

Abstract
CD8+ and CD8− T cell lines expressing the same antigen-specific receptor [the 2C T cell receptor (TCR)] were compared for ability to bind soluble peptide-MHC and to lyse target cells. The 2C TCR on CD8− cells bound a syngeneic MHC (Kb+)-peptide complex 10–100 times less well than the same TCR on CD8+ cells, and the CD8− 2C cells lysed target cells presenting this complex very poorly. Surprisingly, however, the CD8− cells differed little from CD8+ cells in ability to bind an allogeneic MHC (Ld+)-peptide complex and to lyse target cells presenting this complex. The CD8+/CD8− difference provided an opportunity to estimate how long TCR engagements with peptide-MHC have to persist to initiate the cytolytic T cell response.
Antigen-dependent responses by T cells are initiated by reversible interactions between T cell receptors (TCRs) on T cells and peptide-MHC (pepMHC) on antigen-presenting cells (APCs). Both reactants are integral membrane proteins, and analyses of their interactions were greatly hampered until genetic engineering made it possible to obtain them in soluble form. Some studies have examined the binding of soluble pepMHC to TCR on intact T cells, whereas most analyses have been carried out with pepMHC in solution and recombinant TCR molecules immobilized on sensor chips. It has been widely assumed that under these different circumstances kinetic and affinity (equilibrium constant) values are about the same and are equally valid representations of the interactions that take place between live T cells and APCs under physiologic conditions.
Some disparities have become apparent, however, and call this assumption into question. For instance, some pepMHC were found not to bind to a TCR under cell-free conditions, but APCs presenting these complexes were able to stimulate specific cytolytic responses by T cells expressing that TCR (1). Among the many differences between the TCR's microenvironment in buffered salt solution and on the cell membrane, one that has received considerable attention is the presence of coreceptor molecules (CD8 or CD4) on most T cells and their absence in most cell-free systems. This difference could explain the finding that a cognate pepMHC was bound with much higher affinity to the TCR on intact CD8+ cells (2) than to the same receptor on a sensor chip in the absence of CD8 (3). It also would account for the finding that the same pepMHC in the form of streptavidin multimers stained the appropriate CD8+ T cells more intensely than the corresponding CD8− cells (4).
To examine more closely the role of CD8 in TCR–pepMHC interactions and its potential effect on T cell responses, we have here compared the ability of a TCR (the 2C TCR) on CD8+ and CD8− cells to bind soluble pepMHC and the ability of these T cells to lyse target cells that present these complexes. The complexes we examined are SIYRYYGL-Kb (abbreviated hereafter as SYRGL-Kb) and QLSPFPFDL-Ld (called QL9-Ld) (5, 6). They also are referred to as syngeneic and allogeneic complexes because Kb is self or syngeneic with respect to the receptor, and Ld is nonself or allogeneic (7, 8). Both complexes were found to bind strongly and about equally well by the 2C TCR on a CD8+ T cell clone (2, 9). Here we present evidence that the syngeneic complex binds far more weakly to the 2C TCR on CD8− cells than on CD8+ cells, but the allogeneic complex binds almost as well to this receptor on CD8− as on CD8+ cells. This surprising difference was matched by the CD8+ and CD8− 2C cells' functional activity: Target cells presenting the syngeneic complex were lysed far less well by CD8− than by CD8+ 2C cells, whereas target cells presenting the allogeneic complex were lysed virtually as well by the CD8− as by the CD8+ cells. We have taken advantage of the CD8+/CD8− difference to estimate the length of time a TCR–pepMHC engagement has to persist to serve as a “stable engagement,” i.e., one that contributes productively to the initiation of a cytolytic response.
Materials and Methods
T Cells.
Unless otherwise indicated, the T cell clones and lines used here expressed the TCR of the cytotoxic T lymphocyte (CTL) clone called 2C. Clone 2C88 is the original 2C clone (7). Clones G3.1 and L3.100 and the CD8+ 2C cell line were derived from 2C TCR transgenic mice (8). The CD8− 2C T cell line was obtained from 2C TCR transgenic mice that also lack the recombinase-activating gene 1 (2C/RAG mice; ref. 10). The CD8+ 2C CTL cell line and the CD8+ 2C CTL clones (2C88, G3.1, and L3.100) were stimulated weekly with irradiated P815 (Ld+) cells. The CD8− 2C cells were maintained as a line by weekly stimulation with irradiated BALB/c splenocytes (Ld+) supplemented with 100 nM QL9 peptide and 0.5 ng/ml recombinant murine IL-2. Cultured cell lines and CTL clones generally were analyzed 5–7 days after stimulation. No significant differences were found by flow cytometry between the CD8+ and CD8− cell lines in levels of lymphocyte function-associated antigen 1, CD44, and intercellular adhesion molecule (data not shown).
Naïve and memory 2C T cells were isolated from mouse lymph nodes and spleen and purified by negative magnetic sorting to remove other cells as described (11). They were examined within a few hours after they were isolated. Typically, 90% of the naïve cell and 70% of the memory cell preparations were 2C TCR+.
Antibodies and PepMHC.
Anti-CD8 antibody 2.43 (12) was purified from hybridoma cell culture supernatants. Purified clonotypic antibody 1B2, specific for the 2C TCR (7), was digested with immobilized papain (Pierce). Purified Fab fragments (50 μg) were labeled with 2.5 mCi carrier-free Na125I (NEN) using iodobeads (Pierce) in 0.04 M KPO4, pH 7.3, and separated from unbound 125I on a G25 column followed by dialysis against PBS, pH 7.3. The equilibrium constant for the binding of 125I-1B2 Fab to the 2C TCR on intact cells was determined to be KD = 2 nM (25°C).
Soluble Kb was generated in Drosophila melanogaster cells (13) and loaded with a saturating concentration of the SYRGL peptide. For flow cyotometry, biotinylated SYRGL/Kb monomers were obtained from the National Institute of Health's National Institute of Allergy and Infectious Diseases Tetramer Core Facility and incubated at a 1:1 molar ratio with streptavidin-phycoerythrin. QL9-Ld-Ig fusion protein, a kind gift of Sean O'Herrin and Jeffrey Bluestone, University of Chicago (14), was purified from culture supernatants and loaded with an excess of the QL9 peptide.
Peptides (SIYRYYGL, SIINFEKL, and QLSPFPFDL) were synthesized by the t-boc method and purified in the Massachusetts Institute of Technology Biopolymer lab. Peptide concentrations were determined by BCA reaction or amino acid analyses.
Binding Assay.
As described (15), 2.5–5 × 105 2C T cells were incubated with 1.5–2 nM 125I-Fab (1B2) and various concentrations of the SYRGL-Kb complex in 250 μl siliconized microfuge tubes in a total volume of 25–50 μl of PBS (pH 7.3) containing 2% BSA, 0.1% NaN3, and SYRGL (ca. 10−4 M). After gently shaking reaction mixtures for 1 h (ca. 23°C), mixtures were layered over 80% dibutyl phthalate/20% olive oil and centrifuged (ca. 5 sec) to separate bound (cell pellet) and unbound (supernatant) 125I-Fab. Nonspecific binding (the amount of 125I-Fab bound to the cells in the presence of a 50- to 100-fold molar excess of nonradioactive 1B2 antibody) was subtracted from each value. Experimental values were fitted to a competitive binding equation (see equation 7 in ref. 15) using deltagraph to obtain the TCR–pepMHC equilibrium (association) constant.
Flow Cytometry Binding.
A total of 3 × 105 CD8+ and CD8− cells were incubated on ice for 30 min with biotinylated SYRGL-Kb bound to strepavidin-phycoerythrin at a 1:1 molar ratio or with the QL9-Ld-Ig fusion protein, followed by fluorescein-labeled goat anti-mouse IgG. After a single wash, cells were analyzed in a Coulter Epics flow cytometer.
Cytolytic Assays.
51Cr-labeled target cells (T2-Kb or T2-Ld) and peptides were incubated with T cells in round bottom wells of 96-well plates. After 4 h, 51Cr in supernatants was counted. Except for quadruplicate wells to determine spontaneous and maximum 51Cr release, all samples were assayed in duplicate. Specific lysis was calculated as: [(experimental counts − spontaneous counts)/(maximum counts − spontaneous counts)] × 100. Background lysis of the T2-Kb and T2-Ld target cells (in absence of SYRGL or QL9 peptides) by the CD8+ clones and cell lines was negligible (<5%). However, for the CD8− cell line, which had been maintained in culture under different conditions (Materials and Methods), this background varied from 20% to 40%. It was subtracted from the peptide-dependent specific lysis values.
Results
Binding of a Syngeneic and an Allogeneic PepMHC to the TCR on CD8+ and CD8− T Cells.
The CD8+ and CD8− 2C T cell lines expressed approximately the same level of the 2C TCR and various other cell surface markers, except that one expressed CD8 and the other did not (Fig. 1A and data not shown).
Figure 1.

CD8+ and CD8− 2C TCR+ T cell lines and their interactions with soluble pepMHCs. (A) 2C TCR and CD8 levels on the CD8+ and CD8− cell lines. (B) 2C TCR affinity for SYRGL-Kb. Various concentrations of SYRGL-Kb were added to inhibit binding of the 125I-Fab fragment of the 1B2 antibody to the 2C TCR on CD8+ and CD8− cell lines (25°C). B/B0 is the amount of 125I-Fab bound to the cells in the absence (B0) or presence (B) of SYRGL-Kb at the concentrations indicated. Values shown are averages (±SEM) of three titrations, each in duplicate. For the CD8+ cells, the curve is the best fit of experimental values to the competitive-binding equation (Materials and Methods). (C) Homogeneity of the 2C TCR-pepMHC equilibrium constant on cells of the CD8+ T cell line. The log of the ratio of pepMHC-occupied (1-B/B0) to pepMHC-unoccupied (B/B0) TCR sites on CD8+ 2C cells is plotted against the log of the free (total) concentration of pepMHC (SYRGL-Kb). Binding data are from B, where B and B0 are defined. (D) Binding of SYRGL-Kb to 2C TCR on CD8+ and CD8− cells. Biotinylated SYRGL-Kb, bound to streptavidin-phycoerythrin at 1:1 molar ratio, was added at the concentrations shown to CD8+ and CD8− 2C cell lines (4°C). The cells (after one wash) were analyzed by flow cytometry. (E) Binding of QL9-Ld-Ig fusion protein to 2C TCR on CD8+ and CD8− cells. The allogeneic QL9-Ld-Ig fusion protein was added at the concentrations shown to the CD8− 2C T cell line and to CD8+ 2C T cells (clone 2C88) (4°C). The cells (after one wash) were analyzed by flow cytometry, using FITC-labeled goat anti-mouse IgG to detect cell-bound QL9-Ld-Ig.
The binding of soluble SYRGL-Kb to the 2C TCR on CD8+ and CD8− 2C T cells was determined by a competitive binding assay using a fixed amount of 125I-Fab specific to 2C TCR and various concentrations of the SYRGL-Kb complex. Experimental values of cell-bound 125I-Fab were fitted to a competitive binding equation (15) to obtain the TCR–pepMHC equilibrium constant. As shown in Fig. 1B, the SYRGL-Kb complex bound strongly to the CD8+ 2C T cell line, with an equilibrium association constant (affinity) of about 3 × 106 M−1. In contrast, this complex bound very weakly to CD8− cells. We estimate that the affinity of 2C TCR for SYRGL-Kb was about 10–100 times lower on the CD8− cell line than on the CD8+ cell line. The difference between CD8+ and CD8− cells was confirmed qualitatively by flow cytometry using the SYRGL-Kb complex bound in a 1:1 molar ratio with fluorescein-labeled streptavidin (Fig. 1D). The results suggest that the presence of CD8 markedly increases the affinity of 2C TCR for soluble SYRGL-Kb. They can account for the affinity difference for SYRGL-Kb between the 2C TCR on a CD8+ 2C clone and on a sensor chip (2, 3).
The close fit of the binding data for CD8+ cells to the competitive binding curve in Fig. 1B (lower curve) indicates that a single equilibrium constant can account for the 2C TCR-SYRGL-Kb reaction on these cells. To look further for potential equilibrium constant heterogeneity, we plotted the data of Fig. 1B (lower curve) according to the Sips distribution (16). In this plot (Fig. 1C), concentration refers to free SYRGL-Kb (taken as the total pepMHC concentration) and (1-B/Bo)/(B/Bo) refers to the ratio of TCR sites occupied by the pepMHC over unoccupied TCR sites. The slope (a) is a heterogeneity index for the equilibrium association constant (K) for the TCR–pepMHC reaction. When a = 1.0, K is homogeneous. Decreasing values of a indicate increasing diversity of K values. As shown in Fig. 1C, within experimental error the slope (a) is 1.0, indicating that despite considerable variation in CD8 levels (Fig. 1A) they are sufficient to result in a homogenous equilibrium constant for the 2C TCR-SYRGL-Kb interaction on the CD8+ cell line.
We next examined the effect of the presence or absence of CD8 on 2C TCR binding to the allogeneic QL9-Ld complex. Recombinant, soluble Ld was not available, but we were able to examine the binding to these cells of a QL9-Ld-Ig fusion protein generously provided by Sean O'Herrin and Jeffrey Bluestone (14). As shown in Fig. 1E, only an approximately 2-fold higher concentration of this ligand was required to achieve the same level of fluorescence (6-fold above background) on the CD8− cells as on the CD8+ cells. The small difference seen with the QL9-Ld complex in Fig. 1E contrasts with marked difference found in the binding of SYRGL-Kb streptavidin monomer (Fig. 1D) and multimers (4) to CD8+ and CD8− 2C cells.
Functional Differences Between CD8+ and CD8− 2C Cells.
The functional significance of the difference in the CD8 effect on the binding of the two pepMHCs to 2C TCR was examined by testing the ability of CD8+ and CD8− 2C cells to lyse target cells presenting the syngeneic or the allogeneic MHC-peptide complexes. With Kb+ target cells (T2-Kb) and the SYRGL peptide, the cytolytic activity of the CD8+ 2C cell line was indistinguishable from that of an established CD8+ 2C clone (G3.1) (Fig. 2A). The CD8− 2C cells, however, were far less active and required about a 5,000-fold higher peptide (SYRGL) concentration than the CD8+ cells to achieve comparable (half-maximal) lysis of the target cells. In contrast, when the target cells were Ld+ (T2-Ld) and the peptide was QL9, the CD8− 2C cell line was just as effective cytolytically as the CD8+ 2C cell line and a CD8+ 2C clone (G3.1) (Fig. 2B). These results show that a vigorous cytolytic response can occur in the absence of CD8.
Figure 2.

Cytolytic activity of CD8+ and CD8− T cells. T cells were incubated with 51Cr-labeled target cells at a 10:1 ratio for 4 h. T cells were the CD8+ cell line or the CD8− cell line, and a 2C CD8+ T cell clone (G3.1). Target cells were T2 cells expressing either Kb (syngeneic) or Ld (allogeneic). (A) Lysis of syngeneic targets (T2-Kb cells) in the presence of various concentrations of SYRGL peptide. (B) Lysis of allogeneic targets (T2-Ld cells) in the presence of various concentrations of QL9 peptide.
Effect of an Anti-CD8 Antibody.
To establish that the observed differences between CD8+ and CD8− cells was due to CD8 and not to other potential differences between the cell lines, we examined the effect of anti-CD8 antibody (clone 2.43) on cytolytic activity of CD8+ 2C cells. With target cells that presented the allogeneic QL9-Ld complex, the antibody had only a small effect (Fig. 3A), in agreement with the results in Fig. 1E. However, the ability of these cells to lyse target cells expressing the syngeneic SYRGL-Kb complex was completely blocked by the antibody (Fig. 3B). Thus, in the presence of the anti-CD8 blocking antibody, the antigen-mediated responses of the CD8+ 2C clone resembled those of the CD8− cell line. In an unrelated syngeneic reaction, involving a different CD8+ CTL clone (4G3), which recognizes a different syngeneic complex (SIINFEKL-Kb), the anti-CD8 antibody also greatly reduced cytolytic activity (data not shown).
Figure 3.

Effect of an anti-CD8 antibody on the cytolytic activity of a CD8+ 2C CTL clone (L3.100). T cells were incubated with 51Cr-labeled target cells at a 5:1 ratio for 4 h. (A) Lysis of allogeneic target cells (T2-Ld) in the presence of various concentrations of QL9 peptide, in the absence or presence of anti-CD8 antibody 2.43 at 50 μg/ml. (B) Lysis of syngeneic targets cells (T2-Kb) in the presence of various concentrations of SYRGL peptide and in the absence or presence of antibody 2.43 at 50 μg/ml.
Naïve vs. Memory CD8+ T Cells.
Besides the TCR, coreceptor, and pepMHC, many other cell surface structures are involved in T cell–APC interactions, including costimulatory molecules and cell adhesion molecules. To help exclude the involvement of these other molecules in the 2C TCR's affinity for soluble pepMHC on CD8+ and CD8− cells, we examined the binding of soluble SYRGL-Kb to the 2C TCR on CD8+ naïve and memory 2C cells. These naïve and memory cells were used because they express the same number of 2C TCR and CD8 molecules per cell, but differ greatly in their expression of many cell surface markers, including CD44, lymphocyte function-associated antigen 1, IL-2Rβ, and Ly6C (11). Furthermore, memory T cells can respond much faster and to a lower dose of antigen than naïve T cells (17). Accordingly, we measured the equilibrium constant for the binding of SYRGL-Kb complexes to the 2C TCR on CD8+ naïve and memory 2C cells freshly isolated from mice. As shown in Fig. 4, the binding was indistinguishable on the naïve and memory cells and was about the same as on the cultured 2C cell line (Fig. 1B). These results show that besides CD8, other cell surface molecules have no discernable effect on the affinity of 2C TCR for the soluble SYRGL-Kb complex. The ability of memory cells to respond to lower doses of antigen is thus not due to an increase in affinity of TCR for cognate pepMHC.
Figure 4.

Binding of syngeneic (SYRGL-Kb) complexes to naïve and memory CD8+ 2C T cells. The T cells, freshly isolated from mice, were incubated with 125I-Fab of 1B2 antibody and titrated with SYRGL-Kb as in Fig. 1B. Values for naïve and memory 2C cells are averages (±SEM) of three titrations, each in duplicate. A different preparation of isolated cells was used for each titration. Experimental values were curve-fitted to the competitive binding equation as in Fig. 1B. Because the data for naïve and memory cell populations were indistinguishable, the fitted curves were overlaid.
Discussion
Using CD8+ and CD8− T cells expressing the 2C TCR, we show here that the CD8 coreceptor markedly increases the affinity of this receptor for the syngeneic SYRGL-Kb complex. This finding are related to those of Ge et al. (18) who found that adoptively transferred naïve CD8− 2C cells failed to proliferate in syngeneic lymphopenic mice, whereas similarly transferred CD8+ naïve 2C cells proliferated vigorously in these mice and differentiated into memory T cells. Taken together, all of these observations indicate that the affinity of the transferred T cells' TCR for endogenous pepMHCs on APCs of recipient mice is a major determinant of the T cells' proliferative response to lymphopenia.
The finding of a pronounced CD8 effect on the affinity of the 2C TCR for a syngeneic pepMHC agrees with previous studies showing that a photosensitive affinity-labeled peptide reacted more intensely with CD8+ than CD8− cells (19), as did streptavidin multimers of SYRGL-Kb (4). How CD8 increases the equilibrium constant for the 2C TCR-SYRGL-Kb reaction is still not clear despite several extensive studies (e.g., refs. 20–23). Among various suggestions, it has been proposed that in live cells CD8 molecules might be recruited, through association with signaling molecules, to interact with, and somehow stabilize, TCR–pepMHC engagements, as originally suggested for CD4 by Xu and Littman (24). Because we observed the enhancing effect of CD8 on cells under conditions where active recruitment of CD8 to TCR–pepMHC engagements is likely to be limited (25°C, 0.1% azide), it may be that CD8 and TCR molecules exist in such close proximity on the cell surface that extensive recruitment is not essential.
Whatever the reason for the pronounced CD8 effect on the 2C TCR affinity for the SYRGL-Kb, this effect was hardly detectable in the reaction of the receptor with the QL9-Ld complex. Why? One possible explanation arises by considering that if the CD8 effect were as large for the QL9-Ld complex as for SYRGL-Kb the affinity of the 2C TCR for QL9-Ld would have risen to the nanomolar range, a level commonly observed with antibody–antigen reactions. Given such high affinity, pepMHCs might not dissociate fast enough to react successively with TCR molecules, a possible requirement for T cell activation (25). It may now be possible to determine, with the aid of the extremely high-affinity TCR molecules that can be generated by yeast display (26), whether T cells that express TCR with such high affinities can respond effectively to TCR ligation. Stated otherwise, the absence of a significant CD8 effect on the high-affinity reaction with the allogeneic QL9-Ld complex could reflect the potential existence of a TCR affinity ceiling, marked by excessively stable TCR–pepMHC engagements (27).
Because the CD8 effect on the 2C TCR affinity was so pronounced for the syngeneic complex and negligible for the allogeneic complex, we infer that on CD8+ cells, CD8 molecules participate in TCR–pepMHC engagements involving the syngenic complex, but not in those involving QL9-Ld. The recently solved x-ray crystal structures of four TCR–syngeneic pepMHCs, one involving the 2C TCR (28–31), shows in each instance a remarkably similar orientation of the TCR's binding site over the peptide in the MHC's binding cleft. However, from alanine scanning mutagenesis it appears that the interaction of the 2C TCR with QL9-Ld differs from its interaction with SYRGL-Kb in that there is a shift in TCR binding toward the N terminus of the QL9 peptide (32). This shift could hinder optimal association of CD8 with Ld. Whether this shift will be evident in crystallographic studies and extend to other allogeneic pepMHC ligands remains to be seen.
The CD8+/CD8− difference described here provides an opportunity to focus on an elusive determinant of the outcome of T cell-APC encounters in general: namely, the length of time TCR–pepMHC engagements have to persist to trigger a productive T cell response. Much evidence indicates that the outcome of T cell–APC interactions depends, in part, upon (i) the equilibrium constant (affinity) of the TCR–pepMHC reaction, and (ii) the number of copies of the cognate pepMHC per APC (the epitope density). Together, the affinity and epitope density provide an indication of the total number of TCR–pepMHC engagements formed in a T cell–APC encounter (33). However, there is now evidence that the dissociation rate constant (koff) of TCR–pepMHC bonds is also a major factor (34, 35), implying that it is not the total number TCR–pepMHC engagements, but rather the number of stable engagements that is critical. It is not known how long a TCR–pepMHC pair has to remain together to qualify as a “stable engagement,” i.e., one that contributes productively to the initiation of a T cell response.
We suggest that an estimate of the required duration (or residence time) can be derived from the observed differences between CD8+ and CD8− 2C cells in their reactions with soluble SYRGL-Kb and with APC (target cells) presenting this complex. The estimate is based on two main assumptions. First, that the same number of stable TCR–pepMHC engagements (Nt) is required for half-maximal cytolytic activity of the CD8+ and CD8− 2C cells when they react with the same target cell (T2-Kb) and same peptide (SYRGL), as in Fig. 2A. Second, that TCR–pepMHC engagements dissociate (decay) as a first-order process. Thus, using superscripts A and B to refer to values on the CD8+ and the CD8− cell lines, respectively, for the total number of pepMHC engagements (N0) and their dissociation rate constants (k), we can say that
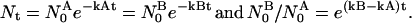 |
If we represent N0B/N0A by R, then
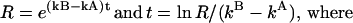 |
t is the residence time that defines a “stable engagement” (for the cytolytic response), R is approximated by [(KB)(TCRB)(EDB)]/[(KA)(TCRA)(EDA)] (33). KA and KB are the equilibrium (association) constants for SYRGL-Kb binding to the 2C TCR on CD8+ and CD8− cells, respectively. From Fig. 1 we take KB/KA to be about 1/100. TCRA and TCRB are free TCR levels on these cells; because TCR levels are about the same (Fig. 1A) and very high (ca. 100,000 TCR per cell), we take TCRB/TCRA to be about 1.0. FEDA and FEDB are free epitope densities (i.e., the number of unengaged SYRGL-Kb complexes per target cell) for CD8+ and CD8− cells, respectively, at half-maximal activity. These values are unknown, and we shall assume that they are proportional to free peptide concentrations at the low concentrations required for this level of cytolytic activity. Thus, from the peptide concentrations needed to achieve half-maximal cytolysis in Fig. 2A, FEDB/FEDA is assumed to be approximately 5,000. R is thus about 50 (i.e., 0.01 × 5,000). kB and kA are the dissociation rate constants for TCR-SYRGL-Kb interactions on CD8− and CD8+ cells. TCR–pepMHC bonds dissociate too rapidly to be measured on intact cells, and we estimate kB and kA by assuming that CD8 affects off-rates but not on-rates of TCR–pepMHC interactions. From equilibrium (association) constants of 3 × 106 and about 3 × 104 M−1 (Fig. 1A) and an assumed on-rate constant (kon) of about 1 × 105 M−1⋅s−1, we estimate kB to be about 3 s−1 and kA about 0.03 s−1 for the CD8− and CD8+ cells, respectively. All of these approximations result in a value for t of about 1 sec.
The foregoing model is based on a number of arguable assumptions. One is that no penalties arise from shorter, nonproductive residence times, as postulated by kinetic proofreading models (36, 37). Another is that CD8 does not affect kon. Our model also neglects a signal transduction role for CD8 in enhancing the cytolytic efficacy of CD8+ cells. While small, this contribution may not be insignificant (see Fig. 3A). Despite all of these limitations the estimated t value may prove useful as a target for future refinement.
Finally, it is well known that in the development of immature T cells destined to become mature CD8+ T cells, CD8 plays an important role in TCR-mediated reactions with syngeneic (self) pepMHCs in the thymus (38, 39). It is thus not surprising that the CD8 effect is prominent in syngeneic reactions by mature CD8+ T cells. But, because there are no parallel constraints on T cell reactions with allogeneic (nonself, foreign) pepMHC, there is no reason for CD8 to have a similar affect on the affinity of TCR for allogeneic complexes (40, 41). It remains to be seen whether the different CD8 effect found here for a syngeneic and an allogeneic reaction is simply a happenstance or applies broadly to antigen recognition by CD8 T cells.
Acknowledgments
We thank Lawrence J. Stern and Jefferson Foote for constructive reviews of the manuscript, Mimi Rasmussen for having derived 2C CTL clones L3.100 and G3.1, Richard Cook and the MIT Biopolymer Laboratory for peptides, Sean O'Herrin for QL9-Ld-Ig, and the National Institutes of Health Tetramer Facility for SYRGL-Kb-SA-PE. This work was supported by National Institutes of Health Grants AI 44477 and CA 60686 (H.N.E.), GM55767 (D.M.K.), and AI 44478 (J.C.), and a Cancer Center Core grant (CA14051 to Richard Hynes).
Abbreviations
- TCR
T cell receptor
- pepMHC
peptide-MHC
- APC
antigen-presenting cell
- CTL
cytotoxic T lymphocyte
References
- 1.Al-Ramadi B K, Jelonek M T, Boyd L F, Margulies D H, Bothwell A L. J Immunol. 1995;155:662–673. [PubMed] [Google Scholar]
- 2.Sykulev Y, Vugmeyster Y, Brunmark A, Ploegh H L, Eisen H N. Immunity. 1998;8:475–483. doi: 10.1016/s1074-7613(00)80631-7. [DOI] [PubMed] [Google Scholar]
- 3.Garcia K C, Tallquist M D, Pease L R, Brunmark A, Scott C A, Degano M, Stura E A, Peterson P A, Wilson I A, Teyton L. Proc Natl Acad Sci USA. 1997;94:13838–13843. doi: 10.1073/pnas.94.25.13838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daniels M A, Jameson S C. J Exp Med. 2000;191:335–346. doi: 10.1084/jem.191.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sykulev Y, Brunmark A, Tsomides T J, Kageyama S, Jackson M, Peterson P A, Eisen H N. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Udaka K, Wiesmuller K H, Kienle S, Jung G, Walden P. J Immunol. 1996;157:670–678. [PubMed] [Google Scholar]
- 7.Kranz D M, Sherman D H, Sitkovsky M V, Pasternack M S, Eisen H N. Proc Natl Acad Sci USA. 1984;81:573–577. doi: 10.1073/pnas.81.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sha W C, Nelson C A, Newberry R D, Pullen J K, Pease L R, Russell J H, Loh D Y. Proc Natl Acad Sci USA. 1990;87:6186–6190. doi: 10.1073/pnas.87.16.6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sykulev Y, Brunmark A, Tsomides T, Kageyama S, Jackson M, Peterson P, Eisen H N. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manning T C, Rund L A, Gruber M M, Fallarino F, Gajewski T F, Kranz D M. J Immunol. 1997;159:4665–4675. [PubMed] [Google Scholar]
- 11.Cho B K, Wang C, Sugawa S, Eisen H N, Chen J. Proc Natl Acad Sci USA. 1999;96:2976–2981. doi: 10.1073/pnas.96.6.2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarmiento M, Glasebrook A L, Fitch F W. J Immunol. 1980;125:2665–2672. [PubMed] [Google Scholar]
- 13.Jackson M R, Song E S, Yang Y, Peterson P A. Proc Natl Acad Sci USA. 1992;89:12117–12121. doi: 10.1073/pnas.89.24.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Herrin S M, Lebowitz M S, Bieler J G, Al-Ramadi B K, Utz U, Bothwell A L, Schneck J P. J Exp Med. 1997;186:1333–1345. doi: 10.1084/jem.186.8.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sykulev Y, Brunmark A, Jackson M, Cohen R J, Peterson P A, Eisen H N. Immunity. 1994;1:15–22. doi: 10.1016/1074-7613(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 16.Karush F. Adv Immunol. 1962;2:1–40. [Google Scholar]
- 17.Cho B, Varada R, Ge Q, Eisen H E, Chen J. J Exp Med. 2000;192:549–556. doi: 10.1084/jem.192.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ge Q, Rao V P, Cho B K, Eisen H N, Chen J. Proc Natl Acad Sci USA. 2001;98:1728–1733. doi: 10.1073/pnas.98.4.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luescher I F, Viver E, Layer A, Mahiou J, Godeau F, Malissen B, Romero P. Nature (London) 1995;373:353–356. doi: 10.1038/373353a0. [DOI] [PubMed] [Google Scholar]
- 20.Wyer J R, Willcox B E, Gao G F, Gerth U C, Davis S J, Bell J I, van der Merwe P A, Jakobsen B K. Immunity. 1999;10:219–225. doi: 10.1016/s1074-7613(00)80022-9. [DOI] [PubMed] [Google Scholar]
- 21.Garcia K C, Scott C A, Brunmark A, Carbone F R, Peterson P A, Wilson I A, Teyton L. Nature (London) 1996;384:577–581. doi: 10.1038/384577a0. [DOI] [PubMed] [Google Scholar]
- 22.Thome M, Germain V, DiSanto J P, Acuto O. Eur J Immunol. 1996;26:2093–2100. doi: 10.1002/eji.1830260920. [DOI] [PubMed] [Google Scholar]
- 23.Delon J, Gregoire C, Malissen B, Darche S, Lemaitre F, Kourilsky P, Abastado J-P, Trautmann A. Immunity. 1998;9:467–173. doi: 10.1016/s1074-7613(00)80630-5. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Littman D R. Cell. 1993;74:633–643. doi: 10.1016/0092-8674(93)90511-n. [DOI] [PubMed] [Google Scholar]
- 25.Valitutti S, Müller S, Cella M, Padovan E, Lanzavecchia A. Nature (London) 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 26.Holler P D, Hollman P O, Shusta E V, O'Herrin S, Wittrup K D, Kranz D M. Proc Natl Acad Sci USA. 2000;97:5387–5392. doi: 10.1073/pnas.080078297. . (First Published April 25, 2000, 10.1073/pnas.080078297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foote J, Eisen H N. Proc Natl Acd Sci, USA. 2000;97:10679–10681. doi: 10.1073/pnas.97.20.10679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garboczi D N, Ghosh P, Utz U, Fan Q R, Biddison W E, Wiley D C. Nature (London) 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 29.Garcia K C, Degano M, Pease L R, Huang M, Peterson P A, Teyton L, Wilson I A. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- 30.Ding Y H, Smith K J, Garboczi D N, Utz U, Biddison W E, Wiley D C. Immunity. 1998;8:403–411. doi: 10.1016/s1074-7613(00)80546-4. [DOI] [PubMed] [Google Scholar]
- 31.Teng M K, Smolyar A, Tse A G, Liu J H, Liu J, Hussey R E, Nathenson S G, Chang H C, Reinherz E L, Wang J H. Curr Biol. 1998;8:409–412. doi: 10.1016/s0960-9822(98)70160-5. [DOI] [PubMed] [Google Scholar]
- 32.Lee P U, Churchill H R, Daniels M, Jameson S C, Kranz D M. J Exp Med. 2000;191:1355–1364. doi: 10.1084/jem.191.8.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sykulev Y, Cohen R, Eisen H N. Proc Natl Acad Sci USA. 1995;92:11990–11992. doi: 10.1073/pnas.92.26.11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyons D S, Lieberman S A, Hampl J, Boniface J J, Chien Y-H, Berg L J, Davis M M. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 35.Kersh G J, Kersh E N, Fremont D H, Allen P M. Immunity. 1998;9:817–826. doi: 10.1016/s1074-7613(00)80647-0. [DOI] [PubMed] [Google Scholar]
- 36.McKeithan T W. Proc Natl Acad Sci USA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rabinowitz J D, Beeson C, Lyons D S, Davis M M, McConnell H M. Proc Natl Acad Sci USA. 1995;93:1401–1405. doi: 10.1073/pnas.93.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fung-Leung W, Wallace V A, Gray D, Sha W C, Pircher H, Teh H-S, Loh D Y, Mak T W. Eur J Immunol. 1993;23:212–216. doi: 10.1002/eji.1830230133. [DOI] [PubMed] [Google Scholar]
- 39.Crooks M E, Littman D R. Immunity. 1994;1:277–285. doi: 10.1016/1074-7613(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 40.Eisen H N, Sykulev Y, Tsomides T. Adv Protein Chem. 1996;49:1–55. doi: 10.1016/s0065-3233(08)60487-8. [DOI] [PubMed] [Google Scholar]
- 41.Zerrahn J, Held W, Raulet D H. Cell. 1997;88:627–636. doi: 10.1016/s0092-8674(00)81905-4. [DOI] [PubMed] [Google Scholar]