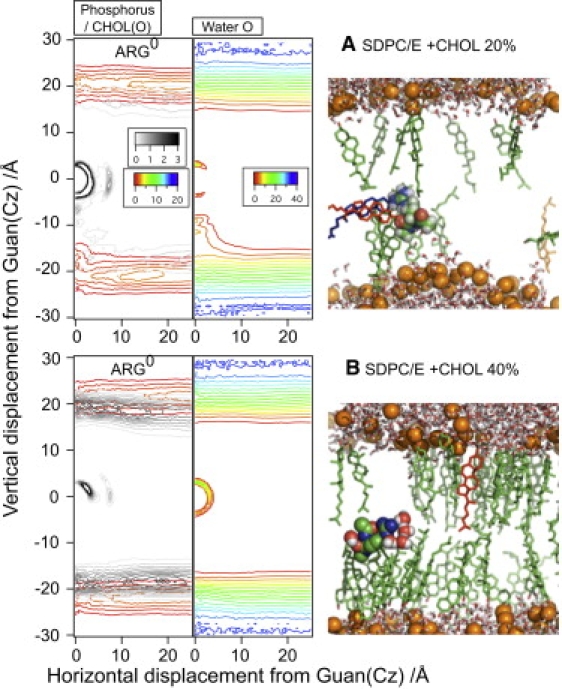
(Color online) Number densities (in nm−3) of lipid phosphorus (rainbow color in the left panel), cholesterol hydroxyl oxygen (gray scale in the left panel), and water oxygen (rainbow color in the right panel) around the neutral Arg at the origin in SDPC/E systems with (A) 20% and (B) 40% (CHOL).
Regarding cholesterol/Arg interaction, we note that although previous MD simulations find that cholesterol molecules predominantly orient themselves to be parallel to the membrane-normal (53), our current simulations find that cholesterol molecules can reorient themselves to be perpendicular to the membrane-normal to stabilize neutral Arg, although the process can take >100 ns. A pair of examples is shown in Fig. S8 (for relevant images, see Fig. S9). This behavior is rather consistent with the fact that cholesterol flip-flop between different leaflets is known to occur at a much faster rate than lipid molecules. For example, recent atomistic and coarse-grained simulations by Bennett et al. (54,55) found that cholesterol undergoes flip-flop on a seconds-timescale in saturated bilayers with high cholesterol content, while the timescale decreases to submicrosecond range in poly-unsaturated bilayers.
It is worth noting that the only significant outlier in all computed free energy derivatives is the neutral (λ = 1) state with 20% cholesterol and a high surface-tension (see Table 1). Analysis of the energy gap data indicates that the total number of oxygen near the neutral Arg is particularly low compared to other simulations (Fig. S4, Fig. S5, and Fig. S6) with either different cholesterol concentration or surface-tension. The origin of this difference is not yet clear. On the one hand, because the membrane is thinner (by ∼3 Å according to the number density plots; compare Fig. 7 and Fig. S6) and the area-per-lipid is larger at the higher surface-tension, the stability of local accumulation of water or cholesterol in the membrane center near a neutral Arg can be reduced. This is related to the observation that permeability of water into bilayers is most intimately correlated with the area-per-lipid (56). Indeed, the free energy derivatives at λ = 1 tend to be consistently higher also at 0 and 40% cholesterol in the high surface-tension simulations, although the difference from the low surface-tension results is less dramatic. However, the difference could be simply a sampling issue despite almost 200 ns of simulations. Much-longer simulations are required for better understanding of this issue, which we do not pursue here, because most results follow a clear trend.
Overall, our analysis highlights that although cholesterol is known to modulate the mechanical properties of lipid membrane, it can also contribute to processes in the membrane (e.g., titration of Arg) through chemical effects, such as by direct hydrogen-bonding interactions or by modulating the local water density to preferentially stabilize a specific chemical state.
Conclusions
Lipid membrane-mediated biological processes are fascinating because the soft nature of biomembrane allows deformations at many different length-scales that conform to the specific biological function. Therefore, although mechanical models are clearly most appropriate at meso/macroscopic scales, it is tempting to invoke mechanics-based considerations to discuss membrane-mediated processes even when the relevant scale approaches a relatively small number of molecules. In practice, precisely when such mechanical considerations start to break down (i.e., gives qualitatively incorrect predictions), it is not always clear; thus, it is fruitful to select specific problems and carefully compare predictions from microscopic and mechanical models.
Regarding stability of a charged species in the membrane center, because local membrane deformation has been shown to be qualitatively (12) and quantitatively (14) important to the discussion of the relevant energetics, it seems logical that modification of the membrane mechanical properties can have an important impact. In particular, one may predict that softening the membrane can better stabilize a charged species in a hydrophobic environment, whereas rigidifying the membrane has the opposite effect—which, in the context of pKa shift of Arg in the membrane center, corresponds to respectively lowering and increasing pKa shifts relative to the bulk value.
Our microscopic simulations, however, lead to rather different predictions. In SDPC (or SDPE) bilayers featuring highly unsaturated lipid tails, which are known to have substantially lower bending modulus than bilayers of saturated lipids (although <10% difference in stretch moduli (31)), much larger pKa shifts are observed than for a DPPC bilayer. The higher flexibility of the poly-unsaturated lipid tails allows them to better bind to the charged state Arg, which leads to smaller water defects despite the higher deformability of the SDPC(/E) bilayer; accordingly, the charged state of Arg is less stabilized, resulting in larger pKa shifts. In the case of cholesterol, which is known to rigidify lipid membranes, predictions from microscopic simulations and mechanical consideration are qualitatively consistent, both pointing to larger pKa shifts. The underlying mechanism that emerges from the microscopic analysis, however, is rather different. Rather than destabilizing the charged state, as the mechanical consideration would predict, the cholesterol molecules influence the pKa of Arg by better stabilizing its neutral state, via either direct hydrogen bonding to or modulating of the local water density around the neutral guanidinium group. Therefore, both sets of examples clearly illustrate the limited applicability of mechanical considerations for a process that involves significant but local deformation of the lipid membrane, further highlighting how the intricate dynamics of membrane components contribute to the energetics of membrane-mediated processes.
From the technical point of view, this work indicates that notable statistical errors remain in the computed pKa values despite samplings at 100–200-ns scale, which underlines the urgent need of developing effective enhanced sampling techniques suitable for membrane-mediated simulations, especially for mixed lipid systems. Nevertheless, the consistent trends observed in a fairly large set of independent simulations in this work suggest that the significantly larger pKa shifts in the presence of poly-unsaturated lipids tails and cholesterol are realistic. Because the charged state of Arg in complex membranes is important to many systems, including those of the voltage-gated ion channels and cell penetration peptides, we encourage further experimental efforts to test our predictions.
Acknowledgments
Computational resources from the National Center for Supercomputing Applications at the University of Illinois and the Center of High Throughput Computing at UW-Madison are greatly appreciated. The authors thank Prof. C. Grosman for the suggestion of looking into the effects of cholesterol, which led to this work.
The research has been supported by the National Institutes of Health (grant No. R01-GM071428).
Supporting Material
References
- 1.Boal D. Cambridge University Press; Cambridge, UK: 2002. Mechanics of the Cell. [Google Scholar]
- 2.Phillips R., Ursell T., Sens P. Emerging roles for lipids in shaping membrane-protein function. Nature. 2009;459:379–385. doi: 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips R., Kondev J., Theriot J. Garland Science Textbooks; London, UK: 2008. Physical Biology of the Cell. [Google Scholar]
- 4.Zimmerberg J., Kozlov M.M. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 5.Khelashvili G., Harries D., Weinstein H. Modeling membrane deformations and lipid demixing upon protein-membrane interaction: the BAR dimer adsorption. Biophys. J. 2009;97:1626–1635. doi: 10.1016/j.bpj.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parsegian A. Energy of an ion crossing a low dielectric membrane: solutions to four relevant electrostatic problems. Nature. 1969;221:844–846. doi: 10.1038/221844a0. [DOI] [PubMed] [Google Scholar]
- 7.Hessa T., Kim H., von Heijne G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 8.Hessa T., Meindl-Beinker N.M., von Heijne G. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature. 2007;450:1026–1030. doi: 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- 9.Andersen O.S. Perspectives on membrane protein insertion, protein-bilayer interactions, and amino-acid side hydrophobicity. J. Gen. Physiol. 2007;129:351–352. [Google Scholar]
- 10.Zhang B., Miller T.F., 3rd Hydrophobically stabilized open state for the lateral gate of the Sec translocon. Proc. Natl. Acad. Sci. USA. 2010;107:5399–5404. doi: 10.1073/pnas.0914752107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johansson A.C.V., Lindahl E. Protein contents in biological membranes can explain abnormal solvation of charged and polar residues. Proc. Natl. Acad. Sci. USA. 2009;106:15684–15689. doi: 10.1073/pnas.0905394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freites J.A., Tobias D.J., White S.H. Interface connections of a transmembrane voltage sensor. Proc. Natl. Acad. Sci. USA. 2005;102:15059–15064. doi: 10.1073/pnas.0507618102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krepkiy D., Mihailescu M., Swartz K.J. Structure and hydration of membranes embedded with voltage-sensing domains. Nature. 2009;462:473–479. doi: 10.1038/nature08542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choe S., Hecht K.A., Grabe M. A continuum method for determining membrane protein insertion energies and the problem of charged residues. J. Gen. Physiol. 2008;131:563–573. doi: 10.1085/jgp.200809959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorairaj S., Allen T.W. On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc. Natl. Acad. Sci. USA. 2007;104:4943–4948. doi: 10.1073/pnas.0610470104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doherty G.J., McMahon H.T. Mediation, modulation, and consequences of membrane-cytoskeleton interactions. Annu. Rev. Biophys. 2008;37:65–95. doi: 10.1146/annurev.biophys.37.032807.125912. [DOI] [PubMed] [Google Scholar]
- 17.Ruta V., Chen J.Y., MacKinnon R. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 2005;123:463–475. doi: 10.1016/j.cell.2005.08.041. [DOI] [PubMed] [Google Scholar]
- 18.Fuchs S.M., Raines R.T. Pathway for polyarginine entry into mammalian cell. Biochemistry. 2004;43:2438–2444. doi: 10.1021/bi035933x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothbard J.B., Jessop T.C., Wender P.A. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J. Am. Chem. Soc. 2004;126:9506–9507. doi: 10.1021/ja0482536. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt N., Mishra A., Wong G.C. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 21.Cymes G.D., Ni Y., Grosman C. Probing ion-channel pores one proton at a time. Nature. 2005;438:975–980. doi: 10.1038/nature04293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li L., Vorobyov I., Allen T.W. Is arginine charged in a membrane? Biophys. J. 2008;94:L11–L13. doi: 10.1529/biophysj.107.121566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li L.B., Vorobyov I., Allen T.W. Potential of mean force and pKa profile calculation for a lipid membrane-exposed arginine side chain. J. Phys. Chem. B. 2008;112:9574–9587. doi: 10.1021/jp7114912. [DOI] [PubMed] [Google Scholar]
- 24.MacCallum J.L., Bennett W.F.D., Tieleman D.P. Partitioning of amino acid side chains into lipid bilayers: results from computer simulations and comparison to experiment. J. Gen. Physiol. 2007;129:371–377. doi: 10.1085/jgp.200709745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacCallum J.L., Bennett W.F.D., Tieleman D.P. Distribution of amino acids in a lipid bilayer from computer simulations. Biophys. J. 2008;94:3393–3404. doi: 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo J., Cui Q. Does arginine remain protonated in the lipid membrane? Insights from microscopic pKa calculations. Biophys. J. 2008;94:L61–L63. doi: 10.1529/biophysj.107.122945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alberts B., Bray D., Watson J.D. Garland Publishing; New York: 1994. Molecular Biology of the Cell. [Google Scholar]
- 28.Sprong H., van der Sluijs P., van Meer G. How proteins move lipids and lipids move proteins. Nat. Rev. Mol. Cell Biol. 2001;2:504–513. doi: 10.1038/35080071. [DOI] [PubMed] [Google Scholar]
- 29.Nezil F.A., Bloom M. Combined influence of cholesterol and synthetic amphiphilic peptides upon bilayer thickness in model membranes. Biophys. J. 1992;61:1176–1183. doi: 10.1016/S0006-3495(92)81926-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagle J.F., Tristram-Nagle S. Structure of lipid bilayers. Biochim. Biophys. Acta Biomembr. 2000;1469:159–195. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rawicz W., Olbrich K.C., Evans E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 2000;79:328–339. doi: 10.1016/S0006-3495(00)76295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y.C., Holst M., McCammon J.A. A nonlinear elasticity model of macromolecular conformational change induced by electrostatic forces. J. Math. Anal. Appl. 2008;340:135–164. doi: 10.1016/j.jmaa.2007.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marrink S.J., Risselada H.J., de Vries A.H. The MARTINI force field: coarse-grained model for biomolecular simulations. J. Phys. Chem. B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 34.Vorobyov I., Li L.B., Allen T.W. Assessing atomistic and coarse-grained force fields for protein-lipid interactions: the formidable challenge of an ionizable side chain in a membrane. J. Phys. Chem. B. 2008;112:9588–9602. doi: 10.1021/jp711492h. [DOI] [PubMed] [Google Scholar]
- 35.Marrink S.J., de Vries A.H., Tieleman D.P. Lipids on the move: simulations of membrane pores, domains, stalks and curves. Biochim. Biophys. Acta Biomembr. 2009;1788:149–168. doi: 10.1016/j.bbamem.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 36.MacKerell A.D.J., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 37.Feller S.E., MacKerell A.D., Jr. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B. 2000;104:7510–7515. [Google Scholar]
- 38.Feller S.E., Gawrisch K., MacKerell A.D., Jr. Polyunsaturated fatty acids in lipid bilayers: intrinsic and environmental contributions to their unique physical properties. J. Am. Chem. Soc. 2002;124:318–326. doi: 10.1021/ja0118340. [DOI] [PubMed] [Google Scholar]
- 39.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 40.Feller S.E., Zhang Y.H., Brooks B.R. Constant-pressure molecular-dynamics simulation - the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 41.Koenig B.W., Strey H.H., Gawrisch K. Membrane lateral compressibility determined by NMR and x-ray diffraction: effect of acyl chain polyunsaturation. Biophys. J. 1997;73:1954–1966. doi: 10.1016/S0006-3495(97)78226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klauda J.B., Venable R.M., Pastor R.W. Considerations for lipid force field development. Curr. Top. Membranes. 2008;60:1–48. [Google Scholar]
- 43.Allen T.W., Andersen O.S., Roux B. Ion permeation through a narrow channel: using gramicidin to ascertain all-atom molecular dynamics potential of mean force methodology and biomolecular force fields. Biophys. J. 2006;90:3447–3468. doi: 10.1529/biophysj.105.077073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antosiewicz J., McCammon J.A., Gilson M.K. Prediction of pH-dependent properties of proteins. J. Mol. Biol. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- 45.Simonson T., Carlsson J., Case D.A. Proton binding to proteins: pKa calculations with explicit and implicit solvent models. J. Am. Chem. Soc. 2004;126:4167–4180. doi: 10.1021/ja039788m. [DOI] [PubMed] [Google Scholar]
- 46.Lide D.R., editor. CRC Handbook Chemistry and Physics. 85th Ed. CRC Press; Boca Raton, FL: 2005. [Google Scholar]
- 47.Ghosh N., Cui Q. pKa of residue 66 in Staphylococcal nuclease. I. Insights from QM/MM simulations with conventional sampling. J. Phys. Chem. B. 2008;112:8387–8397. doi: 10.1021/jp800168z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng L.Q., Chen M.G., Yang W. Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc. Natl. Acad. Sci. USA. 2008;105:20227–20232. doi: 10.1073/pnas.0810631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaminski G.A., Friesner R.A., Jorgensen W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- 50.Evans E., Needham D. Physical-properties of surfactant bilayer-membranes—thermal transitions, elasticity, rigidity, cohesion, and colloidal interactions. J. Phys. Chem. 1987;91:4219–4228. [Google Scholar]
- 51.Lundbaek J.A., Birn P., Andersen O.S. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. doi: 10.1021/bi952250b. [DOI] [PubMed] [Google Scholar]
- 52.Levy R.M., Belhadj M., Kitchen D.B. Gaussian fluctuation formula for electrostatic free-energy changes in solution. J. Chem. Phys. 1991;95:3627–3633. [Google Scholar]
- 53.Olsen B.N., Schlesinger P.H., Baker N.A. Perturbations of membrane structure by cholesterol and cholesterol derivatives are determined by sterol orientation. J. Am. Chem. Soc. 2009;131:4854–4865. doi: 10.1021/ja8095224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bennett W.F., MacCallum J.L., Tieleman D.P. Thermodynamic analysis of the effect of cholesterol on dipalmitoylphosphatidylcholine lipid membranes. J. Am. Chem. Soc. 2009;131:1972–1978. doi: 10.1021/ja808541r. [DOI] [PubMed] [Google Scholar]
- 55.Bennett W.F., MacCallum J.L., Tieleman D.P. Molecular view of cholesterol flip-flop and chemical potential in different membrane environments. J. Am. Chem. Soc. 2009;131:12714–12720. doi: 10.1021/ja903529f. [DOI] [PubMed] [Google Scholar]
- 56.Mathai J.C., Tristram-Nagle S., Zeidel M.L. Structural determinants of water permeability through the lipid membrane. J. Gen. Physiol. 2008;131:69–76. doi: 10.1085/jgp.200709848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schiferl S.K., Wallace D.C. Statistical errors in molecular dynamics averages. J. Chem. Phys. 1985;83:5203–5209. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.