

Abstract
Dehaloperoxidase (DHP) from the annelid Amphitrite ornata is a catalytically active hemoglobin-peroxidase that possesses a unique internal binding cavity in the distal pocket above the heme. The previously published crystal structure of DHP shows 4-iodophenol bound internally. This led to the proposal that the internal binding site is the active site for phenol oxidation. However, the native substrate for DHP is 2,4,6-tribromophenol, and all attempts to bind 2,4,6-tribromophenol in the internal site under physiological conditions have failed. Herein, we show that the binding of 4-halophenols in the internal pocket inhibits enzymatic function. Furthermore, we demonstrate that DHP has a unique two-site competitive binding mechanism in which the internal and external binding sites communicate through two conformations of the distal histidine of the enzyme, resulting in nonclassical competitive inhibition. The same distal histidine conformations involved in DHP function regulate oxygen binding and release during transport and storage by hemoglobins and myoglobins. This work provides further support for the hypothesis that DHP possesses an external binding site for substrate oxidation, as is typical for the peroxidase family of enzymes.
Introduction
The two dehaloperoxidase (DHP) hemoglobins (Hbs) from Amphitrite ornate, DHP A and DHP B, are the first characterized Hbs that have natural peroxidase function (1–6). Hbs, including DHP, are readily identified by their characteristic 3/3 α-helical protein structure. Although Hbs are primarily associated with O2 storage and transport, the characteristic globin fold actually encodes a diversity of protein functions. In addition to allosteric regulation of oxygen uptake, globins minimize the autooxidation rate of the heme iron, discriminate against CO binding, and carry out other natural functions such as NO binding and oxidation to nitrate. DHP brings this functional diversity to a new level by combining the seemingly contradictory functions of reversible oxygen binding (globin) and hydrogen peroxide activation (peroxidase).
Although Hbs and heme peroxidases are structurally distinct and perform different functions, they have a common heme cofactor and iron-binding site, with a histidine residue positioned on each side (Fig. 1). The proximal histidine is coordinated to the heme iron and provides a charge relay that supports either the ferrous (Fe2+) or ferric (Fe3+) iron oxidation state (7,8) depending on the local environment of globins or peroxidases, respectively. In myoglobins (Mbs) and Hbs, the distal histidine stabilizes diatomic oxygen during uptake and transport, whereas in peroxidases, the distal histidine serves as the acid-base catalyst necessary for heterolytic O-O bond cleavage, which constitutes the activation of bound hydrogen peroxide. Allostery in Hb is vital for the proper uptake of oxygen in the lungs and release of oxygen to respiring tissues (9). Whereas allostery may modify a binding constant or facilitate communication between multimers in cooperative proteins such as Hb, allosteric inhibition in heme peroxidases is an off-switch that can cause the enzyme to become completely inactive (10–12). The significance of both allostery and inhibition is more complex in a dual-function protein like DHP because the regulation involves not only each individual function, but also the switch between functions.
Figure 1.
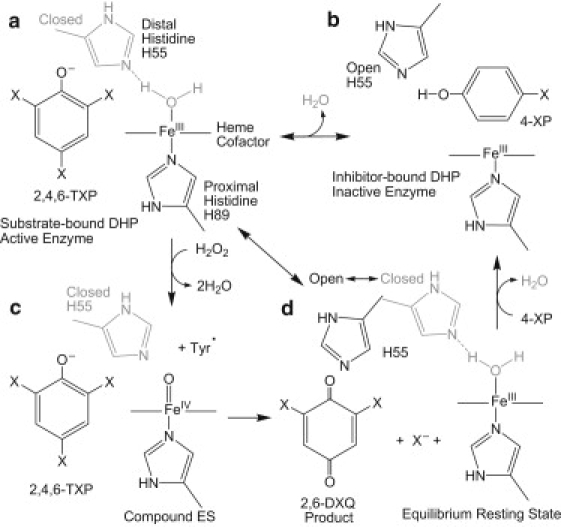
Reaction scheme emphasizing the conformation of the distal histidine, H55, in response to binding of the substrate, 2,4,6-TXP, and inhibitor, 4-XP. (a) Active enzyme: DHP with TXP substrate bound external to the heme pocket. The protein is 6cHS (aquo) with the distal H55 in a closed position. (b) When an inhibitor (4-XP) binds in the internal pocket of DHP, H2O is displaced (5cHS) and the distal H55 is pushed to the open position. The resulting conformation leads to inactivation of the enzyme. (c) Addition of H2O2 leads to the formation of compound ES (39), the high-valent iron-oxo protein (Tyr) radical intermediate, which can lead to formation of the product 2,6-DXQ by two-electron oxidation. Compound ES cannot be formed in the inhibitor-bound state because the FeIII site is blocked by 4-XP binding in the distal cavity. (d) In the resting state of DHP the distal histidine exists in two conformations, known as open (black) and closed (gray). H2O is bound to the heme iron only in the closed conformation.
The first x-ray crystal structures of DHP A (Protein Data Bank (PDB) 1EW6 and 1EWA), obtained at room temperature, showed two features that are unique in the Hb superfamily (1). First, the distal histidine was observed in two conformations at pH 6, identified as open or closed. The closed conformation is the commonly observed conformation shown in Fig. 2 a, in which the histidine is in the distal pocket and interacts with a ligand coordinated to the heme iron (13). In the open conformation (Fig. 2 b), the distal histidine (H55) has swung out to a solvent-exposed position (14). Although the open conformation is known in sperm whale Mb, it is only observed below pH 4.5 when the distal histidine is protonated (15). We previously showed by a comparison of x-ray crystal structures (3DR9 and 2QFK) at 100 K that the open and closed conformations in DHP A are correlated with the 5-coordinate (5c) and 6-coordinate (6c) forms of the heme iron, respectively (13,14). Furthermore, the unique flexibility of the distal histidine has been shown to play an important role in heme-coordinated ligand stabilization (16). The second unprecedented observation in the initial x-ray crystal structure of DHP A was the presence of a substrate analog, 4-iodophenol, in a well-defined position in the distal pocket of the globin but not coordinated to the heme iron (1). This unusual mode of binding in an Hb led to the suggestion that the internal binding site is the substrate-binding site (1). We have systematically investigated this hypothesis and found that the 4-halogenated phenols (4-XP) that bind internally are inhibitors rather than substrates. The active site for oxidation of substrates such as 2,4,6-tribromophenol (2,4,6-TBP), 2,4,6,-trichlorophenol (2,4,6-TCP), and 2,4,6,-trifluorophenol (2,4,6-TFP) is external (17). The possibility that an external substrate-binding site is located on DHP A was recently established via backbone NMR experiments (18). Given the extensive data available on DHP A relative to the more recently characterized DHP B, the remainder of this study will focus on DHP A, which will be referred to as DHP for brevity.
Figure 2.
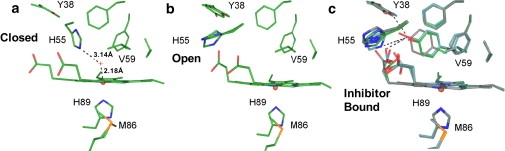
X-ray crystal structures of DHP at 100 K. (a) The metaquo form shows a hydrogen-bond interaction between the distal histidine (H55) and the water molecule coordinated to the ferric heme iron (PDB 2QFK; closed conformation). (b) The deoxy form has a pentacoordinate heme iron. Two rotamers of H55 are observed in which H55 appears exclusively in a solvent-exposed conformation (PDB 3DR9). This His conformer corresponds to the open form observed in Mb at pH 4.5 (15). (c) The three structures of 4-IP (3LB1, green), 4-BP (3LB2, light blue), and 4-CP (3LB3, gray) bound in the distal pocket of DHP are superimposed. 4-FP (3LB4) is not shown in the figure (see text).
In this work, we provide detailed evidence that internal binding of 4-XPs inhibits the peroxidase function of DHP. This result is contradictory to the previous hypothesis that the distal pocket binding site for 4-XP is the substrate-binding site for phenol oxidation. This work builds on a number of observations that support the existence of distinct binding sites for 4-BP and 2,4,6-TBP, and demonstrates that these distinct sites are involved in inhibition of a competitive nature. Normally, competitive inhibition implies that the substrate and inhibitor compete for the same binding site. However, the structural and kinetic evidence presented below suggests that DHP exhibits a form of competitive inhibition (formally known as allosteric or nonclassical competitive inhibition (19–22)) in which the inhibitor binds remote to the active site and creates a conformational change in the enzyme that prevents the substrate from binding. The x-ray crystallographic, resonance Raman (RR), and kinetic data presented here are consistent with competitive inhibition between the internal and external sites mediated by the distal histidine (H55). The proposed functional role for the distal histidine, as the switch that leads to peroxidase inhibition (open) and activation (closed), underlies the regulation of oxygen-binding affinity by the same histidine in the open (low affinity) and closed (high affinity) conformations traditionally observed in globins (15,23,24).
Materials and Methods
Materials
Buffer salts were purchased from Fisher Scientific (Waltham, MA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification.
Crystallization, data collection, and processing
Recombinant wild-type (WT) protein was expressed in E. coli, purified, and characterized as previously described (13). To obtain crystals of DHP complexed with substrate analogs, the protein, at a concentration of 8 mg/mL dissolved in 10 mM Na cacodylate pH 6.5, was incubated on ice for 30 min with the parahalogenated phenols (1.5 mM 4-IP, 10 mM 4-BP, or 10 mM 4-CP, respectively) and crystallized using the hanging drop vapor diffusion method, with the reservoir solution containing unbuffered 0.2 M ammonium sulfate and 32% PEG 4000 (w/v) as described previously (13,14). The crystals were cryoprotected in a solution containing 0.2 M ammonium sulfate, 35% PEG 4000 (w/v), and 15% PEG 400 as the cryoprotectant. Data were collected at 100 K on the SER-CAT 22-ID beamline at the APS synchrotron facility using a wavelength of 1 Å for crystals derivatized with 4-chloro- and 4-fluorophenol, 0.91942 Å for crystals derivatized with 4-bromophenol, and 1.5 Å for crystals derivatized with 4-iodophenol. The latter two wavelengths were chosen so that two data sets could be collected in a single-wavelength anomalous dispersion mode to correctly orient the halogenated phenol in its electron density. The collected diffraction data sets were processed using the HKL2000 program suite (25). The new crystals belong to the same space group (P212121) as the ferric water-ligated (metaquo) form (PDB entry 2QFK), and the structures were solved by molecular replacement using 2QFK coordinates as a starting model in the Phaser molecular replacement program (26). Structure determination and refinement calculations were performed using the CCP4 suite of programs (27,28), and visualization and manual model building were conducted using Coot model building software (29). Waters were placed with the Coot routine Find Waters using 2Fo-Fc contoured at the 1σ level, and Fo-Fc maps at the 3σ level. The occupancies were refined manually until no residual Fo-Fc density remained. Final models were obtained by iterative cycles of model building in Coot using 2Fo-Fc (contoured at the 1σ level) and Fo-Fc electron density maps (contoured at the 3σ level), and positional and anisotropic B factor structure refinement using Refmac5 (30) in the CCP4 suite of programs (31) and CNS (32). Simulated annealing and composite omit maps were constructed with the CNS program. All of the figures were prepared using VMD (33). The refinement statistics of the four x-ray crystal structures (3LB1, 3LB2, 3LB3, and 3LB4) are given in Table 1.
Table 1.
Data collection and refinement statistics
| I∗ | Br∗ | Cl∗ | F∗ | |
|---|---|---|---|---|
| PDB code | 3LB1 | 3LB2 | 3LB3 | 3LB4 |
| Space group | P212121 | P212121 | P212121 | P212121 |
| Unit-cell parameters | ||||
| a (Å) | 57.84 | 58.01 | 58.20 | 59.78 |
| b (Å) | 67.25 | 67.36 | 67.21 | 67.49 |
| c (Å) | 69.13 | 69.42 | 68.72 | 67.84 |
| Data collection | ||||
| Temperature (K) | 100 | 100 | 100 | 100 |
| Wavelength (Å) | 1.5000 | 0.91942 | 1.0000 | 1.0000 |
| Resolution (Å) | 35.0–1.76 | 35.0–1.06 | 35.0–1.85 | 35.0–1.56 |
| (1.81–1.76)† | (1.09–1.06)† | (1.90–1.85)† | (1.60–1.56)† | |
| Unique reflections | 25737(1737)† | 121970(8490)† | 22277(15730)† | 33583(2740)† |
| Completeness (%) | 94.24(87.82)† | 98.67(93.80)† | 94.25(92.91)† | 94.03(95.34)† |
| Rmerge‡ (%) | 4.1(9.8) | 5.2(31.8) | 10.1(32.2) | 6.1(38.3) |
| I/σI | 37.9(13.2)† | 22.5(3.8)† | 7.9(3.4)† | 10.9(3.9)† |
| Redundancy | 3.5(3.4)† | 4.0(3.3)† | 3.4(3.4)† | 3.1(3.3)† |
| Refinement | ||||
| Rwork/Rfree (%)§ | 18.6/22.7 | 16.8/18.3 | 18.7/24.3 | 19.5/23.5 |
| Average B factor (Å2) | ||||
| All atoms | 12.73 | 10.16 | 19.13 | 17.19 |
| Protein | 11.81 | 8.98 | 18.55 | 15.19 |
| Water | 21.59 | 20.21 | 30.35 | 28.75 |
| No. of atoms | ||||
| Protein | 2658 | 3122 | 2192 | 2849 |
| Water | 268 | 356 | 213 | 299 |
| RMSD from ideal | ||||
| Bond length (Å) | 0.008 | 0.006 | 0.008 | 0.011 |
| Bond angle (deg) | 1.124 | 1.170 | 1.157 | 1.377 |
| Ramachandran plot¶ (%) | ||||
| Most favored | 94.4 | 94.4 | 94.4 | 94.8 |
| Allowed | 5.6 | 5.6 | 5.6 | 5.2 |
Crystal containing 4-halophenol in complex with WT DHP; the letter specifies the halogen substituent.
Values in parentheses are the highest-resolution shell.
Rmerge= (ΣhΣi{} × 100%, where Ii is the ith measurement and <I(h)> is the weighted mean of all measurements of I(h).
Rwork = Σ|Fo − Fc|/ΣFo × 100%, where Fo and Fc are observed and calculated structure factors, respectively; Rfree is R factor for the subset (5%) of reflections selected before, and not included in the refinement.
Calculated using PROCHECK.
Electronic absorption spectroscopy and kinetic assays
Recombinant his-tagged WT protein was expressed in Escherichia coli and purified as previously described (17,18). Initial inhibition experiments were conducted in 100 mM potassium phosphate buffer at pH 7 using an Agilent 8453 UV-vis spectrometer equipped with a temperature control and Hewlett Packard UV-Visible Chemstation software set to kinetics mode. The concentration of DHP in each sample was ∼2.4 μM and the temperature was equilibrated to 20°C. A 100-fold excess of H2O2 (240 μM) was added to the cuvette to initiate the assay. Electronic absorption spectra were taken every 2 s for 2 min, monitoring the 273 nm peak of the 2,6-dichloro-1,4-dibenzoquinone (2,4-DCQ) product. The assays were repeated with the addition of 250 μM 4-bromophenol in each sample to demonstrate the inhibition effect.
Inhibition assays for Michaelis-Menten analysis were conducted on a Cary 100 UV-vis equipped with a Cary temperature control system and an Applied Photophysics RX2000 rapid kinetics spectrometer accessory. The Applied Photophysics premixing chambers were temperature-controlled with a Fisher Scientific Isotemp 3006S set to 25°C. Instruments were controlled by Cary WinUV software in kinetics mode set to monitor the 273 2,4-DCQ peak every 0.1 s for 60 s. Assay conditions were 2 μM DHP initiated with 2 mM H2O2 in 100 mM potassium phosphate buffer at pH 7. Eight assays were completed at each of the 2,4,6-TCP concentrations. The assays were then repeated at each of the 2,4,6-TCP concentrations with the presence of 125, 250, and 500 μM 4-BP. The initial velocity, Vo, of enzyme turnover was obtained for each concentration of the 2,4,6-TCP substrate and the 2,4,6-TCP substrate with inhibitor. The Vo versus 2,4,6-TCP concentration data were fit independently to the Michaelis-Menten equation using nonlinear least squares in Igor Pro 5.0.
RR spectroscopy
All protein samples used in the RR experiments were purified as above and maintained in 150 mM potassium phosphate buffer, pH 6. The final protein concentration for all RR samples was 100 μM. Parahalogenated substrate analogs were introduced to final concentrations of 8 mM for 4-BP, 4-CP, 4-FP, and phenol, and to 1 mM for 4-IP (the concentration of 4-IP is limited by its low solubility at 25°C). The samples were placed into 5-mm-diameter glass NMR tubes and stored on ice until used.
RR spectra were obtained by Soret band excitation using a Coherent Mira 900 titanium sapphire (Ti:sapphire) laser. The Ti:sapphire laser was pumped using a Coherent Verdi 10 frequency-doubled diode-pumped Nd:vanadate laser generating 10 W at 532 nm. The beam generated from the Ti:sapphire is tunable through ∼700–1000 nm, and was sent through a Coherent 5-050 doubler to generate a normal working range of 400–430 nm for Soret band excitation. The beam was collimated and cylindrically focused to a vertical line of ∼0.5 mm on the sample. Scattered light was collected with a Spex 1877 triple spectrometer (2400 grooves/mm final stage grating) equipped with an ISA SPEX liquid nitrogen-cooled CCD at ∼1.7 cm−1 resolution. Computer acquisition of the data was accomplished with SpectraMax 2.0 software. The spectra were calibrated using known peaks from indene, toluene, and carbon tetrachloride standards.
Binding isotherm analysis
A titration data set for each inhibitor binding to DHP was collected via a series of RR spectra. Each sample contained 100 μM DHP in 150 mM potassium phosphate buffer at pH 6, with concentrations of the inhibitor from 0 to 8 mM (0–1 mM for 4-IP only). Singular value decomposition (SVD) was performed on the spectral data set with the use of Igor Pro 5.0. The SVD analysis yields one-dimensional column and row eigenvectors, with columns corresponding to changes with respect to wavenumber, and rows corresponding to changes with respect to concentration. The SVD row eigenvectors representing the intensity changes and the peak shifts of the Raman data set were fit using nonlinear least-squares to the single-site binding equation
to determine apparent substrate dissociation constants, Kd, where θ is the fraction 5c high spin (5cHS) protein and [I] is the concentration of inhibitor.
Results
Crystallography
Previously published x-ray crystal structures (13,14) and spectroscopic data (16) strongly suggest a role for distal histidine flexibility in DHP. Fig. 2, a (closed) and b (open), shows PDB structures 2QFK and 3DR9, respectively. As mentioned above, in the metaquo form, the distal His is stabilized in the closed conformation by hydrogen bonding to the heme-coordinated water molecule (Fig. 2 a), and the heme iron is 6c high spin (6cHS). However, unlike other Hb structures (34–36), in the 5c deoxy form, the His is observed in the open conformation (Fig. 2 b). Therefore, the open and closed conformations in DHP are correlated with the 5c and 6c forms of the heme iron. Fig. 2 c shows an overlay of the new heme pocket structures of DHP cocrystallized with 4-IP (3LB1), 4-BP (3LB2), and 4-CP (3LB3) following established protocols (13). The 4-XPs bind in a conformation close to that originally reported for 4-IP (1). The occupancy of the 4-IP, 4-BP, and 4-CP molecules is >90% in all three structures. The structure of DHP with 4-FP (3LB4) is not shown, due to its low occupancy (<50%) and for clarity of the figure. Upon binding of these molecules in the internal site, the heme-coordinated water molecule is displaced and the histidine is pushed into the open conformation; thus, the iron is 5cHS (see also Fig. 1 for a schematic). The secondary structure of DHP A exhibits remarkably little change when 4-XPs bind in the distal pocket. The backbone root mean-square deviations (RMSDs) from the metaquo structure are ∼≤0.4 Å, and the pairwise main-chain differences between the complexed structures are on the order of 0.1–0.2 Å. On the other hand, superposition of the structures shows that as the size of the parahalogen atom increases, the position of the 4-XP molecules bound in the distal pocket shifts slightly toward the heme-7-propionate and the solvent-exposed distal histidine.
Binding of parahalogenated phenols
In this study, the x-ray crystal structures provided meaningful insight into DHP in the solid state, whereas RR spectroscopy revealed the solution-state properties of halophenol binding. Fig. 3 a compares the RR spectra of WT-DHP with those obtained upon addition of phenol, and the 4-XP molecules (X = F, Cl, Br, I). The 5cHS core size marker band frequencies (ν3 at 1494 cm−1, ν2 at 1568 cm−1, and ν10 at 1632 cm−1) systematically become more intense at the expense of the aquo 6cHS heme state (ν3 at 1481 cm−1, ν2 at 1562 cm−1, and ν10 at 1611 cm−1) for the series of 4-XP bound DHP relative to WT-DHP. Similar systematic changes are also observed in the corresponding electronic absorption spectra. The Soret maximum undergoes a systematic blue shift as the substrate halogen is changed and follows the halogen series (see Fig. S1 in the Supporting Material). Therefore, in agreement with the x-ray crystal structures, binding of 4-XP in the internal pocket is consistent with the loss of the 6cHS population and subsequent movement of the distal His to the open, solvent-exposed position. Fig. 3 b shows that 4-halophenols bind in the distal pocket with a binding affinity that follows the trend I > Br > Cl > F > H, with apparent dissociation constants of 0.536, 1.15, 1.78, 3.72, and 10.0 mM, respectively. We use the term “apparent dissociation constant” because the binding isotherms represent the fraction of enzyme that is converted to 5cHS, which does not necessarily reflect total binding to the enzyme. The relative binding affinity of 4-FP reflects its low occupancy in the crystal structure. The binding isotherms were determined using the change in relative intensities and the frequency shifts of the core size heme vibrational modes measured by RR spectroscopy and obtained from the data shown in Fig. 3 a by SVD (as shown in Fig. S2).
Figure 3.
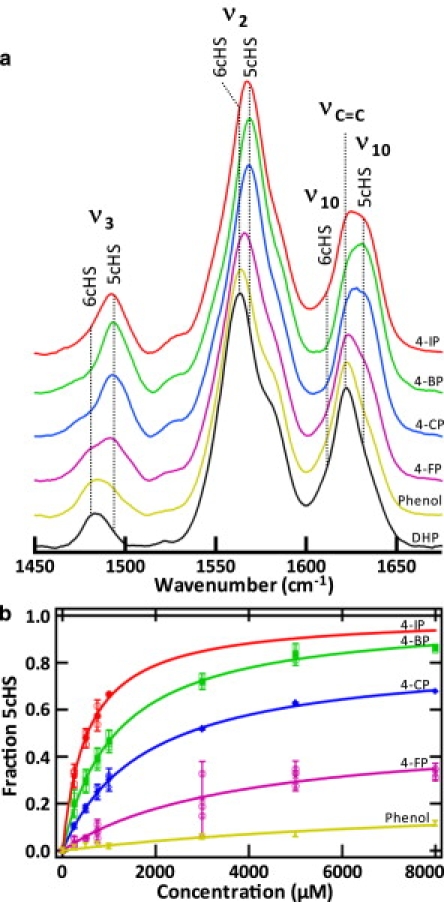
Determination of internal binding affinity. (a) RR core size marker band region for WT-DHP (black), DHP with phenol (yellow), DHP with 4-FP (purple), DHP with 4-CP (blue), DHP with 4-BP (green), and DHP with 4-IP (red). The ν2, ν3, and ν10 frequencies are consistent with the increase of a 5cHS iron upon addition of the inhibitors. The final concentration of 4-IP was 1 mM (maximum solubility), and the final concentration of 4-BP, 4-CP, 4-FP, and phenol was 8 mM. Protein concentration: 100 μM; excitation wavelength: 406 nm; resolution: 1.7 cm−1; laser power at the sample: 60 mW; acquisition time: 300 s. (b) Binding isotherms for 4-IP (red), 4-BP (green), 4-CP (blue), 4-FP (purple), and phenol (yellow). The isotherms clearly establish that the affinity of DHP for 4-XP inhibitors follows the pattern 4-IP > 4-BP > 4-CP > 4-FP, > phenol at pH 6.
Binding of trihalogenated phenols
In contrast to the binding of 4-XPs, binding of 2,4,6-TXP substrates produces an increase of the 6cHS species. Fig. 4 shows the change of the RR core size marker bands, indicating the formation of a predominantly 6cHS heme (ν3 at 1481 cm−1, ν2 at 1562 cm−1, and ν10 at 1611 cm−1) when 2,4,6-TBP and 2,4,6-TCP bind to DHP. Although the binding of 2,4,6-TFP also produces predominantly 6cHS heme, an appreciable amount of 5cHS (ν3 at 1494 cm−1, ν2 at 1568 cm−1, and ν10 at 1632 cm−1) is also formed, indicating that, unlike 2,4,6-TCP or 2,4,6-TBP, 2,4,6-TFP binds either externally or internally. This result is in agreement with previous studies of DHP showing that 2,4,6-TFP binds internally at cryogenic temperatures (16,37,38) but binds nonspecifically to both sites at room temperature (16,18). The corresponding electronic absorption spectra of 2,4,6-TXP binding (Fig. S3) change accordingly. Of greater importance, we can control the distal histidine and force DHP to adopt either the 5cHS or 6cHS state by changing the concentrations of 4-BP and 2,4,6-TCP present in solution. Fig. S4 shows that the same ratio of 5cHS/6cHS heme is achieved by first adding DHP to 2,4,6-TCP and then adding 4-BP, or by first adding DHP to 4-BP and then adding 2,4,6-TCP. The result demonstrates that 2,4,6-TCP and 4-BP compete for their respective binding locations until an equilibrium between the two states is attained. The x-ray crystal structures show the mutual exclusivity of the closed conformation of the distal histidine and the binding of 4-XP in the distal pocket. Since binding of 2,4,6-TCP forces the distal histidine into the closed position, it must also effectively remove 4-BP from the distal pocket. Thus, by correlation of the x-ray crystal structures and the RR data, we conclude that binding at the internal site and binding at the external site are mutually exclusive events.
Figure 4.
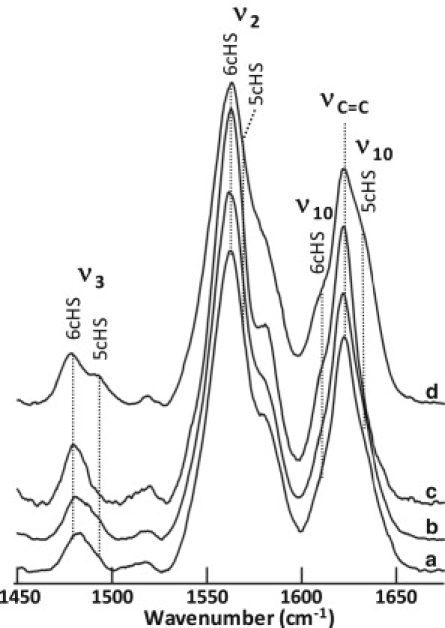
Evidence of external binding. RR core size marker band region for WT-DHP (a), DHP with 2,4,6-TBP (b), DHP with 2,4,6-TCP (c), and DHP with 2,4,6-TFP (d). The ν2, ν3, and ν10 frequencies shift to predominantly 6cHS upon addition of the TXP substrates. The 5cHS population observed upon addition of 2,4,6-TFP may indicate that 2,4,6-TFP does enter the distal pocket to some degree, in agreement with cryogenic experiments (16,37,38). The final concentration of 2,4,6-TBP was 200 μM, and the final concentrations of 2,4,6-TCP and 2,4,6-TFP were 4 mM in 150 mM potassium phosphate buffer, pH 6. Excitation wavelength: 406 nm; resolution: 1.7 cm−1; laser power at the sample: 60 mW; acquisition time: 300 s.
Enzyme inhibition by parahalogenated phenols
Davis et al. (18) confirmed the existence of two distinct binding sites for 2,4,6-TCP and 4-BP, and showed that DHP has little activity toward 4-BP compared to 2,4,6-TCP. We therefore tested the effect of internal binding of 4-XP on the turnover of 2,4,6-TCP. Fig. 5 provides kinetic evidence that the oxidation of 2,4,6-TCP is in fact inhibited by internal binding of 4-halophenols. The substrate (2,4,6-TCP) is readily converted to product (2,6-DCQ) when DHP is activated with excess H2O2 (Fig. 5 a). However, in the presence of even a 2:1 ratio of 4-XP/2,4,6-TCP, little turnover of 2,4,6-TCP to 2,6-DCQ is observed (Fig. 5, b–e). In fact, the only appreciable turnover is observed in the presence of 4-FP, in agreement with the binding isotherms from Fig. 2 b that suggest only a small fraction of 4-FP binds internally. Since the 4-XP molecules bind internally and force the distal histidine to adopt the open conformation, it is likely that they prevent the formation of compound ES (39), the H2O2 activated protein radical form of DHP that is analogous to compound I of cytochrome c peroxidase (40) (see Fig. 1 c). Indeed, Fig. 5 f shows that increasing the concentration of 4-BP systematically blocks the formation of compound ES.
Figure 5.
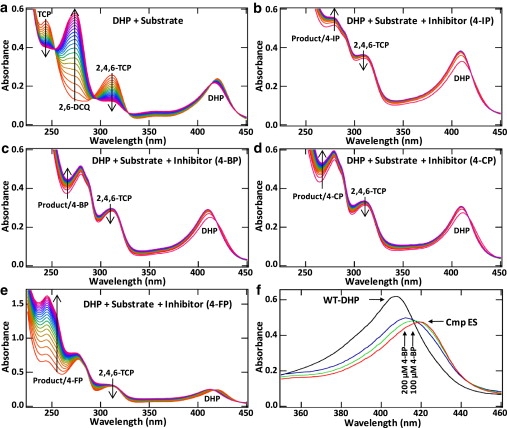
Kinetic assays showing inhibition by internally bound 4-XPs. (a–e) Time-dependent UV-Vis spectra from 0 s (red) to 120 s (purple). In the absence of 4-XP (a), the TCP substrate (312 nm) is converted to the DCQ product (273 nm). In the presence of 4-IP, 4-BP, and 4-CP (b–d), little product is formed, with no significant decrease in the substrate band. In the presence of 4-FP (e) some turnover is observed, in agreement with the lower affinity of 4-FP to bind internally. (f) Inhibition of compound ES formation (λmax (Soret) = 420 nm) in ferric DHP at pH 7 due to increasing concentrations of 4-BP.
Michaelis-Menten inhibition/kinetic analysis
Since the unique internal binding of 4-XPs inhibits the peroxidase function of DHP at a remote external site, it is of interest to determine the type of inhibition that occurs between the two sites. The solubility of the native substrate, 2,4,6-TBP, is relatively low (∼200 μM), which limits its usefulness for kinetic studies. However, 2,4,6-TCP is an excellent substrate and has higher solubility than 2,4,6-TBP. Therefore, we used 2,4,6-TCP rather than 2,4,6-TBP for the subsequent kinetic studies to demonstrate enzymatic inhibition by 4-BP (Fig. 6). The data obtained with and without 4-BP were fit independently to a Michaelis-Menten kinetic model. The fit parameters for the kinetic data are summarized in Table 2. KM increases as the concentration of inhibitor increases, but Vmax is essentially unchanged within the limits of the fitting errors. Ideally, one would use substrate concentrations several times greater than KM, i.e., [S]max >> KM, but even when 2,4,6-TCP is used, the substrate solubility remains the limiting factor. Thus, errors in the fit data arise as KM becomes greater than [S]max. Although we recognize this limitation, the relative effect of the inhibitor is clearly observed. Based on these data, the substrate/inhibitor pair exhibits competitive inhibition in DHP. Although results of this type cannot be demonstrated for 2,4,6-TBP because of its limited solubility, kinetic assays of 2,4,6-TBP with 4-BP under conditions identical to those used for 2,4,6-TCP with 4-BP yield the same complete inhibition effect (Fig. S5).
Figure 6.
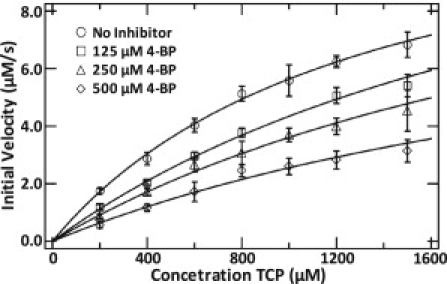
Michaelis-Menten analysis of inhibition, showing the initial reaction velocity versus substrate (2,4,6-TCP) concentration. Assays were conducted on WT-DHP without the presence of inhibitor, WT-DHP with 125 μM 4-BP, WT-DHP with 250 μM 4-BP, and WT-DHP with 500 μM 4-BP. The Michaelis-Menten fit parameters are given in Table 2.
Table 2.
Mechaelis-Menten fit parameters
| Vmax (μM/s) | KM (mM) | |
|---|---|---|
| No inhibitor | 13.1 ± 1.32 | 1.32 ± 2.11 |
| 125 μM 4-BP | 15.3 ± 2.94 | 2.53 ± 6.11 |
| 250 μM 4-BP | 13.7 ± 4.12 | 2.80 ± 1.06 |
| 500 μM 4-BP | 10.4 ± 4.58 | 3.08 ± 1.71 |
Discussion
The shifts in the relative position of 4-XPs bound in the distal pocket of DHP suggest that there are two driving forces that stabilize the molecule. First, the parahalogen atom fills a cavity in the protein. Previous studies have examined cavities in Mb by determining Xe binding sites using x-ray crystallography (41). The cavity within DHP that is filled by the parahalogen is surrounded by hydrophobic residues (Fig. S6) and resembles the Xe4 binding site in sperm whale Mb (41). The second factor is the interaction of the hydroxyl group with Y38, heme-7-propionate, and, to a lesser extent, H55 (Fig. 2 c). As the atomic radius of the halogen decreases, the hydroxyl group moves into closer contact with H55, Y38, and the heme propionate. Therefore, as shown in Figs. 2 and 3, a single atom, the parahalogen X in 4-XP, determines the binding affinity of the molecule in the distal pocket. Since the bound 4-XP displaces the water coordinated to the heme iron, one can also expect that it prevents the coordination of H2O2 and thereby inhibits peroxidase activity. The finding that increased concentrations of 4-BP systematically block the formation of the radical enzymatic intermediate (compound ES (39); see Fig. 1) in ferric DHP at pH 7 supports this hypothesis. Moreover, in accord with this hypothesis, the H55V mutation effectively eliminates the enzymatic activity of DHP (42).
On the basis of these results, and in agreement with the known mechanism for the peroxidase family members, we propose that enzymatic oxidation of substrates such as 2,4,6-TXP (X = I, Br, Cl, F) occurs at an external site. Recent 1H-15N HSQC experiments on 13C/15N-labeled DHP clearly indicate different binding interactions between 4-BP inhibitor and 2,4,6-TCP substrate (18). 4-BP binding causes deviations in the internal binding pocket residues (F24, F35, F21, H55, and V59; see Fig. S7), whereas 2,4,6-TCP binding affects the distal H55 and amino acids residing at the protein dimer interface (R122, G1, S129, and L76; see Fig. S8) (18). Of interest, the common amino acid that is affected by either binding event, as observed by NMR, is the distal H55. The combination of RR and NMR strongly supports the existence of an external substrate-binding site. Consistent with these observations, every attempt to infuse 2,4,6-TXPs into the distal pocket in crystals of DHP under conditions identical to those used for the 4-XP x-ray structures resulted in no observable binding.
It appears from the RR data in Fig. 4 that 2,4,6-TFP binds both externally and internally, which implies that it acts as substrate and inhibitor, respectively. It is not possible to directly measure whether 2,4,6-TFP inhibits itself (i.e., whether autoinhibition is occurring). However, 2,4,6-TFP is a poor substrate for DHP compared with 2,4,6-TCP or 2,4,6-TBP, which may be in part due to its propensity to bind as an inhibitor. Fig. S9 provides a kinetic summary of the inhibition of 2,4,6-TCP by 2,4,6-TFP. It is clear from the data that 2,4,6-TFP does in fact inhibit 2,4,6-TCP oxidation. The sigmoidal shape of the 2,6-DCQ product formation suggests that 2,4,6-TFP is oxidized until its concentration is reduced such that it no longer acts as an inhibitor to 2,4,6-TCP oxidation. Several previous binding studies (16,37,38) used 2,4,6-TFP as a model for the native substrate because of its high solubility and similar substitution pattern.
The proposed external active site is consistent with pH-dependent studies of enzyme activity. The greatest activity of DHP for oxidation of 2,4,6-TCP was observed at pH 7.5 (43). Since at this pH, 2,4,6-TCP (pKa 6.4 (44)) is in the phenolate form, it is unlikely that it would be able to enter the distal pocket. On the other hand, the pKa of 4-BP is 9.3 (45), so the inhibitor would be protonated and hence neutral at pH 7.5. It is well known that buried charges are not stable in proteins. Attempts to place a buried charge in Mb by the mutation V68D resulted in ligation of the negatively charged carboxylate to the ferric heme iron, thus neutralizing the charge (46). Hence, neither the structural nor the functional observations of enzymatic activity are consistent with substrate binding in the distal pocket, but the same considerations are consistent with the internal binding of 4-bromophenol as an inhibitor. In line with this reasoning, the pKa of 2,4,6-TFP is ∼7.2 (47), the highest of all of the TXPs studied. Thus 2,4,6-TFP would have the highest percentage of the phenol form at physiological pH, and therefore the greatest propensity to enter the distal cavity of the enzyme.
The correlation of the x-ray structural and RR data elucidates the key role played by the flexibility of the distal histidine. The x-ray crystal structure of Lebioda et al. (1) (PDB 1EW6) shows that H55 is in equilibrium between the open and closed conformations at room temperature. Nicoletti et al. (16) showed that the ν3 band of the RR spectrum displays an ∼40:60 ratio of 5cHS and 6cHS forms. Binding of 4-XP to DHP forces the distal H55 and H2O out of the distal pocket (Figs. 1 b, 2 c, and 3 a). Accordingly, the RR spectra in the presence of 4-XP are typical of 5cHS heme, indicating that the water molecule has been expelled. Conversely, the RR spectroscopic data in Fig. 4 and the UV-vis data in Fig. S3 show that binding of 2,4,6-TXP increases the population of aquo 6cHS heme. Thus, external binding of a 2,4,6-TXP substrate forces the distal H55 into the closed position, stabilizing the heme iron-coordinated water molecule (Fig. 1 a). Substrate binding, then, enforces the closed H55 conformation, which strengthens H2O2 coordination to the heme iron and positions H55 to serve as the acid-base catalyst in the peroxidase mechanism. Furthermore, we have demonstrated that the 5cHS/6cHS ratio can be driven to either extreme by varying the concentrations of inhibitor and substrate (Fig. S4), indicating mutual exclusivity between their respective binding sites.
The kinetic data presented in Fig. 6 and Table 2 demonstrate the competitive nature of the inhibition that occurs between the substrate-inhibitor pair. The structural and spectroscopic data presented herein indicate that this unique mechanism involves two independent but concerted competitive binding events. The first arises from the steric interactions of 4-XP binding in the distal pocket preventing ligation of H2O2 to the heme iron. The second is allosteric communication by movement of the distal histidine resulting in two-site competitive binding between the substrate and inhibitor pair. Thus, inhibition by 4-XP appears to involve three simultaneous effects: 1), H2O2 coordination to the heme iron is impeded; 2), the distal H55, which is the acid-base catalyst in the peroxidase mechanism (48–50), is displaced out of the cavity; and 3), the external binding site is subsequently rearranged by the solvent-exposed (open) distal H55 conformation. Therefore, DHP exhibits a nonclassical two-site competitive inhibition in which the inhibitor and substrate have mutually exclusive binding interactions. When the substrate is bound, the distal histidine is in the internal, or active, conformation (6cHS), and the inhibitor cannot access the internal site (see Fig. 1 a). On the contrary, when the inhibitor is bound, the distal histidine is in the solvent-exposed, or inactive, conformation (5cHS), and the substrate cannot access the external site (see Fig. 1 b). The two-site competitive inhibition is driven by the same open/closed conformational change that has been studied for many years in Mb (15,24).
Conclusions
Since the x-ray crystal structure of DHP showing the substrate analog 4-IP bound in the distal pocket (PDB 1EWA) was the first observation of internal binding, it was reasonable to consider that the internal site may serve as the active site (1). However, structural, functional, and spectroscopic studies have repeatedly contradicted that assumption (17,18,37,38,43,51). The finding that 4-BP and 2,4,6-TBP act as an inhibitor and a substrate, respectively, for DHP is noteworthy because both molecules are present in benthic ecosystems (52). However, since 4-BP is not an oxidation product of 2,4,6-TBP, this inhibitor-substrate pair is not part of a feedback system for this enzyme. Organisms such as Notomastus lobatus (among many others) synthesize 4-BP and 2,4,6-TBP, but this does not appear to be the case for A. ornata (3,53). Instead, DHP, which is the most abundant protein in A. ornata, oxidizes 2,4,6-TBP to 2,6-dibromo-1,4-benzoquinone (Fig. 1). The substrate 2,4,6-TBP acts as both a repellent, protecting marine organisms from predators, and a potentially lethal toxin. Therefore, the degradation of 2,4,6-TBP must be a protective function that minimizes the concentration of the highly toxic molecule in A. ornata (52). Although the reason for the inhibition of 2,4,6-TBP oxidation by 4-BP is not known, it is clear that DHP is a finely tuned enzyme that has an unusual mechanism for inhibitor specificity.
Acknowledgments
We thank R. Ghiladi for a technical critique of the manuscript, and E. Ison for use of the Cary 100.
This work was supported by the U.S. Army Research Office (grant 52278-LS) and Italian grant ex60%.
The atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data Bank under accession codes 3LB1, 3LB2, 3LB3, and 3LB4.
Supporting Material
References
- 1.Lebioda L., LaCount M.W., Woodin S.A. An enzymatic globin from a marine worm. Nature. 1999;401:445. doi: 10.1038/46728. [DOI] [PubMed] [Google Scholar]
- 2.Weber R.E., Mangum C., Bonaventura J. Hemoglobins of two terebellid polychaetes: Enoplobranchus sanguineus and Amphitrite ornata. Comp. Biochem. Physiol. Comp. Physiol. 1977;56:179–187. doi: 10.1016/0300-9629(77)90182-7. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y.P., Woodin S.A., Lovell C.R. An unusual dehalogenating peroxidase from the marine terebellid polychaete Amphitrite ornata. J. Biol. Chem. 1996;271:4609–4612. [PubMed] [Google Scholar]
- 4.Reference deleted in proof.
- 5.Han K., Woodin S.A., Ely B. Amphitrite ornata, a marine worm, contains two dehaloperoxidase genes. Mar. Biotechnol. 2001;3:287–292. doi: 10.1007/s10126-001-0003-8. [DOI] [PubMed] [Google Scholar]
- 6.de Sorrano V., D'Antonio J., Ghiladi R.A. Crystal structure of dehaloperoxidase B at 1.58 A and characterization of the A/B dimer from Amphitrite ornata. Acta Crystallogr. D Biol. Crystallogr. 2010;66:529–538. doi: 10.1107/S0907444910004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodin D.B., McRee D.E. The Asp-His-Fe triad of cytochrome c peroxidase controls the reduction potential, electronic structure, and coupling of the tryptophan free radical to the heme. Biochemistry. 1993;32:3313–3324. [PubMed] [Google Scholar]
- 8.Spiro T.G., Smulevich G., Su C. Probing protein structure and dynamics with resonance Raman spectroscopy: cytochrome c peroxidase and hemoglobin. Biochemistry. 1990;29:4497–4508. doi: 10.1021/bi00471a001. [DOI] [PubMed] [Google Scholar]
- 9.Perutz M.F., Wilkinson A.J., Dodson G.G. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu. Rev. Biophys. Biomol. Struct. 1998;27:1–34. doi: 10.1146/annurev.biophys.27.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y.R., Deterding L.J., Mason R.P. Nature of the inhibition of horseradish peroxidase and mitochondrial cytochrome c oxidase by cyanyl radical. Biochemistry. 2000;39:4415–4422. doi: 10.1021/bi992652+. [DOI] [PubMed] [Google Scholar]
- 11.Zatón A.M., Ochoa de Aspuru E. Horseradish peroxidase inhibition by thiouracils. FEBS Lett. 1995;374:192–194. doi: 10.1016/0014-5793(95)01088-v. [DOI] [PubMed] [Google Scholar]
- 12.Ziemys A., Kulys J. Heme peroxidase clothing and inhibition with polyphenolic substances revealed by molecular modeling. Comput. Biol. Chem. 2005;29:83–90. doi: 10.1016/j.compbiolchem.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 13.de Serrano V., Chen Z., Franzen S. X-ray crystal structural analysis of the binding site in the ferric and oxyferrous forms of the recombinant heme dehaloperoxidase cloned from Amphitrite ornata. Acta Crystallogr. D Biol. Crystallogr. 2007;63:1094–1101. doi: 10.1107/S0907444907043417. [DOI] [PubMed] [Google Scholar]
- 14.Chen Z., de Serrano V., Franzen S. Distal histidine conformational flexibility in dehaloperoxidase from Amphitrite ornata. Acta Crystallogr. D Biol. Crystallogr. 2009;65:34–40. doi: 10.1107/S0907444908036548. [DOI] [PubMed] [Google Scholar]
- 15.Yang F., Phillips G.N., Jr. Crystal structures of CO-, deoxy- and met-myoglobins at various pH values. J. Mol. Biol. 1996;256:762–774. doi: 10.1006/jmbi.1996.0123. [DOI] [PubMed] [Google Scholar]
- 16.Nicoletti F.P., Thompson M.K., Smulevich G. New insights into the role of distal histidine flexibility in ligand stabilization of dehaloperoxidase-hemoglobin from Amphitrite ornata. Biochemistry. 2010;49:1903–1912. doi: 10.1021/bi9020567. [DOI] [PubMed] [Google Scholar]
- 17.Davis M.F., Gracz H., Franzen S. Different modes of binding of mono-, di-, and trihalogenated phenols to the hemoglobin dehaloperoxidase from Amphitrite ornata. Biochemistry. 2009;48:2164–2172. doi: 10.1021/bi801568s. [DOI] [PubMed] [Google Scholar]
- 18.Davis M.F., Bobay B.G., Franzen S. Determination of separate inhibitor and substrate binding sites in the dehaloperoxidase-hemoglobin from Amphitrite ornata. Biochemistry. 2010;49:1199–1206. doi: 10.1021/bi9018576. [DOI] [PubMed] [Google Scholar]
- 19.Tippett P.S., Neet K.E. An allosteric model for the inhibition of glucokinase by long chain acyl coenzyme A. J. Biol. Chem. 1982;257:12846–12852. [PubMed] [Google Scholar]
- 20.Hu D.D., White C.A., Smith J.W. A new model of dual interacting ligand binding sites on integrin αIIbβ3. J. Biol. Chem. 1999;274:4633–4639. doi: 10.1074/jbc.274.8.4633. [DOI] [PubMed] [Google Scholar]
- 21.Tran K.L., Aronov P.A., Morisseau C. Lipid sulfates and sulfonates are allosteric competitive inhibitors of the N-terminal phosphatase activity of the mammalian soluble epoxide hydrolase. Biochemistry. 2005;44:12179–12187. doi: 10.1021/bi050842g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horton H.R., Morton L.A., Rawn D. Pearson Prentice Hall; Upper Saddle River, NJ: 2006. Principles of Biochemistry. [Google Scholar]
- 23.Johnson J.B., Lamb D.C., Young R.D. Ligand binding to heme proteins. VI. Interconversion of taxonomic substates in carbonmonoxymyoglobin. Biophys. J. 1996;71:1563–1573. doi: 10.1016/S0006-3495(96)79359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian W.D., Sage J.T., Champion P.M. Investigations of ligand association and dissociation rates in the “open” and “closed” states of myoglobin. J. Mol. Biol. 1993;233:155–166. doi: 10.1006/jmbi.1993.1491. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z., Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.McCoy A.J., Grosse-Kunstleve R.W., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krissinel E.B., Winn M.D., Emsley P. The new CCP4 Coordinate Library as a toolkit for the design of coordinate-related applications in protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2250–2255. doi: 10.1107/S0907444904027167. [DOI] [PubMed] [Google Scholar]
- 28.Potterton L., McNicholas S., Noble M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Collaborative Computational Project, Project Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 32.Brünger A.T., Adams P.D., Warren G.L. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 33.Humphrey W., Dalke A., Schulten K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 34.Fermi G., Perutz M.F., Fourme R. The crystal structure of human deoxyhaemoglobin at 1.74 A resolution. J. Mol. Biol. 1984;175:159–174. doi: 10.1016/0022-2836(84)90472-8. [DOI] [PubMed] [Google Scholar]
- 35.Harrington D.J., Adachi K., Royer W.E., Jr. The high resolution crystal structure of deoxyhemoglobin S. J. Mol. Biol. 1997;272:398–407. doi: 10.1006/jmbi.1997.1253. [DOI] [PubMed] [Google Scholar]
- 36.Schlichting I., Berendzen J., Sweet R.M. Crystal structure of photolysed carbonmonoxy-myoglobin. Nature. 1994;371:808–812. doi: 10.1038/371808a0. [DOI] [PubMed] [Google Scholar]
- 37.Nienhaus K., Deng P., Nienhaus G.U. Spectroscopic study of substrate binding to the carbonmonoxy form of dehaloperoxidase from Amphitrite ornata. J. Phys. Chem. B. 2006;110:13264–13276. doi: 10.1021/jp060278z. [DOI] [PubMed] [Google Scholar]
- 38.Smirnova T.I., Weber R.T., Franzen S. Substrate binding triggers a switch in the iron coordination in dehaloperoxidase from Amphitrite ornata: HYSCORE experiments. J. Am. Chem. Soc. 2008;130:2128–2129. doi: 10.1021/ja0772952. [DOI] [PubMed] [Google Scholar]
- 39.Feducia J., Dumarieh R., Ghiladi R.A. Characterization of dehaloperoxidase compound ES and its reactivity with trihalophenols. Biochemistry. 2009;48:995–1005. doi: 10.1021/bi801916j. [DOI] [PubMed] [Google Scholar]
- 40.Sivaraja M., Goodin D.B., Hoffman B.M. Identification by ENDOR of Trp191 as the free-radical site in cytochrome c peroxidase compound ES. Science. 1989;245:738–740. doi: 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- 41.Tilton R.F., Jr., Kuntz I.D., Jr., Petsko G.A. Cavities in proteins: structure of a metmyoglobin-xenon complex solved to 1.9 A. Biochemistry. 1984;23:2849–2857. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- 42.Franzen S., Belyea J., Lommel S.A. Proximal cavity, distal histidine, and substrate hydrogen-bonding mutations modulate the activity of Amphitrite ornata dehaloperoxidase. Biochemistry. 2006;45:9085–9094. doi: 10.1021/bi060020z. [DOI] [PubMed] [Google Scholar]
- 43.Franzen S., Gilvey L.B., Belyea J.L. The pH dependence of the activity of dehaloperoxidase from Amphitrite ornata. Biochim. Biophys. Acta. 2007;1774:121–130. doi: 10.1016/j.bbapap.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 44.Dodgson K.S., Smith J.N., Williams R.T. Studies in detoxication. 29. The orientation of glucuronic acid conjugation in chloroquinol. Biochem. J. 1950;46:124–128. doi: 10.1042/bj0460124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kortum G., Vogel W., Andrussow K. Dissociation constants of organic acids in aqueous solution. Pure Appl. Chem. 1961;1:190–536. [Google Scholar]
- 46.Varadarajan R., Zewert T.E., Boxer S.G. Effects of buried ionizable amino acids on the reduction potential of recombinant myoglobin. Science. 1989;243:69–72. doi: 10.1126/science.2563171. [DOI] [PubMed] [Google Scholar]
- 47.Gilvey, L. B. 2006. Kinetic studies of dehaloperoxidase-hemoglobin from Amphitrite ornata. MS thesis. North Carolina State University, Raleigh, North Carolina.
- 48.Dawson J.H. Probing structure-function relations in heme-containing oxygenases and peroxidases. Science. 1988;240:433–439. doi: 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- 49.Poulos T.L. Heme enzyme crystal structures. Adv. Inorg. Biochem. 1988;7:1–36. [PubMed] [Google Scholar]
- 50.Poulos T.L., Kraut J. The stereochemistry of peroxidase catalysis. J. Biol. Chem. 1980;255:8199–8205. [PubMed] [Google Scholar]
- 51.Belyea J., Gilvey L.B., Franzen S. Enzyme function of the globin dehaloperoxidase from Amphitrite ornata is activated by substrate binding. Biochemistry. 2005;44:15637–15644. doi: 10.1021/bi051731k. [DOI] [PubMed] [Google Scholar]
- 52.Lincoln D.E., Fielman K.T., Woodin S.A. Bromophenol accumulation and sediment contamination by the marine annelids Notomastus lobatus and Thelepus crispus. Biochem. Syst. Ecol. 2005;33:559–570. [Google Scholar]
- 53.Chen Y.P., Lincoln D.E., Lovell C.R. Purification and properties of a unique flavin-containing chloroperoxidase from the capitellid polychaete Notomastus lobatus. J. Biol. Chem. 1991;266:23909–23915. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.