

SUMMARY
Animals employ two systems for the de novo biosynthesis of fatty acids: a megasynthase complex in the cytosol (type I) that produces mainly palmitate, and an ensemble of freestanding enzymes in the mitochondria (type II) that produces mainly octanoyl moieties. The acyltransferases responsible for initiation of fatty acid biosynthesis in the two compartments are distinguished by their different substrate specificities: the type I enzyme transfers both the acetyl primer and the malonyl chain extender, whereas the type II enzyme is responsible for translocation of only the malonyl substrate. Crystal structures for the type I and II enzymes, supported by in silico substrate docking studies and mutagenesis experiments that alter their respective specificities, reveal that although the two enzymes adopt a similar overall fold, subtle differences at their catalytic centers account for their different specificities.
Keywords: fatty acid synthase, acyltransferase, malonyltransferase, acetyltransferase, substrate docking, X-ray structure
INTRODUCTION
The de novo biosynthesis of fatty acids is accomplished employing an ensemble of iteratively-acting enzymes that elongate an acetyl precursor by the sequential addition of two-carbon units derived from malonyl moieties. The constituent enzymes are found in two distinct architectural forms. In prokaryotes and plant chloroplast, the catalytic components are freestanding monofunctional proteins, whereas in the cytosol of fungi and animals they are covalently linked in megasynthase complexes. The two architectural forms are known as type II and type I, respectively. Surprisingly, in recent years the simple association of architectural type with phylogenetic status has had to be revised as it has become clear that fungi and animals make fatty acids using both the type I and the type II systems (Schweizer and Hofmann, 2004; Zhang et al., 2005). In animals, the type I cytosolic megasynthase is responsible for the synthesis of the bulk of long-chain fatty acids used for energy storage or lipid synthesis, particularly in the ‘lipogenic tissues’ such as liver and adipose (Clarke, 1993). The type II system, however, is located in the mitochondrial matrix and closely resembles counterparts found in prokaryotes and plant plastids (White et al., 2005). Although a full appreciation of the physiological importance of the mitochondrial system in animals has yet to be established, one important role appears to be the generation of eight-carbon acyl chains that are the precursors of lipoyl moieties utilized for the posttranslational modification of several mitochondrial proteins (Gueguen et al., 2000; Schweizer and Hofmann, 2004; Witkowski et al., 2007). In both the type I and II systems, fatty acyl moieties are assembled on a phosphopantetheine thiol template that is covalently linked to an acyl carrier protein (ACP) component. Both systems also utilize an acyltransferase to translocate the chain extender substrate, typically a malonyl moiety, from CoA to ACP. However, the acyltransferase domain (MAT) associated with the type I animal FAS is unusual in that it is also responsible for loading the primer substrate, usually an acetyl moiety. We report here the crystal structures of the type I and type II acyltransferases associated with the cytosolic and mitochondrial fatty acid synthase systems in humans. Supported by mutagenesis and in silico substrate docking experiments, the study reveals the location of the substrate binding sites of the two acyltransferases.
RESULTS
Overall Structure of MT and MAT
The MAT domain crystallized in an asymmetric unit consisting of four molecules (a–d) that could be superimposed with an rmsd of 0.39 Å for 397 Cα atoms (Figure S1A); hence the molecules can be considered to be structurally identical. The structure was solved to a resolution of 2.81 Å. The MT crystallized with two molecules in the asymmetric unit that could be superimposed with an rmsd of 0.36 Å for 308 Cα atoms (Figure S1B). This structure was solved to a resolution of 1.55 Å. Both MAT and MT are active as monomeric enzymes (Rangan et al., 1991; Zhang et al., 2003), so the arrangement of molecules in the crystal lattice has no functional significance. Although the sequence identity between them is only 18%, the two structures are strikingly similar (Figure 1) and 308 Cα atoms can be superimposed with an rmsd of 1.98 Å. Both enzymes are composed of two subdomains. The larger of these exhibits a typical α/β hydrolase fold with a multi-stranded parallel β-sheet (four strands in MAT, five in MT,) surrounded by α-helices. The smaller domain has a ferredoxin-like fold containing a multi-stranded (three in MAT, four in MT) anti-parallel β-sheet packed against two distal α-helices. The MAT domain of the human FAS more closely resembles its counterparts in the modular polyketide synthases than it does the human mitochondrial enzyme. Thus the enzyme shares 26% sequence identity with the acyltransferase domain from module 5 of the 6-deoxyerythonolide B synthase (DEBS) (Tang et al., 2006) and the Cα atoms of the two structures can be superimposed with a rmsd of 1.86 Å (297 Cα atoms, linkers excluded). The human mitochondrial MT, on the other hand, is more closely related to the malonyltransferases involved in de novo fatty acid biosynthesis in prokaryotes (Figure S2). Indeed, the amino acid sequence is 33%, 27% and 23% identical with its Escherichia coli, Streptomyces coelicolor and Helicobacter pylori counterparts, respectively (Keatinge-Clay et al., 2003; Serre et al., 1995; Zhang et al., 2007) and the Cα atoms of the structures can be superimposed with an rmsd of 1.5 Å (286 Cα atoms for E. coli, Figure S3).
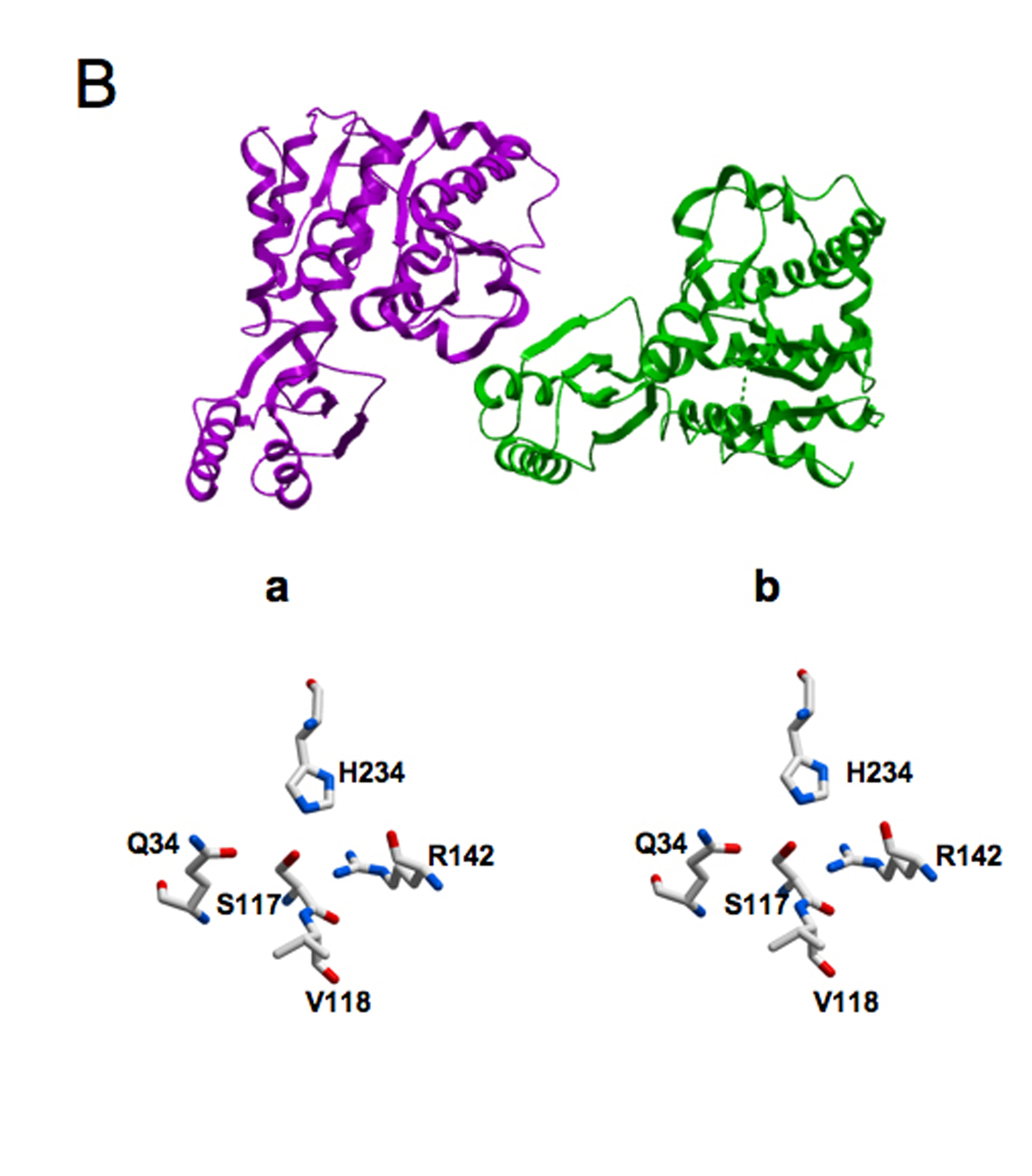
Figure 1.


Comparison of the Three-dimensional Structures of the Acyltransferase Components of the Human Type I Cytosolic and Type II Mitochondrial Fatty Acid Synthase Systems The two structures were first superimposed and then displayed side-by-side for clarity. A: The structure of MAT includes the flanking aminoterminal (yellow) and carboxyterminal (grey) linkers that connect this domain to the adjacent domains in the megasynthase, β-ketoacyl synthase and dehydratase, respectively. B: The structure of the processed MT component of the mitochondrial fatty acid synthase with the mitochondrial targeting sequence removed. In both figures, the active site residues, histidine, serine and arginine are shown in green. C: Superimposition of the Active Sites of MAT (orange) and MT (grey). The region of MAT around Met499 was optimized as described in the Experimental Procedures. D: Primary and secondary structure alignments of the MAT and MT enzymes. The amino- and carboxyterminal linkers are colored yellow and grey, respectively and the three key active-site residues are marked with asterisks
The MAT Linker Regions
The MAT domain contains two additional structural elements corresponding to the amino- and carboxy-terminal linkers that connect the enzyme to its adjacent catalytic domains in the complete FAS structure (Figure 1). Although the aminoterminal linker is not particularly well conserved in 17 animal FAS sequences (Figure S2), it is well structured and defined by a three-stranded β-sheet packed against two helices on the distal side. The carboxyterminal linker, which is moderately well conserved in animal FASs, consists of two helices and a coiled region that folds back and makes contacts with the proximal side of the aminoterminal linker region. The highly structured nature of the flanking linker regions suggests that they do not act merely as flexible tethers, but serve to position the domain in a precise orientation with respect to its neighbors in the megasynthase.
Active Sites
In both the MAT and MT, the active site is located in a gorge between the two subdomains where three key residues are positionally conserved: Ser581, His683 and Arg606 in the human MAT and Ser117, His234 and Arg142 in the human mitochondrial MT (Figure 1). However, the channel leading to the active site of the crystallographic structure of MAT (PDB code 2JFD) was found to be blocked when compared to other structural homologs. In the open unobstructed structures a deep and well-defined channel leads the substrate to the active site. Superimposition of the MAT structure with MT (PDB code 2C2N) and E. coli FabD (PDB code 2G2Z) helped in the identification of Met499 as the residue responsible for the blockage. This region could be reliably remodelled, since there are several homologous structures (e.g. 2C2N, 2G2Z and 2VZ8) with an open channel that could be used as templates. Remodelling of the main-chain conformation around Met499 created a gorge similar to that observed in all other structural homologs. The binding pocket of the remodeled MAT extended further into the gorge between the two subdomains than did the MT substrate-binding pocket and consequently was considerably larger (938 versus 587 Å3). The arrangement of key residues around the active site of the two enzymes is compared in Figure 1C.
As in all acyltransferases, regardless of specificity, the active-site serine residue benefits from its position at the aminoterminus of an α-helix where its nucleophilicity is enhanced by a helix dipole effect. In addition, the conserved histidine residue is positioned within hydrogen-bonding distance of the active-site serine where it can abstract the proton from the hydroxyl and potentiate the nucleophilic character of that residue. This role for the active-site histidine has been validated by inhibitor and mutagenesis experiments with the isolated MAT domain (Rangan and Smith, 1996). An arginine residue is positionally conserved in sequence alignments of all acyltransferases that utilize malonyl moieties and the two crystal structures reveal that this residue is located in the active-site with the guanidinium side-chain directed toward the serine nucleophile (Figure 1C). Earlier mutagenesis experiments with the isolated MAT domain suggested that the arginine residue played a critical role in recognition of the malonyl substrate by interaction with the negatively charged 3-carboxylate (Rangan and Smith, 1997).
Typically in serine active-site acyltransferases, on formation of the tetrahedral intermediate, the negatively charged oxygen of the substrate carbonyl is accommodated in an oxyanion hole. The new crystal structures suggest that the oxyanion holes for the two acyltransferases likely are formed by backbone amides of Met499 and Leu582 in MAT and Gln34 and Val118 in MT (Figure 1C). Met499 is part of a Gly-Met-Gly motif that is positionally conserved in all animal type I FASs, whereas Gln34 is part of a Gly-Gln-Gly motif that is universally conserved in type II MTs (Figure S2).
The essential role of Arg142 in malonyl binding to the MT is illustrated by replacing this residue with glutamine, glycine or alanine (Table 1). All three mutants exhibited significantly reduced catalytic activity and lowered affinity for malonyl-CoA; consequently, catalytic efficiency (kcat/ KM) was reduced by more than four orders of magnitude (Table 1). Clearly, a positively charged residue is necessary for the binding of the malonyl group, as proposed earlier for MAT (Rangan and Smith, 1997). The Arg to Ala substitution also increased both the affinity of the MAT enzyme for its alternative substrate, acetyl-CoA, and overall catalytic activity (Table 1). Unlike the MAT enzyme, wild-type mitochondrial MT is completely inactive toward acetyl-CoA in the standard kinetic assay (Table I) and, in the absence of an acceptor, is not radiolabeled when incubated with [1-14C] acetyl-CoA (details not shown). Surprisingly, replacement of Arg142 in MT actually induces activity toward acetyl-CoA, so that all three mutants exhibited higher catalytic efficiencies with acetyl- rather than malonyl-CoA (Table 1). In this respect, the mitochondrial MT appears to differ from its prokaryotic type II counterparts, since active-site arginine mutants of the Streptomyces coelicolor malonyltransferase are inactive toward acetyl-CoA (Koppisch and Khosla, 2003). Compared to the activity of the wild-type MT toward its natural malonyl-CoA substrate, or the activity of the MAT toward acetyl-CoA, however, the activity of the mitochondrial MT mutants toward acetyl-CoA was low. An additional difference between the bacterial and mitochondrial malonyltransferases is that whereas acetyl-CoA is a weak competitive inhibitor of the bacterial enzyme (Joshi and Wakil, 1971), even in 100-fold molar excess, acetyl-CoA does not inhibit malonyl transfer by the mitochondrial enzyme (details not shown).
Table 1.
Catalytic Properties wild-type MT Enzyme and Mutants
| Enzyme | Mutation | Malonyl CoA | Acetyl CoA | ||||
|---|---|---|---|---|---|---|---|
| KM (µM) | kcat (min−1) | kcat/ KM | KM (µM) | kcat (min−1) | kcat/ KM | ||
| MTa | WT | 3.1 ± 0.6 | 65.1 ± 6.1 | 21 | - | 0 | - |
| R142Q | 38.6 ±9.6 | 0.13 ± 0.01 | 0.003 | 9.1 ± 1.25 | 0.37 ± 0.03 | 0.04 | |
| R142G | 71.2 ± 21 | 0.17 ± 0.02 | 0.0023 | 32.2 ± 6.9 | 0.40 ± 0.05 | 0.012 | |
| R142A | 1060 ± 99 | 0.37 ± 0.02 | 0.00035 | 32.9 ± 5.7 | 0.29 ± 0.04 | 0.009 | |
| MATb | WT | 1.9 ± 0.23 | 72 ± 0.5 | 37.9 | 3.9 ± 0.4± | 114 ± 3.6 | 29.2 |
| R606A | 16.2 ±2.4 | 0.6 ± 0.048 | 0.037 | 1.8 ± 0.28 | 852 ±12 | 473 | |
Assayed at 20 °C.
Assayed on the rat enzyme at 0 °C; data from {Rangan, 1997 #7104}.
Substrate Docking Simulations
In order to optimize performance of the docking experiments, the region around Met499 in MAT was remodelled as described earlier. The CoA thioester substrates could be docked into the active sites of the two acyltransferases in positions compatible with catalysis. The substrates were positioned with the malonyl and/or acetyl moiety inserted deep into the gorge between the two subdomains and the CoA moiety on the surface at the entrance to the binding pocket. In both the malonyl-CoA/MAT and malonyl-CoA/MT structures, the carbonyl oxygen is positioned in the oxyanion hole with the phosphopantetheine sulfur atom positioned close to the active-site serine side-chain oxygen and the backbone amide nitrogen atoms of Met499 in MAT, Gln34 in MT. In both enzymes, the malonyl moiety was positioned such that the 3-carboxylate formed a salt bridge with a terminal nitrogen of the guanidinium side-chain at Arg606 in MAT and Arg142 in MT (Figure 2A and C, respectively).
Figure 2. Stereo Models of the Acyltransferases with Substrates Docked Non-covalently in the Active-sites.

A: Malonyl-CoA docked in MAT. B: Acetyl CoA docked in MAT. C: Malonyl-CoA docked in MT. D. Superimposed substrate binding pockets of MAT (orange) and MT (grey).
Acetyl-CoA could also be docked into the MAT with the cysteamine part of the substrate similarly positioned as in the malonyl-CoA/MAT structure (Figure 2B). However, in the absence of a direct interaction with the Arg606 side-chain, the position of the acetyl methyl group appears to be determined primarily through hydrophobic interactions with the side-chains of neighboring Phe682 and Phe553. A partial density for the Met499 side-chain was tentatively located in only one of the four MAT molecules crystallized in the asymmetric unit (2JFD), so it is likely that this residue enjoys considerable freedom of movement. It too, could contribute to acetyl binding through hydrophobic interaction with the substrate methyl group. Indeed, in the mitochondrial MT and all other malonyl-specific, type II acyltransferases, this methionine is replaced by glutamine, a more polar residue.
Although, in the various simulated MAT and MT enzyme-substrate complexes, the CoA moiety was always positioned at the entrance to the gorge between the two subdomains, the exact location of the nucleotide portion, at the entrance to the substrate-binding pocket, was quite variable and only the cysteamine portion was similarly located. This result implies that the CoA nucleotide moiety may be unconstrained and does not play an important role in substrate binding and is consistent with the earlier finding that malonyl-N-hexanoylcysteamine and malonyl-CoA are equally good substrates for this enzyme (Smith, 1982). Indeed, docking simulations with S-malonyl-N-hexylcysteamine and malonyl-CoA positioned the malonyl moiety in identical locations (Figure S4).
The results of the docking experiments clearly support the proposed essential role for the guanidinium side-chains of Arg606 and Arg142 in the binding of malonyl moieties by the two enzymes. Nevertheless, earlier mutagenesis experiments had shown that the arginine side-chain at position 606 in MAT is not required for acetyl binding. In fact, rather surprisingly, these studies revealed that replacement of the active-site residue Arg606 with alanine dramatically increased activity of the enzyme toward acetyl and longer chain-length substrates (Rangan and Smith, 1997). Thus activity toward octanoyl and decanoyl thioester substrates was increased by 650- and 4760-fold, respectively. In an attempt to identify possible cryptic binding-sites for these longer acyl chains, we performed docking experiments of an Arg606Ala mutant with various CoA thioesters. Malonyl-CoA could not be satisfactorily accommodated in the active-site in the absence of the guanidinium side-chain, consistent with the observation that the catalytic efficiency of this mutant toward malonyl-CoA is reduced by three orders of magnitude (Table I), and further emphasizing the importance of the guanidinium group for malonyl binding. In contrast, docking of acetyl-CoA into MAT was not impeded in the Arg606Ala mutant. In fact CoA thioesters containing up to 10 carbon atoms in the acyl chain could be docked into the active site of this mutant with their acyl chains extending deep into the gorge between the two subdomains. Thus, these medium chain-length acyl moieties are accommodated in the active site where the Arg606 side-chain would be located in the wild-type enzyme (Figure 3A). In the absence of the guanidinium side chain at position 606 the substrate binding pocket extends into a predominantly hydrophobic region defined by residues Phe553, Leu582, Val585, Ala602, Ala603, Phe682, Leu694, Leu698, Val736 and Leu739 (Figure 3B). Again, these findings are consistent with earlier biochemical experiments that showed that acyl moieties containing up to 10 carbon atoms were good substrates for the Arg606Ala mutant, but not for the wild-type enzyme (Rangan and Smith, 1997).
Figure 3. Model of MAT R606A Mutant with Covalently-docked Decanoyl Moiety.

A. The decanoyl group is in thioester linkage with Ser 581. Replacement of Arg by Ala allows binding of the decanoyl chain. Location of Arg606 in the WT enzyme, which obstructs entry of long acyl chains, is also shown for comparison. B. Interaction of the decanoyl chain (yellow) with hydrophobic side chains (green).
DISCUSSION
Structural analyses of the acyltransferase components of the human cytosolic and mitochondrial FAS systems reveal that, despite their association with radically different architectural forms of FAS, these type I and type II enzymes adopt a similar fold and utilize the same three conserved active-site residues, a serine nucleophile and its supporting histidine and an arginine essential for binding the malonyl substrate. The universal conservation of these three residues in all type I and type II fatty acid and polyketide synthases indicates that all of these enzymes likely share the same catalytic mechanism. Nevertheless, the human MAT and MTs are distinguished by their different substrate specificities in that the type I enzyme exhibits dual specificity for both malonyl and acetyl moieties. Our results suggest a possible structural basis for the different specificities. In the simulated substrate dockings with MAT, the malonyl substrate binds with the 3-carboxylate engaged in an ionic interaction with the guanidinium side chain of the active-site arginine residue, whereas the acetyl moiety is stabilized by hydrophobic interactions with Phe553, Phe682 and likely Met499. Malonyl moieties bind to the mitochondrial MT in a similar manner with Arg142 playing an essential role. However, in MT, the active-site region is less hydrophobic with polar glutamine residues (Gln34 and Gln85) replacing two of the residues implicated in acetyl binding in MAT (Phe553 and Met499) (Figure 2D). The absence of these hydrophobic residues in MT likely accounts for the inability of MT to accept acetyl CoA as a substrate. The substrate binding pocket of MAT potentially extends deeper into the gorge between the two subdomains than does that of the MT and, when the normal Arg606 side-chain is replaced with alanine, the overall size of the binding pocket is extended from 938 to 1233 Å3 allowing access to acyl chains containing as many as 10 carbon atoms in the extended, hydrophobic cavity.
The fatty acid megasynthases found in the cytosol of fungi also contain acyltransferases with different specificities. One acyltransferase is dedicated to loading the acetyl primer whereas the other has dual specificity for malonyl and palmitoyl moieties (Schweizer and Hofmann, 2004). This enzyme is responsible both for loading of the malonyl substrate and unloading of the palmitoyl product, by transfer back to a CoA acceptor. In contrast, the animal FAS unloads the palmitoyl product as a free fatty acid through the action of a resident thioesterase (Smith and Tsai, 2007).
Despite the overall similarity with the animal type I megasynthases in terms of the chemical reactions catalyzed, the fungal FASs are organized along completely different architectural lines. Whereas the animal FASs are open, flexible structures, the fungal FASs are rigid barrel-shaped structures in which the constituent, covalently linked enzymes are embedded in the interior walls (Smith, 2006). This difference is illustrated by comparison of the human FAS MAT and the Thermomyces lanuginosus malonyl/palmitoyl transferase (MPT) (Figure S5A). The two catalytic domains adopt essentially the same fold with the active-site serine, histidine and arginine residues identically positioned. A striking difference is that the fungal enzyme includes several structural appendages (shown in grey) that, in the whole megasynthase, are responsible for anchoring the enzyme in the wall of the barrel. Additional differences in the topology of the substrate binding pockets account for the different specificities of the two enzymes. In MPT, the substrate-binding pocket is located in a long groove at the subdomain interface with a hydrophobic patch positioned such that only acyl chains with 16–18 carbon atoms can bind productively (Jenni et al., 2007; Leibundgut et al., 2007; Lomakin et al., 2007). In MAT, the binding pocket is relatively short and in the Arg606Ala mutant can accommodate acyl chains with a maximum of 10 carbon atoms. The fungal acetyl-specific transferase (AT) also adopts a similar fold to other acyltransferases except that the binding pocket is much shorter and the typical conserved arginine is replaced by an isoleucine (Jenni et al., 2007; Leibundgut et al., 2007; Lomakin et al., 2007). An additional residue close to the serine nucleophile, Ile494, projects into the binding cavity, whereas in the MAT structure, the positionally equivalent residue, Leu739, does not (Fig S5B). These modifications may preclude binding of malonyl moieties and longer chain-length acyl moieties in the fungal acetyltransferase.
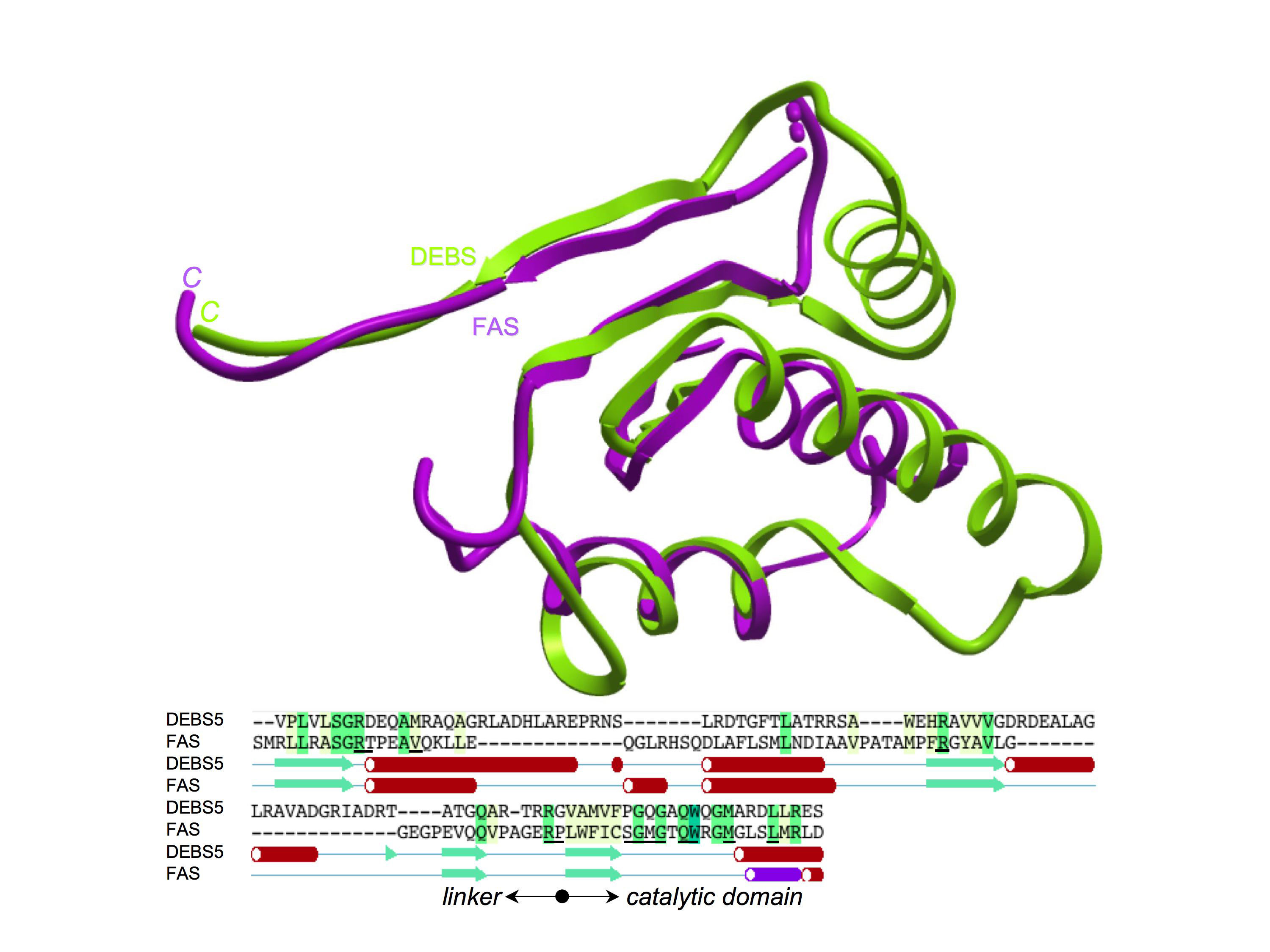
The MAT structure includes extensions at the amino- and carboxytermini that form part of the linkers that connect the enzyme to its neighboring β-ketoacyl synthase and dehydratase domains, respectively, in the cytosolic FAS. The structures of these linker regions are remarkably similar to those flanking the acyltransferase counterpart in module 5 of the 6-deoxyerythronolide B synthase polyketide synthase, as revealed in the crystal structure of the KS~AT didomain (Tang et al., 2006) (Figure S6). The aminoterminal linker structures of the FAS and DEBS acyltransferases can be aligned with an rmsd of 2.39 Å. The only difference is the presence of an additional helix that appears to represents an insertion common to all six 6-deoxyerythronolide B synthase modules but absent from all documented FAS sequences. This additional helix is located on the MAT side of the linker and thus does not appear to make direct contact with the adjacent β-ketoacyl synthase domain. The DEBS acyltransferase crystal structure, which includes the adjacent β-ketoacyl synthase domain, reveals that both the amino- and carboxyterminal linkers interact directly with the β-ketoacyl synthase domain in an arrangement that likely would preclude significant movement of the two domains relative to one another.
The properties of human mitochondrial FAS enzymes described thus far are remarkably similar to those of their type II counterparts found in prokaryotes, consistent with the generally accepted endosymbiotic theory of mitochondrial origin, and these common features should be taken into account when evaluating the effectiveness of antibacterial agents that target type II FAS enzymes. Nevertheless, the prokaryotic and eukaryotic type II systems do have distinguishing features. Whereas the prokaryotic systems use three different β-ketoacyl synthases with overlapping specificities that range from 2-carbon to 16-carbon substrates (White et al., 2005), the mitochondrial system uses only one and has limited ability to elongate beyond the 14-carbon stage (Zhang et al., 2005). The prokaryotic type II system has a unique β-ketoacyl synthase III that catalyzes the chain initiation step, using acetyl-CoA as substrate (Tsay et al., 1992). In contrast, the mitochondrial type II system, appears to generate the acetyl primer by decarboxylation of malonyl moieties at the β-ketoacyl synthase (Christensen et al., 2007; Yasuno et al., 2004; Zhang et al., 2005; Zhang et al., 2003).
While this manuscript was under review, a 3.2 Å-resolution crystal structure was reported for the type I porcine FAS that includes the entire MAT domain and its flanking linkers (Maier et al., 2008). The porcine MAT structure is essentially identical to that of the human counterpart reported here and the authors also conclude that the unusual dual specificity of this enzyme is due to the combined presence of the conserved arginine residue and the hydrophobic nature of the active site.
SIGNIFICANCE
In Metazoans, both the type I cytosolic and type II mitochondrial FASs, translocation of the substrates from CoA thioester linkage to the acyl carrier protein is catalyzed by acyltransferases. The type I acyltransferase transfers both the acetyl primer and the malonyl chain extender, whereas the type II enzyme is responsible for translocation of only the malonyl substrate. Crystal structures and mutagenesis studies of the human type I and II acyltransferases have revealed the structural basis for their different specificities. Both rely on the presence of guanidinium side chain in the active site to anchor the 3-carboxylate of the malonyl substrate. However, whereas binding of the acetyl substrate by the type I enzyme is facilitated by an adjacent hydrophobic patch, acetyl binding in the type II enzyme is precluded by the intrusion of two glutamine side-chains. Unlike the type I system, the type II mitochondrial FAS system lacks an acetyl transferase and therefore cannot elongate an acetyl primer derived directly from acetyl-CoA. Instead this system relies on the decarboxylation of the chain extending malonyl substrate to generate the primer. In mitochondria, the concentration of acetyl-CoA is typically ~ 1mM (Lopaschuk, 2000; Roberts et al., 2005) and likely is much higher than that of malonyl-CoA, so that this arrangement may ensure that acetyl moieties do not block the pathway by saturating the available ACP pool.
EXPERIMENTAL PROCEDURES
Cloning, expression, mutagenesis and purification of mitochondrial MT and FAS-MAT
cDNAs coding for human MT and MAT(FAS) were obtained from the MGC clone collection, and subcloned into a T7 based pET vector (pNIC28-Bsa4, O. Gileadi, SGC Oxford). Site-directed mutagenesis of MT was carried out using the QuikChange® site-directed mutagenesis kit (Stratagene) and primers were from Elim Biopharmaceuticals (Hayward, CA). Authenticity of mutants was confirmed by DNA sequencing. Plasmids encoding either wild type or mutant enzymes were transformed into E. coli strain BL21(DE3). Cells were grown for ~24 hrs at 30 °C in 1L of Overnight™ Expression Instant TB Medium (Novagen) in the presence of kanamycin (50 mg/mL). Cell pellets were collected by centrifugation and stored at −80 °C. Frozen cells were resuspended in 30 mL Buffer A (25 mM Tris-HCl pH 7.8, 0.3 M NaCl, 2 mM Tris[2-carboxyethyl] phosphine, 10% glycerol at pH 7.8) containing 5 mM imidazole. Lysis of cells was carried out at 4 °C in the presence of protease inhibitors (leupeptin 5 µg/ml, trans-epoxysuccinyl-LGB 10 µM, pepstatin 1 µg/ml, and antitrypsin 5 µg/ml) using a microfluidiser. The cell lysate was centrifuged at 12,000 × g for 1 hour at 4 °C, the supernatant passed through a 0.45 μ filter and applied to a 1mL HiTrap™ Chelating HP column (Amersham Biosciences). The column was washed with Buffer A containing 50 mM imidazole then bound proteins were eluted with 500 mM imidazole and concentrated using a 10,000 MWCO Amicon device. All enzymes for kinetic analysis were stored in aliquots at −80 °C in 25 mM Tris-HCl buffer (pH 7.8), 10 % glycerol, 0.3 M NaCl, 2 mM DTT and 2 mM EDTA. For crystallization experiments, proteins after IMAC were subjected to size-exclusion chromatography (Hiload 16/60 Superdex 200, GE Healthcare) in 10 mM HEPES, pH 7.5, 0.5 mM TECP, 5% (MAT) or 10% (MT) glycerol and 300 mM (MT) or 500 mM (MAT) NaCl. Correct masses of the purified wild-type proteins were confirmed by ESI-TOF mass spectrometry.
Measurement of Human Mitochondrial MT Activity
The standard assay was carried out at 20 °C in 83 mM potassium phosphate buffer, pH 6.8 (Zhang et al., 2003). Briefly, the reaction was initiated by the addition of enzyme (0.05–10 µg, depending on enzyme activity) to buffer containing 20 µM mitochondrial holo-ACP and 20 µM [2-14C] malonyl-CoA (Moravek Biochemicals and Radiochemicals) and continued for 2 min. The reaction was stopped by the addition of trichloroacetic acid. The precipitate of protein was washed three times and used for determination of radioactivity. For evaluating activity towards acetyl-CoA, 20 µM [1-14C] acetyl-CoA was used as a substrate. To measure binding of acetyl moieties in the absence of an acceptor, ACP was omitted from the reaction mixture.
MAT and MT Structure Determination
Selection of suitable constructs for crystallization experiments was achieved by screening panels of MAT and MT constructs having different amino- and carboxytermini. A TEV-protease cleavable hexahistidine tag was encoded at the aminoterminus of both the MAT and MT constructs. The MAT construct chosen for crystallization encompassed FAS residues 422–823. This includes the complete catalytic domain (residues 488–809) (Rangan et al., 1997), together with the aminoterminal linker region beginning at residue 422, and a short carboxyterminal extension to residue 823. The crystallized mitochondrial MT construct consisted of residues 69–375 and approximated the mature, processed form of the protein with the aminoterminal targeting sequence removed (Zhang et al., 2003), except that residues 376–390 were also eliminated from the carboxyterminus.
Initial screens set up with native MAT yielded several hits. However, to decrease crystal degradation by crystal handling a condition was selected that provided sufficient cryoprotection After optimization, diffraction quality rod-like crystals were obtained from 20% PEG10K, 0.04 M Na/KPO4 and 25% glycerol with average dimensions 0.6*0.2*0.2 mm3, and were mounted with a loop and flash-frozen by plunging into liquid nitrogen. Datasets were collected at the PXII beamline at the Swiss Light Source using a MAR225 detector at a wavelength of 0.9793 Å. Selenomethionine-labelled protein could be crystallized using the same crystallization condition, and crystals were analogous to the native in morphology and size but degraded after one week and had to be mounted immediately after they appeared. A SAD dataset was collected at the PXII beamline at the SLS, using the selenium peak wavelength (0.9789 Å, determined from a fluorescence scan). The diffraction pattern for both the native and selenomethionine-labelled protein was highly anisotropic, but images could be processed with XDS. Different scans were scaled together using XSCALE. Due to severe anisotropy the anomalous signal obtained from the selenomethione-labelled crystal was significant to 4.5 Å only. However, this proved to be sufficient for SHELXD to locate 40 selenium positions. These were refined by SOLVE and solvent-flattened by RESOLVE to yield an interpretable map. NCS operators could be derived from selenium positions, but NCS averaging did not improve maps, possibly due to translational-only symmetry. Although an initial model could be built, the sequence could not be assigned and refinement stalled. Phases obtained from this model were therefore used as external phase information in SHARP, which allowed the completion of the anomalous substructure by finding minor selenium sites. Phases were solvent-flattened and the model was rebuilt using the new map. Phase restraints (solvent-flattened phases expressed as Hendrickson-Lattman coefficients) proved very useful to keep refinement stable and improved maps significantly. After several rounds of rebuilding and refinement, the new model was used to solve the higher resolution native dataset, and was refined to convergence. Data processing and refinement statistics are given in Table 2.
Table 2.
Crystallographic Analysis
| MAT | MT | |
|---|---|---|
| PDB ID | 2JFD | 2C2N |
| Space group | P212121 | P21 |
| Unit cell | a=85.98 Å, b=92.69 Å, c=159.87 Å | a=52.57 Å, b=84.83 Å, c=74.56 Å, β=93.43° |
| Resolution | 2.81 Å (2.90 Å − 2.81 Å) | 1.55 Å (1.65 Å − 1.55 Å) |
| Completeness | 98.4% (90.5%) | 99.4% (96.5%) |
| Redundancy | 4.0 (3.4) | 4.0 (2.4) |
| I/σ(I) | 15.8 (2.4) | 12.5 (2.3) |
| Rint | 0.050 (0.410) | 0.080 (0.280) |
| Number of atoms | 11392 | 5479 |
| Number of unique reflections | 39706 | 92079 |
| Rwork | 0.212 | 0.178 |
| Rfree | 0.273 | 0.218 |
| R.m.s.d bond lengths | 0.011 Å | 0.016 Å |
| R.m.s.d bond angles | 1.23° | 1.65° |
Values in parentheses are for data in highest resolution shell.
Crystals of MT were obtained using the sitting drop vapor diffusion method at room temperature. Diffraction quality crystals grew from 32% PEG4K, 0.25 M Li2SO4 in Tris/HCl at pH 8.5. Data were collected at the PXII beamline at the Swiss Light Source using a MAR225 detector at a wavelength of 1.0038 Å. Frames were integrated and scaled with MOSFLM and SCALA. Initial phases were calculated by molecular replacement using Phaser and the structures of E. coli and Streptomyces coelicolor malonyl-CoA ACP transacylases (1MLA and 1NM2, respectively) as an ensemble search model. Iterative rounds of model building with COOT and refinement with refmac5 converged to the final model for which statistics are given in Table 2.
Structural remodelling of MAT
Structure modelling was performed using ICM software. Superimposition of the structures to be modelled (MAT PDB code 2JFD) and the templates (PDB codes 2C2N, 2G2Z, 2VZ8) was performed showing agreement in the backbone conformation for all the templates. Tethers were set for atoms of Met499 (2JFD, to be modelled) and corresponding atoms of Met499 (2VZ8, template). Main-chain torsion angles were set free for Met499 (2JFD) and two flanking residues and then energy minimised with the inclusion of the tethers parameter, which was overweighted (tzWeight parameter in ICM =100.00) to allow fast convergence. The resulting main-chain conformation of the loop was again energy minimised for Met499 and two flanking residues, now without the inclusion of the tethered parameter to solve energy strains created by the tethered modelling step. Finally, the side-chain of Met499 was manually optimised to adopt a rotamer similar to that of the 3D equivalents Gln11 (2G2Z) and Gln34 (2C2N). This was followed by a local energy minimisation of the side-chains in the vicinity to ensure the absence of steric clashes.
Structural Analysis Methods
Calculation of the rmsd for Cα alignments was performed on-line using MSDfold (SSM) at http://www.ebi.ac.uk/msd-srv/ssm/ssmstart.html. The size of substrate binding pockets was estimated using CASTp (Dundas et al., 2006).
Substrate Docking in silico
The ICM software (Molsoft LLC, La Jolla, CA) was used to perform docking experiments. The substrates (malonyl-coenzyme A, MLZ and acetyl-coenzyme A, ACO) were retrieved from the Hetero-compound Information Centre Uppsala (release 12.1) (Kleywegt et al., 2003) converted to ICM internal format and processed (protonation, charge assignment and 3D energy minimisation). Docking receptor structures (MT, PDB code 2C2N and MAT, modified model from 2JFD; see above) were also loaded into ICM, converted to ICM internal format, processed (protonation, charge assignment) and regularised (global optimisation of bond lengths, angles, hydrogen bond network, removal of steric clashes). The ligands were then docked into the receptors according to the docking protocol implemented in ICM (Abagyan et al., 2008; Abagyan et al., 1994) without positional restraints. At least three docking experiments were performed for each combination of receptor and ligand. The resulting poses were visually inspected to identify those with the malonyl or acyl moiety of the substrate positioned in a catalytically competent way (i.e. close to the catalytic serine and histidine residues and the oxyanion hole). Once identified, the poses were refined by allowing both the side-chains of the receptor and the substrate to freely rotate and optimise the binding mode.
Supplementary Material
A: Four molecules of MAT with their corresponding active sites shown below. Two side chains were missing from some structures: M499 in b, c and d and R606 in d. B: Two molecules of MT with their corresponding active-sites shown below.
{kind=link}
{kind=link}
The alignments were performed using Clustal W with default parameters. Identical residues are shown in red (*), strongly similar residues in green (:) and weakly similar residues in blue (.). Common names are shown for animal species. The numbering shown refer to the human enzymes. A: Type I malonyl/acetyl transferases. C. is Caenorhabditis. B: Type II malonyltransferases. MT sequences above the dotted line are mitochondrial, those below are prokaryotic. The full scientific names for microorganisms are: Neurospora crassa, Mycobacterium leprae, Escherichia coli, Streptomyces coelicolor, Salmonella enterica, Yersinia enterocolitica, Haemophilus influenzae, Pseudomonas fluorescens, Rickettsiella grylli, Neisseria meningitides.
The human mitochondrial (blue) and E. coli (2g2z, yellow) malonyltransferases are superimposed with active site residues, serine, histidine and arginine shown in red (human) and green (E. coli).
{kind=link}
The substrates S-malonyl-N-hexanoyl cysteamine (grey) and malonyl-CoA (green) were docked into the enzyme using ICM software. Parenth = cysteamine ester.
{kind=link}
A. Superimposition of human MAT (pink) with fungal (T. lanuginosus) MPT (green). Structural domains of MPT are in grey. The active site residues are in indigo (MAT) and yellow (MPT). Hydrophobic residues are in red (MAT) and cyan (MPT).
B. Superimposition of human MAT (pink) with fungal (T. lanuginosus) acetyltransferase (green). Structural domains are in grey, active site residues in indigo (MAT), and yellow (AT).
{kind=link}
The beginning of the catalytic domain, not shown in the structures, is marked on the sequence alignment. The linkers superimpose with an rmsd of 2.39 Å; 108 Cα atoms for DEBS and 91 for FAS. In the DEBS/FAS sequence alignment, residues universally conserved in FASs are shown in red.
{kind=link}
ACKNOWLEDGEMENTS
These studies were supported by NIH grants DK16073 and GM69717 to SS. The Structural Genomics Consortium is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust. We thank Dr. Andrew Orry of Molsoft for his advice on the use of the ICM software.
Footnotes
ACCESSION NUMBERS
Atomic coordinates have been deposited in the Protein Data Bank under ID codes 2C2N (human mitochondrial malonyltransferase) and 2JFD (human fatty acid synthase, malonyl/acetyl transferase domain).
REFERENCES
- Abagyan R, Orry A, Raush E, Budagyan L, Totrov M. ICM Graphical User Interface Guide v3.0. 2008 [Google Scholar]
- Abagyan R, Totrov M, Kuznetsov DA. A new method for protein modelling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15:488–506. [Google Scholar]
- Christensen CE, Kragelund BB, von Wettstein-Knowles P, Henriksen A. Structure of the human beta-ketoacyl [ACP] synthase from the mitochondrial type II fatty acid synthase. Protein Sci. 2007;16:261–272. doi: 10.1110/ps.062473707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SD. Regulation of fatty acid synthase gene expression: an approach for reducing fat accumulation. J Anim Sci. 1993;71:1957–1965. doi: 10.2527/1993.7171957x. [DOI] [PubMed] [Google Scholar]
- Dundas J, Ouyang Z, Tsemg J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucl Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguen V, Macherel D, Jaquinod M, Douce R, Bourguignon J. Fatty acid and lipoic acid biosynthesis in higher plant mitochondria. J Biol Chem. 2000;275:5016–5025. doi: 10.1074/jbc.275.7.5016. [DOI] [PubMed] [Google Scholar]
- Jenni S, Leibundgut M, Boehringer D, Frick C, Mikolasek B, Ban N. Structure of fungal fatty acid synthase and implications for iterative substrate shuttling. Science. 2007;316:254–261. doi: 10.1126/science.1138248. [DOI] [PubMed] [Google Scholar]
- Joshi VC, Wakil SJ. Studies on the mechanism of fatty acid synthesis - XXVI. Purification and properties of malonyl-coenzyme A—acyl carrier protein transacylase of Escherichia coli. Arch Biochem Biophys. 1971;143:493–505. doi: 10.1016/0003-9861(71)90234-7. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay AT, Shelat AA, Savage DF, Tsai SC, Miercke LJ, O'Connell JD, 3rd, Khosla C, Stroud RM. Catalysis, specificity, and ACP docking site of Strep to myces coelicolor malonyl-CoA:ACP transacylase. Structure (Camb) 2003;11:147–154. doi: 10.1016/s0969-2126(03)00004-2. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Henrick K, Dodson EJ, van Aalten DM. Pound-wise but penny-foolish: How well do micromolecules fare in macromolecular refinement? Structure. 2003;11:1051–1059. doi: 10.1016/s0969-2126(03)00186-2. [DOI] [PubMed] [Google Scholar]
- Koppisch AT, Khosla C. Structure-based mutagenesis of the malonyl-CoA:acyl carrier protein transacylase from Streptomyces coelicolor. Biochemistry. 2003;42:11057–11064. doi: 10.1021/bi0349672. [DOI] [PubMed] [Google Scholar]
- Leibundgut M, Jenni S, Frick C, Ban N. Structural basis for substrate delivery by acyl carrier protein in the yeast fatty acid synthase. Science. 2007;316:288–290. doi: 10.1126/science.1138249. [DOI] [PubMed] [Google Scholar]
- Lomakin IB, Xiong Y, Steitz TA. The crystal structure of yeast fatty acid synthase, a cellular machine with eight active sites working together. Cell. 2007;129:319–332. doi: 10.1016/j.cell.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Lopaschuk G. Regulation of carbohydrate metabolism in ischemia and reperfusion. Am Heart J. 2000;139:S115–S119. doi: 10.1067/mhj.2000.103919. [DOI] [PubMed] [Google Scholar]
- Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–1322. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- Rangan VS, Serre L, Witkowska HE, Bari A, Smith S. Characterization of the malonyl-/acetyltransacylase domain of the multifunctional animal fatty acid synthase by expression in Escherichia coli and refolding in vitro. Protein Eng. 1997;10:561–566. doi: 10.1093/protein/10.5.561. [DOI] [PubMed] [Google Scholar]
- Rangan VS, Smith S. Expression in Escherichia coli and refolding of the malonyl/acetyl transferase domain of the multifunctional animal fatty acid synthase. J Biol Chem. 1996;271:31749–31755. doi: 10.1074/jbc.271.49.31749. [DOI] [PubMed] [Google Scholar]
- Rangan VS, Smith S. Alteration of the substrate specificity of the malonyl-CoA/acetyl-CoA:ACP S-acyltransferase domain of the multifunctional fatty acid synthase by mutation of a single arginine residue. J Biol Chem. 1997;272:11975–11978. doi: 10.1074/jbc.272.18.11975. [DOI] [PubMed] [Google Scholar]
- Rangan VS, Witkowski A, Smith S. Isolation of a functional transferase component from the rat fatty acid synthase by limited trypsinization of the subunit monomer. J Biol Chem. 1991;266:19180–19185. [PubMed] [Google Scholar]
- Roberts PA, Loxham SJ, Poucher SM, Constantin-Teodosiu D, Greenhaff PL. Acetyl-CoA provision and the acetyl group deficit at the onset of contraction in ischemic canine skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E327–E334. doi: 10.1152/ajpendo.00441.2003. [DOI] [PubMed] [Google Scholar]
- Schweizer E, Hofmann J. Microbial type I fatty acid synthases (FAS): major players in a network of cellular FAS systems. Microbiol Mol Biol Rev. 2004;68:501–517. doi: 10.1128/MMBR.68.3.501-517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre L, Verbree EC, Dauter Z, Stuitje AR, Derewenda ZS. The Escherichia coli malonyl-CoA:ACP transacylase at 1.5 Å resolution. J Biol Chem. 1995;270:12961–12964. doi: 10.1074/jbc.270.22.12961. [DOI] [PubMed] [Google Scholar]
- Smith S. The effect of CoA and structurally related thiols on the mammalian fatty acid synthase. Arch Biochem Biophys. 1982;218:249–253. doi: 10.1016/0003-9861(82)90343-5. [DOI] [PubMed] [Google Scholar]
- Smith S. Structural biology. Architectural options for a fatty acid synthase. Science. 2006;311:1251–1252. doi: 10.1126/science.1125411. [DOI] [PubMed] [Google Scholar]
- Smith S, Tsai SC. The type I fatty acid and polyketide synthases: a tale of two megasynthases. Nat Prod Rep. 2007;24:1041–1072. doi: 10.1039/b603600g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Kim CY, Mathews, Cane DE, Khosla C. The 2.7-Angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci U S A. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay J-T, Oh W, Larson TJ, Jackowski S, Rock CO. Isolation and characterization of the b-ketoacyl-acyl carrier protein synthase III gene (fabH) from Escherichia coli K-12. J Biol Chem. 1992;267:6807–6814. [PubMed] [Google Scholar]
- White SW, Zheng J, Zhang Y-M, Rock CO. The structural biology of type II fatty acid biosynthesis. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- Witkowski A, Joshi AK, Smith S. Coupling of the de novo fatty acid biosynthesis and lipoylation pathways in mammalian mitochondria. J Biol Chem. 2007;282:14178–14185. doi: 10.1074/jbc.M701486200. [DOI] [PubMed] [Google Scholar]
- Yasuno R, von Wettstein-Knowles P, Wada H. Identification and molecular characterization of the beta-ketoacyl-[acyl carrier protein] synthase component of the Arabidopsis mitochondrial fatty acid synthase. J Biol Chem. 2004;279:8242–8251. doi: 10.1074/jbc.M308894200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Joshi AK, Hofmann J, Schweizer E, Smith S. Cloning, expression, and characterization of the human mitochondrial beta-ketoacyl synthase Complementation of the yeast CEM1 knock-out strain. J Biol Chem. 2005;280:12422–12429. doi: 10.1074/jbc.M413686200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Joshi AK, Smith S. Cloning, expression, characterization and interaction of two components of a human mitochondrial fatty acid synthase: malonyl transferase and acyl carrier protein. J Biol Chem. 2003;278:40067–40074. doi: 10.1074/jbc.M306121200. [DOI] [PubMed] [Google Scholar]
- Zhang L, Liu W, Xiao J, Hu T, Chen J, Chen K, Jiang H, Shen X. Malonyl-CoA: acyl carrier protein transacylase from Helicobacter pylori: Crystal structure and its interaction with acyl carrier protein. Protein Sci. 2007;16:1184–1192. doi: 10.1110/ps.072757307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Four molecules of MAT with their corresponding active sites shown below. Two side chains were missing from some structures: M499 in b, c and d and R606 in d. B: Two molecules of MT with their corresponding active-sites shown below.
The alignments were performed using Clustal W with default parameters. Identical residues are shown in red (*), strongly similar residues in green (:) and weakly similar residues in blue (.). Common names are shown for animal species. The numbering shown refer to the human enzymes. A: Type I malonyl/acetyl transferases. C. is Caenorhabditis. B: Type II malonyltransferases. MT sequences above the dotted line are mitochondrial, those below are prokaryotic. The full scientific names for microorganisms are: Neurospora crassa, Mycobacterium leprae, Escherichia coli, Streptomyces coelicolor, Salmonella enterica, Yersinia enterocolitica, Haemophilus influenzae, Pseudomonas fluorescens, Rickettsiella grylli, Neisseria meningitides.
The human mitochondrial (blue) and E. coli (2g2z, yellow) malonyltransferases are superimposed with active site residues, serine, histidine and arginine shown in red (human) and green (E. coli).
The substrates S-malonyl-N-hexanoyl cysteamine (grey) and malonyl-CoA (green) were docked into the enzyme using ICM software. Parenth = cysteamine ester.
A. Superimposition of human MAT (pink) with fungal (T. lanuginosus) MPT (green). Structural domains of MPT are in grey. The active site residues are in indigo (MAT) and yellow (MPT). Hydrophobic residues are in red (MAT) and cyan (MPT).
B. Superimposition of human MAT (pink) with fungal (T. lanuginosus) acetyltransferase (green). Structural domains are in grey, active site residues in indigo (MAT), and yellow (AT).
The beginning of the catalytic domain, not shown in the structures, is marked on the sequence alignment. The linkers superimpose with an rmsd of 2.39 Å; 108 Cα atoms for DEBS and 91 for FAS. In the DEBS/FAS sequence alignment, residues universally conserved in FASs are shown in red.