

Summary
We report a 28-year-old-female who presented with primary amenorrhoea, absence of puberty, obesity and normal stature. The subject was clearly short as a child, with a height more than 2 SD below normal until the age of 15 years. The pubertal growth spurt failed to develop. She continued growing at a prepubertal rate until growth ceased at the age of 20 years, reaching her final adult height of 157 cm (SDS −0.86) without hormonal treatment. A combined pituitary hormone stimulation test of anterior pituitary function showed deficiencies of GH, LH and FSH, and low normal serum levels of TSH and PRL. Magnetic resonance imaging revealed a hypoplastic pituitary with markedly reduced pituitary height. In addition, a whole body dual energy X-ray absorptiometry scan showed high levels of body fat (54%). Combined pituitary hormone deficiencies with a hypoplastic pituitary suggested the diagnosis of a Prophet of Pit-1 (PROP1) gene mutation. Normal stature in this case, however, confounded this diagnosis. Sequencing of PROP1 revealed homozygosity for a single base-pair substitution (C to T), resulting in the replacement of an Arg by a Cys at codon 120 (R120C) in the third helix of the homeodomain of the Prop-1 protein. To our knowledge, this is the first report of a patient with a mutation in the PROP1 gene that attained normal height without hormonal treatment, indicating a new variability in the PROP1 phenotype, with important implications for the diagnosis of these patients. We suggest that this can be explained by (i) the presence of low levels of GH in the circulation during childhood and adolescence; (ii) the lack of circulating oestrogen delaying epiphyseal fusion, resulting in growth beyond the period of normal growth; and (iii) fusion of the epiphyseal plates, possibly as a result of circulating oestrogens originating from peripheral conversion of androgens by adipose tissue.
Defects in development of the anterior pituitary can result in combined pituitary hormone deficiency (CPHD), defined as impaired production of GH and one or more of the other anterior pituitary-derived hormones. Genetic analysis of CPHD patients resulted in the isolation and characterization of genes encoding transcription factors involved in pituitary development. The Pit-1 transcription factor is critical for the differentiation of somatotroph, thyrotroph and lactotroph cells in both mice and humans, but does not affect corticotroph or gonadotroph cells (Parks & Brown, 1999). The Prophet of Pit-1 gene (PROP1), encoding a paired-like homeodomain protein necessary for PIT1 gene expression, is also involved in the differentiation and function of somatotroph, thyrotroph and lactotroph cells in mice (Sornson et al., 1996). In humans, mutations of the PROP1 gene cause a more extensive phenotype, with a dramatic decrease in gonadotroph function, in addition to the absence of Pit-1-dependent cells.
To date, eight distinct mutations have been identified in the human PROP1 gene: (i) 301–302delAG (Cogan et al., 1998; Fofanova et al., 1998b; Wu et al., 1998; Deladoëy et al., 1999; Mendonca et al., 1999; Nogueira et al., 1999; Rosenbloom et al., 1999; Pernasetti et al., 2000); (ii) R120C (Flück et al., 1998; Wu et al., 1998); (iii) F117I (Wu et al., 1998; Deladoëy et al., 1999); (iv) 149delAG (Fofanova et al., 1998a; Deladoëy et al., 1999); (v) codon50delA (Krzisnik et al., 1999); (vi) R73C (Duquesnoy et al., 1998; Deladoëy et al., 1999); (vii) 343-2A > T (Deladoëy et al., 1999); and (viii) F88S (Osorio et al., 2000) (Table 1). These mutations result in deficiencies in GH, PRL and TSH, as well as LH and FSH. The phenotype associated with PROP1 mutations is variable with respect to the severity of pituitary hormone deficiency, puberty onset and adrenal function. However, the most consistent presenting feature for these PROP1 patients is short height, usually observed during childhood.
Table 1.
CPHD caused by PROP1 gene mutations
| Reference | No. of patients | Phenotype |
Genotype |
|||
|---|---|---|---|---|---|---|
| Stature | Spontaneous puberty | Hormonal status | DNA | Protein | ||
| Wu et al. (1998) | 3 | Short | No | CPHD | 358 C > T | R120C |
| 6 | Short | No | CPHD | 301–302delAG | S109X | |
| 1 | Short | No | CPHD | 349T > A/301–302delAG | F117I/S109X | |
| Cogan et al. (1998) | 9 | Short | No | CPHD | 301–302delAG | S109X |
| Flück et al. (1998) | 5 | Short | Yes | CPHD | 358 C > T | R120C |
| Fofanova et al. (1998a) | 5 | Short | No | CPHD | 149delGA/301–302delAG | S109X |
| Fofanova et al. (1998b) | 3 | Short | No | CPHD | 301–302delAG | S109X |
| Duquesnoy et al. (1998) | 2 | Short | No | CPHD | 217 C > T | R73C |
| 4 | Short | No | CPHD | 343–2 A > T | ||
| 2 | Short | No | CPHD | 301–302delAG | S109X | |
| Krzisnik et al. (1999) | 6 | Short | No | CPHD | codon50delA | Arg50Asp |
| Nogueira et al. (1999) | 4 | Short | No | CPHD | 301–302delAG | S109X |
| Rosenbloom et al. (1999) | 8 | Short | No | CPHD | 301–302delAG | S109X |
| Deladoëy et al. (1999) | 35 | Short | No | CPHD | 149delGA | S109X |
| 301–302delAG | S109X | |||||
| 358 C > T | R120C | |||||
| 349T > A | F117I | |||||
| 217 C > T | R73C | |||||
| Mendonca et al. (1999) | 2 | Short | No | CPHD | 301–302delAG | S109X |
| Pernasetti et al. (2000) | 10 | Short | No | CPHD | 301–302delAG | S109X |
| Osorio et al. (2000) | 1 | Short | NA* | CPHD | 263T > C | F88S |
| Present study | 1 | Normal | No | CPHD | 358 C > T | R120C |
In this study, we report the case of a 28-year-old Mexican-American female who came to our attention because of primary amenorrhoea. Pituitary combined stimulation test revealed low basal LH and FSH levels, with no response to GnRH stimulation. Although the patient presented with normal height, her basal GH levels were low, and did not increase upon GHRH stimulation. Molecular analysis of the PROP1 gene of this patient showed that she was homozygous for the R120C mutation. To our knowledge, this is the first report of an untreated PROP1 patient with normal height in adulthood.
Case report
A 28-year-old Mexican-American female was referred to the University of California San Diego Reproductive Endocrinology clinic with primary amenorrhoea and lack of secondary sexual development. The patient was born by spontaneous vaginal delivery at term at the University of Southern California in Los Angeles (Fig. 1). At birth, she weighed 3.2 kg (25th percentile) and her length was normal by mother’s report. The patient’s height and weight were obtained from school, clinic, personal and family history. Between the ages of 2 and 11 years, she was short (SDS for height in childhood ranged from −2.9 to −2.2); however, she grew normally along a line parallel and below the 3rd percentile (Fig. 2a, Table 2). Thereafter, the pubertal growth spurt failed to develop and growth continued below the 50th percentile at a prepubertal rate, until she reached her final adult height of 157 cm (17th percentile, SDS −0.86) at the age of 20 years. Psychosocial development was normal. She graduated from high school at 18 years. During puberty, her weight was around the 50th percentile. Thereafter, at 18 years, her weight increased to approximately the 90th percentile reaching the 95th percentile (83 kg) at 28 years (Fig. 2b). She reported normal eating behaviour during this period. At 25 years of age, she was treated with birth control pills for 6 months, developing monthly withdrawal bleeds. After 6 months of treatment, the contraceptive pills were discontinued secondary to headaches. She is single, and currently works as a clerk in a grocery store.
Fig. 1.

Genealogical tree showing the affected patient [black, (homozygous) (5)] and nonaffected individuals [white (wild-type) and grey (heterozygous)]. Dotted symbols represent individuals not available for this study. Squares represent males and circles represent females. The broken line symbol represents a divorce.
Fig. 2.

(a) Growth chart representing the height of the patient between the ages of 0 and 28 years, based on the data in Table 2 (closed circles). The curves for 97th, 50th and 3rd percentile for girls are shown for comparison (Kuczmarski et al., 2000). (b) Weight curve of the patient between the ages of 0 and 28 years, based on data obtained from school records, clinical, personal and family history. The curves for 97th, 90th 50th and 3rd percentile for girls are shown for comparison.
Table 2.
Evaluation of the patient’s height
| Age | Height (cm) | Height (SDS) |
|---|---|---|
| 2 | 76 | −2.9 |
| 5 | 96 | −2.3 |
| 9 | 115 | −2.6 |
| 11 | 127 | −2.2 |
| 13 | 137 | −2.6 |
| 15 | 147 | −2.4 |
| 17 | 152 | −1.7 |
| 18 | 155 | −1.2 |
| 29 | 157 | −0.86 |
The patient’s parents were born and raised in two independent nonconsanguineous families, originating from two different states in Mexico (Sinaloa and Sonora). The father (1) of the affected patient (5) had a first marriage with one child (3) (Fig. 1). His second marriage, with individual 2, resulted in three female siblings [4, 5 (affected patient) and not-available]. The mother of the affected patient had two subsequent marriages, each resulting in one male sibling (6 and 7, respectively) (Fig. 1). The heights of the family members studied were normal: sister 4 [167 cm (75th percentile)], half-sister 3 [160 cm (35th percentile)] and half-brothers 6 and 7 [180 cm (70th percentile); 183 cm (77th percentile), respectively], patient’s mother 2 [162 cm (36th percentile] and father 1 [173 cm (27th percentile)]. Sisters 3 and 4 and brother 7 were married, and had offspring without medical assistance. Informed written consent was obtained from all participating adults, and this study was approved by the Human Subjects Committee of the University of California, San Diego.
Physical examination showed that the patient’s height was 157 cm (17th percentile) and weight was 83 kg (95th percentile). Arm span minus height was approximately 4 cm, upper/lower body ratio was 1.01. Breast development, pubic and axillary hair growth was Tanner stage I. On pelvic examination, the external genitalia, cervix, uterus and adnexa were normal. On transvaginal ultrasound, the uterus measured 4.9 × 1.4 × 2.4 cm, the right ovary measured 1.2 × 1.0 × 1.6 cm, and the left ovary measured 1.9 × 1.4 × 1.8 cm. The karyotype was 46, XX.
Methods
Anthropometric measurements
The SD scores for height (cm) and height-age were estimated based on WHO growth charts (WHO, 1986). The body mass index (BMI) was calculated as weight in kg/m2. BMI percentile for height-age and for bone age were determined based on US population data for children (Hammer et al., 1991) and adults (Cronk & Roche, 1982). Arm span was evaluated with the patient leaning against the wall with arms extended horizontally. Bone age was evaluated with hand and wrist X-rays compared to the standards of Greulich and Pyle (1959). Arm span minus height (AS – Ht) for normal chronological, statural, and bone age were determined based on Wilkins (1965).
Percent body fat and bone mineral density were determined using a Hologic QDR-2000 pencil-beam dual energy X-ray absorptiometry (DEXA) scanner (Hologic Inc., Waltham, MA, USA) performed at the General Clinical Research Center, University of California, San Diego.
Pituitary magnetic resonance scans were performed for sagittal and coronal imaging using a 1.5 Tesla Unit (Siemens Symphony, Germany). Images were obtained before and after contrast. The pituitary maximal height was measured perpendicular to the sella turcica and compared with that of normal values (Suzuki et al., 1990).
Hormone measurements
Hormonal determinations were performed by Quest Diagnostics, Inc. (Nichols Institute, San Juan Capistrano, CA, USA). GH was measured by immunochemiluminometric assay (ICMA), using a goat polyclonal antihuman GH antibody; the sensitivity level of the assay is 0.04 mU/l. LH and FSH concentrations were also measured using ICMA assays (Ciba Corning ACS-180, Bayer, Tarrytown, NY, USA), the sensitivity of which is 0.3 IU/l. Oestradiol and testosterone were measured using Celite chromatography purification with a specific radioimmunosay (RIA) using rabbit anti-oestradiol and rabbit anti-testosterone antiserum, the sensitivity of which are 11.0 pmol/l and < 0.04 nmol/l, respectively. PRL was also measured by ICMA (Ciba Corning ACS-180) using a monoclonal mouse anti-PRL antibody, the sensitivity of which is 6 mU/l. TSH was also measured using ICMA containing acridinium ester-labelled antibody and a biotin-coupled antibody linked to an avidin-coated poly-styrene bead, the sensitivity of which is 0.01 mU/l. Serum T4 was measured by RIA using a polyclonal anti-T4, the sensitivity of which is 12.87 nmol/l. Free T4 was measured by ICMA. Total T3 was determined by RIA; the sensitivity of which is 0.38 nmol/l. Cortisol and ACTH were both measured by RIA, with sensitivities of 27.6 nmol/l and 2.2 pmol/l, respectively. IGF-I and IGF-II concentrations were measured by RIA; the sensitivities of which are 0.1 and 0.2 μg/l, respectively. The IGF-binding proteins, IGFBP-1, IGFBP-2 and IGFBP-3, were measured by RIA, the sensitivities of which are 0.4, 0.1 and 3.1 μg/l, respectively.
DHEA-S, androstenedione and insulin assays were performed by Associate Regional l University Pathology Incorporated (ARUP) (Salt Lake City, UT, USA). Cholesterol, HDL, LDL, triglycerides and glucose assays were all performed by Clinical Laboratories (Thornton Hospital, University of California, San Diego, CA, USA).
Stimulation tests
The combined pituitary stimulation test was performed by simultaneous i.v. administration of 100 g of GnRH (Wyeth Ayerst, Philadelphia, PA, USA), 200 μg of TRH (Ferring Pharmaceuticals, Tarrytown, NY, USA), 1 μg/kg of GHRH (Geref, Serono Laboratories, Randolph, MA, USA) and 1 μg/kg of CRH (Ferring Pharmaceuticals, Tarrytown, NY, USA). Blood samples were collected at −15, 0, 15, 30, 60, 90 and 120 minutes before and after injection of hypothalamic neuropeptides, for measurement of serum LH, FSH, TSH, PRL, GH and ACTH.
DNA analysis
DNA was extracted from blood samples using Chelex® (BioRad Labs, Hercules, CA, USA) 100 following the manufacturer’s protocol (Walsh et al., 1991). 10–20 μl of the extracted genomic DNA were used as a template in a final volume of 50 μl. Three exons and two introns of the PROP1 gene were amplified by polymerase chain reaction (PCR) using a 5′ sense primer (5′-CGAACATTCAGAGACAGAGTCCCAGA-3′) and a 3′-antisense primer (5′-GAATTCACCATGATCTCCCA-3′) to generate a 3.5-kb fragment. PCR of these long fragments was carried out using the Extender PCR system (Stratgene, La Jolla, CA, USA). The reaction was performed for 1 minute at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 56 °C and 6 minutes at 68 °C. The PCR products were verified on 0.8% agarose gel and purified using the Wizard® PCR Preps DNA Purification Systems (Promega, Madison, WI, USA), following the manufacturer’s protocol. Direct sequencing of the double-stranded PCR fragments was carried out at the UCSD Center for Aids Research Molecular Biology Core using an Applied Biosystems 373 Automated DNA sequencer (Perkin Elmer Applied Biosystems Inc., Foster City, CA, USA).
Results
Basal serum concentrations of GH, LH and FSH, were low, and ACTH, PRL and TSH were modestly low (Table 3). Upon stimulation, the responses of GH, FSH and LH were absent. For ACTH, PRL and TSH, the responses were markedly attenuated relative to the normal range. Cortisol response was similarly low (Table 3). These results are consistent with CPHD.
Table 3.
Evaluation of the anterior pituitary by combined stimulation test*
| GH (mU/l) |
FSH (IU/l) |
LH (IU/l) |
Prolactin (mU/l) |
TSH (mU/l) |
ACTH (pmol/l) |
Cortisol (nmol/l) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B | P | B | P | B | P | B | P | B | P | B | P | B | P | |
| Patient | < 0.2 | 0.2 | < 0.5 | 1.4 | 0.5 | 1.6 | 80.0 | 244.0 | 1.63 | 6.02 | < 4.4 | 6.6 | 228.9 | 469 |
| Normal | – | (64–76) | (2.5–10.2) | (5.5–13.2) | (1.9–12.5) | (20–120) | (70–620) | (210–1860) | (0.35–5.5) | (6–30) | (3.5–14) | (8.5–25) | (220–358) | (469–689) |
The details of the assays are given in the Methods section. B, Basal; P, peak after pituitary stimulation.
GHRH 100 μg, CRH 100 μg, GnRH 100 μg, TRH 200 μg, i.v. bolus.
Sex steroid serum concentrations were abnormally low (Table 4). Serum thyroid levels were in the low normal range. IGF hormone levels in peripheral blood were all below the normal range. While fasting and 2 h post glucose tolerance test glucose levels were normal, this was accompanied by fasting hyperinsulinaemia (Table 4). The fasting glucose/insulin ratio was 3.3, consistent with insulin resistance (ratio < 6). Lipids were within the normal range.
Table 4.
Basal hormone concentrations
| Hormone | Value | Normal range |
|---|---|---|
| TT4 | 69.5 nmol/l | 57.9–160.8 nmol/l |
| TT3 | 1.4 nmol/l | 0.92–2.78 nmol/l |
| FT4 | 10.7 pmol/l | 10.7–23.1 pmol/l |
| Oestradiol | 36.7 pmol/l | 73.4–1468.4 pmol/l |
| Testosterone | 0.3 nmol/l | 0.4–3.1 nrnol/l |
| DHEA-S | 0.52 μmol/l | 1.2–7.32 μmol/l |
| Androstenedione | 0.013 nmol/l | 0.017–0.094 nmol/l |
| IGF-1 | 40 μg/l | 128–470 μg/l |
| IGF-11 | 238 μg/l | 405–1085 μg/l |
| IGFBP-1 | < 5 μg/l | 13–73 μg/l |
| IGFBP-2 | 45 μg/l | 55–240 μg/l |
| IGFBP-3 | 1.1 rng/l | 2–4 mg/l |
| Fasting glucose | 5.3 mmol/l | 4.2–6.4 mmol/l |
| 2 h Glucose tolerance test | 6.9 mmol/l | < 7.8 mmol/l |
| Fasting insulin | 207.9 pmol/l | 10.7–146.9 prnol/l |
| Cholesterol | 5.1 mmol/l | < 5.2 nmol/l |
| HDL-cholesterol | 1.2 mmol/l | 0.59–1.9 mmol/l |
| LDL-cholesterol | 3.3 mmol/l | 1.7–4.8 mmol/l |
| Triglyceride | 1.3 mmol/l | 0 1–1.69 mmol/l |
Magnetic resonance imaging of the pituitary revealed a partially empty sella and normal pituitary stalk. The pituitary volume was 48 mm3 (normal volume 290 ± 68 mm3), maximal pituitary height, measured perpendicular to the sella turcica floor, was 2 mm (normal pituitary height, for females between 20 and 29 year of age, 6.1 mm) (Fig. 3) (Suzuki et al., 1990). No abnormality of the neurohypophysis was evident.
Fig. 3.
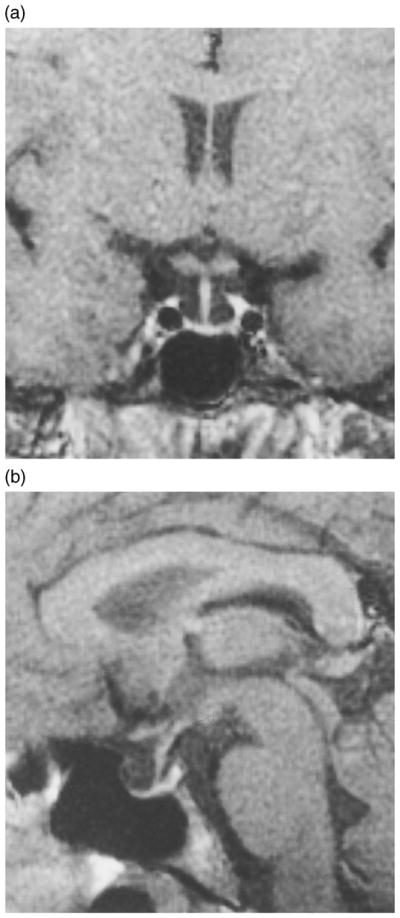
Coronal (a) and sagittal (b) images obtained by magnetic resonance imaging showing the hypoplastic pituitary gland of the patient.
X-ray of the right wrist revealed complete epiphyseal closure. Bone mineral density of the lumbar spine (L2–L4) was 0.989 g/cm2, where normal for age-matched controls is 1.087 g/cm2 (T score −1.55), indicating osteopenia (T between −1.0 and −2.5) (Blake et al., 1999) by DEXA.
The patient BMI was 34 kg/m2, which is consistent with obesity (defined by a BMI > 30 kg/m2), while the patient’s mother’s BMI was 28 kg/m2. Patient’s BMI for both height and age was greater than the 95th percentile. Her percent body fat was evaluated at 54.5% by DEXA (normal for age and gender matched group is 12–35%) (Blake et al., 1999).
Genomic analysis of the PROP1 gene
We analysed the sequence of the PROP1 alleles of our patient (5), her nonaffected siblings (3, 4, 6 and 7) and her mother (2) (Fig. 1). The DNA sequences revealed that the patient was homozygous for a single base-pair substitution (C to T), resulting in the substitution of Arg for Cys at codon 120 (amino acid 52) in the third helix of the homeodomain (R120C). Siblings 3, 4, 7 and the patient’s mother (2) were all heterozygous for the same mutation, while sibling 6 was homozygous wild-type (Fig. 1). The fact that sibling 3, who is the patient’s half-sister from the paternal side, was heterozygous for this mutation, suggesting that the father carried at least one allele with the PROP1 R120C mutation, and that the homozygosity observed in our patient is most probably genetically inherited from both parents, and not due to spontaneous mutation or gene conversion.
Discussion
Over one hundred patients with CPHD caused by PROP1 mutations have been reported following the first case reported by Wu et al. (1998). The most striking phenotypic characteristic observed in patients with mutations in the PROP1 gene is growth impairment (Table 1). Here we report the first case of an untreated PROP1 patient who, although short during childhood, achieved normal final adult height. In this patient, we found a normal prepubertal growth rate, failure of the pubertal growth spurt, followed by growth beyond the normal growth period, and fusion of the epiphyseal plates (Fig. 2).
Children with GH gene, GH receptor or PIT1 gene mutations generally have severe growth failure (Rosenfeld et al., 1994; Parks et al., 1999). In contrast, patients with PROP1 mutations, GH responses to GHRH stimulation are more variable, and adult height is often not as severely affected as in patients with PIT1 mutations (Parks & Brown, 1999). Pituitaries of Snell mice (PIT1 mutations) lack functional somatotrophs; in contrast, the pituitaries of Ames mice (PROP1 mutations) have a small number of functional somatotrophs (Parks et al., 1999). Previous molecular studies of the R120C mutated Prop-1 protein revealed that the impairment of both the transcriptional activation and DNA binding capacities of this protein is not as dramatic as the impairment observed in all other Prop-1 mutants studied (Wu et al., 1998). This suggests that residual activity of the R120C Prop-1 protein supports differentiation of certain pituitary cell-lineages in patients carrying this mutation. Therefore, clinical, animal and molecular observations suggest that the pituitary gland of our patient would be capable of sustaining secretion of GH at low but physiologically active levels during childhood.
Although the mechanism involved in pituitary degeneration occurring in PROP1 patients is unclear, the progressive decrease of pituitary hormone levels suggests that pituitary cells may enter an apoptotic program. Apoptosis might occur due to lack of proper formation of the pituitary gland and/or absence of paracrine and autocrine signalling. It has been recently shown by several groups that IGF-I can inhibit apoptosis in susceptible cells by regulating the balance between pro- and anti-apoptotic proteins, such as p53 and Bcl-2, respectively, at the molecular level (Leri et al., 1999; Pugazhenthi et al., 1999; Adams et al., 2000). Thus, reduced levels of IGF-1, such as those observed in our patient, could also be involved in this process.
The lack of the pubertal growth spurt and growth beyond the normal period of human growth observed in our patient, is likely to be due to delayed epiphyseal fusion resulting from oestrogen deficiency. The growth trajectory of our patient (Fig. 2) is similar to that observed in untreated idiopathic hypogonadotrophic hypogonadism (IHH). Similar to our patient, these patients experience a normal prepubertal growth rate, fail to develop a growth spurt, grow beyond the period of normal growth and fail to fuse their epiphyses (Uriarte et al., 1992). The key role for oestrogen in epiphyseal fusion in both women and men was confirmed recently with the recognition of oestrogen deficiency due to mutations in the aromatase gene and oestrogen resistance due to mutations in the oestrogen receptor-alpha gene (Smith et al., 1994; Morishima et al., 1995; MacGillivray et al., 1998). In both mutations, the epiphyseal plate fails to fuse, no pubertal growth is observed and growth persists into adulthood.
At the time of presentation, our patient’s epiphysis were completely fused. The epiphysis likely fused around the age of 20 years when she reached her final adult height (Fig. 2a). At this same age, our patient’s weight approached the 90th percentile, consistent with obesity (Fig. 2b). Aromatase expression in adipose tissue accounts for the extraglandular formation of oestrogen and its levels increase with body weight (Bulun et al., 1999). In patients who are prematurely exposed to oestrogen (i.e. precocious puberty), the growth plates fuse prematurely resulting in short stature (Chemaitilly et al., 2001). Obese girls were described to have earlier menarche and faster growth and to be shorter than nonobese girls, suggesting that extraglandular oestrogen may play a role both in pubertal growth spurt and epiphysis fusion (Jaruratanasirikul et al., 1997). Similarly, final height in GH deficient (GHD) patients who underwent spontaneous puberty is lower than that in those GHD patients who did not have spontaneous puberty (Hibi et al., 1989). These data suggest that epiphyseal fusion in our patient most likely occurred secondary to increased oestrogen levels around the age of 20 years, possibly as a result of peripheral conversion of adrenal androgens to oestrogens from increasing total body fat developed between the ages of 18–20 years (Fig. 2b).
Although the cause of obesity in our patient remains unclear, it is possibly associated with severe GHD. The onset of obesity correlates temporally with cessation of growth and presumably reduction of GH levels (Fig. 2b). In addition, she developed hyperinsulinaemia (insulin 29 IU/l; normal 20 IU/l) in adulthood. These observations suggest that GHD and/or the combination of pituitary hormone deficiencies resulted in the metabolic abnormalities observed in our patient. Growth hormone deficiency in children and adults is associated with obesity and insulin resistance (Sorgo et al., 1982; Blethen et al., 1993). However, the reason for these metabolic abnormalities in GHD is not known. Interestingly, increased body weight is also associated with CPHD. Children with CPHD have higher BMI than normal children (Baars et al., 1998). Recently, several groups have described obesity in patients with PROP1 mutations. Krzisnik et al. (1999) reported obesity in four out of six patients carrying the codon 50delA PROP1 mutation. In addition, Rosenbloom et al. (1999) reported one out of eight patients with a BMI at 24.7 kg/m2 at the upper limit of normal and Pernasetti et al. (2000) reported two out of 10 patients with BMI greater than 25 kg/m2. GHD and PROP1 patients may have similar mechanisms causing obesity and insulin resistance.
Contrary to what we observed, five PROP1 patients from Switzerland with the same mutation (R120C) presented with short stature during childhood (Flück et al., 1998), all below the 3rd percentile on the respective growth curves. Despite the fact that all five patients received GH treatment during childhood, four out of five did not reach an adult height above the 3rd percentile. The absence of spontaneous menarche observed in our patient is another difference in phenotype compared to the Swiss R120C patients who entered puberty spontaneously, with normal gonadotrophin levels observed before puberty (Flück et al., 1998). This finding further supports that distinct genetic backgrounds could account for the differences in the phenotype of patients with the same PROP1 mutation.
In summary, this case demonstrates that patients with PROP1 mutations may achieve normal adult height. We suggest that this can be explained by (i) the presence of GH, although at low levels in the circulation during childhood and adolescence; (ii) the lack of circulating oestrogen delaying epiphyseal fusion, resulting in growth beyond the period of normal growth, and (iii) fusion of the epiphyseal plates, possibly as a result of circulating oestrogens originating from peripheral conversion of androgens by adipose tissue. This case further illustrates that combined pituitary hormone deficiency caused by PROP1 mutations is a disorder comprising a spectrum of clinical phenotypes, even among patients with the same PROP1 gene mutation. The relationship between clinical phenotype and loss-of-function PROP1 mutations will become clearer as more phenotypes and mutations are identified. Clinically, this diagnosis should be considered in patients presenting with normal stature, combined pituitary hormone deficiency and absent puberty.
Acknowledgments
We thank Diane M. Claflin and Cecilia M. Echon of the UCSD General Clinic Research Center for assistance with the DEXA scan and the combined pituitary stimulation test, respectively. We thank Fumio Otsuka, Sergio P.A. Toledo and Cesar Y. Hayashida for their helpful discussions and suggestions. This work was supported by the following grants: NIH R37 HD2037 (P.L.M.), Women’s Reproductive Health Research Career Centers HD-99–001 (A.A.), an American Heart Association Postdoctoral Fellowship and a Lalor Postdoctoral Fellowship (F.P.) and the training grant, NIH T32 DK07541 (V.V.V.).
References
- Adams TE, Epa VC, Garrett TP, Ward CW. Structure and function of the type 1 insulin-like growth factor receptor. Cellular and Molecular Life Sciences. 2000;57:1050–1093. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baars J, Van den Broeck J, le Cessie S, Massa G, Wit JM. Body mass index in growth hormone deficient children before and during growth hormone treatment. Hormone Research. 1998;49:39–45. doi: 10.1159/000023124. [DOI] [PubMed] [Google Scholar]
- Blake GM, Wahner HW, Forgelman I. The Evaluation of Osteoporosis: Dual Energy X-Ray Absormetry and Ultrasound in Clinical Practice. 2. Martin Dunitz Ltd; London: 1999. [Google Scholar]
- Blethen SL, Compton P, Lippe BM, Rosenfeld RG, August GP, Johanson A. Factors predicting the response to growth hormone (GH) therapy in prepubertal children with GH deficiency. Journal of Clinical Endocrinology and Metabolism. 1993;76:574–579. doi: 10.1210/jcem.76.3.8445013. [DOI] [PubMed] [Google Scholar]
- Bulun SE, Zeitoun K, Sasano H, Simpson ER. Aromatase in aging women. Seminars in Reproductive Endocrinology. 1999;17:349–358. doi: 10.1055/s-2007-1016244. [DOI] [PubMed] [Google Scholar]
- Chemaitilly W, Trivin C, Adan L, Gall V, Sainte-Rose C, Brauner R. Central precocious puberty: clinical and laboratory features. Clinical Endocrinology. 2001;54:289–294. doi: 10.1046/j.1365-2265.2001.01229.x. [DOI] [PubMed] [Google Scholar]
- Cogan JD, Wu W, Phillips JA, III, Arnhold IJ, Agapito A, Fofanova OV, Osorio MG, Bircan I, Moreno A, Mendonca BB. The PROP1 2-base pair deletion is a common cause of combined pituitary hormone deficiency. Journal of Clinical Endocrinology and Metabolism. 1998;83:3346–3349. doi: 10.1210/jcem.83.9.5142. [DOI] [PubMed] [Google Scholar]
- Cronk CE, Roche AF. Race-and sex-specific reference data for triceps and subscapular skinfolds and weight/stature2. American Journal of Clinical Nutrition. 1982;35:347–354. doi: 10.1093/ajcn/35.2.347. [DOI] [PubMed] [Google Scholar]
- Deladoëy J, Flück C, Büyükgebiz A, Kuhlmann BV, Eblé A, Hindmarsh PC, Wu W, Mullis PE. ‘Hot spot’ in the PROP1 gene responsible for combined pituitary hormone deficiency. Journal of Clinical Endocrinology and Metabolism. 1999;84:1645–1650. doi: 10.1210/jcem.84.5.5681. [DOI] [PubMed] [Google Scholar]
- Duquesnoy P, Roy A, Dastot F, Ghali I, Teinturier C, Netchine I, Cacheux V, Hafez M, Salah N, Chaussain JL, Goossens M, Bougnères P, Amselem S. Human Prop-1: cloning, mapping, genomic structure. Mutations in familial combined pituitary hormone deficiency. FEBS Letters. 1998;437:216–220. doi: 10.1016/s0014-5793(98)01234-4. [DOI] [PubMed] [Google Scholar]
- Flück C, Deladoëy J, Rutishauser K, Eblé A, Marti U, Wu W, Mullis PE. Phenotypic variablity in familial combined pituitary hormone deficiency caused by a PROP1 gene mutation resulting in the substitutions of Arg-Cys at codon 120 (R120C) Journal of Clinical Endocrinology and Metabolism. 1998;83:3727–3734. doi: 10.1210/jcem.83.10.5172. [DOI] [PubMed] [Google Scholar]
- Fofanova O, Takamura N, Kinoshita E, Parks JS, Brown MR, Peterkova VA, Evgrafov OV, Goncharov NP, Bulatov AA, Dedov I, Yamashita S. Compound heterozygous deletion of the PROP1 gene in children with combined pituitary hormone deficiency. Journal of Clinical Endocrinology and Metabolism. 1998a;7:2601–2604. doi: 10.1210/jcem.83.7.5094. [DOI] [PubMed] [Google Scholar]
- Fofanova OV, Takamura N, Kinoshita E, Parks JS, Brown MR, Peterkova VA, Evgrafov OV, Goncharov NP, Bulatov AA, Dedov I, Yamashita S. A mutational hot spot in the Prop-1 gene in Russian children with combined pituitary hormone deficiency. Pituitary. 1998b;1:45–49. doi: 10.1023/a:1009918924945. [DOI] [PubMed] [Google Scholar]
- Greulich WW, Pyle SI. Radiographic Atlas of Skeleton Development of the Hands and Wrist. 2. Stanford University Press; Stanford: 1959. [Google Scholar]
- Hammer LD, Kraemer HC, Wilson DM, Ritter PL, Dornbusch SM. Standardized percentile curves of body-mass index for children and adolescents. American Journal of Diseases of Children. 1991;145:259–263. doi: 10.1001/archpedi.1991.02160030027015. [DOI] [PubMed] [Google Scholar]
- Hibi I, Tanaka T, Tanae A, Kagawa J, Hashimoto N, Yoshizawa A, Shizume K. The influence of gonadal function and the effect of gonadal suppression treatment on final height in growth hormone (GH)-treated GH-deficient children. Journal of Clinical Endocrinology and Metabolism. 1989;69:221–226. doi: 10.1210/jcem-69-2-221. [DOI] [PubMed] [Google Scholar]
- Jaruratanasirikul S, Mo-suwan L, Lebel L. Growth pattern and age at menarche of obese girls in a transitional society. Journal of Pediatric Endocrinology and Metabolism. 1997;10:487–490. doi: 10.1515/JPEM.1997.10.5.487. [DOI] [PubMed] [Google Scholar]
- Krzisnik C, Kolacio Z, Battelino T, Brown M, Parks JS, Laron Z. The ‘Little People’ of the Island of Krk-revisited etiology of hypopituitarism revealed. The Journal of Endocrine Genetics. 1999;1:9–19. [Google Scholar]
- Kuczmarski RG, Ogden CL, Grummer-Strawn LM, Fligal KM, Guo SS, Wei R, Mei Z, Curtin LR, Roche AF, Johnson CL. Centers for disease control (CDC) growth charts: United States. Vital and Health Statistics. 2000;314:1–27. [PubMed] [Google Scholar]
- Leri A, Liu Y, Wang X, Kajstura J, Malhotra A, Meggs LG, Anversa P. Overexpression of insulin-like growth factor-1 attenuates the myocyte renin-angiotensin system in transgenic mice. Circulation Research. 1999;84:752–762. doi: 10.1161/01.res.84.7.752. [DOI] [PubMed] [Google Scholar]
- MacGillivray MH, Morishima A, Conte F, Grumbach M, Smith EP. Pediatric endocrinology update: an overview. The essential roles of estrogens in pubertal growth, epiphyseal fusion and bone turnover: lessons from mutations in the genes for aromatase and the estrogen receptor. Hormone Research. 1998;49 (Suppl 1):2–8. doi: 10.1159/000053061. [DOI] [PubMed] [Google Scholar]
- Mendonca BB, Osorio MGF, Vatronico AC, Estefan V, Lo LSS, Arnhold JP. Longitudinal hormonal and pituitary imaging changes in two females with combined pituitary hormone deficiency due to deletion of A301, G302 in the PROP1 gene. Journal of Clinical Endocrinology and Metabolism. 1999;84:942–945. doi: 10.1210/jcem.84.3.5537. [DOI] [PubMed] [Google Scholar]
- Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. Journal of Clinical Endocrinology and Metabolism. 1995;80:3689–3698. doi: 10.1210/jcem.80.12.8530621. [DOI] [PubMed] [Google Scholar]
- Nogueira CR, Sabacan L, Jameson JL, Medeiros-Neto G, Kopp P. Combined pituitary hormone deficiency in an inbred Brazilian kindred associated with a mutation in the PROP-1 gene. Molecular Genetics and Metabolism. 1999;67:58–61. doi: 10.1006/mgme.1999.2841. [DOI] [PubMed] [Google Scholar]
- Osorio MJF, Kopp P, Marui S, Latronico AC, Mendonca BB, Arnhold IV. Combined pituitary hormone deficiency caused by a novel mutation of a highly conserved residue (F88S) in the homeodomain of PROP-1. Journal of Clinical Endocrinology and Metabolism. 2000;85:2779–2785. doi: 10.1210/jcem.85.8.6744. [DOI] [PubMed] [Google Scholar]
- Parks JS, Brown MR. Molecular Basis of Multiple Pituitary Hormone Deficiency. 1. Humana Press Inc; Totowa, NJ: 1999. [Google Scholar]
- Parks JS, Brown MR, Hurley DL, Phelps CJ, Wajnrajch MP. Heritable disorders of pituitary development. Journal of Clinical Endocrinology and Metabolism. 1999;84:4362–4370. doi: 10.1210/jcem.84.12.6209. [DOI] [PubMed] [Google Scholar]
- Pernasetti F, Toledo SP, Vasilyev VV, Hayashida CY, Cogan JD, Ferrari C, Lourenço DM, Mellon PL. Impaired adrenocorticotropin-adrenal axis in combined pituitary hormone deficiency caused by a two-base pair deletion (301–302delAG) in the prophet of Pit-1 gene. Journal of Clinical Endocrinology and Metabolism. 2000;85:390–397. doi: 10.1210/jcem.85.1.6324. [DOI] [PubMed] [Google Scholar]
- Pugazhenthi S, Boras T, O’Connor D, Meintzer MK, Heidenreich KA, Reusch JE. Insulin-like growth factor I-mediated activation of the transcription factor cAMP response element-binding protein in PC12 cells. Involvement of p38 mitogen-activated protein kinase-mediated pathway. Journal of Biological Chemistry. 1999;274:2829–2837. doi: 10.1074/jbc.274.5.2829. [DOI] [PubMed] [Google Scholar]
- Rosenbloom AL, Almonte AS, Brown MR, Fisher DA, Baumbach L, Parks JS. Clinical and biochemical phenotype of familial anterior hypopituitarism from mutation of the PROP1 gene. Journal of Clinical Endocrinology and Metabolism. 1999;84:50–57. doi: 10.1210/jcem.84.1.5366. [DOI] [PubMed] [Google Scholar]
- Rosenfeld RG, Rosenbloom AL, Guevara-Aguirre J. Growth hormone (GH) insensitivity due to primary GH receptor deficiency. Endocrine Reviews. 1994;15:369–390. doi: 10.1210/edrv-15-3-369. [DOI] [PubMed] [Google Scholar]
- Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, Williams TC, Lubahan DB, Korach KS. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. New England Journal of Medicine. 1994;331:1056–1061. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- Sorgo W, Zachmann M, Tassinari D, Fernandez F, Prader A. Longitudinal anthropometric measurements in patients with growth hormone deficiency. Effect of human growth hormone treatment. European Journal of Pediatrics. 1982;138:38–45. doi: 10.1007/BF00442326. [DOI] [PubMed] [Google Scholar]
- Sornson MW, Wu W, Dasen JS, Flynn SE, Norman DJ, O’Connell SM, Gukovsky I, Carriere C, Ryan AK, Miller AP, Zuo L, Gleiberman AS, Andersen B, Beamer WG, Rosenfeld MG. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature. 1996;384:327–333. doi: 10.1038/384327a0. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Takashima T, Kadoya M, Konishi H, Kameyama T, Yoshikawa J, Gabata T, Arai K, Tamura S, Yamamoto T, et al. Height of normal pituitary gland on MR imaging: age and sex differentiation. Journal of Computer Assisted Tomography. 1990;14:36–39. doi: 10.1097/00004728-199001000-00006. [DOI] [PubMed] [Google Scholar]
- Uriarte MM, Baron J, Garcia HB, Barnes KM, Loriaux DL, Cutler GB., Jr The effect of pubertal delay on adult height in men with isolated hypogonadotropic hypogonadism. Journal of Clinical Endocrinology and Metabolism. 1992;74:436–440. doi: 10.1210/jcem.74.2.1449545. [DOI] [PubMed] [Google Scholar]
- Walsh PS, Metzger DA, Higuchi R. Chelex100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 1991;10:506–513. [PubMed] [Google Scholar]
- WHO Working Group. Use and interpretation of anthropometric indicators of nutritional status. Bulletin of the World Health Organization. 1986;64:929–941. [PMC free article] [PubMed] [Google Scholar]
- Wilkins L. The Diagnosis and Treatment of Endocrine Disorders in Childhood and Adolescence. 3. Thomas; Springfield: 1965. [Google Scholar]
- Wu W, Cogan JD, Pfäffle RW, Dasen JS, Frisch H, O’Connell SM, Flynn SE, Brown MR, Mullis PE, Parks JS, Phillips JA, 3rd, Rosenfeld MG. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nature Genetics. 1998;18:147–149. doi: 10.1038/ng0298-147. [DOI] [PubMed] [Google Scholar]