

Abstract
Multifunctional macrophage inhibitory cytokine-1, MIC-1, is a member of the transforming growth factor-β (TGF-β) superfamily that plays key roles in the prenatal development and regulation of the cellular responses to stress signals and inflammation and tissue repair after acute injuries in adult life. The stringent control of the MIC-1 expression, secretion, and functions involves complex regulatory mechanisms and the interplay of other growth factor signaling networks that control the cell behavior. The deregulation of MIC-1 expression and signaling pathways has been associated with diverse human diseases and cancer progression. The MIC-1 expression levels substantially increase in cancer cells, serum, and/or cerebrospinal fluid during the progression of diverse human aggressive cancers, such as intracranial brain tumors, melanoma, and lung, gastrointestinal, pancreatic, colorectal, prostate, and breast epithelial cancers. Of clinical interest, an enhanced MIC-1 expression has been positively correlated with poor prognosis and patient survival. Secreted MIC-1 cytokine, like the TGF-β prototypic member of the superfamily, may provide pleiotropic roles in the early and late stages of carcinogenesis. In particular, MIC-1 may contribute to the proliferation, migration, invasion, metastases, and treatment resistance of cancer cells as well as tumor-induced anorexia and weight loss in the late stages of cancer. Thus, secreted MIC-1 cytokine constitutes a new potential biomarker and therapeutic target of great clinical interest for the development of novel diagnostic and prognostic methods and/or cancer treatment against numerous metastatic, recurrent, and lethal cancers.
The transforming growth factor-β (TGF-β) superfamily comprises a wide number of structurally and functionally related growth and differentiation factors that provide critical roles in tissue patterning during embryogenesis and the maintenance of tissue homeostasis and repair after injuries in adult life (Piek et al., 1999; Blobe et al., 2000; Massague and Wotton, 2000; Massague et al., 2000; de Caestecker, 2004; Feng and Derynck, 2005; Bernabeu et al., 2009; Soderberg et al., 2009; Trombly et al., 2009). Human macrophage inhibitory cytokine-1 (MIC-1) has attracted much attention because of its remarkable multifunctional roles in controlling numerous physiological and pathological processes. MIC-1, also designated as prostate-derived factor (PDF), placental TGF-β (PTGF-β), placental bone morphogenetic protein (PLAB), and non-steroidal anti-inflammatory drug-activated gene-1 (NAG-1), or its murine ortholog known as growth/differentiation factor-15 (GDF-15), may participate in the stringent regulation of the expression of specific target gene products in response to diverse external stimuli and tissue damage (Bootcov et al., 1997; Hromas et al., 1997; Lawton et al., 1997; Paralkar et al., 1998; Bottner et al., 1999; Moore et al., 2000; Strelau et al., 2000, 2009; Kempf et al., 2006; Xu et al., 2006; Zimmers et al., 2006; Van Huyen et al., 2008; Ding et al., 2009; Ago et al., 2010). The functions mediated by secreted MIC-1 include the control of embryonic, osteogenic, and hematopoietic development and embryo implantation and pregnancy (Paralkar et al., 1998; Detmer et al., 1999; Moore et al., 2000). MIC-1 also plays important roles in the regulation of the cellular stress and immune responses, cartilage and bone formation, and adipose tissue function and body fat mass (Paralkar et al., 1998; Ding et al., 2009). MIC-1 can also suppress the inflammation through the inhibition of macrophage activation, inhibit the proliferation of primitive hematopoietic progenitors, and participate in the repair of the brain, bone, heart, liver, lung, kidney, and other tissues after severe injuries (Bootcov et al., 1997; Hromas et al., 1997; Bottner et al., 1999; Blobe et al., 2000; Moore et al., 2000; Schober et al., 2001; Xu et al., 2006; Zimmers et al., 2006; Van Huyen et al., 2008). In addition, MIC-1 can act as a potent survival and anti-apoptotic factor and display protective roles for a developing fetus and diverse cell types such as midbrain dopaminergic and serotonergic neurons and cardiomyocytes (Blobe et al., 2000; Moore et al., 2000; Strelau et al., 2000; Tong et al., 2004; Kempf et al., 2006; Xu et al., 2006; Ago et al., 2010). Hence, the modulation of MIC-1 expression and functions represents a potential therapeutic strategy for the treatment of diverse human disorders such as obesity, miscarriage, and neurodegenerative and cardiovascular disorders.
In counterbalance, intense cellular stress and inflammation and/or genetic alterations, leading to an enhanced MIC-1 expression and aberrant activation of the MIC-1-mediated signaling cascades, may result in an enhanced risk of developing diverse diseases such as thalassemia, and congenital dyserythropoietic anemia (CDA), characterized by ineffective erythropoiesis and increased iron absorption (Tanno et al., 2007; Tamary et al., 2008). Moreover, enhanced MIC-1 levels may also contribute to cancer progression and tumor-associated weight loss (Brown et al., 2002; Bauskin et al., 2006; Levy and Hill, 2006; Johnen et al., 2007; Boyle et al., 2009; Karan et al., 2009). Especially, an increase of MIC-1 expression level frequently occurs during the progression of numerous aggressive cancers, including brain, melanoma, oral squamous cell carcinoma and lung, thyroid, gastrointestinal, colorectal, pancreatic, prostate, breast, and cervical epithelial cancers (Brown et al., 2003, 2006, 2009; Karan et al., 2003; Lee et al., 2003; Nakamura et al., 2003; Welsh et al., 2003; Cheung et al., 2004; Koopmann et al., 2004; Wollmann et al., 2005; de Wit et al., 2005; Rasiah et al., 2006; Chen et al., 2007; Shnaper et al., 2009; Zhang et al., 2009; Zhao et al., 2009; Xue et al., 2010). Like the TGF-β prototype of superfamily members, MIC-1 can play dual roles during cancer development by negatively or positively modulating cancer cell behavior. MIC-1 may influence the proliferation, differentiation, survival, migration, invasion, and/or metastatic spread of cancer cells in a manner depending on cell type and context. In general, MIC-1 displays anti-tumoral activities in the early stages of cancer development, while this cytokine can rather promote the invasion and metastases of cancer cells at distant tissues in the late stages of cancer. In this regard, we reviewed the structural organization and expression pattern of mature MIC-1 ligand and the potential signal transduction elements involved in the mediation of the cellular effects of this secreted cytokine in physiological and pathological conditions. The molecular mechanisms regulating the expression, secretion, and multiple cellular functions of MIC-1 and the pathological consequences resulting from their deregulation in the development of diverse human diseases and cancers are discussed. The emphasis is on the pleiotropic roles played by secreted MIC-1 cytokine in the carcinogenesis process and its therapeutic implications in the development of novel diagnostic and prognostic methods and cancer therapies.
MIC-1 Structural Organization, Expression Pattern, and Signal Transduction Mechanisms
MIC-1 structural organization, processing, and secretion
The human MIC-1 gene maps to chromosome 19 in the region p13.1-13.2 and consists of a DNA sequence of 2,746 base pairs that encompass two exons separated by a single intron. MIC-1 is synthesized under a 308-amino acid (aa) protein precursor composed of a 29-aa signal peptide, a 167-aa propeptide, and a 112-aa mature region (Fig. 1) (Bootcov et al., 1997; Bauskin et al., 2000). The studies carried out by deletion mutagenesis indicated that the N-terminal 28-aa of the propeptide may be involved in the proteasomal degradation of misfolded pro-MIC-1 monomer (Bootcov et al., 1997; Bauskin et al., 2000). During proteolytic processing, the N-terminal hydrophobic signal peptide sequence is removed from the pro-MIC-1 precursor. After disulfide-linked dimerization and correct folding in the endoplasmic reticulum, the dimeric pro-MIC-1 precursor is further cleaved at a typical furin-like RRAR processing site at the position 196 by a proprotein convertase. The proteolytic cleavage generates a N-terminal propeptide and a C-terminal polypeptide fragment that constitutes the mature and biologically active portion of the molecule, which is then secreted in the extracellular medium (Fig. 1) (Bootcov et al., 1997; Bauskin et al., 2000, 2005, 2006). Unlike other TGF-β superfamily members, propeptide is not required for the proper folding and secretion of a mature and biologically active MIC-1 dimer (Bauskin et al., 2000, 2005, 2006; Fairlie et al., 2001). During the secretion process, the propeptide of pro-MIC-1 precursor may be N-glycosylated at the 70-aa position (Bauskin et al., 2000). The mature MIC-1 also contains seven cysteine residues forming a cysteine knot, a structural hallmark that is highly conserved among the different members of the TGF-β superfamily. Nevertheless, although the mature MIC-1 polypeptide may be secreted as a disulfide-linked homodimer after intracellular processing, the unprocessed pro-MIC-1 precursor molecules may also be secreted and associated with the extracellular matrix (ECM) components of cancer cells via an interaction mediated through the propeptide (Fig. 1) (Bauskin et al., 2005, 2006). It has been proposed that the association of unprocessed pro-MIC-1 precursor with ECM components may contribute to its latent storage in stroma, and thereby modulate its local bioavailability, cellular functions, and serum concentration (Bauskin et al., 2005, 2006). In fact, the presence of proprotein convertases within ECM at the cell surface could contribute to the extracellular processing of the pro-MIC-1 precursor under specific conditions (Tsuji et al., 2003; Bauskin et al., 2005, 2006).
Fig. 1.
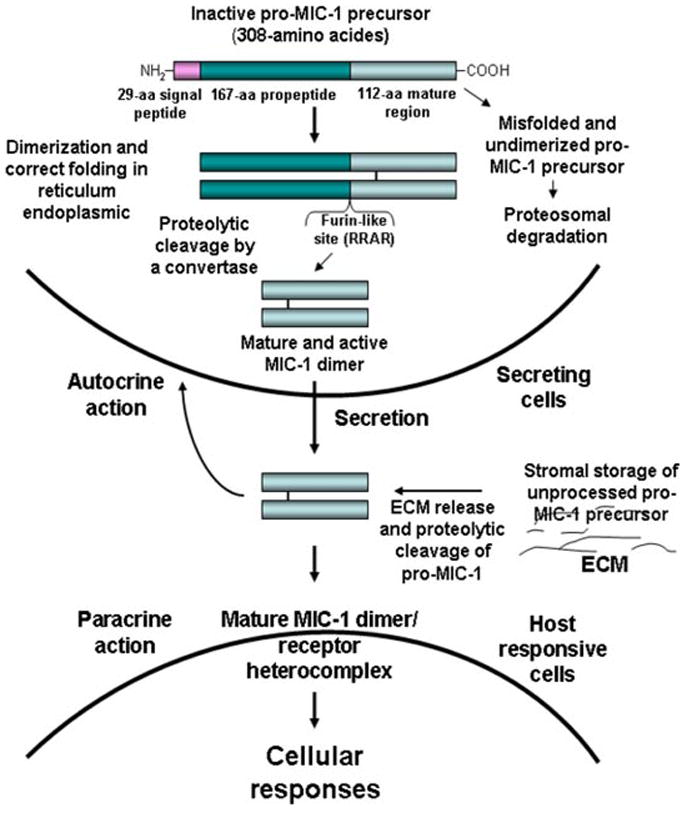
Molecular mechanisms associated with the cellular MIC-1 processing, secretion, storage, and its autocrine and paracrine actions. The scheme shows the molecular mechanisms associated with the cellular processing of inactive pro-MIC-1 precursor via the formation of dimeric molecules followed by their proteolytic cleavage at a furin-like site catalyzed by a convertase in reticulum endoplasmic, which results in there lease of N-terminal propeptide and C-terminal fragment constituting the mature and active form. The secretion of dimeric MIC-1 protein into extracellular compartment as well as the storage of unprocessed pro-MIC-1 precursor in extracellular matrix (ECM) is also shown. The potential autocrine or paracrine actions of mature MIC-1 dimer on secreting and neighboring responsive cells are also illustrated. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
MIC-1 expression pattern and regulatory mechanisms
A variety of signaling pathways may contribute to the stringent regulation of endogenous MIC-1 expression, secretion, and stromal storage, and thereby modulate its functions in physiological and pathological conditions (Fig. 2). MIC-1 expression level is usually low in resting cells but may be substantially increased following an adaptive response to diverse cellular stress signals, such as a partial or complete oxygen deprivation termed as hypoxia and anoxia, inflammation, short-wavelength light exposure, acute tissue injuries, and during cancer progression (Albertoni et al., 2002; Zimmers et al., 2006; Akiyama et al., 2009; Ding et al., 2009; Ago et al., 2010; Krieg et al., 2010). In this regard, MIC-1 was first isolated by a subtraction cloning approach using a cDNA library enriched for macrophage activation-associated genes prepared from U937 myelomonocytic cells (Bootcov et al., 1997). It has been observed that the MIC-1 expression in human U937 and KG-1 monocytoid cell lines was greatly enhanced by a treatment with a differentiation agent, trans-retinoic acid (RA) followed by activation with phorbol 12-myristate 13-acetate (PMA) (Bootcov et al., 1997). More specifically, the induction of MIC-1 mRNA expression in activated macrophages by secreted pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α ), interleukin (IL)-1β and IL-6, macrophage colony-stimulating factor (M-CSF) and TGF-β, suggests that MIC-1 may act through an autocrine loop as an inhibitory factor in the late phases of the macrophage activation during inflammatory responses (Bootcov et al., 1997; Fairlie et al., 1999). In this regard, the macrophages in the adrenal gland and tissue-infiltrating activated macrophages recruited after acute tissue injuries strongly express high amounts of MIC-1/GDF-15 mRNA transcript and mature product (Bottner et al., 1999; Fairlie et al., 1999; Schober et al., 2001). Interestingly, it has also been observed that the blue- and near-UV light exposure also induced MIC-1 expression in human dermal fibroblasts, suggesting that certain functions of this cytokine could be controlled by the light signals (Akiyama et al., 2009).
Fig. 2.

Cellular events and signaling elements involved in the regulation of the MIC-1 expression levels. The increase of MIC-1 expression induced via cellular stress signals, light signal, inflammation, cancer progression, and chemotherapeutic drugs is indicated. The potential cellular signaling elements involved in the regulation of MIC-1 expression as well as the cells expressing high levels of MIC-1, such as activated macrophage, normal cells, cancer cells, and tumor host cells are also shown. The potential to assess the increase of MIC concentration in serum or cerebrospinal fluid as diagnostic and prognostic biomarkers of cancers and the molecular targeting of MIC-1 for improving the current cancer treatment is also indicated. α-DHT, α-dihydrotestosterone; H2O2, hydrogen peroxide; HIF-α1, hypoxia inducible factor-α1; IL, interleukin; MIC-1, macrophage inhibitory cytokine-1; NF-κB, nuclear factor-κB; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
In addition, the results from in situ hybridization and immunohistochemical analyses have also revealed that MIC-1 and its murine ortholog GDF-15 show a comparable expression pattern in adult tissues in normal physiological conditions. MIC-1/GDF-15 is detected at high levels in placenta and, to a lower extent, in epithelial cells of mammary and prostate glands, lung, liver, gastrointestinal tract, pancreas, and kidney (Lawton et al., 1997; Paralkar et al., 1998; Bottner et al., 1999; Fairlie et al., 1999; Li et al., 2000; Schober et al., 2001; Kim et al., 2002; Marjono et al., 2003). The epithelium of the choroid plexus of all ventricles and ependyma in the adult brain also express MIC-1 which is secreted into the cerebral spinal fluid (Strelau et al., 2000; Schober et al., 2001). A significant increase of human MIC-1 concentration in the serum has also been detected by using a sensitive enzyme-linked immunosorbent assay (ELISA) in pregnant women as well as individuals with an elevated risk for developing stroke and myocardial infarction as compared to the basal serum level of this cytokine (Moore et al., 2000; Brown et al., 2002). In contrast, low MIC-1 serum concentration has been detected before miscarriage, suggesting possible predictive and causative roles of the low expression of this cytokine in pregnancy complications and the therapeutic potential to up-regulate MIC-1 to prevent the miscarriage (Tong et al., 2004).
Importantly, numerous lines of experimental evidence have also revealed that MIC-1 can act as a downstream mediator of the cell growth arrest and/or apoptosis induced following the DNA damages and the activation of p53/p21WAF1/CIP1 pathway in response to cellular stress signals and inflammation (Li et al., 2000; Tan et al., 2000; Albertoni et al., 2002; Yang et al., 2003; Modlich et al., 2004; Chenau et al., 2009; Kelly et al., 2009). Consistently, it has been shown that the MIC-1 gene promoter, which contains two consensus p53-binding sites, may be activated by the wild-type p53 tumor suppressor protein but not by the transcriptionally inactive p53 mutants (Li et al., 2000; Tan et al., 2000; Yang et al., 2003). Also, MIC-1 expression was significantly up-regulated by TAp63α and involved in keratinocyte differentiation (Ichikawa et al., 2008). Furthermore, several non-steroidal anti-inflammatory drugs (NSAIDs), peroxisome proliferator-activated receptor-γ ligands, dietary compounds such as genistein, retinoids, and resveratrol as well as chemotherapeutic drugs, including etoposide and doxorubicin, have been shown to increase MIC-1 expression levels in a p53-dependent or -independent manner in cancer cells in vitro and in xenograft models established in mice in vivo (Li et al., 2000; Tan et al., 2000; Yang et al., 2003; Yamaguchi et al., 2004; Baek et al., 2005). It has also been reported that MIC-1 is markedly up-regulated in biopsies from breast cancer patients after chemotherapeutic treatment with the combination of epirubicin and cyclophosphamide or epirubicin and taxol (Modlich et al., 2004). Thereby, these therapeutic agents may trigger anti-tumorigenic mechanisms leading to a decreased rate of cell proliferation and/or altered cell survival. For instance, the expression of endogenous MIC-1 was remarkably induced by chemotherapeutic drug, etoposide in p53 wild-type cancer cell lines but not in p53-deficient cells (Li et al., 2000; Tan et al., 2000).
On the other hand, other transcription factors, such as EGR-1 tumor suppressor protein, nuclear factor-κB (NF-κB), and hypoxia-inducible factor-1α (HIF-1α ), can also induce the MIC-1 expression in certain cell types, including p53 wild-type and p53-mutant cancer cells (Fig. 2) (Baek et al., 2005; Shim and Eling, 2005; Krieg et al., 2010). More specifically, it has been observed that the treatment of LNCaP prostate cancer cells with 12-O-tetradecanoyl-phorbol-13-acetate (TPA) resulted in an up-regulation of MIC-1 expression via the activation of protein kinase C and an increase of transcriptional activity of NF-κB that specifically interacts with the MIC-1 promoter (Shim and Eling, 2005). Moreover, it has been reported that the induction of HIF-1α under hypoxic conditions prevalent within the tumor microenvironment may result in the activation of histone demethylase, JMJD1A that, in turn, may decrease the promoter histone methylation, and thereby enhance the hypoxic gene expression including MIC-1 (Krieg et al., 2010). The Akt activation also up-regulated the MIC-1 expression level in breast cancer cell lines, including MCF-7 cells through the Sp1-binding site found in the MIC-1 promoter (Wollmann et al., 2005). The treatment of rats with α-dihydrotestosterone (α-DHT) has also been observed to increase the MIC-1/PDF expression in the prostate suggesting that the androgens may control the expression of this cytokine in the normal prostate gland (Paralkar et al., 1998). It has also been reported that androgens may contribute to the regulation of MIC-1 expression in prostate cancer cell lines (Karan et al., 2003; Liu et al., 2003; Kakehi et al., 2004). The MIC-1 mRNA expression may also be up-regulated by ultraviolet (UV) irradiation in melanocytes and via the stimulation of the MAPK pathway and microphthalmia-associated transcription factor (MITF) in melanoma cell lines (Boyle et al., 2009). Hence, the MIC-1 expression level and secretion will influence the cellular functions mediated by this cytokine in a given cell type.
Analogies between MIC-1 signal transduction mechanisms and other TGF-β superfamily members
Major progress has been made over the past years to define the molecular mechanisms at the basis of the observed autocrine and paracrine effects mediated through different TGF-β superfamily members (Piek et al., 1999; Massague and Wotton, 2000; de Caestecker, 2004; Feng and Derynck, 2005; Levy and Hill, 2006). In general, TGF-β superfamily members, including TGF-β1, 2, and 3, GDFs, bone morphogenetic proteins (BMPs), activins, nodal, inhibins, myostatins, and anti-Müllerian hormone (AMH) mediate their biological effects, at least in part, by activating the heteromeric transmembrane receptor complex comprised of two type I and two type II receptor serine/threonine protein kinases and intracellular downstream effectors, Smad proteins (Figs. 1 and 3) (Piek et al., 1999; Feng and Derynck, 2005; Levy and Hill, 2006). The specificity and versatility of the biological effects mediated by each ligand of the TGF-β superfamily may be achieved through the activation of different combinations of type I and II receptor serine/threonine kinases at the cell surface and the formation of different intracellular Smad protein complexes or other signaling effectors (Fig. 3). More specifically, seven type I receptor kinases termed as activin-like receptors (ALKs) 1–7 have been identified in mammalians and shown to form a specific complex with five different type II receptors designated as TGF-βR-II, ActR-II, ActR-IIB, bone morphogenetic protein receptor-II (BMPR-II), and anti-Müllerian hormone receptor-II receptor (AMHR-II) (Piek et al., 1999; Gouedard et al., 2000; Oh et al., 2000; Reissmann et al., 2001; Levy and Hill, 2006). There are two principal Smad pathways, the activated type I receptors ALKs 1, 2, 3, and 6 can phosphorylate the Smad members 1, 5, and 8 while Smads 2 and 3 are substrates for ALKs 4, 5, and 7 (Massague and Wotton, 2000; Levy and Hill, 2006).
Fig. 3.
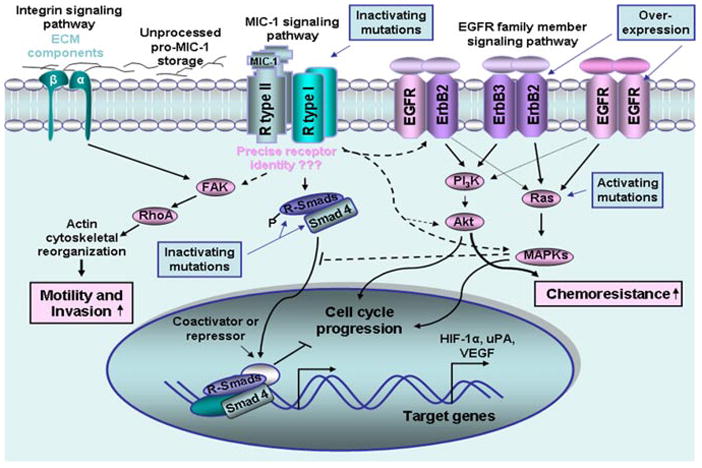
Potential signal transduction mechanisms involved in the mediation of cellular responses induced by secreted MIC-1 cytokine in certain cancer cells. The scheme shows the potential signaling transduction elements mediating the cellular responses induced by MIC-1 in certain cancer cell types and interactive cross-talks with other oncogenic signaling pathways. In analogy with other TGF-β superfamily members, MIC-1 can interact with type I and II receptor serine/threonine kinases, which remain to be identified more precisely in a given cell type. Thereby, MIC-1 can induce the formation of heteromeric receptor complex that, in turn, may stimulate different R-Smad proteins by phosphorylation and their association with their Smad4 co-partner. The R-Smad/Smad4 complexes may translocate to the nucleus, where they may participate in co-operation with other factors, such as co-activators or co-repressors, in the transcriptional regulation of the expression of gene products that mediate specific cellular responses. Moreover, MIC-1 can also induce its cellular effects through the stimulation of other signaling elements and cross-talks with intracellular pathways initiated by distinct growth factors. Particularly, the activation of the EGFR family members, EGFR, ErbB2, and ErbB3 via their phosphorylation which can be induced by MIC-1, is shown. The activation of the FAK/RhoA, PI3K/Akt, and MAPK signaling elements induced by MIC-1, which may contribute to the acquisition of more malignant behavior by cancer cells, including an increase in their migratory and invasive abilities, is also illustrated. In addition, the frequent deregulations in the MIC-1 signaling network that may promote its oncogenic effects are also indicated. Moreover, the cytoprotective effect induced by MIC-1 via the stimulation of the PI3K/Akt survival pathway, which may contribute to the resistance of certain cancer cell types, to current chemotherapeutic treatment is also indicated. EGFR, epidermal growth factor receptor; FAK, focal adhesion kinase; HIF-1α, hypoxia inducible factor-1α; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3′ kinase; R-Smads, receptor regulated Smads; VEGF, vascular endothelial growth factor; uPA, urokinase type-plasminogen activator. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Although the specific receptors activated by secreted MIC-1 have not been precisely identified, it has been suggested that secreted MIC-1 cytokine, like all other TGF-β superfamily members, can mediate certain cellular responses via the stimulation of TGF-β receptors type I and II and intracellular Smad signal transduction protein complexes (Fig. 3) (Tan et al., 2000; Xu et al., 2006; Johnen et al., 2007; Soto-Cerrato et al., 2007; Ago et al., 2010). More specifically, it has been observed that MIC-1 can activate certain TGF-β-responsive promoters and pure recombinant or transfected MIC-1-induced growth inhibition in cancer cell lines with intact TGF-β signaling (Tan et al., 2000). In contrast, neither TGF-β1 ligand nor conditioned medium from MIC-1 expressing cells exhibited the growth inhibitory effect on TGF-β receptor type I or II mutant cells or Smad 4 null cells (Tan et al., 2000). It has also been reported that the prodigiosin-mediated p21WIF1/CIP1 expression in MCF-7 breast cancer cells was blocked by a selective inhibitor of the TGF-β type I receptor kinases ALK5, ALK4, and ALK7 and MIC-1/NAG-1, which is overexpressed in response to prodigiosin was co-localized with TGF-β receptor type I at the cell surface, suggesting a possible interaction between them (Soto-Cerrato et al., 2007). In addition, the MIC-1/GDF-15 expression in cultured neonatal cardiomyocytes also induced an anti-hypertrophic response through the activation of Smad2/3 signaling elements, whereas an overexpression of the inhibitory proteins, Smad6/7, reversed the anti-hypertrophic effects of MIC-1/GDF-15 (Xu et al., 2006).
Thus, in analogy with other TGF-β superfamily members, a simplified view of potential signal transduction mechanisms of MIC-1 may involve the ligand binding to a constitutively active type II receptor kinase that recruits and phosphorylates a type I receptor (Fig. 3). The activated type I receptor kinase, in turn, may phosphorylate the downstream signaling effectors, designated as receptor-regulated Smads (R-Smads), which can bind the common co-Smad partner, Smad4 protein. The activated R-Smad/Smad4 complexes then translocate from cytoplasm into nucleus where they act as transcription factors in interacting with the DNA recognition sequence, CAGAC, found in the regulatory elements of target genes (Shi et al., 1998; Massague and Wotton, 2000). Other nuclear DNA-binding co-factors may also co-operate with Smad proteins in the regulation of the expression of numerous target genes (Fig. 3) (Tsukazaki et al., 1998; Massague and Wotton, 2000). Moreover, the nuclear Smad multi-subunit complexes may recruit other transcriptional factors, acting as the co-activators or co-repressors, and which may influence the specific cellular response mediated by heteromeric Smad complexes in a given cell type (Fig. 3).
In addition, secreted MIC-1 cytokine, like other TGF-β superfamily members, can also stimulate other intracellular signaling elements, such as Ras/MAPKs and PI3K/Akt/mTOR signaling pathways (Massague and Wotton, 2000). Moreover, a variety of positive and negative regulatory molecules may interfere with the expression, subcellular localization, secretion, storage, and stability of MIC-1 and/or the activities of its receptors and intracellular signaling elements, and thereby control its functions. The cross-talks between MIC-1 signaling with other growth factor cascades have been undiscovered, which may promote or attenuate the Smad-dependent- and independent-mediated responses (Massague and Wotton, 2000). Hence, the activation of the MIC-1 pathway and its integration within the intracellular signaling network will determine the final outcome on the target gene expression and responses in a given cell type.
Physiological and Pathological Functions of Secreted MIC-1 Cytokine
Numerous studies on cell culture and animal models have revealed that the signal transduction pathways and cellular responses induced by secreted MIC-1 cytokine during tissue patterning, embryonic, and fetal development as well as its pleiotropic effects on normal and cancer cells in adult life are highly dependent on cell type and context (Bootcov et al., 1997; Hromas et al., 1997; Lawton et al., 1997; Paralkar et al., 1998; Bottner et al., 1999; Moore et al., 2000; Strelau et al., 2000, 2009; Kempf et al., 2006; Xu et al., 2006; Zimmers et al., 2006; Van Huyen et al., 2008; Ding et al., 2009; Ago et al., 2010). Moreover, MIC-1 can mediate certain biological effects via the modulation of other signaling cascades initiated by different growth factors through their cognate receptors (Fig. 3). Since the identity of MIC-1 receptors are not well defined, the molecular mechanisms at the basis of the observed physiological and cellular effects of endogenous MIC-1 as well as its implications in the development of diverse human diseases remain to be established more precisely. In this regard, we discussed the postulated/potential functions of MIC-1 in physiological and pathological conditions and their therapeutic implications for the development of novel therapies to treat diverse human disorders and aggressive cancers.
MIC-1 functions during embryonic and fetal development and adult life and its therapeutic applications
Numerous accumulating lines of evidence obtained on human subjects and using transgenic mouse models in which the MIC-1 expression is manipulated or pure recombinant MIC-1 has been systemically administrated to animal models have indicated that this secreted cytokine may control different developmental and physiological processes and tissue homeostasis and repair in adult life. For instance, it has been observed that the subcutaneous implantation of pure recombinant MIC-1/PDF to rat embryos induced cartilage formation and the early stages of endochondral bone formation (Paralkar et al., 1998). A substantial rise of MIC-1 level in the sera of pregnant women has also been observed to progressively occur during placental gestation (Moore et al., 2000). In fact, MIC-1 has been detected, in large amounts, in amniotic fluid and placental extracts. More specifically, it has been observed that the placental trophoblastic cell line constitutively expresses the MIC-1 transcript and secretes large amounts of this cytokine (Moore et al., 2000). Hence, the secreted MIC-1 by placental trophoblast could promote fetal survival by suppressing the production of maternally derived pro-inflammatory cytokines within the uterus (Moore et al., 2000).
On the other hand, it has also been observed that the homozygous GDF-15−/− deficient mice were viable and fertile, but the weight of adult female GDF-15-deficient mice was increased as compared to the wild-type mice (Strelau et al., 2009). Importantly, GDF-15−/− null mice, however, exhibited progressive postnatal losses of about 20% at the age of 6 months of spinal, facial, and trigeminal motoneurones which was accompanied by losses of motor axons and a significant impairment of motor skills as well as sensory neurons in the dorsal root ganglia (Strelau et al., 2009). It has been proposed that GDF-15 may be secreted by Schwann cells and act in a paracrine manner as a neurotropic factor by promoting the survival of axotomized facial neurons, cultured neurons, sensory and sympathic neurons (Strelau et al., 2009). Consistent with this, GDF-15 also induced the neurotropic and neuroprotector effects on cultured dopaminergic and serotonic neurons (Strelau et al., 2000). Hence, these data suggest that the secreted MIC-1/GDF-15 cytokine can supply critical roles in the adult central nervous system (CNS), by acting as a postnatal survival factor for the motor, sensory, and dopaminergic sympathetic neurons. These observations support the potential applications of MIC-1 to develop new strategies to treat diverse devastating neurodegenerative disorders such as Parkinson’s disease.
Importantly, it has also been reported that the MIC-1 mRNA expression was negatively associated with body mass index and fat mass in human subjects and MIC-1 secreted by adipocytes could supply a paracrine role in the regulation of adipose tissue function (Ding et al., 2009). In this regard, the transgenic mice engineered to overexpress MIC-1 or normal mice treated with systemic recombinant MIC-1 also exhibited the symptoms of hypophagia and a marked reduction of body weight and total fat mass as compared to normal and untreated mice used as group control (Johnen et al., 2007). These observations suggest then that an up-regulation of endogenous MIC-1 or systemic application of exogenous MIC-1 could represent a potential therapeutic approach to treat certain cases of obesity.
Although secreted MIC-1/GDF-15 cytokine is not expressed at a significant level in the normal adult heart, it may be induced in response to diverse pathophysiological stimuli such as hypertension and ischemic heart diseases that promote hypertrophic growth of myocardium, dilated cardiomyopathy, and heart failure (Kempf et al., 2006; Xu et al., 2006; Ago et al., 2010). In fact, the secreted MIC-1/GDF-15 protein can display the cardioprotective effects and act as an anti-hypertrophic regulatory factor in the heart, and thereby this cytokine can antagonize the hypertrophic response and improve the ventricular performance (Kempf et al., 2006; Xu et al., 2006; Ago et al., 2010). Consistently, it has been shown that the transgenic mice with cardiac-specific overexpression of mouse GDF-15 were normal but partially resistant to pressure overload-induced hypertrophy (Xu et al., 2006). Moreover, the expression of mouse GDF-15 by adenovirus-based gene transfer in neonatal cardiomyocyte cultures antagonized phenylephrine plus angiotensin II-induced cardiac hypertrophy in vitro (Xu et al., 2006). The intravenous adenoviral delivery of GDF-15 outside the heart or the injection of human recombinant MIC-1 protein also attenuated the ventricular dilation and heart failure in the muscle LIM protein (MLP) gene-targeted mice through an endocrine effect (Xu et al., 2006). Conversely, it has been observed that homozygous GDF-15−/− null mice generated by gene targeting were viable but exhibited an enhanced cardiac hypertrophic growth following a pressure overload stimulation and a pronounced loss in the ventricular performance after a pressure overload stimulation as compared to the wild-type mice (Xu et al., 2006). These data support the interest to up-regulate the MIC-1 expression in the heart to attenuate the cardiac hypertrophy, and thereby prevent ischemia/reperfusion injury and heart failure, which are among the main causes of morbidity and mortality in humans.
MIC-1 functions in cancer and treatment resistance
MIC-1 expression levels during cancer progression and its therapeutic implications as diagnostic and prognostic biomarker
Many efforts have been made to establish the MIC-1 expression levels and define its specific roles during the early and late stages of cancer progression. It has been observed that MIC-1 expression levels and its secreted form markedly enhance in malignant tissues, established cancer cells, and plasma during the transition of numerous localized cancers to invasive and metastatic disease stages as compared to non-malignant tissues, normal cells, and basal MIC-1 concentration detected in the serum (Brown et al., 2003, 2006, 2009; Karan et al., 2003; Lee et al., 2003; Nakamura et al., 2003; Welsh et al., 2003; Cheung et al., 2004; Koopmann et al., 2004; Wollmann et al., 2005; de Wit et al., 2005; Rasiah et al., 2006; Chen et al., 2007; Selander et al., 2007; Shnaper et al., 2009; Zhang et al., 2009; Zhao et al., 2009; Xue et al., 2010). More specifically, an increase in MIC-1 levels has been observed during the progression of melanoma, oral squamous cell carcinomas, and gastrointestinal, colorectal, pancreatic, prostate, breast, and cervical epithelial cancers. Of clinical interest, high MIC-1 levels in serum samples from cancer patients have been associated with a poor outcome and patient survival (Brown et al., 2003, 2009; Zhao et al., 2009). Consequently, secreted MIC-1 cytokine constitutes a novel potential diagnostic and prognostic biomarker of great clinical interest for a better risk assessment of disease progression of cancer patients. As a matter of fact, it has been reported that the MIC-1 levels in serum progressively increase during the transition of premalignant colonic lesions to cancer initiation and progression and might represent an independent prognostic marker of colon relapse-free and overall survival of patients (Brown et al., 2003). Moreover, it has been noted that the allelic histidine 6-to-aspartate (H6D) polymorphic variation in the MIC-1 DNA sequence was an independent predictor of the presence of metastases at the time of diagnosis (Brown et al., 2003). The establishment of serum MIC-1 level combined with the current diagnostic marker, prostate-specific antigen (PSA), has also been reported to significantly improve the diagnostic specificity (Brown et al., 2006). Moreover, it has been noted that the serum MIC-1 concentration was correlated with the presence of bone metastases in prostate cancer patients (Selander et al., 2007). Consequently, it appears that the combined use of serum MIC-1 level and current biomarkers associated with prostate cancer could represent the potential diagnostic and prognostic methods to reduce the number of unnecessary diagnostic biopsies and surgeries and predict the risk of bone metastases for PC patients. In the same way, it has also been reported that the determination of serum MIC-1 level and cancer antigen 19-9 (CA 19-9) significantly improved diagnostic accuracy of the patients with pancreatic ductal adenocarcinoma (Koopmann et al., 2004). Interestingly, the measurement of MIC-1 expression levels in the cerebrospinal fluid samples from 94 patients with intracranial tumors has also revealed a significant increase of MIC-1 concentration in glioblastoma patients as compared to the patient’s samples with non-neoplastic diseases (Shnaper et al., 2009). The enhanced MIC-1 expression was also correlated with a shorter patient survival (Shnaper et al., 2009). These observations support the interest in considering the assessment of the MIC-1 level in serum in combination with other current available tumor biomarkers for earlier and accurate diagnosis and prognosis and the management of cancer patients in the clinics.
Pleiotropic functions of MIC-1 during cancer progression and its therapeutic implications as a molecular target
Secreted MIC-1 cytokine has been implicated in both the inhibition and promotion of cancer progression and tumor-induced anorexia and weight loss in the late stages of cancer (Li et al., 2000; Baek et al., 2001; Albertoni et al., 2002; Lee et al., 2003; Liu et al., 2003; Levy and Hill, 2006; Chen et al., 2007; Johnen et al., 2007; Kim et al., 2008; Boyle et al., 2009; Senapati et al., 2010; Zhang et al., 2009). More specifically, MIC-1 can induce various pleiotropic effects during cancer progression by negatively or positively modulating cell proliferation, differentiation, apoptosis, invasion, and metastases in a manner dependent of cancer cell types, disease stage, and tumor microenvironment. In general, MIC-1 can act as a tumor suppressor protein by inhibiting tumor growth and inducing apoptosis in the early stages of cancer, while this secreted cytokine rather can promote the proliferation, migration, invasion, and metastases at distant tissues of cancer cells in advanced stages of disease. In this regard, the genetic and/or epigenetic alterations in the signaling elements involved in the mediation of MIC-1 effects on cancer cells or modulators of these pathways may occur during cancer progression, and thereby influence the final cellular response induced by this cytokine in a given cell type (Fig. 3). For instance, MIC-1 overexpression in epithelial cancer cells, such as HCT116 colon cancer cells and breast cancer cell lines, including MDA-MB-468 and MCF-7, resulted in a decreased cell viability in vitro and induced the growth inhibitory effect on tumor xenograft established from HCT-116 cells in vivo (Li et al., 2000; Baek et al., 2001). Moreover, it has also been observed that the ectopic overexpression of MIC-1 in the LN-Z308 glioblastoma cell line completely inhibited its tumorigenic property in nude mice in vivo, while the proliferation of these cancer cells established by in vitro assays was not attenuated (Albertoni et al., 2002). These data suggest that secreted MIC-1 cytokine can display its anti-tumorigenic properties at least in part, via a paracrine mechanism mediated by host cells found in the tumor microenvironment in the animal model (Albertoni et al., 2002).
In contrast, secreted MIC-1 may contribute to the acquisition of a more malignant behavior and enhanced invasive and metastatic abilities by cancer cells and co-operate with other oncogenic growth factors in late stages of carcinogenesis. In support with this, it has been observed that the MIC-1 down-regulation by short hairpin RNA (shRNA) in melanoma cell lines inhibited the tumor growth in an in vivo mouse xenograft mouse model (Boyle et al., 2009). Moreover, MIC-1 overexpression or treatment with purified recombinant MIC-1 of gastric cancer cell lines significantly increased their invasive ability in vitro through the activation of extracellular signal-regulated kinase-1/2 (ERK-1/2)-dependent pathway and an up-regulation of the urokinase type-plasminogen-activator (uPA) activity (Lee et al., 2003). The pure recombinant MIC-1 also induced the activation of basal and estrogen-stimulated ERK-1 phosphorylation in ERα-positive MCF-7 breast cancer cells, suggesting the potential role of this cytokine in breast cancer progression under specific conditions (Wollmann et al., 2005). Importantly, purified recombinant MIC-1 has also been shown to activate ERK1/2 and Akt via a transactivation of the ErbB2 receptor tyrosine kinase concomitant with an increase of tyrosine phosphorylation of EGFR (ErbB1), ErbB2, and ErbB3 in SK-BR-3 breast cancer cells and SNU-216 gastric cancer cells in vitro, and thereby promote their invasive potential (Fig. 3) (Kim et al., 2008). It has also been noticed that MIC-1 increased the expression levels of ErBb2 target genes, such as vascular endothelial growth factor (VEGF) and HIF-1α in SK-BR-3 cells through the phosphorylation of mammalian target of rapamycin (mTOR) signaling pathway and its downstream targets p70S6K and 4E-BP1 (Kim et al., 2008). In the same way, it has been reported that the small interference RNA (siRNA) directed against MIC-1 inhibited the proliferation and colony formation of Tca3118 oral squamous cell carcinoma suggesting this cytokine could mediate the oncogenic functions during OSCC development (Zhang et al., 2009). The gain-and loss-functional studies of MIC-1 in androgen-dependent LNCaP-C33 and its highly metastatic variant, androgen-independent LNCaP-LN3 cell line have also revealed that MIC-1 can promote the proliferation of androgen receptor (AR+)-positive LNCaP cells via the stimulation of ERK-1/2 signal pathway, while Smad2/3 was not activated in these cells expressing a high MIC-1 level (Chen et al., 2007). Moreover, the MIC-1 overexpression in androgen-independent AR− PC3 prostate cancer cells (PC3-MIC-1) enhanced their migratory and invasive abilities in vitro at least in part by the activation of focal adhesion kinase (FAK)–RhoA signaling pathway-mediated actin reorganization (Fig. 3) (Senapati et al., 2010). Importantly, it has also been observed that the intraprostatic orthotopically implanted PC3-MIC-1 cells in nude mice developed the metastases at near and distant tissues, such as lymph nodes, liver, and kidney, while no metastasis was seen for the animal group implanted with a PC3 vector (Senapati et al., 2010). No significant difference, however, was noted between the intratumoral growth of the PC3-MIC-1 cells and the PC3 vector in this orthotopic animal model (Senapati et al., 2010). Importantly, it has also been observed that the parental MIC-1 non-expressing DU145 prostate cancer cells and their derivative DU145-MIC-1 cells engineered to overexpress MIC-1 induced mixed sclerotic and osteolytic bone lesions (Wakchoure et al., 2009). An increase of the osteolytic components of tumors, however, was seen for DU145-MIC-1 cells grown in bone as compared to parental MIC-1 non-expressing DU145 cells (Wakchoure et al., 2009). This suggests that the MIC-1 can also play an important role in the pathophysiology of bone metastases by inducing the osteoclast formation and promoting osteolytic lesions which constitute a major cause of severe bone pain in prostate cancer patients.
In addition to its effect on cancer cells, MIC-1 has also been implicated in the mediation of tumor-induced anorexia and weight loss occurring generally in the late stages of cancer, and which may contribute to morbidity and mortality in cancer patients (Johnen et al., 2007; Senapati et al., 2010; Wakchoure et al., 2009). Consistently, it has been reported that the MIC-1 concentration in serum samples from patients with advanced prostate cancer was associated with a weight loss (Johnen et al., 2007). In subcutaneously or orthotopically implanted prostate cancer cell models, the elevated MIC-1 level was also related with a marked body weight, fat, and lean tissue loss (Johnen et al., 2007; Senapati et al., 2010; Wakchoure et al., 2009). It has been proposed that the promoting effect of MIC-1 on cancer-associated anorexia and weight loss may be induced through its central action on appetite control (Johnen et al., 2007). This cachectic effect of MIC-1 may be mediated via its specific interaction with the hypothalamic TGF-β receptor II, stimulation of ERK-1/2 and anorexigenic pro-opiomelanocortin, and inhibition of orexigenic neuropeptide Y (Johnen et al., 2007). Of therapeutic interest, it has also been noticed that the decreased food intake of mice could be reversed by administration of an antibody directed against MIC-1, suggesting that MIC-1 down-regulation may constitute a potential strategy to prevent the cancer-associated anorexia and weight loss in cancer patients at the late stages of disease (Johnen et al., 2007).
MIC-1 functions in the resistance to current cancer therapies
Numerous gene products altered during cancer progression may contribute to the acquisition of survival advantages by cancer cells, and thereby enhance their resistance to the current therapeutic therapies. In this regard, MIC-1 overexpression in certain cancer cell types, such as lung, colon, prostate, and breast cancer cells, has been shown to supply important cytoprotective roles and resistance to different clinical chemotherapeutic drugs (Campbell et al., 2001; Whiteside et al., 2004; Huang et al., 2007; Proutski et al., 2009; Zhao et al., 2009). For instance, cDNA microarray analyses of changes in gene expression in NCI-H226 and NCI-H2170 lung cancer cells after treatment with cisplatin have revealed that the increase in MIC-1 expression correlated with their resistance to cisplatin (Whiteside et al., 2004). The results from a phase II clinical trial with patients diagnosed with high-risk localized prostate cancer treated with neoadjuvant chemotherapy consisting of docetaxel and mitoxantrone followed by prostatectomy have revealed that the MIC-1 transcript level was significantly enhanced in prostate cancer cells surviving after chemotherapy. This suggests that MIC-1 may contribute to docetaxel and mitoxantrone resistance (Huang et al., 2007). Consistently, it has been observed that MIC-1/GDF-15 overexpression in metastatic PC3 and DU145 prostate cancer cell lines or their exposure to exogenous recombinant MIC-1/GDF-15 protein enhanced their resistance to the current chemotherapeutic drugs, docetaxel and mitoxantrone in vitro (Huang et al., 2007). Of clinical interest, it has also been observed the MIC-1 serum levels detected in patients with hormone-refractory prostate cancer was enhanced after docetaxel treatment and significantly associated with cancer progression and shorter patient survival after chemotherapeutic treatment (Zhao et al., 2009). Moreover, the MIC-1 level was also up-regulated in PC3-Rx cells made resistance to docetaxel as compared to parental PC3 cells and the down-regulation of endogenous MIC-1 by siRNA method sensibilized the PC3-Rx cells to the cytotoxic effects induced by docetaxel (Zhao et al., 2009). In the same way, it has also been reported that the Akt overexpression in breast cancer cells expressing estrogen receptor α (ERα) led to the activation of ERα in the absence of estrogen and the induction of ERα target genes pS2, Bcl-2, and MIC-1 (Campbell et al., 2001). These molecular events conferred resistance of breast cancer cells to tamoxifen-induced apoptosis (Campbell et al., 2001). The exposure of colon cancer cell lines to current clinical chemotherapeutic drugs such as oxaliplatin, 5-fluorouracil, and SN38, which is the active metabolite of the widely used cancer drug irinotecan, has also been observed to result in an increase in MIC-1/PDF mRNA levels (Proutski et al., 2009). The MIC-1 overexpression in colon cancer cells, in turn, contributed to their chemoresistance through the activation of the PI3K/Akt survival pathway (Proutski et al., 2009). Interestingly, it has also been shown that the MIC-1/PDF gene silencing did not induce a significant apoptotic effect on colon cancer cell lines while MIC-1/PDF down-regulation before chemotherapeutic treatment significantly sensitized colon cancer cells expressing wild-type p53 but not p53-null or p53-mutant cells to apoptotic effects induced by chemotherapeutic drugs (Proutski et al., 2009). Thus, in view of these observations, it appears that the MIC-1 may supply an important protective role against the cytotoxic effects induced by diverse current chemotherapeutic drugs in cancer cells. Therefore, the MIC-1 down-regulation may represent a potential therapeutic strategy of great clinical interest for reversing chemoresistance and improving cytotoxic effects induced by different current chemotherapeutic drugs in certain cancer cell types and prevent disease relapse.
Conclusions and Perspectives
Together these recent investigations have revealed that secreted MIC-1 cytokine provides critical functions in the maintenance of tissue homeostasis and repair after intense injuries by controlling cell behavior and more particularly modulating adaptive responses to cellular stress signals and inflammation. The deregulation of the molecular mechanisms involved in the stringent regulation of endogenous expression of MIC-1 and/or signal transduction mediating its cellular effects, however, may lead to diverse pathological disorders and promote the progression of numerous aggressive cancers to invasive and metastatic disease stages.
Although this is an importance advance, the precise physiological and pathological roles of MIC-1 are still not well understood, due in part to the pleiotropic effects of this cytokine on different normal and cancer cell types and complex cellular mechanisms regulating the endogenous MIC-1 expression and signal transduction process. The lack of precise information on the MIC-1 receptor types I and II and other regulatory factors of signal transduction pathways induced by this cytokine underlines the urgent need to further establish their identities and implications in mediating the specific cellular responses induced by secreted MIC-1 in a given cell type. It will be especially important to define the molecular mechanisms that regulate enhanced MIC-1 expression levels and its dual functions during cancer progression. The determination of how MIC-1 signaling deregulation may induce tumor growth inhibition at early stages or promote the proliferation, invasion, and/or metastases of cancer cells at the late stage of disease is of immense importance. The establishment of the potential functions of the transmembrane protein co-receptors, such as endoglin and betaglycan, which are known to modulate the actions of TGF-β superfamily members in diverse human cancers (Bernabeu et al., 2009), in the mediation of cellular responses induced by MIC-1 is also of particular interest. Future studies are also required to determine the potential implications of MIC-1 in the acquisition of more malignant phenotypes and behavior by cancer stem/progenitor cells versus their differentiated progenies during cancer progression as well as in their resistance to current cancer therapies.
These additional studies should lead to a better understanding of the molecular mechanisms at the basis of the pleiotropic effects of MIC-1 during cancer etiopathogenesis and progression as well as shed light on its functional divergences and analogies with other TGF-β superfamily members. Hence, this work should help researchers to develop new therapeutic approaches against a variety of human pathological disorders and aggressive, metastatic, and recurrent cancers that remain incurable with the treatments currently available.
Acknowledgments
Contract grant sponsor: U.S. Department of Defense;
Contract grant numbers: PC04502, PC074289.
Contract grant sponsor: National Institutes of Health;
Contract grant numbers: CA78590, CA111294, CA133774, CA131944.
We thank Kristi L. Berger for editing the article. The authors on this work are supported by grants from the U.S. Department of Defense (PC04502 and PC074289) and National Institutes of Health (CA78590, CA111294, CA133774, and CA131944).
Footnotes
The authors declare no conflict of interest.
Literature Cited
- Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama M, Okano K, Fukada Y, Okano T. Macrophage inhibitory cytokine MIC-1 is upregulated by short-wavelength light in cultured normal human dermal fibroblasts. FEBS Lett. 2009;583:933–937. doi: 10.1016/j.febslet.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Albertoni M, Shaw PH, Nozaki M, Godard S, Tenan M, Hamou MF, Fairlie DW, Breit SN, Paralkar VM, de Tribolet N, Van Meir EG, Hegi ME. Anoxia induces macrophage inhibitory cytokine-1 (MIC-1) in glioblastoma cells independently of p53 and HIF-1. Oncogene. 2002;21:4212–4219. doi: 10.1038/sj.onc.1205610. [DOI] [PubMed] [Google Scholar]
- Baek SJ, Kim KS, Nixon JB, Wilson LC, Eling TE. Cyclooxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol Pharmacol. 2001;59:901–908. [PubMed] [Google Scholar]
- Baek SJ, Kim JS, Moore SM, Lee SH, Martinez J, Eling TE. Cyclooxygenase inhibitors induce the expression of the tumor suppressor gene EGR-1, which results in the up-regulation of NAG-1, an antitumorigenic protein. Mol Pharmacol. 2005;67:356–364. doi: 10.1124/mol.104.005108. [DOI] [PubMed] [Google Scholar]
- Bauskin AR, Zhang HP, Fairlie WD, He XY, Russell PK, Moore AG, Brown DA, Stanley KK, Breit SN. The propeptide of macrophage inhibitory cytokine (MIC-1), a TGF-beta superfamily member, acts as a quality control determinant for correctly folded MIC-1. EMBO J. 2000;19:2212–2220. doi: 10.1093/emboj/19.10.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauskin AR, Brown DA, Junankar S, Rasiah KK, Eggleton S, Hunter M, Liu T, Smith D, Kuffner T, Pankhurst GJ, Johnen H, Russell PJ, Barret W, Stricker PD, Grygiel JJ, Kench JG, Henshall SM, Sutherland RL, Breit SN. The propeptide mediates formation of stromal stores of PROMIC-1: Role in determining prostate cancer outcome. Cancer Res. 2005;65:2330–2336. doi: 10.1158/0008-5472.CAN-04-3827. [DOI] [PubMed] [Google Scholar]
- Bauskin AR, Brown DA, Kuffner T, Johnen H, Luo XW, Hunter M, Breit SN. Role of macrophage inhibitory cytokine-1 in tumorigenesis and diagnosis of cancer. Cancer Res. 2006;66:4983–4986. doi: 10.1158/0008-5472.CAN-05-4067. [DOI] [PubMed] [Google Scholar]
- Bernabeu C, Lopez-Novoa JM, Quintanilla M. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim Biophys Acta. 2009;1792:954–973. doi: 10.1016/j.bbadis.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, Walsh BJ, Nicholson RC, Fairlie WD, Por SB, Robbins JM, Breit SN. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci USA. 1997;94:11514–11519. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottner M, Suter-Crazzolara C, Schober A, Unsicker K. Expression of a novel member of the TGF-beta superfamily, growth/differentiation factor-15/macrophage-inhibiting cytokine-1 (GDF-15/MIC-1) in adult rat tissues. Cell Tissue Res. 1999;297:103–110. doi: 10.1007/s004410051337. [DOI] [PubMed] [Google Scholar]
- Boyle GM, Pedley J, Martyn AC, Banducci KJ, Strutton GM, Brown DA, Breit SN, Parsons PG. Macrophage inhibitory cytokine-1 is overexpressed in malignant melanoma and is associated with tumorigenicity. J Invest Dermatol. 2009;129:383–391. doi: 10.1038/jid.2008.270. [DOI] [PubMed] [Google Scholar]
- Brown DA, Breit SN, Buring J, Fairlie WD, Bauskin AR, Liu T, Ridker PM. Concentration in plasma of macrophage inhibitory cytokine-1 and risk of cardiovascular events in women: A nested case-control study. Lancet. 2002;359:2159–2163. doi: 10.1016/S0140-6736(02)09093-1. [DOI] [PubMed] [Google Scholar]
- Brown DA, Ward RL, Buckhaults P, Liu T, Romans KE, Hawkins NJ, Bauskin AR, Kinzler KW, Vogelstein B, Breit SN. MIC-1 serum level and genotype: Associations with progress and prognosis of colorectal carcinoma. Clin Cancer Res. 2003;9:2642–2650. [PubMed] [Google Scholar]
- Brown DA, Stephan C, Ward RL, Law M, Hunter M, Bauskin AR, Amin J, Jung K, Diamandis EP, Hampton GM, Russell PJ, Giles GG, Breit SN. Measurement of serum levels of macrophage inhibitory cytokine 1 combined with prostate-specific antigen improves prostate cancer diagnosis. Clin Cancer Res. 2006;12:89–96. doi: 10.1158/1078-0432.CCR-05-1331. [DOI] [PubMed] [Google Scholar]
- Brown DA, Lindmark F, Stattin P, Balter K, Adami HO, Zheng SL, Xu J, Isaacs WB, Gronberg H, Breit SN, Wiklund FE. Macrophage inhibitory cytokine 1: A new prognostic marker in prostate cancer. Clin Cancer Res. 2009;15:6658–6664. doi: 10.1158/1078-0432.CCR-08-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: A new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- Chen SJ, Karan D, Johansson SL, Lin FF, Zeckser J, Singh AP, Batra SK, Lin MF. Prostate-derived factor as a paracrine and autocrine factor for the proliferation of androgen receptor-positive human prostate cancer cells. Prostate. 2007;67:557–571. doi: 10.1002/pros.20551. [DOI] [PubMed] [Google Scholar]
- Chenau J, Michelland S, de Fraipont F, Josserand V, Coll JL, Favrot MC, Seve M. The cell line secretome, a suitable tool for investigating proteins released in vivo by tumors: Application to the study of p53-modulated proteins secreted in lung cancer cells. J Proteome Res. 2009;8:4579–4591. doi: 10.1021/pr900383g. [DOI] [PubMed] [Google Scholar]
- Cheung PK, Woolcock B, Adomat H, Sutcliffe M, Bainbridge TC, Jones EC, Webber D, Kinahan T, Sadar M, Gleave ME, Vielkind J. Protein profiling of microdissected prostate tissue links growth differentiation factor 15 to prostate carcinogenesis. Cancer Res. 2004;64:5929–5933. doi: 10.1158/0008-5472.CAN-04-1216. [DOI] [PubMed] [Google Scholar]
- de Caestecker M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15:1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- de Wit NJ, Rijntjes J, Diepstra JH, van Kuppevelt TH, Weidle UH, Ruiter DJ, van Muijen GN. Analysis of differential gene expression in human melanocytic tumour lesions by custom made oligonucleotide arrays. Br J Cancer. 2005;92:2249–2261. doi: 10.1038/sj.bjc.6602612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer K, Steele TA, Shoop MA, Dannawi H. Lineage-restricted expression of bone morphogenetic protein genes in human hematopoietic cell lines. Blood Cells Mol Dis. 1999;25:310–323. doi: 10.1006/bcmd.1999.0259. [DOI] [PubMed] [Google Scholar]
- Ding Q, Mracek T, Gonzalez-Muniesa P, Kos K, Wilding J, Trayhurn P, Bing C. Identification of macrophage inhibitory cytokine-1 in adipose tissue and its secretion as an adipokine by human adipocytes. Endocrinology. 2009;150:1688–1696. doi: 10.1210/en.2008-0952. [DOI] [PubMed] [Google Scholar]
- Fairlie WD, Moore AG, Bauskin AR, Russell PK, Zhang HP, Breit SN. MIC-1 is a novel TGF-beta superfamily cytokine associated with macrophage activation. J Leukoc Biol. 1999;65:2–5. doi: 10.1002/jlb.65.1.2. [DOI] [PubMed] [Google Scholar]
- Fairlie WD, Zhang HP, Wu WM, Pankhurst SL, Bauskin AR, Russell PK, Brown PK, Breit SN. The propeptide of the transforming growth factor-beta superfamily member, macrophage inhibitory cytokine-1 (MIC-1), is a multifunctional domain that can facilitate protein folding and secretion. J Biol Chem. 2001;276:16911–16918. doi: 10.1074/jbc.M010000200. [DOI] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Gouedard L, Chen YG, Thevenet L, Racine C, Borie S, Lamarre I, Josso N, Massague J, di Clemente N. Engagement of bone morphogenetic protein type IB receptor and Smad1 signaling by anti-Mullerian hormone and its type II receptor. J Biol Chem. 2000;275:27973–27978. doi: 10.1074/jbc.M002704200. [DOI] [PubMed] [Google Scholar]
- Hromas R, Hufford M, Sutton J, Xu D, Li Y, Lu L. PLAB, a novel placental bone morphogenetic protein. Biochim Biophys Acta. 1997;1354:40–44. doi: 10.1016/s0167-4781(97)00122-x. [DOI] [PubMed] [Google Scholar]
- Huang CY, Beer TM, Higano CS, True LD, Vessella R, Lange PH, Garzotto M, Nelson PS. Molecular alterations in prostate carcinomas that associate with in vivo exposure to chemotherapy: Identification of a cytoprotective mechanism involving growth differentiation factor 15. Clin Cancer Res. 2007;13:5825–5833. doi: 10.1158/1078-0432.CCR-07-1037. [DOI] [PubMed] [Google Scholar]
- Ichikawa T, Suenaga Y, Koda T, Ozaki T, Nakagawara A. TAp63-dependent induction of growth differentiation factor 15 (GDF15) plays a critical role in the regulation of keratinocyte differentiation. Oncogene. 2008;27:409–420. doi: 10.1038/sj.onc.1210658. [DOI] [PubMed] [Google Scholar]
- Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, Bauskin AR, Wu L, Pankhurst G, Jiang L, Junankar S, Hunter M, Fairlie WD, Lee NJ, Enriquez RF, Baldock PA, Corey E, Apple FS, Murakami MM, Lin EJ, Wang C, During MJ, Sainsbury A, Herzog H, Breit SN. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat Med. 2007;13:1333–1340. doi: 10.1038/nm1677. [DOI] [PubMed] [Google Scholar]
- Kakehi Y, Segawa T, Wu XX, Kulkarni P, Dhir R, Getzenberg RH. Down-regulation of macrophage inhibitory cytokine-1/prostate derived factor in benign prostatic hyperplasia. Prostate. 2004;59:351–356. doi: 10.1002/pros.10365. [DOI] [PubMed] [Google Scholar]
- Karan D, Chen SJ, Johansson SL, Singh AP, Paralkar VM, Lin MF, Batra SK. Dysregulated expression of MIC-1/PDF in human prostate tumor cells. Biochem Biophys Res Commun. 2003;305:598–604. doi: 10.1016/s0006-291x(03)00823-4. [DOI] [PubMed] [Google Scholar]
- Karan D, Holzbeierlein J, Thrasher JB. Macrophage inhibitory cytokine-1: Possible bridge molecule of inflammation and prostate cancer. Cancer Res. 2009;69:2–5. doi: 10.1158/0008-5472.CAN-08-1230. [DOI] [PubMed] [Google Scholar]
- Kelly JA, Lucia MS, Lambert JR. p53 controls prostate-derived factor/macrophage inhibitory cytokine/NSAID-activated gene expression in response to cell density, DNA damage and hypoxia through diverse mechanisms. Cancer Lett. 2009;277:38–47. doi: 10.1016/j.canlet.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, Heineke J, Kotlarz D, Xu J, Molkentin JD, Niessen HW, Drexler H, Wollert KC. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res. 2006;98:351–360. doi: 10.1161/01.RES.0000202805.73038.48. [DOI] [PubMed] [Google Scholar]
- Kim KS, Baek SJ, Flake GP, Loftin CD, Calvo BF, Eling TE. Expression and regulation of nonsteroidal anti-inflammatory drug-activated gene (NAG-1) in human and mouse tissue. Gastroenterology. 2002;122:1388–1398. doi: 10.1053/gast.2002.32972. [DOI] [PubMed] [Google Scholar]
- Kim KK, Lee JJ, Yang Y, You KH, Lee JH. Macrophage inhibitory cytokine-1 activates AKT and ERK-1/2 via the transactivation of ErbB2 in human breast and gastric cancer cells. Carcinogenesis. 2008;29:704–712. doi: 10.1093/carcin/bgn031. [DOI] [PubMed] [Google Scholar]
- Koopmann J, Buckhaults P, Brown DA, Zahurak ML, Sato N, Fukushima N, Sokoll LJ, Chan DW, Yeo CJ, Hruban RH, Breit SN, Kinzler KW, Vogelstein B, Goggins M. Serum macrophage inhibitory cytokine 1 as a marker of pancreatic and other periampullary cancers. Clin Cancer Res. 2004;10:2386–2392. doi: 10.1158/1078-0432.ccr-03-0165. [DOI] [PubMed] [Google Scholar]
- Krieg AJ, Rankin EB, Chan D, Razorenova O, Fernandez S, Giaccia AJ. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 alpha enhances hypoxic gene expression and tumor growth. Mol Cell Biol. 2010;30:344–353. doi: 10.1128/MCB.00444-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton LN, Bonaldo MF, Jelenc PC, Qiu L, Baumes SA, Marcelino RA, de Jesus GM, Wellington S, Knowles JA, Warburton D, Brown S, Soares MB. Identification of a novel member of the TGF-beta superfamily highly expressed in human placenta. Gene. 1997;203:17–26. doi: 10.1016/s0378-1119(97)00485-x. [DOI] [PubMed] [Google Scholar]
- Lee DH, Yang Y, Lee SJ, Kim KY, Koo TH, Shin SM, Song KS, Lee YH, Kim YJ, Lee JJ, Choi I, Lee JH. Macrophage inhibitory cytokine-1 induces the invasiveness of gastric cancer cells by up-regulating the urokinase-type plasminogen activator system. Cancer Res. 2003;63:4648–4655. [PubMed] [Google Scholar]
- Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Li PX, Wong J, Ayed A, Ngo D, Brade AM, Arrowsmith C, Austin RC, Klamut HJ. Placental transforming growth factor-beta is a downstream mediator of the growth arrest and apoptotic response of tumor cells to DNA damage and p53 overexpression. J Biol Chem. 2000;275:20127–20135. doi: 10.1074/jbc.M909580199. [DOI] [PubMed] [Google Scholar]
- Liu T, Bauskin AR, Zaunders J, Brown DA, Pankhurst S, Russell PJ, Breit SN. Macrophage inhibitory cytokine 1 reduces cell adhesion and induces apoptosis in prostate cancer cells. Cancer Res. 2003;63:5034–5040. [PubMed] [Google Scholar]
- Marjono AB, Brown DA, Horton KE, Wallace EM, Breit SN, Manuelpillai U. Macrophage inhibitory cytokine-1 in gestational tissues and maternal serum in normal and pre-eclamptic pregnancy. Placenta. 2003;24:100–106. doi: 10.1053/plac.2002.0881. [DOI] [PubMed] [Google Scholar]
- Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Blain SW, Lo RS. TGF beta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Modlich O, Prisack HB, Munnes M, Audretsch W, Bojar H. Immediate gene expression changes after the first course of neoadjuvant chemotherapy in patients with primary breast cancer disease. Clin Cancer Res. 2004;10:6418–6431. doi: 10.1158/1078-0432.CCR-04-1031. [DOI] [PubMed] [Google Scholar]
- Moore AG, Brown DA, Fairlie WD, Bauskin AR, Brown PK, Munier ML, Russell PK, Salamonsen LA, Wallace EM, Breit SN. The transforming growth factor-ss superfamily cytokine macrophage inhibitory cytokine-1 is present in high concentrations in the serum of pregnant women. J Clin Endocrinol Metab. 2000;85:4781–4788. doi: 10.1210/jcem.85.12.7007. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Scorilas A, Stephan C, Yousef GM, Kristiansen G, Jung K, Diamandis EP. Quantitative analysis of macrophage inhibitory cytokine-1 (MIC-1) gene expression in human prostatic tissues. Br J Cancer. 2003;88:1101–1104. doi: 10.1038/sj.bjc.6600869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, Ten DP, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paralkar VM, Vail AL, Grasser WA, Brown TA, Xu H, Vukicevic S, Ke HZ, Qi H, Owen TA, Thompson DD. Cloning and characterization of a novel member of the transforming growth factor-beta/bone morphogenetic protein family. J Biol Chem. 1998;273:13760–13767. doi: 10.1074/jbc.273.22.13760. [DOI] [PubMed] [Google Scholar]
- Piek E, Heldin CH, Ten DP. Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13:2105–2124. [PubMed] [Google Scholar]
- Proutski I, Stevenson L, Allen WL, McCulla A, Boyer J, McLean EG, Longley DB, Johnston PG. Prostate-derived factor—A novel inhibitor of drug-induced cell death in colon cancer cells. Mol Cancer Ther. 2009;8:2566–2574. doi: 10.1158/1535-7163.MCT-09-0158. [DOI] [PubMed] [Google Scholar]
- Rasiah KK, Kench JG, Gardiner-Garden M, Biankin AV, Golovsky D, Brenner PC, Kooner R, O’Neill GF, Turner JJ, Delprado W, Lee CS, Brown DA, Breit SN, Grygiel JJ, Horvath LG, Stricker PD, Sutherland RL, Henshall SM. Aberrant neuropeptide Y and macrophage inhibitory cytokine-1 expression are early events in prostate cancer development and are associated with poor prognosis. Cancer Epidemiol Biomarkers Prev. 2006;15:711–716. doi: 10.1158/1055-9965.EPI-05-0752. [DOI] [PubMed] [Google Scholar]
- Reissmann E, Jornvall H, Blokzijl A, Andersson O, Chang C, Minchiotti G, Persico MG, Ibanez CF, Brivanlou AH. The orphan receptor ALK7 and the activin receptor ALK4 mediate signaling by nodal proteins during vertebrate development. Genes Dev. 2001;15:2010–2022. doi: 10.1101/gad.201801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober A, Bottner M, Strelau J, Kinscherf R, Bonaterra GA, Barth M, Schilling L, Fairlie WD, Breit SN, Unsicker K. Expression of growth differentiation factor-15/macrophage inhibitory cytokine-1 (GDF-15/MIC-1) in the perinatal, adult, and injured rat brain. J Comp Neurol. 2001;439:32–45. doi: 10.1002/cne.1333. [DOI] [PubMed] [Google Scholar]
- Selander KS, Brown DA, Sequeiros GB, Hunter M, Desmond R, Parpala T, Risteli J, Breit SN, Jukkola-Vuorinen A. Serum macrophage inhibitory cytokine-1 concentrations correlate with the presence of prostate cancer bone metastases. Cancer Epidemiol Biomarkers Prev. 2007;16:532–537. doi: 10.1158/1055-9965.EPI-06-0841. [DOI] [PubMed] [Google Scholar]
- Senapati S, Rachagani S, Chaudhary K, Johansson SL, Singh RK, Batra SK. Overexpression of macrophage inhibitory cytokine-1 induces metastasis of human prostate cancer cells through the FAK-RhoA signaling pathway. Oncogene. 2010;9:1293–1302. doi: 10.1038/onc.2009.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: Insights on DNA binding in TGF-beta signaling. Cell. 1998;94:585–594. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- Shim M, Eling TE. Protein kinase C-dependent regulation of NAG-1/placental bone morphogenic protein/MIC-1 expression in LNCaP prostate carcinoma cells. J Biol Chem. 2005;280:18636–18642. doi: 10.1074/jbc.M414613200. [DOI] [PubMed] [Google Scholar]
- Shnaper S, Desbaillets I, Brown DA, Murat A, Migliavacca E, Schluep M, Ostermann S, Hamou MF, Stupp R, Breit SN, de Tribolet N, Hegi ME. Elevated levels of MIC-1/GDF15 in the cerebrospinal fluid of patients are associated with glioblastoma and worse outcome. Int J Cancer. 2009;125:2624–2630. doi: 10.1002/ijc.24639. [DOI] [PubMed] [Google Scholar]
- Soderberg SS, Karlsson G, Karlsson S. Complex and context dependent regulation of hematopoiesis by TGF-beta superfamily signaling. Ann N Y Acad Sci. 2009;1176:55–69. doi: 10.1111/j.1749-6632.2009.04569.x. [DOI] [PubMed] [Google Scholar]
- Soto-Cerrato V, Vinals F, Lambert JR, Perez-Tomas R. The anticancer agent prodigiosin induces p21WAF1/CIP1 expression via transforming growth factor-beta receptor pathway. Biochem Pharmacol. 2007;74:1340–1349. doi: 10.1016/j.bcp.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Strelau J, Sullivan A, Bottner M, Lingor P, Falkenstein E, Suter-Crazzolara C, Galter D, Jaszai J, Krieglstein K, Unsicker K. Growth/differentiation factor-15/macrophage inhibitory cytokine-1 is a novel trophic factor for midbrain dopaminergic neurons in vivo. J Neurosci. 2000;20:8597–8603. doi: 10.1523/JNEUROSCI.20-23-08597.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelau J, Strzelczyk A, Rusu P, Bendner G, Wiese S, Diella F, Altick AL, von Bartheld CS, Klein R, Sendtner M, Unsicker K. Progressive postnatal motoneuron loss in mice lacking GDF-15. J Neurosci. 2009;29:13640–13648. doi: 10.1523/JNEUROSCI.1133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamary H, Shalev H, Perez-Avraham G, Zoldan M, Levi I, Swinkels DW, Tanno T, Miller JL. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood. 2008;112:5241–5244. doi: 10.1182/blood-2008-06-165738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Wang Y, Guan K, Sun Y. PTGF-beta, a type beta transforming growth factor (TGF-beta) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-beta signaling pathway. Proc Natl Acad Sci USA. 2000;97:109–114. doi: 10.1073/pnas.97.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, Moroney JW, Reed CH, Luban NL, Wang RH, Eling TE, Childs R, Ganz T, Leitman SF, Fucharoen S, Miller JL. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- Tong S, Marjono B, Brown DA, Mulvey S, Breit SN, Manuelpillai U, Wallace EM. Serum concentrations of macrophage inhibitory cytokine 1 (MIC 1) as a predictor of miscarriage. Lancet. 2004;363:129–130. doi: 10.1016/S0140-6736(03)15265-8. [DOI] [PubMed] [Google Scholar]
- Trombly DJ, Woodruff TK, Mayo KE. Roles for transforming growth factor beta superfamily proteins in early folliculogenesis. Semin Reprod Med. 2009;27:14–23. doi: 10.1055/s-0028-1108006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji A, Sakurai K, Kiyokage E, Yamazaki T, Koide S, Toida K, Ishimura K, Matsuda Y. Secretory proprotein convertases PACE4 and PC6A are heparin-binding proteins which are localized in the extracellular matrix. Potential role of PACE4 in the activation of proproteins in the extracellular matrix. Biochim Biophys Acta. 2003;1645:95–104. doi: 10.1016/s1570-9639(02)00532-0. [DOI] [PubMed] [Google Scholar]
- Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- Van Huyen JP, Cheval L, Bloch-Faure M, Belair MF, Heudes D, Bruneval P, Doucet A. GDF15 triggers homeostatic proliferation of acid-secreting collecting duct cells. J Am Soc Nephrol. 2008;19:1965–1974. doi: 10.1681/ASN.2007070781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakchoure S, Swain TM, Hentunen TA, Bauskin AR, Brown DA, Breit SN, Vuopala KS, Harris KW, Selander KS. Expression of macrophage inhibitory cytokine-1 in prostate cancer bone metastases induces osteoclast activation and weight loss. Prostate. 2009;69:652–661. doi: 10.1002/pros.20913. [DOI] [PubMed] [Google Scholar]
- Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, Bauskin AR, Ward RL, Hawkins NJ, Quinn DI, Russell PJ, Sutherland RL, Breit SN, Moskaluk CA, Frierson HF, Jr, Hampton GM. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc Natl Acad Sci USA. 2003;100:3410–3415. doi: 10.1073/pnas.0530278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside MA, Chen DT, Desmond RA, Abdulkadir SA, Johanning GL. A novel time-course cDNA microarray analysis method identifies genes associated with the development of cisplatin resistance. Oncogene. 2004;23:744–752. doi: 10.1038/sj.onc.1207164. [DOI] [PubMed] [Google Scholar]
- Wollmann W, Goodman ML, Bhat-Nakshatri P, Kishimoto H, Goulet RJ, Jr, Mehrotra S, Morimiya A, Badve S, Nakshatri H. The macrophage inhibitory cytokine integrates AKT/PKB and MAP kinase signaling pathways in breast cancer cells. Carcinogenesis. 2005;26:900–907. doi: 10.1093/carcin/bgi031. [DOI] [PubMed] [Google Scholar]
- Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, Hewett TE, Breit SN, Molkentin JD. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. 2006;98:342–350. doi: 10.1161/01.RES.0000202804.84885.d0. [DOI] [PubMed] [Google Scholar]
- Xue H, Lu B, Zhang J, Wu M, Huang Q, Wu Q, Sheng H, Wu D, Hu J, Lai M. Identification of serum biomarkers for colorectal cancer metastasis using a differential secretome approach. J Proteome Res. 2010;9:545–555. doi: 10.1021/pr9008817. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K, Lee SH, Eling TE, Baek SJ. Identification of nonsteroidal anti-inflammatory drug-activated gene (NAG-1) as a novel downstream target of phosphatidylinositol 3-kinase/AKT/GSK-3beta pathway. J Biol Chem. 2004;279:49617–49623. doi: 10.1074/jbc.M408796200. [DOI] [PubMed] [Google Scholar]
- Yang H, Filipovic Z, Brown D, Breit SN, Vassilev LT. Macrophage inhibitory cytokine-1: A novel biomarker for p53 pathway activation. Mol Cancer Ther. 2003;2:1023–1029. [PubMed] [Google Scholar]
- Zhang L, Yang X, Pan HY, Zhou XJ, Li J, Chen WT, Zhong LP, Zhang ZY. Expression of growth differentiation factor 15 is positively correlated with histopathological malignant grade and in vitro cell proliferation in oral squamous cell carcinoma. Oral Oncol. 2009;45:627–632. doi: 10.1016/j.oraloncology.2008.07.017. [DOI] [PubMed] [Google Scholar]
- Zhao L, Lee BY, Brown DA, Molloy MP, Marx GM, Pavlakis N, Boyer MJ, Stockler MR, Kaplan W, Breit SN, Sutherland RL, Henshall SM, Horvath LG. Identification of candidate biomarkers of therapeutic response to docetaxel by proteomic profiling. Cancer Res. 2009;69:7696–7703. doi: 10.1158/0008-5472.CAN-08-4901. [DOI] [PubMed] [Google Scholar]
- Zimmers TA, Jin X, Hsiao EC, Perez EA, Pierce RH, Chavin KD, Koniaris LG. Growth differentiation factor-15: Induction in liver injury through p53 and tumor necrosis factor-independent mechanisms. J Surg Res. 2006;130:45–51. doi: 10.1016/j.jss.2005.07.036. [DOI] [PubMed] [Google Scholar]