

Abstract
Little is known about the molecular mechanisms of androgen regulation of the FSHβ gene; however, studies suggest that it consists of a complex feedback loop that involves multiple mechanisms acting at both the level of the hypothalamus and the pituitary. In the present study, we address androgen regulation of the FSHβ gene in immortalized gonadotrope cells and investigate the roles of activin and GnRH in androgen action. Using transient transfection assays in the FSHβ-expressing mouse gonadotrope cell line, LβT2, we demonstrate that androgens stimulate expression of an ovine FSHβ reporter gene in a dose-dependent manner. Mutation of either of two conserved androgen response elements at −245/−231 and −153/−139 within the proximal region of the ovine FSHβ gene promoter abolishes this stimulation, and androgen receptor binds directly to the −244 ARE in vitro. Androgen induction of the FSHβ reporter gene is also dependent upon the activin autocrine loop present in the LβT2 cells, as well as an activin-response element at −138/−124 of the FSHβ gene. However, activin regulation of other genes remains unaffected by androgens. In addition, androgens stimulate expression of a mouse GnRH receptor reporter gene, and thus may indirectly augment the response of the FSHβ gene to GnRH. Taken together, these data demonstrate that, in mouse gonadotropes, androgens act directly on the ovine FSHβ gene to stimulate expression by a mechanism that is dependent upon activin, as well as acting indirectly, potentially through a second mechanism that may be dependent upon induction of GnRH receptor.
Androgens are a class of sex steroids that play important roles in sexual development and reproduction in both males and females. Androgen action at both the hypothalamus and the anterior pituitary regulates synthesis and secretion of the heterodimeric gonadotropin hormones LH and FSH. At the level of the hypothalamus, androgens reduce steady-state levels of GnRH mRNA (1, 2) and GnRH peptide (3, 4) and modulate processing of the GnRH prohormone (5) in rats, all resulting in dampened GnRH stimulation of LH and FSH. At the level of the pituitary gland, androgen action differs in a species-and gonadotropin subunit gene-specific manner. Androgens repress both α-subunit of the glycoprotein hormones (α-GSU) and LHβ-subunit gene expression in GnRH antagonist-treated rats (6) and rat primary pituitary cell cultures (7, 8). Recently, two different molecular mechanisms by which androgen receptor (AR) represses LHβ gene expression have been elucidated using the immortalized mouse gonadotrope cell line, LβT2 (9, 10). Both studies show that repression is dependent upon GnRH stimulation and that the mechanisms are indirect, requiring protein-protein interactions with Sp1 (9) or steroidogenic factor 1 (10), rather than through AR binding to the LHβ gene.
In contrast to LHβ regulation, the mechanisms through which androgens modulate FSHβ expression at the level of the pituitary are unknown. A growing body of evidence indicates that androgens act at the level of the pituitary to stimulate, rather than repress, FSHβ gene transcription in rodents. Previous studies demonstrated that testosterone (T) increases FSHβ mRNA levels by 2-fold in GnRH antagonist-treated rats, whereas α-GSU and LHβ-subunit mRNA levels are decreased (11, 12). Furthermore, T selectively induces FSHβ mRNA in both male and female primary rat pituitary cell cultures (7, 8, 13). In addition to these actions of androgen on FSHβ transcription, androgens have been shown to modulate levels of follistatin (FS) in the rat pituitary in vivo and both activin and FS in cultured rat pituitary cells (8, 14, 15), indicating that the mechanism of androgen action on FSHβ might be indirect, through modulation of the activin/FS system in the pituitary. Furthermore, GnRH receptor (GnRH-R) within the pituitary of rodents is modulated by androgens (16–20), which in turn may alter responsiveness of FSHβ to GnRH, and thus act as another indirect mechanism through which androgens regulate FSHβ expression at the level of the pituitary. Because androgens stimulate GnRH-R expression in mouse pituitary (9, 21), in contrast to the inhibitory effect most commonly observed in rats (18–20), androgens may stimulate mouse FSHβ gene expression through multiple mechanisms, including via enhancing responsiveness of FSHβ to GnRH.
Historically, the lack of an appropriate gonadotrope cell culture model has impeded efforts to characterize the molecular mechanisms of steroid regulation of FSHβ gene expression. In this study, the LβT2 mouse gonadotrope cell line serves as a model system to discriminate androgen action at the level of the isolated gonadotrope from that of the whole pituitary gland by eliminating the variables associated with whole-animal studies or complex primary pituitary cultures. The LβT2 cells express GnRH, androgen, and activin receptors, and produce activin and FS, providing a model that allows investigation of the interactions of these hormones on the expression of gonadotropin genes (22, 23). We find that androgens act directly within gonadotropes to stimulate FSHβ gene transcription independent of GnRH, and that this androgen stimulation occurs through two conserved androgen response elements (AREs) within the FSHβ gene, at least one of which specifically binds to AR in vitro. Furthermore, we demonstrate that androgen regulation is dependent upon the activin autocrine loop in the gonadotrope and specifically on an element in the FSHβ gene that is also required for activin responsiveness. However, androgens do not alter the activin responsiveness of an activin response element from the GnRH-R gene, indicating that the mechanism of androgen action is not through global modulation of the activin/FS system. Furthermore, activin and androgen act synergistically to induce FSHβ gene expression, as do androgen and GnRH. Finally, androgens stimulate mouse GnRH-R gene expression in gonadotropes, revealing a potential mechanism by which androgens might enhance the response of the ovine FSHβ gene to GnRH. Thus, we demonstrate that androgen stimulation of FSHβ gene transcription at the level of the gonadotrope is dependent upon activin action and requires both AREs and elements conferring activin response, providing insight into the mechanisms of androgen regulation of FSH.
RESULTS
Androgens Stimulate FSHβ Transcription in a Dose-Dependent Manner
The ovine FSHβ gene regulatory region drives gene expression and is hormonally regulated in the LβT2 mouse gonadotrope cell line (22). Furthermore, androgen regulation of the rat and bovine LHβ subunit genes has been studied in this model (9, 10). Here, we have used this cell line model system to address the question of whether androgens stimulate FSHβ gene expression by acting directly and solely within gonadotropes. We chose to utilize the ovine (o) FSHβ promoter for three reasons. First, it is the longest mammalian FSHβ promoter clone available (−4.7 kb). Second, the accuracy of the targeting and hormonal responses of the ovine gene has been demonstrated in transgenic mice (24). Finally, the ovine gene is regulated by activin and GnRH in LβT2 cells (22), and the signal transduction of the GnRH response has been elucidated (25).
LβT2 cells were transiently transfected with a lucif-erase reporter gene in which expression was driven by approximately 5.5 kb of the ovine FSHβ gene regulatory region with the first exon and the first intron (oFSHβ-Luc), deprived of steroids [10% charcoal-stripped fetal bovine serum (FBS)] for 24 h and then treated with 0.1% ethanol vehicle or T at a range of concentrations for 24 h. T treatment stimulates FSHβ promoter activity in a dose-dependent manner, with statistically significant induction starting at 100 pM T (P = 0.006) and a maximal 2-fold stimulation (P < 0.0001) at 100 nM (Fig. 1). The nonaromatizable androgen, dihydrotestosterone (DHT), similarly stimulates oFSHβ-Luc transcriptional activity when administered for 24 h, with a significant induction at 1 nM (P < 0.05), and peak induction at 10 nM DHT (P < 0.0001; Fig. 1). These data demonstrate that androgens are capable of stimulating FSHβ gene transcription in a dose-dependent manner by acting solely and directly upon gonadotropes. Furthermore, the stimulation of FSHβ is likely due to androgenic rather than estrogenic activity, because both the nonaromatizable (DHT) and the more physiological, aromatizable (T) androgens have identical stimulatory effects on FSHβ promoter activity. Taken together, these data indicate that physiologically relevant concentrations of androgens stimulate FSHβ gene expression by acting directly within gonadotropes. This androgen regulation of FSHβ occurs in a specific and saturable, dose-dependent manner typical of classic steroid hormone action.
Fig. 1. Androgen Stimulates oFSHβ Transcription in LβT2 Cells.

LβT2 cells were transfected with 4.7 kb oFSHβ-luciferase reporter plasmid, deprived of steroids for 24 h, and then treated with either ethanol vehicle or various concentrations of T (top panel) or DHT (bottom panel) for 24 h. Data were pooled from three independent experiments (n = 3 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase/CMV-β-galactosidase activity, normalized to the vehicle-treated mean. * Indicates a statistically significant difference (P < 0.05) from vehicle-treated mean. ** Indicates a highly significant difference (P < 0.0001) from the vehicle-treated mean.
Two AREs in the Proximal Promoter Are Required for Androgen Response
To further elucidate the mechanism(s) through which androgens stimulate the FSHβ gene, we mapped regions of the oFSHβ gene promoter that confer androgen response using truncation-deletion analysis. LβT2 cells were transiently transfected with a series of truncations of the oFSHβ gene regulatory region ranging in length from 982–105 bp upstream of the transcription start site, and the ability of 100 nM DHT, administered for 24 h, to induce FSHβ gene transcription was tested (Fig. 2). DHT significantly induced reporter gene activity by 1.9- (P < 0.0001), 1.7- (P < 0.0001), and 1.8-fold (P < 0.0001), respectively, in transfections of reporter genes containing 982, 561, or 401 bp upstream of the oFSHβ gene mRNA start site. The degree of DHT stimulation of these three promoters was identical to that of the 4.7-kb flanking region of the oFSHβ gene (see Fig. 1). Further truncation to −105 resulted in a loss of the ability to respond to DHT (P = 1.0). These data demonstrate that androgen response is conferred by the 296 bp region of the oFSHβ gene-proximal promoter between −401 and −105 bp.
Fig. 2. Localization of the Androgen-Responsive Region in the oFSHβ Regulatory Region.

LβT2 cells were transfected with a series of truncated oFSHβ luciferase reporter plasmids (−982, −561, −401, and −105), deprived of steroids for 24 h, and then treated with ethanol vehicle or 100 nM DHT for 24 h. Data were pooled from five independent experiments (n = 4–6 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase to CMV-β-galactosidase activity, normalized to the vehicle-treated mean within the same plasmid group. * Indicates a statistically significant difference (P < 0.0001) from the vehicle-treated mean within the same plasmid group.
In examining the −401 to −105 region of the oFSHβ gene promoter for candidate AREs, homology searches revealed three candidate sites, located at −245/−231, −212/−198, and −153/−139 (Fig. 3A). These three elements have homology to known AREs (Fig. 3A), including a shared consensus ARE/glucocorticoid response element (GRE)/progesterone response element (PRE) from the mouse mammary tumor virus (MMTV) long terminal repeat (26, 27), consensus AREs from various androgen responsive genes (reviewed in Ref. 28), and an experimentally deduced sequence proven to be highly selective and specific for AR (hasp-ARE) (29). Of the three candidate AREs, only the one at −245/−231 contains a perfect consensus half-site (TGTTCT). Overall, the −245/−231 and −153/−139 putative AREs exhibit the highest homology to previously characterized AREs (Fig. 3A). In contrast, the putative ARE at −212/−198 has higher homology to the shared ARE/GRE/PRE of MMTV (71%) than do either the −245/−231 (47%) or the −153/−139 (47%) sites, and a lower homology (66%) than either the −245/−231 (73%) or −153/−139 (80%) sites to the hasp-ARE. These data implicate these three sites as potential targets for AR action and suggest the −245/−231 and −153/−139 sites may have higher specificity for AR and thus are most likely to be AREs.
Fig. 3. Sequence Comparison to Consensus AREs and Evolutionary Conservation of Three Regions in the oFSHβ Promoter.

A, The three putative ARE motifs within the proximal region of the oFSHβ gene promoter were examined for sequence homology to known ARE motifs from three different androgen-responsive genes, as well as a computer-generated high-affinity and specific consensus ARE (hasp-ARE). The ARE motif of the MMTV gene is a shared ARE/GRE/PRE, whereas the PSA, C3, and hasp-ARE motifs are more specific for the AR than the related GR and PR. Boxes outline the critical conserved C and G bases of the consensus AREs. Percent homology of each ARE is compared at right. B, The proximal promoter region of the oFSHβ gene from −248 to −137 nucleotides was aligned to sequences from the FSHβ gene of various mammalian species, and the relative conservation of sequence homology was determined. Conserved sequences with ARE motifs (ARE 1–3) are shadowed in gray. Minus symbols (−) indicate a deletion compared with alignment with the oFSHβ gene. Asterisks indicate the site of insertion of an additional G nucleotide, only present in the mouse and rat genes. An inverted triangle indicates the site of insertion of a trinucleotide repeat (CAT), present only in the mouse, rat, and human genes.
This entire 211-bp proximal region of the oFSHβ gene (−248 to −137), encompassing all three putative ARE motifs, is remarkably well conserved among numerous mammalian species (Fig. 3B). The −245 and −153 AREs are the most highly conserved sites, and the C/G bases known to be critical for function of the consensus ARE/GRE/PRE are perfectly conserved in all species examined. The less than perfect overall conservation homology of the middle ARE (−212/−198) site is due to insertions of a single guanidine base in FSHβ of rodent species and of a CAT duplication in the mouse, rat, and human FSHβ genes compared with the sheep, cow, and pig FSHβ genes (Fig. 3B). Previously, Webster et al. (30) identified these three same sites within the oFSH proximal promoter to be PREs that bind PR in vitro and confer progesterone response when present as multimers on a heterologous promoter. We hypothesized that one or more of these three PREs may also act as AREs, because both AR and PR are known to bind the same DNA elements within certain genes, including the long-terminal repeat of the MMTV gene promoter (26).
To determine the role of these putative regulatory elements in androgen stimulation of FSHβ, we examined the ability of DHT treatment (24 h) to stimulate promoter activity when the sequences from the −245/−231, −212/−198, or −153/−139 candidate ARE motifs were disrupted by site-directed mutagenesis (mutations shown in Fig. 4A). As shown in Fig. 2, expression of the wild-type −982 FSHβ reporter was stimulated 1.9-fold by DHT (P < 0.0001). A 2-bp mutation (GT to CC) within the conserved −236/−231 half-site of the putative ARE at −245/−231 completely abolished DHT stimulation of reporter gene expression in the context of the −985 FSHβ promoter (P = 1.0) (Fig. 4B). Similarly, a 2-bp mutation (GA to CC) of the −144/−139 half-site of the putative ARE at −153/−139 also completely abolished the ability of DHT to stimulate the FSHβ promoter (P = 1.0). In contrast, mutation of 2 critical C/G base pairs within the putative ARE at −212/−198, one in each half-site, had no deleterious effect on the ability of DHT to induce FSHβ promoter activity (1.65-fold compared with 1.9-fold induction of β985 FSHβ; P = 0.453). Similarly, a CA to CC mutation of the −202/−198 half-site of the putative ARE at −212/−198 also had no effect on DHT stimulation (data not shown). These data indicate that the −245/−231 and −153/−139 ARE motifs, but not the −212/−198 ARE, are crucial for androgen response. The lack of activity from the central ARE is consistent with its lack of evolutionary conservation. Thus, we have identified two functional AREs within the oFSHβ subunit gene, the sequences of which are conserved in other mammalian species and thus are likely to be functional in those species as well.
Fig. 4. Mutagenesis of AREs in the oFSHβ Promoter.

A, Three candidate AREs identified within the −401 to −105 bp fragment of the oFSHβ promoter were individually disrupted by site-directed mutagenesis. Base pairs in the region of the consensus are in uppercase and others are in lowercase. Mutated bases are underlined. Because the C nucleotide chosen for mutation in the −245 and −153 sites was not conserved with the consensus within the −212 site, two different base pairs, one in each half-site, which were conserved with the consensus, were mutated as shown. B, LβT2 cells were transfected with the wild-type −982 oFSHβ-luciferase reporter plasmid or one of three mutant oFSHβ-luciferase reporter plasmids with mutations in the −245 ARE, −212 ARE, or −153 ARE, deprived of steroids for 24 h, and then treated with vehicle or 100 nM DHT for 24 h. Data were pooled from three to five independent experiments (n = 4–6 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase to CMV-β-galactosidase activity, normalized to the vehicle-treated mean. * Indicates a statistically significant difference (P < 0.0001) from the vehicle-treated mean within the same plasmid group.
AR Binds to the −245 ARE
To determine whether AR can bind directly to an ARE in the oFSHβ subunit gene, we obtained full-length human AR protein by overexpressing it in baculovirus-infected Sf9 insect cells using a Flag-tagged AR baculovirus (kindly provided by E. Wilson). Cells were treated with androgens (1 mM DHT final concentration) for 24 h before harvest. Soluble whole-cell extract was incubated with a 29-bp probe to the conserved −245 oFSHβ ARE and analyzed by EMSA (Fig. 5). The resulting complex was supershifted by anti-AR antibody but not by IgG, showed self-competition, failed to compete with a mutant probe containing the same mutations noted in Fig. 4, and was competed by an ARE consensus sequence (Materials and Methods). We conclude that AR can bind directly and specifically to the −245 ARE element in the oFSHβ subunit gene. However, under these conditions, we were unable to demonstrate AR binding to the −153/−139 ARE in EMSA. This might be due to the fact noted above, that only the −245/−231 ARE contains a perfect consensus half-site (TGTTCT) for AR binding.
Fig. 5. Ligand-Bound AR Binds Specifically to an Oligonucleotide Containing the −245 ARE in the oFSHβ Promoter.
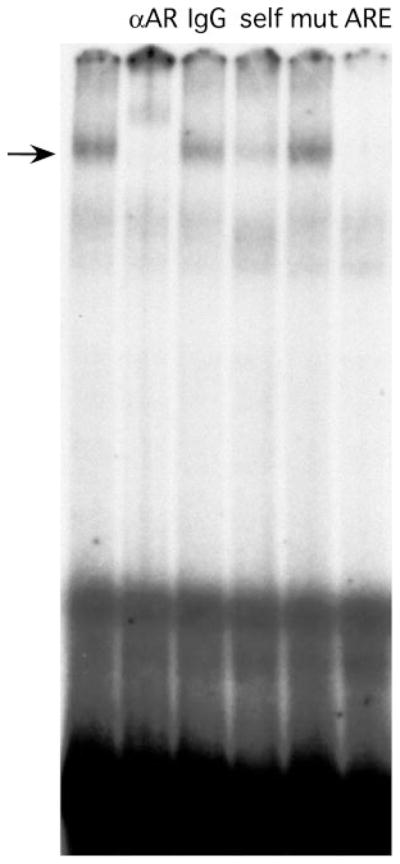
Extracts of AR-baculovirus-infected insect cells were incubated with the −245 probe and tested for complex formation in EMSA. The AR-DNA complex is apparent in lane 1, anti-AR antibody supershift (lane 2), IgG control (lane 3), self-competition (lane 4), mutant competition (mutation as in Fig. 4) (lane 5), and ARE consensus competition (Materials and Methods) (lane 6).
Androgen Stimulation is Dependent on the Activin Autoregulatory Loop
In addition to elucidating the mechanism through which androgens act within gonadotropes to directly stimulate the oFSHβ gene, we also examined whether androgens act within the gonadotrope through additional mechanisms to indirectly regulate FSHβ gene expression. Previous studies in rats in vivo and in cultured rat pituitary cells have shown that androgens can modulate the levels of activin and FS in the pituitary (8, 14, 15, 31, 32). To determine whether the activin autocrine loop, the key regulatory system of FSH synthesis and secretion, plays a role in androgen stimulation of FSHβ transcription, we examined the effects of coadministration of FS or activin on androgen stimulation of FSHβ expression. LβT2 cells were transiently transfected with the 4.7 kb oFSHβ-Luc reporter, incubated overnight in DMEM containing 10% charcoal-treated FBS and 250 ng/ml FS to block endogenous activin action, followed by treatment with vehicle or 100 nM DHT ± 250 ng/ml FS or 10 ng/ml activin-A for 24 h (Fig. 6). As expected, treatment with either DHT or activin alone induced FSHβ promoter activity by 1.5-fold (P = 0.0015) and 11-fold (P = 0.000003), respectively. Interestingly, addition of FS abolished the ability of DHT to stimulate FSHβ promoter activity, whereas addition of activin with DHT resulted in induction of FSHβ promoter activity by 19-fold, significantly greater than either DHT (P = 0.00007) or activin treatment alone (P = 0.0039). Using the methodology as described by Slinker (33), analysis of the data by two-way ANOVA reveals a synergistic increase upon activin and androgen cotreatment that is significantly (P < 0.0001) different than an additive effect. Therefore, these data indicate that there is a synergistic interaction between androgen and the activin/FS system in the regulation of the FSHβ promoter as well as a complete dependence upon activin as a permissive agent for androgen action. Thus, the endogenous activin secretion by the gonadotrope plays a crucial role in androgen regulation of FSHβ gene expression.
Fig. 6. Androgen Induction of the oFSHβ Gene Is Blocked by FS and Synergistic with Activin (Act).
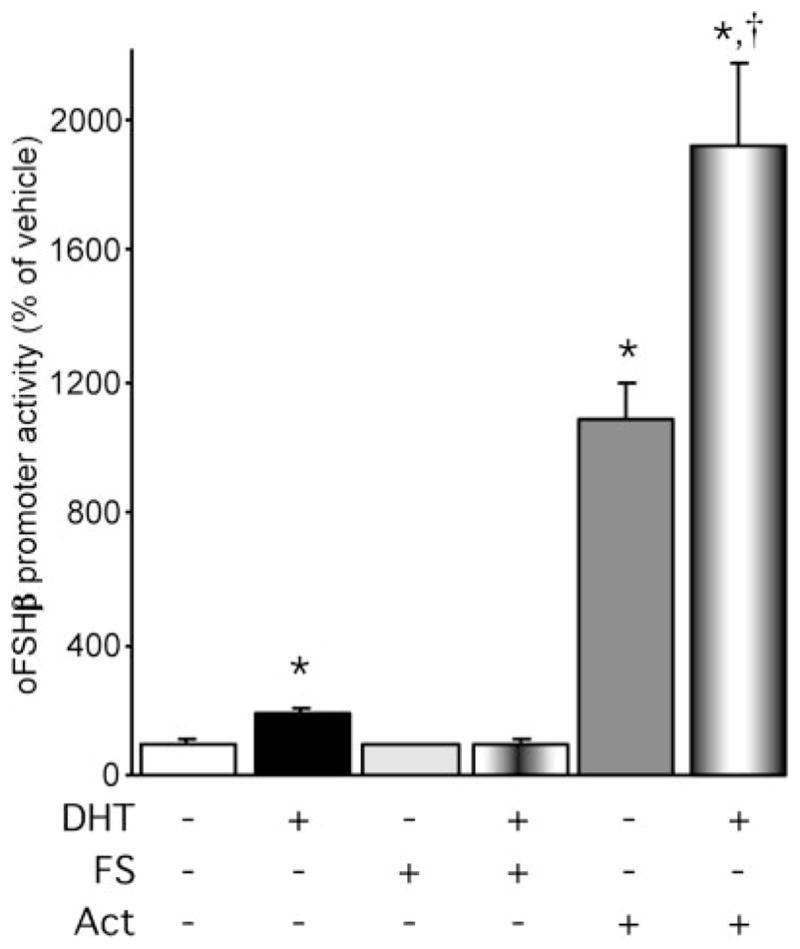
LβT2 cells were transfected with 4.7 kb oFSHβ-Luc reporter plasmid, incubated overnight in DMEM containing 10% charcoal-treated FBS and 250 ng/ml FS, and treated for 24 h with vehicle or 100 nM DHT ± 250 ng/ml FS or 10 ng/ml activin A. Data were pooled from four independent experiments (n = 4 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase to RSV-β-galactosidase activity, normalized to the vehicle-treated mean. * Indicates a statistically significant difference (P < 0.001) from the vehicle-treated mean. † Indicates a statistically significant difference (P < 0.001) from the activin only-treated mean.
The Activin Autoregulatory Loop Does Not Affect Androgen Action Globally, nor Vice Versa
To determine whether the interaction between androgen and activin is specific to the FSHβ gene or involves a general effect of the activin system on AR function or vice versa, we examined both the effect of FS on androgen induction of the highly androgen-sensitive MMTV promoter and the effect of androgen on FS or activin regulation of the highly activin-sensitive GnRH receptor-activating sequence (GRAS) element of the mouse GnRH-R gene promoter (34). In the first series of experiments, LβT2 cells were transiently transfected with the MMTV-Luc reporter plasmid, pre-treated overnight with DMEM containing 10% charcoal-treated FBS and 250 ng/ml FS, and treated with vehicle or 100 nM DHT ± 250 ng/ml FS for 24 h (Fig. 7A). DHT stimulated MMTV promoter activity by 2.56-fold (P = 0.024). FS treatment did not inhibit MMTV promoter activity, nor did FS diminish the ability of DHT to stimulate the MMTV promoter. These data indicate that FS blockade of androgen action is gene specific, rather than due to a global perturbation of AR function in the gonadotrope.
Fig. 7. Activin (Act) Does Not Globally Affect Androgen Action and Vice Versa.

A, LβT2 cells were transfected with the androgen-sensitive MMTV-Luc reporter plasmid, deprived of steroids for 24 h, and then treated with ethanol vehicle or 100 nM DHT ± 250 ng/ml FS for 24 h. Data were pooled from three independent experiments (n = 4 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase/RSV-β-galactosidase activity, normalized to the vehicle-treated mean within the same plasmid group. B, LβT2 cells were transfected with the activin-sensitive GRAS-Luc reporter plasmid, incubated overnight in DMEM containing 10% charcoal-treated FBS and 250 ng/ml FS, and treated for 24 h with vehicle or 100 nM DHT ± 250 ng/ml FS ± 10 ng/ml activin A. Data were pooled from seven independent experiments (n = 4 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase to RSV-β-galactosidase activity, normalized to the vehicle-treated mean. * Indicates a statistically significant difference (P < 0.001) from the vehicle-treated mean within the same plasmid group.
A route for androgen regulation of FSHβ gene expression in pituitary that has been previously suggested is androgen regulation of activin system components such as FS (14, 15). To determine whether androgens were globally affecting activin action, LβT2 cells were transiently transfected with the activin-sensitive GRAS-Luc reporter plasmid containing the activin response element from the GnRH-R gene (34, 35), pretreated overnight with DMEM containing 10% charcoal-treated FBS and 250 ng/ml FS, and treated with vehicle or 100 nM DHT ± 10 ng/ml activin-A ± 250 ng/ml FS for 24 h (Fig. 7B). FS treatment inhibited GRAS-Luc reporter activity by 70% (P < 0.00000001), whereas activin treatment stimulated reporter activity by 6-fold (P = 0.0000000001). DHT alone had no effect and DHT in combination with FS or activin had no effect on either FS repression or activin stimulation of GRAS promoter activity. These data suggest that the stimulatory effect of androgens on FSHβ gene expression is not due to modulating bioavailable levels or activity of activin, its receptors, FS, or other activin-regulatory system components present in gonadotropes. Taken together, these data show that androgen regulation of FSHβ gene expression is not due to an indirect action on the activin/FS system, but likely involves interaction with the activin system specifically within the context of the FSHβ gene promoter.
Androgen Regulation Is Dependent upon an Activin-Responsive Element in the FSHβ Gene
To investigate potential interactions of the androgen-and activin-regulatory systems on the FSHβ gene, we addressed the potential role of an activin response element in the FSHβ gene in androgen action. Recent data from our laboratory indicate that a putative Sma-and Mad-related protein (Smad) binding element (SBE) located at −138/−124 of the oFSHβ gene promoter is crucial for activin regulation of transcription (35a). In the current study, we used an activin-insensitive −138/−124 mutant oFSHβ-Luc reporter to further elucidate the molecular mechanism of interaction between androgen and the activin loop in the regulation of FSHβ transcription. LβT2 cells were transiently transfected with either the −982 truncation of oFSHβ-Luc, the −982 truncation of oFSHβ-Luc with the mutation in the −245/−231 ARE, or the −982 truncation of oFSHβ-Luc with a 2-bp mutation in the putative SBE half-site (Fig. 8A), pretreated overnight with 10% charcoal-treated FBS and treated with vehicle or 100 nM DHT ± 10 ng/ml activin-A for 24 h. These experiments were performed in serum-free conditions, and we did not include a FS pretreatment to lower the endogenous activin; hence, the magnitude of the activin induction is smaller than in Fig. 6. This is not due to differences in the activin responsiveness of the −982 vs. the −4.7-kb oFSHβ promoter (35a). As shown in Fig. 8B, DHT or activin treatment alone each stimulated activity of the −982 oFSHβ-Luc reporter by approximately 2-fold (P = 0.001 and P < 0.000001, respectively), and combined DHT plus activin treatment stimulated reporter activity by 4-fold (P < 0.000001). The oFSHβ-Luc reporter with the ARE mutation was not affected by DHT alone (P < 0.64) and was induced 1.7-fold by activin alone (P = 0.00004), and the cotreatment of DHT with activin did not significantly increase reporter activity above that of activin treatment alone (P = 0.28; 1.9-fold vs. 1.7-fold). As expected, the oFSHβ-Luc reporter in which the −138/−124 putative SBE half-site has been disrupted by a double-point mutation was not significantly induced by activin treatment alone (1.2-fold; P = 0.213). Interestingly, the −138/−124 activin nonresponsive mutant of the oFSHβ-Luc reporter was completely insensitive to DHT treatment alone (0.98-fold vs. vehicle; P = 0.11) or DHT enhancement of activin induction (0.90-fold vs. vehicle and activin; P = 0.446) of FSHβ reporter gene activity. Therefore, androgen induction of FSHβ requires both the −245/−231 ARE and −138/−124 SBE sites, whereas activin response requires the −138/−124 SBE site, but may be slightly dampened by loss of the −245/−231 ARE. These data provide strong evidence in support of the activin dependence of androgen stimulation demonstrated in Fig. 7. Moreover, they suggest that an interaction between AR and the activin-regulatory system occurs through at least two distinct cis elements in the proximal oFSHβ promoter.
Fig. 8. Androgen Induction of the FSHβ Gene Is Dependent upon an Activin-Responsive Element.

A, Base pairs in the region of the consensus sequences for the −245 ARE and −138 SBE are in uppercase and others are in lowercase. Mutated bases are underlined. B, LβT2 cells were transfected with the wild-type, ARE mutant, and SBE mutant FSHβ-Luc reporter plasmids, incubated overnight in DMEM containing 10% charcoal-treated FBS, and treated for 24 h with vehicle or 100 nM DHT ± 10 mg/ml activin A. This simplified protocol does not include the FS pretreatment as compared with the protocols for Figs. 6 and 7, but the activin stimulation remains significant. Data were pooled from seven independent experiments (n = 3–4 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase to RSV-β-galactosidase activity, normalized to the vehicle-treated mean. * Indicates a statistically significant difference (P < 0.05) from the vehicle-treated mean. † Indicates a statistically significant difference (P < 0.05) from the activin only-treated mean. mut, Mutant.
Androgen Enhances GnRH Stimulation of FSHβ Gene Expression
In addition to modulating the activin/FS system in the pituitary, androgens have been shown to regulate GnRH sensitivity via alterations in the level of GnRH-Rs (16–20). In fact, in the case of the LHβ gene, GnRH stimulation was required to observe androgen repression in the LβT2 cell model (9, 10). We hypothesized that androgens may act upon the gonadotrope to alter the responsiveness of the FSHβ gene to GnRH and thereby indirectly regulate its expression. To test this hypothesis, LβT2 cells were transfected with the 4.7-kb oFSHβ-luciferase reporter and the ability of 10−12 to 10−8 M DHT administered for 24 h to affect GnRH (10 nM for 6 h) induction of oFSHβ promoter activity was examined (Fig. 9). Treatment for 24 h with 10 nM DHT induced FSHβ promoter activity by 1.6-fold (P = 0.032), and 6 h treatment with 10 nM GnRH induced promoter activity by 1.7-fold (P < 0.015). These results were expected because both androgens (Fig. 1) and GnRH (22, 25) are each able to independently stimulate FSHβ expression in LβT2 cells. Interestingly, when increasing concentrations of DHT were coadministered with 10 nM GnRH, the oFSHβ promoter was increasingly stimulated in a DHT dose-dependent manner. The 1 nM DHT concentration, when coadministered with GnRH, resulted in a 2.75-fold induction of FSHβ promoter activity, which was significantly greater than either 10 nM DHT (P < 0.0001) or 10 nM GnRH (P < 0.001) treatment alone. The maximal stimulation of 3-fold was observed in cells cotreated with 10 nM DHT and GnRH. FSHβ promoter activity of this treatment group was also significantly greater than that of cells treated with either DHT (P < 0.0001) or GnRH (P < 0.0001) alone. Analysis of the data by two-way ANOVA reveals a synergistic increase upon cotreatment with GnRH and androgen that is significantly (P = 0.001) different than an additive effect (33). These data suggest that androgens, in addition to directly stimulating FSHβ expression through novel AREs, may also be enhancing the ability of GnRH to stimulate FSHβ expression. To test this hypothesis, we examined whether androgens are able to modulate responsiveness of gonadotropes to GnRH by inducing the GnRH-R gene.
Fig. 9. DHT and GnRH Act Synergistically to Stimulate oFSHβ Expression.
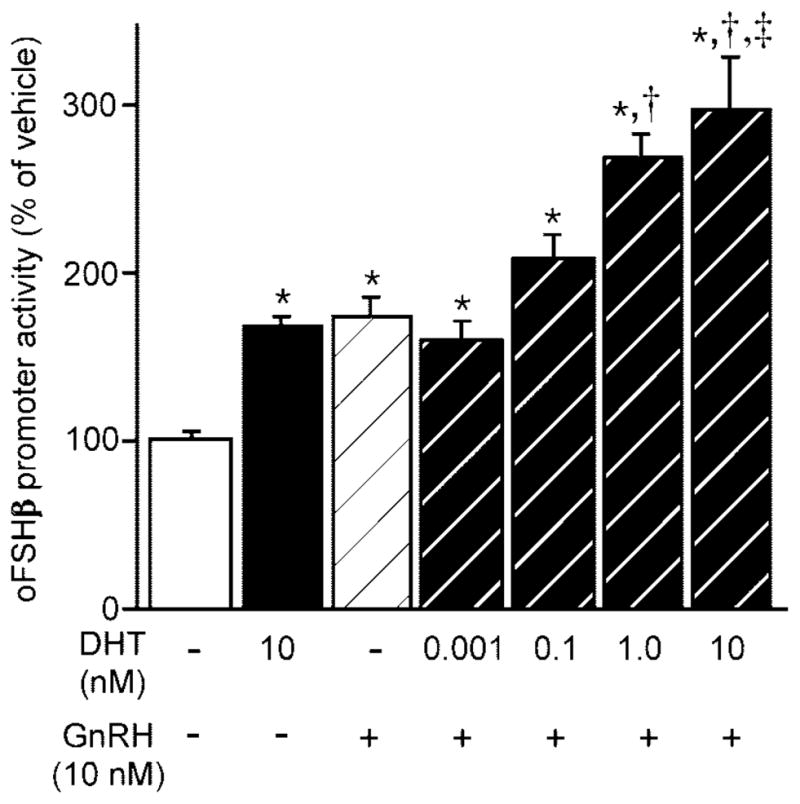
LβT2 cells were transfected with 4.7 kb oFSHβ-luciferase reporter plasmid, deprived of serum for 20 h, psretreated for 18 h with vehicle or various concentrations of DHT, and treated an additional 6 h with the same concentration of DHT in the presence or absence of 10 nM GnRH. Data were pooled from six independent experiments (n = 3–4 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase/TK-β-galactosidase activity, normalized to the vehicle-treated mean. * Indicates a statistically significant difference (P < 0.05) from the vehicle-treated mean. † Indicates a statistically significant difference (P < 0.0001) from the DHT only-treated mean. ‡ Indicates a statistically significant difference (P < 0.0001) from the GnRH only-treated mean.
Androgen Stimulates GnRH-R Gene Expression in a Time- and Dose-Dependent Manner
One mechanism through which androgens could enhance response of gonadotropes to GnRH is by stimulating expression of the GnRH-R. We examined the effects of androgen treatment on expression of the mouse GnRH-R gene in LβT2 cells transfected with a luciferase reporter plasmid the expression of which was driven by 1.2 kb of the mouse GnRH-R gene promoter. LβT2 cells were deprived of steroids for 24 h, and then incubated for 24 h with media containing either 0.1% ethanol vehicle alone or increasing concentrations of DHT. Treatment for 24 h with DHT resulted in a gradual dose-dependent increase in mouse GnRH-R promoter activity (Fig. 10A). Statistically significant induction of GnRH-R gene expression was observed at 1 (P = 0.002), 10 (P < 0.0001), and 100 nM (P < 0.0001) DHT concentrations, with maximal stimulation at 10 nM. These data indicate that at doses physiologically relevant to adult male mice, androgen stimulates expression of the mouse GnRH-R gene in a dose-dependent manner.
Fig. 10. The GnRH-R Gene Is Induced by Androgens.

A, LβT2 cells were transfected with a 1.2-kb mouse GnRH-R-luciferase reporter plasmid, deprived of steroids for 20 h, and then treated with vehicle or various concentrations of DHT for 24 h. Data were pooled from five independent experiments (n = 5–6 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase to RSV-β-galactosidase activity, normalized to the vehicle-treated mean. * Indicates a statistically significant difference (P < 0.0001) from the vehicle-treated mean. B, LβT2 cells were transfected with the mouse GnRH-R-luciferase expression vector, deprived of steroids for 20 h, and then treated with 10 nM DHT for 6, 12, 24, or 48 h. Data were pooled from three independent experiments (n = 4–6 replicates per treatment group for each experiment) and expressed as the mean ± SEM of the ratio of luciferase to RSV-β-galactosidase activity, normalized to the vehicle-treated mean of the same treatment time. * Indicates a statistically significant difference (P < 0.0001) from the vehicle-treated mean of the same treatment time.
To further characterize the mechanism of androgen stimulation of GnRH-R gene expression, we treated transfected LβT2 cells with either ethanol vehicle or 10 nM DHT for 6, 12, 24, or 48 h (Fig. 10B). DHT (10 nM) treatment for 6 h had no effect on GnRH-R promoter activity. As the length of exposure to DHT was increased, the induction of promoter activity increased. DHT treatment for 12 h resulted in a modest 1.2-fold induction of promoter activity but was not statistically significant (P = 0.15). As expected from the results of Fig. 10A, 24 h of 10 nM DHT resulted in a statistically significant 1.4-fold (P < 0.0001) induction of GnRH-R promoter activity. DHT treatment for 48 h resulted in an even greater 1.9-fold (P < 0.0001) induction of GnRH-R reporter gene expression. These data indicate that a physiologically relevant dose of androgen stimulates GnRH-R gene expression in a time-dependent manner.
Androgen Enhancement of GnRH Stimulation Persists after Abrogation of Direct Androgen Stimulation of FSHβ Expression
We have demonstrated that androgen stimulates GnRH-R gene expression in the mouse gonadotrope; however, these data do not discount that the effect of DHT and GnRH treatment in the stimulation of FSHβ promoter activity might still be due to cross-talk between the androgen and GnRH signaling pathways rather than due to androgen causing sensitization of the gonadotrope to GnRH. To test this alternate hypothesis, we examined the ability of DHT and GnRH to stimulate promoter activity of a 4.7-kb oFSHβ-luciferase expression plasmid that had been rendered insensitive to direct androgen stimulation. The −245/−231 ARE was mutated in the same manner as in Fig. 4, but in the context of the 4.7-kb oFSHβ 5′-flanking region. As described in Fig. 4, this same mutation completely abolished androgen stimulation in the context of the −982 oFSHβ promoter. As expected, 24 h of 10 nM DHT failed to result in stimulation of reporter gene activity in LβT2 cells transiently transfected with the 4.7-kb oFSHβ-luciferase plasmid containing the ARE mutation (P = 0.85; Fig. 11). This indicates that mutation of this ARE alone is sufficient to block androgen induction even in the context of the −4.7 kb promoter. GnRH (10 nM) for 6 h induced mutant FSHβ promoter activity by 2.2-fold compared with vehicle-treated controls (P < 0.0001). Interestingly, when 10 nM DHT was coadministered with GnRH, the activity of the ARE mutant oFSHβ promoter was still significantly increased above that of GnRH treatment alone by 1.78-fold (P < 0.0001). These data indicate that androgens act indirectly through GnRH to induce FSHβ promoter activity, even when androgens are no longer able to directly induce FSHβ expression. Taken together with the data from Figs. 9 and 10, these results support the hypothesis that androgens also act in the gonadotrope to indirectly promote transcription of the FSHβ gene by stimulating GnRH-R expression and thereby enhancing responsiveness of the gonadotrope and FSHβ gene to GnRH. Alternatively, it is also possible that androgen could be affecting the sensitivity of the LβT2 cell to GnRH by augmenting the signal transduction pathways through which GnRH is acting on the FSHβ gene.
Fig. 11. DHT Enhances GnRH Stimulation of an Androgen-Insensitive Mutant of FSHβ.

The −245/−231 ARE was disrupted in the 4.7 kb oFSHβ-luciferase reporter plasmid by site-directed mutagenesis, and the ability of 10 nM DHT for 24 h, 10 nM GnRH for 6 h, or the combination of both DHT (24 h) and GnRH (final 6 h) treatments to stimulate the mutant FSHβ promoter activity was examined. Data were pooled from four independent experiments (n = 4 per group in each experiment) and expressed as the mean ± SEM of the ratio of luciferase to TK-β-galactosidase, normalized to the vehicle-treated mean. * Indicates a statistically significant difference (P < 0.0001) from the vehicle-treated mean. † Indicates a statistically significant difference (P < 0.0001) from GnRH only-treated mean.
DISCUSSION
Accumulating evidence indicates that the steroid hormone feedback regulation of gonadotropin synthesis involves multiple mechanisms, with distinct actions at the levels of both the hypothalamus and pituitary gland. Very little is known about how androgenic steroids regulate the FSHβ gonadotropin subunit gene at the level of the pituitary gland, the molecular mechanisms involved, or even whether the gonadotrope itself is the direct target of androgen. The key impediment to such investigations has been the lack of an appropriate FSHβ-expressing gonadotrope model system. Recent studies using highly sensitive RT-PCR (22), immunohistochemistry (36), and RNase protection assays (37) have demonstrated that the LβT2 mouse gonadotrope cell line expresses endogenous FSHβ, and that expression of FSHβ in LβT2 cells is regulated by the same factors known to regulate FSHβ in vivo, such as GnRH, activin, and FS (22). Previous studies had reported that LβT2 cells, like normal gonadotropes, express ER (38) and AR (23). The data presented in the current study confirm that endogenous AR is functional and active in LβT2 cells, demonstrating the relevance of the LβT2 cell line as a model system to study the action of androgenic steroid hormones.
Previous literature has demonstrated that androgen regulation of FSHβ gene expression appears to be species specific and involves actions at both the hypothalamus and pituitary gland. In rats, for example, androgens act through at least two opposing mechanisms to regulate FSH synthesis, i.e. an inhibitory action at the hypothalamic level that is GnRH dependent and a stimulatory action at the pituitary level that is independent of GnRH (7, 11–13). In primates, as in rats, androgens act both at the hypothalamic level in a GnRH-dependent manner and at the pituitary level in a GnRH-independent manner. However, unlike the rat, both mechanisms of androgens appear to be inhibitory to FSHβ expression in primates (39, 40). In the current study, we elucidate the activin-dependent, GnRH-independent, molecular mechanism through which androgens stimulate expression of the oFSHβ gene at the level of the pituitary gonadotrope. Furthermore, we identify a GnRH-dependent mechanism of androgen stimulation that potentially functions through the induction of GnRH-R gene expression.
Several investigators have demonstrated, using either GnRH antagonist-treated rats or primary cultures of rat pituitary cells, that at the level of the pituitary gland, androgens increase steady-state levels of FSHβ mRNA (7, 8, 11–13) and primary transcript (41) and enhance stability of FSHβ transcripts (11) through unknown mechanisms independent of GnRH. These studies, however, were not able to address whether androgen regulation of FSHβ expression occurs by direct actions on the FSH-producing gonadotrope or is indirectly mediated by one of the other many distinct populations of the anterior pituitary. This is an important distinction because gonadotropes comprise only a small minority (5–15%) of the total secretory cell types within the anterior pituitary (42), and several anterior pituitary cell types express AR in vivo (43–45). Moreover, the different pituitary cell populations communicate with each other through paracrine, juxtacrine, and endocrine mechanisms (46). The data presented herein specifically address whether the androgen regulation of FSHβ expression observed at the level of the whole pituitary gland also occurs at the level of the gonadotrope. Taken together, these data indicate that physiologically relevant concentrations of androgens stimulate FSHβ gene expression by acting directly within gonadotropes. This androgen regulation of FSHβ occurs in a specific and saturable, dose-dependent manner typical of classic steroid hormone action. Furthermore, androgen induction of FSHβ transcription occurs within 24 h, a physiologically relevant time frame when compared with the diurnal changes in androgen levels that occur in many different animal genera, including rodents (47, 48).
Our finding that two of the three sites within the proximal oFSHβ promoter shown to act as PREs by Webster et al. (30) also act as AREs, demonstrates that PR and AR likely use distinct mechanisms with shared components. Because disruption of the −212/−198 PRE does not interrupt androgen stimulation of FSHβ, it must not be crucial to the mechanism of androgen regulation. Although the necessity of the three PREs for progesterone stimulation was not examined in the studies by Webster et al. (30), all three PREs were shown to specifically bind PR in gel shift assays and confer progesterone response when multimerized on a heterologous promoter, demonstrating their sufficiency as PREs. In contrast, we have shown that two of these elements (−245/−231 and −153/−139) are individually required for androgen induction of the oFSHβ gene, i.e. that neither is sufficient when the other is mutated. Notably, only one of these elements could be demonstrated to bind to AR protein in vitro (−245/−231). This may indicate that the −153/−139 element is low affinity despite the fact that its mutation ablates androgen responsiveness. Perhaps the −153/−139 element is required to coordinate accessory or interacting proteins that provide context for the action of the AR binding at the −245/−231 element or perhaps other binding proteins present in the LβT2 cell nuclei are required to allow AR to bind to the −153/−139 sequence.
Interestingly, two of the three PRE/ARE sequences within the oFSHβ gene proximal promoter are highly conserved among mammalian species, including pigs, cows, rats, mice, and humans (Fig. 4). The high degree of conservation of these sequences would suggest that they play important roles in androgen and/or progesterone regulation of FSHβ in a wide variety of species. However, the significance of this high degree of sequence conservation in mammals is complicated by observations of androgen inhibition of FSH synthesis in primate model systems. In castrated rhesus macaques treated with GnRH antagonist, administration of T significantly reduced the levels of serum FSH, suggesting an inhibitory role of androgen in FSH synthesis at the level of the pituitary gland in nonhuman primates (39). Similarly, in primary pituitary cell cultures from hypogonadal (hpg), GnRH-deficient, human FSHβ promoter-containing transgenic mice, administration of testosterone propionate or DHT for 24 h reduced steady-state levels of human FSHβ transgene mRNA (40). The authors postulated that differences in the FSHβ gene between humans and rats were a possible reason for the contradiction of their data to that observed in rats. Our finding that both sequences responsible for androgen action within the oFSHβ gene are extremely well conserved among mammals does not support such a hypothesis, and therefore, the relative differences in the responses of the rat and sheep gene to androgen (stimulatory) compared with that of the human gene (inhibitory) are probably not due to sequence differences. We cannot, however, disregard the possibility that subtle sequence differences in regions flanking the highly conserved 110-bp region of the proximal FSHβ promoter examined could affect sensitivity to androgens. Alternatively, the two ARE sequences identified in the current study may be crucial to the actions of AR in FSHβ expression for both sheep and humans, but interaction of AR with different combinations of coactivators or corepressors on the FSHβ gene may ultimately determine whether androgens will stimulate or repress expression in a given species.
The dependence of androgen activation of the oFSHβ gene on the activin autocrine loop indicates a potential mechanism for species differences. For example, both Smad 3 and Smad 4 are known to bind AR in vitro, and overexpression of AR affects expression of Smad 3/4-sensitive genes and vice versa (49–51). This is of physiological importance because Smads are downstream mediators of the activin receptor. Activin is a key physiological regulator of FSH synthesis and secretion and is believed to be a principal mechanism through which FSH is regulated differentially from LH (52). Thus, the dependence of androgen induction on the presence of endogenous activin indicates that activin acts as a permissive agent for the regulation of this gene by androgen.
Androgens have been hypothesized to act in the anterior pituitary through modulation of the components of the activin and FS system (8, 14, 15, 31, 32). Our finding that FS blocks the action of androgens on the FSHβ promoter might seem to support this hypothesis. However, the further demonstration that androgens have no effect on activin induction of an activin-response element from the GnRH-R gene indicates that the role that the activin/FS system plays in androgen action is gene specific, not a global alteration in the bioavailability of activin or FS or in the level or responsiveness of the activin receptors. Furthermore, the removal of activin by FS tends to increase, rather than decrease, the response of MMTV to androgens, indicating that activin is not necessary for the action of AR on expression of other genes. Finally, mutation of only one ARE in the FSHβ promoter prevents androgen regulation despite the presence of the intact activin-response elements and activin responsiveness indicating that androgen is not regulating this promoter by altering the levels of activin or FS. This finding is supported by a recent study by Burger et al. (41) in which androgen was shown to induce FSHβ primary transcript independent of pituitary FS mRNA regulation. The mechanism of the activin dependence is further elucidated by the demonstration that mutation of an element required for activin action on the oFSHβ gene abrogates androgen induction despite the presence of both intact AREs. This indicates the potential for an interaction between these elements perhaps through AR/SMAD protein-protein interactions, as has been demonstrated in other systems (49–51).
In addition to species-specific and activin-dependent effects of direct androgen action on FSHβ, the current study also supports a species-specific effect of indirect androgen action on FSHβ. Studies using rats have most commonly demonstrated that the levels of GnRH-R and GnRH-R gene expression are reduced by androgen treatment (18–20). However, this does not appear to be the case in the mouse. Naik et al. (21) determined by radioligand binding studies that castration reduced the number of GnRH binding sites (GnRH-Rs) within the pituitary gland of male mice by approximately 50%, and that T replacement at the time of castration completely prevented the castration-mediated decline in the number of GnRH-Rs. More recently, Curtin et al. (9) demonstrated that the levels of GnRH-R mRNA are increased approximately 1.7-fold by 24 h of 1 nM DHT in LβT2 cells. Our data confirm the findings of these earlier studies that androgens stimulate GnRH-R gene expression in the mouse gonadotrope. Furthermore, our demonstration of the dose and time dependency of androgen stimulation of a mouse GnRH-R reporter gene in mouse gonadotropes indicates the specificity of this apparent species-specific androgen action. Although our data demonstrate that androgens synergistically enhance GnRH stimulation of FSHβ gene expression, and that this same regimen of androgen treatment induces GnRH-R gene expression, these data do not prove that the mechanism through which androgens act synergistically with GnRH to stimulate FSHβ gene expression depends on an increase in GnRH-R number. It remains possible that this synergism and the remaining effects of androgen on the ARE-mutated FSHβ gene are due to androgen effects on the GnRH signaling cascade. Gonadotropes possess a reservoir of spare GnRH-Rs (53), and increasing the number of cell-surface GnRH-Rs does not necessarily increase the responsiveness to GnRH (54, 55). Furthermore, studies have shown that GnRH can regulate the level of FS (56, 57) in rat pituitary, providing another possible mechanism for the interaction of GnRH and androgen on the FSHβ gene. However because GnRH was shown to induce FS levels (56, 57), this would be counter to our finding of GnRH induction of the FSHβ gene. Nevertheless, our data are strongly coincidental, and the hypothesis that the two androgen-regulated events are linked is an alluring one.
The physiological significance of the opposing mechanisms of androgen-positive and -negative feedback regulation of FSHβ at the pituitary and hypothalamic levels, respectively, may seem paradoxical or counterproductive. However, these two opposing mechanisms of androgen feedback regulation of FSHβ have potential benefits. First, the contrary effects of androgen at the pituitary and hypothalamic levels provide a means through which a single hormone, androgen, may differentially regulate synthesis of the two gonadotropin hormones, LH and FSH, by acting differently at only one hypothalamic-pituitary-gonadal (HPG) axis site (gonadotrope) without requiring different actions on the other axis sites (hypothalamus and gonads). Thus, expression of the two gonadotropin genes could be altered differentially in a subtle yet complex manner using relatively simple mechanisms. This is further supported by our suggestion that androgens may stimulate responsiveness of the gonadotrope to GnRH and, in turn, further enhance FSHβ transcription. In this manner, androgens could selectively stimulate FSHβ gene transcription by two mechanisms occurring at the level of the gonadotrope. Additionally, the dependence on activin of androgen regulation of the FSHβ further distinguishes the responses of the two gonadotropin genes. Another advantage of opposing mechanisms of androgen regulation of FSH is that in males, which have sufficient concentrations of androgen to stimulate FSH expression, the opposing mechanisms of androgen action in the context of the entire HPG axis would result in relatively stable expression of FSH at a time of dramatic and precipitous decline in LH. Androgen acts at the hypothalamus to reduce GnRH availability, which by itself results in reduction of both LH and FSHβ transcription (due to loss of GnRH stimulation). Simultaneously at the level of the gonadotrope, androgen stimulates the FSHβ gene directly and may also increase the number of GnRH-Rs, which would result in each gonadotrope being more sensitive to the GnRH that is still available. When the results of each mechanism within the context of the entire HPG axis are added together, they balance each other out so that net FSHβ expression is unchanged or perhaps even modestly increased. The increased expression of GnRH-R would not, however, enhance GnRH induction of LH, because androgen interferes with the binding of downstream GnRH-R-induced signal transducers to the LHβ gene itself (9, 10). Because AR repression of LHβ expression at the level of the gonadotrope appears to be downstream of the GnRH-R, increasing GnRH-R does not alter the inhibitory effect of androgen on LH, resulting in inhibition of GnRH stimulation of LH by androgen. The physiological relevance of these combined opposing mechanisms are applicable both to nonseasonal animals like rats, which would require steady FSH levels to maintain spermatogenesis throughout the year, and to seasonal animals like sheep, in which seasonally increasing androgen levels could result in seasonal net increases in FSH and thereby dramatically increase spermatogenesis during the breeding season. Interestingly, FSH levels in male rats do not fluctuate substantially once maturity is reached, whereas both androgen and LH levels surge diurnally (48, 58, 59). In male sheep, androgen and FSH levels rise simultaneously at the beginning of the breeding season (60). The physiological significance of the findings of the current study, that androgens act directly through AR binding to ARE(s) on FSHβ in an activin-dependent manner, and may act indirectly through stimulation of GnRH-R expression at the level of the gonadotrope to stimulate FSHβ gene expression, is likely relevant to the underlying physiological mechanisms of steroid hormone feedback regulation of reproduction in a variety of both seasonal and nonseasonal breeding animal species.
MATERIALS AND METHODS
Hormones
T and DHT were purchased from Sigma Chemical Co. (St. Louis, MO). All steroid stock solutions were prepared by dissolving crystalline hormone in 100% ethanol at a 10 mM concentration and stored in lightproof borosilicate glass vials at 4 C. Before each experiment, fresh steroid treatment preparations were made by diluting the 10 mM stock in 100% ethanol to 103-fold higher concentration than the final target concentration, and subsequently, 1 μl of diluted steroid was added to 1 ml total volume of media to achieve the final target concentration of steroid in 0.1% ethanol vehicle. The human recombinant activin A was obtained from Calbiochem (San Diego, CA). Recombinant mouse FS 288 was kindly provided by Shunichi Shimasaki. Both were resuspended in PBS with 0.1% BSA. GnRH was obtained from Sigma.
Hormone Treatments
For all transient transfection experiments, 8 h after transfection, the LβT2 cells were preincubated in steroid-free DMEM supplemented with 10% charcoal/dextran-treated FBS for 20 h, followed by the appropriate treatment protocol. In the steroid experiments, fresh DMEM with 10% charcoal/dextran-treated FBS containing T or DHT was added, and the cells were incubated for 6–48 h as indicated in the figure legends. For the activin/FS experiments in Figs. 6 and 7, the preincubation media was DMEM with 10% charcoal/dextran-treated FBS containing 250 ng/ml (final concentration) recombinant human FS. After 20 h of steroid-free FS pretreatment, fresh DMEM with 10% charcoal/dextran-treated FBS containing 100 nM DHT and/or 250 ng/ml FS or 10 ng/ml recombinant human activin-A was added, and the cells were incubated for 24 h. In the activin/DHT experiments in Fig. 8, the transfection and treatment procedure was the same as that of Figs. 6 and 7, except that DMEM supplemented with 10% charcoal/dextran-treated FBS without FS was used for the 20-h preincubation before treatment for 24 h with 100 nM DHT and/or 10 ng/ml activin A. In the GnRH/DHT experiments, steroid-free DMEM with 10% charcoal/dextran-treated FBS was used as preincubation media. After 20 h of steroid deprivation, the media were changed to serum-free DMEM supplemented with 0.1% BSA and 5 ng/ml transferrin containing DHT, and the cells were incubated for 24 h. Six hours before harvest, additional serum-free DMEM (with BSA/transferrin supplement) containing DHT and/or GnRH was added, and the cells were incubated for the remaining 6 h until harvest. The cells were then harvested, and lucif-erase and β-galactosidase assays were performed.
Construction of Plasmids for Transfection
A 5.5-kb region of the oFSHβ gene (oFSHβ) encompassing 4741 bp of the promoter and 759 bp downstream from the +1 transcription start site was subcloned into the pGL3-Basic luciferase promoter plasmid (Promega Life Science, Madison, WI) as described previously (22). The cloning of the −982 truncation was described previously (25). Cloning of the −750 truncation was performed by inserting a SacI to SalI fragment of oFSHβ into the SacI to XhoI sites of the pGL3-Basic vector. Cloning of the −561 truncation was performed by inserting a NdeI (filled in with Klenow fragment enzyme) to BglII fragment of oFSHβ into the SmaI to BglII sites of the pGL3-Basic vector. Cloning of the −105 truncation was performed by inserting a HindIII fragment of oFSHβ into the HindIII site of the pGL3-Basic vector. The −401 truncation was made by PCR amplification using a 5′-primer: 5′-CTCT GCTA GTTTT TCAA TCTA CC- 3′ and a 3′-primer: 5′-CTGC AGCA GATT GCTCTCC-3′, and the amplified fragment was cloned into the SmaI site of the pGL3-Basic vector. Mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. All plasmids were sequenced to confirm the fidelity of the sequence, the junctions, and the mutations.
Plasmid DNA was prepared from overnight bacterial cultures using DNA plasmid columns according to the manufacturer’s protocol (QIAGEN, Chatsworth, CA) or a cesium chloride protocol adapted from Sambrook et al. (61).
The MMTV-pGL3 basic luciferase promoter plasmid was kindly provided by Jeff Miner of Ligand Pharmaceuticals. The GRAS-luciferase reporter gene contains four repeats of the GRAS element (−340 to −315 from the mouse GnRH-R gene) upstream of a minimal −81 thymidine kinase (TK) promoter in pGL3. The cytomegalovirus (CMV)-β-galactosidase, TK-β-galactosidase, and Rous sarcoma virus (RSV)-β-galactosidase reporter plasmids were prepared as described previously (22, 25, 62, 63).
Cell Culture and Transient Transfection
LβT2 cells were maintained in 100-mm diameter dishes in DMEM (Cellgro, Mediatech, Inc., Herndon, VA) supplemented with 10% FBS (Omega Scientific, Inc., Tarzana, CA) at 37 C with 5% CO2. Charcoal-treated FBS was also obtained from Omega Scientific, Inc. When cells reached 70–80% confluency, they were trypsinized and 2 × 105 cells were plated per well into 24-well plates (Nunclon) in DMEM supplemented with 10% FBS. Transient transfections were performed using FuGENE 6 transfection reagent (Roche Molecular Biochemicals, Indianapolis, IN), following the manufacturer’s protocol. Unless otherwise noted in the figure legends, CMV-β-galactosidase was used as an internal transfection efficiency control for androgen experiments, RSV-β-galactosidase was used for an internal control in activin/androgen cotreatment experiments, and TK-β-galactosidase was used as an internal control for GnRH/androgen cotreatment experiments. None of the hormone treatments had significant effects on expression of the internal control plasmids (data not shown).
Luciferase and β-Galactosidase Assays
LβT2 cells were washed once with 1× PBS and then 40 βl of lysis solution (Galacto-light assay system, Tropix, Bedford, MA) was added to each well of the 24-well plates. Cells were then incubated at room temperature on a plate shaker for 5 min to detach and lyse cells. The contents of the wells were then transferred to microcentrifuge tubes on ice and centrifuged at 12,300 rpm for 8 min at 4 C. Lysed sample (20 μl) was assayed for luciferase activity, and 10 μl were aliquoted, incubated at 48 C for 1 h to heat inactivate endogenous β-galactosidase, and then assayed for β-galactosidase activity from the reporter gene. Luciferase and β-galactosidase activity were measured using an EG&G Berthold Microplate Luminometer (PerkinElmer Corp., Norwalk, CT) as described previously (22).
EMSA
Full-length, human AR containing a Flag epitope tag was overexpressed in Sf9 cells via a baculovirus expression system (64). The Sf9 cells were inoculated with virus at a multiplicity of infection of 1.0 and allowed to grow for an additional 48 h at 27 C. They were treated for the last 24 h before harvest with 1 mM DHT (final concentration). Cells were harvested by centrifugation at 1500 rpm for 15 min, washed once in TG buffer (10 mM Tris-HCl, pH 8.0; and 10% glycerol) and frozen as a pellet at −80 C. The Sf9 cells were lysed in a homogenization buffer (20 mM Tris-HCl, pH 7.5; 350 mM NaCl, 1 mM dithiothreitol, 10% glycerol, 0.5 μg/ml leupeptin, 10 μg/ml bacitracin, 2 μg/ml aprotinin, 1 μg/ml pepstatin). All procedures were done at 0–4 C. The cell lysate was centrifuged at 40,000 rpm for 30 min, and the supernatant was taken as a soluble whole-cell extract. The ability of AR to bind an oligonucleotide from the oFSHβ promoter was determined by EMSA. AR was incubated with 1 fmol of 32P-labeled oligonucleotide at 4 C in a DNA binding buffer (10 mM HEPES, pH 7.8; 50 mM KCl, 5 mM MgCl2, 0.1% Nonidet P-40, 1 mM dithiothreitol, 2 μg poly(dI-dC), and 10% glycerol) in the absence or presence of the C-19x polyclonal antibody to AR (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), rabbit IgG control, or 1000-fold excess of the −245, −245 mutant or ARE consensus oligonucleotides. The 245 oligo is 5′-CAAG-GTAAAGGAGTGGGTGTTCTACTATA-3ʹ; the 245 mutant is 5ʹ-CAAGGTAAAGGAGTGGGTCCTCTACTATA-3ʹ; and the ARE consensus is 5ʹ-ACGGGTGGAACGCGGTGTTCTTTT-GGC-3ʹ. The oligonucleotide was end-labeled with T4 DNA polymerase and [γ-32P]ATP. After 30 min, the DNA binding reactions were electrophoresed on a 5% polyacrylamide gel (40:1 acrylamide-bisacrylamide) containing 2.5% glycerol in a 0.25− TBE buffer (20 mM Trisborate, pH 8; 20 mM boric acid; 0.5 mM EDTA).
Acknowledgments
We thank Djurdjica Coss, Suzanne B. R. Jacobs, Mark A. Lawson, Flavia Pernasetti, and Vyacheslav Vasilyev for helpful discussions, providing plasmids, and/or reading the manuscript. We thank Shunichi Shimasaki for the gift of FS. We thank Laura Neely and Rachel White for technical assistance. We also thank Elizabeth Wilson for providing the Flag-tagged AR baculovirus and the University of Colorado Cancer Center Tissue Culture Core facility for baculovirus production.
This work was supported by National Institute of Child Health and Human Development/National Institutes of Health through cooperative agreement (U54 HD12303) as part of the Specialized Cooperative Centers Program in Reproduction Research (P.L.M.). This work was also supported by NIH Grant R37 HD20377 (to P.L.M.). T.J.S. was supported by NIH NRSA F32 HD08686 and NIH T32 DK07044; R.S. was supported by a Summer Medical Student Fellowship from NIH T32 HL07491; V.G.T. was supported by NIH T32 HD07203; L.E. was supported in part by a Howard Hughes Medical Institute Summer Fellowship; and J.S.B. was supported in part by NIH T32 GM08666.
Abbreviations
- AR
Androgen receptor
- ARE
androgen response element
- CMV
cytomegalovirus
- DHT
dihydrotestosterone
- FBS
fetal bovine serum
- FS
follistatin
- GnRH-R
GnRH receptor
- GRAS
GnRH receptor-activating sequence
- GRE
glucocorticoid response element
- α-GSU
α-subunit of the glycoprotein hormones
- HPG
hypothalamic-pituitarygonadal
- MMTV
mouse mammary tumor virus
- PRE
progesterone response element
- RSV
Rous sarcoma virus
- SBE
Smad binding element
- Smad
Sma- and Mad-related protein
- T
testosterone
- TK
thymidine kinase
References
- 1.Toranzo D, Dupont E, Simard J, Labrie C, Couet J, Labrie F, Pelletier G. Regulation of pro-gonadotropin-releasing hormone gene expression by sex steroids in the brain of male and female rats. Mol Endocrinol. 1989;3:1748–1756. doi: 10.1210/mend-3-11-1748. [DOI] [PubMed] [Google Scholar]
- 2.Selmanoff M, Shu C, Petersen SL, Barraclough CA, Zoeller RT. Single cell levels of hypothalamic messenger ribonucleic acid encoding luteinizing hormone-releasing hormone in intact, castrated, and hyperprolactinemic male rats. Endocrinology. 1991;128:459–466. doi: 10.1210/endo-128-1-459. [DOI] [PubMed] [Google Scholar]
- 3.Gross DS. Effect of castration and steroid replacement on immunoreactive gonadotropin-releasing hormone in hypothalamus and preoptic area. Endocrinology. 1980;106:1442–1450. doi: 10.1210/endo-106-5-1442. [DOI] [PubMed] [Google Scholar]
- 4.Kalra PS, Kalra SP. Modulation of hypothalamic luteinizing hormone-releasing hormone levels by intracranial and subcutaneous implants of gonadal steroids in castrated rats: effects of androgen and estrogen antagonists. Endocrinology. 1980;106:390–397. doi: 10.1210/endo-106-1-390. [DOI] [PubMed] [Google Scholar]
- 5.Roselli CE, Kelly MJ, Ronnekleiv OK. Testosterone regulates progonadotropin-releasing hormone levels in the preoptic area and basal hypothalamus of the male rat. Endocrinology. 1990;126:1080–1086. doi: 10.1210/endo-126-2-1080. [DOI] [PubMed] [Google Scholar]
- 6.Wierman ME, Gharib SD, LaRovere JM, Badger TM, Chin WW. Selective failure of androgens to regulate follicle stimulating hormone β messenger ribonucleic acid levels in the male rat. Mol Endocrinol. 1988;2:492–498. doi: 10.1210/mend-2-6-492. [DOI] [PubMed] [Google Scholar]
- 7.Winters SJ, Ishizaka K, Kitahara S, Troen P, Attardi B. Effects of testosterone on gonadotropin subunit messenger ribonucleic acids in the presence or absence of gonadotropin-releasing hormone. Endocrinology. 1992;130:726–734. doi: 10.1210/endo.130.2.1370794. [DOI] [PubMed] [Google Scholar]
- 8.Leal AM, Blount AL, Donaldson CJ, Bilezikjian LM, Vale WW. Regulation of follicle-stimulating hormone secretion by the interactions of activin-A, dexamethasone and testosterone in anterior pituitary cell cultures of male rats. Neuroendocrinology. 2003;77:298–304. doi: 10.1159/000070896. [DOI] [PubMed] [Google Scholar]
- 9.Curtin D, Jenkins S, Farmer N, Anderson AC, Haisenleder DJ, Rissman E, Wilson EM, Shupnik MA. Androgen suppression of GnRH-stimulated rat LHβ gene transcription occurs through Sp1 sites in the distal GnRH-responsive promoter region. Mol Endocrinol. 2001;15:1906–1917. doi: 10.1210/mend.15.11.0723. [DOI] [PubMed] [Google Scholar]
- 10.Jorgensen JS, Nilson JH. AR suppresses transcription of the LHβ subunit by interacting with steroidogenic factor-1. Mol Endocrinol. 2001;15:1505–1516. doi: 10.1210/mend.15.9.0691. [DOI] [PubMed] [Google Scholar]
- 11.Paul SJ, Ortolano GA, Haisenleder DJ, Stewart JM, Shupnik MA, Marshall JC. Gonadotropin subunit messenger RNA concentrations after blockade of gona-dotropin-releasing hormone action: testosterone selectively increases follicle-stimulating hormone β-subunit messenger RNA by posttranscriptional mechanisms. Mol Endocrinol. 1990;4:1943–1955. doi: 10.1210/mend-4-12-1943. [DOI] [PubMed] [Google Scholar]
- 12.Dalkin AC, Paul SJ, Haisenleder DJ, Ortolano GA, Yasin M, Marshall JC. Gonadal steroids effect similar regulation of gonadotrophin subunit mRNA expression in both male and female rats. J Endocrinol. 1992;132:39–45. doi: 10.1677/joe.0.1320039. [DOI] [PubMed] [Google Scholar]
- 13.Gharib SD, Leung PC, Carroll RS, Chin WW. Androgens positively regulate follicle-stimulating hormone β-subunit mRNA levels in rat pituitary cells. Mol Endocrinol. 1990;4:1620–1626. doi: 10.1210/mend-4-11-1620. [DOI] [PubMed] [Google Scholar]
- 14.Kaiser UB, Chin WW. Regulation of follistatin messenger ribonucleic acid levels in the rat pituitary. J Clin Invest. 1993;91:2523–2531. doi: 10.1172/JCI116488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bilezikjian LM, Corrigan AZ, Blount AL, Vale WW. Pituitary follistatin and inhibin subunit messenger ribonucleic acid levels are differentially regulated by local and hormonal factors. Endocrinology. 1996;137:4277–4284. doi: 10.1210/endo.137.10.8828487. [DOI] [PubMed] [Google Scholar]
- 16.Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Regulation of rat pituitary gonadotropin-releasing hormone receptor mRNA levels in vivo and in vitro. Endocrinology. 1993;133:931–934. doi: 10.1210/endo.133.2.8393779. [DOI] [PubMed] [Google Scholar]
- 17.Marchetti B, Reeves JJ, Pelletier G, Labrie F. Modulation of pituitary luteinizing hormone releasing hormone receptors by sex steroids and luteinizing hormone releasing hormone in the rat. Biol Reprod. 1982;27:133–145. doi: 10.1095/biolreprod27.1.133. [DOI] [PubMed] [Google Scholar]
- 18.Jegou B, Brekke I, Naess O, Torjesen P, Hansson V. Properties and regulation of GnRH receptors in the anterior pituitary and the testis of the rat: different response of Leydig cell LH and GnRH receptors to hormonal treatments. Arch Androl. 1985;14:161–170. doi: 10.3109/01485018508988293. [DOI] [PubMed] [Google Scholar]
- 19.Conne BS, Scaglioni S, Lang U, Sizonenko PC, Aubert ML. Pituitary receptor sites for gonadotropin-releasing hormone: effect of castration and substitutive therapy with sex steroids in the male rat. Endocrinology. 1982;110:70–79. doi: 10.1210/endo-110-1-70. [DOI] [PubMed] [Google Scholar]
- 20.Duncan JA, Dalkin AC, Barkan A, Regiani S, Marshall JC. Gonadal regulation of pituitary gonadotropin-releasing hormone receptors during sexual maturation in the rat. Endocrinology. 1983;113:2238–2246. doi: 10.1210/endo-113-6-2238. [DOI] [PubMed] [Google Scholar]
- 21.Naik SI, Young LS, Charlton HM, Clayton RN. Pituitary gonadotropin-releasing hormone receptor regulation in mice. I. Males. Endocrinology. 1984;115:106–113. doi: 10.1210/endo-115-1-106. [DOI] [PubMed] [Google Scholar]
- 22.Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang HJ, Miller WL, Mellon PL. Cell-specific transcriptional regulation of FSHβ by activin and GnRH in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- 23.Lawson MA, Li D, Glidewell-Kenney CA, Lopez FJ. Androgen responsiveness of the pituitary gonadotrope cell line LβT2. J Endocrinol. 2001;170:601–607. doi: 10.1677/joe.0.1700601. [DOI] [PubMed] [Google Scholar]
- 24.Huang HJ, Sebastian J, Strahl BD, Wu JC, Miller WL. The promoter for the ovine follicle-stimulating hormone-β gene (FSHβ) confers FSHβ-like expression on luciferase in transgenic mice: regulatory studies in vivo and in vitro. Endocrinology. 2001;142:2260–2266. doi: 10.1210/endo.142.6.8202. [DOI] [PubMed] [Google Scholar]
- 25.Vasilyev VV, Pernasetti F, Rosenberg SB, Barsoum MJ, Austin DA, Webster NJ, Mellon PL. Transcriptional activation of the ovine follicle-stimulating hormone-β gene by gonadotropin-releasing hormone involves multiple signal transduction pathways. Endocrinology. 2002;143:1651–1659. doi: 10.1210/endo.143.5.8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ham J, Thomson A, Needham M, Webb P, Parker M. Characterization of response elements for androgens, glucocorticoids and progestins in mouse mammary tumour virus. Nucleic Acids Res. 1988;16:5263–5276. doi: 10.1093/nar/16.12.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darbre P, Page M, King RJ. Androgen regulation by the long terminal repeat of mouse mammary tumor virus. Mol Cell Biol. 1986;6:2847–2854. doi: 10.1128/mcb.6.8.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang C, Saltzman A, Yeh S, Young W, Keller E, Lee HJ, Wang C, Mizokami A. Androgen receptor: an overview. Crit Rev Eukaryot Gene Expr. 1995;5:97–125. doi: 10.1615/critreveukargeneexpr.v5.i2.10. [DOI] [PubMed] [Google Scholar]
- 29.Nelson CC, Hendy SC, Shukin RJ, Cheng H, Bruchovsky N, Koop BF, Rennie PS. Determinants of DNA sequence specificity of the androgen, progesterone, and glucocorticoid receptors: evidence for differential steroid receptor response elements. Mol Endocrinol. 1999;13:2090–2107. doi: 10.1210/mend.13.12.0396. [DOI] [PubMed] [Google Scholar]
- 30.Webster JC, Pedersen NR, Edwards DP, Beck CA, Miller WL. The 5′-flanking region of the ovine follicle-stimulating hormone-β gene contains six progesterone response elements: three proximal elements are sufficient to increase transcription in the presence of progesterone. Endocrinology. 1995;136:1049–1058. doi: 10.1210/endo.136.3.7867558. [DOI] [PubMed] [Google Scholar]
- 31.Bohnsack BL, Szabo M, Kilen SM, Tam DH, Schwartz NB. Follistatin suppresses steroid-enhanced follicle-stimulating hormone release in vitro in rats. Biol Reprod. 2000;62:636–641. doi: 10.1095/biolreprod62.3.636. [DOI] [PubMed] [Google Scholar]
- 32.Miyake T, Irahara M, Shitukawa K, Yasui T, Aono T. Interaction of activin A and gonadal steroids on FSH secretion from primary cultured rat anterior pituitary cells. Biochem Biophys Res Commun. 1993;194:413–419. doi: 10.1006/bbrc.1993.1835. [DOI] [PubMed] [Google Scholar]
- 33.Slinker BK. The statistics of synergism. J Mol Cell Cardiol. 1998;30:723–731. doi: 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- 34.Duval DL, Ellsworth BS, Clay CM. Is gonadotrope expression of the gonadotropin releasing hormone receptor gene mediated by autocrine/paracrine stimulation of an activin response element? Endocrinology. 1999;140:1949–1952. doi: 10.1210/endo.140.4.6780. [DOI] [PubMed] [Google Scholar]
- 35.Duval DL, Nelson SE, Clay CM. The tripartite basal enhancer of the gonadotropin-releasing hormone (GnRH) receptor gene promoter regulates cell-specific expression through a novel GnRH receptor activating sequence. Mol Endocrinol. 1997;11:1814–1821. doi: 10.1210/mend.11.12.0020. [DOI] [PubMed] [Google Scholar]
- 35a.Bailey JS, Rave-Harel N, McGillivray SM, Coss D, Mellon PL. Activin regulation of the follicle-stimulating hormone. β-subunit gene involves Smads and the TALE homeodomain proteins Pbx1 and Prep1. doi: 10.1210/me.2003-0442. Mol Endocrinol first published February 5, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobs SBR, Coss D, McGillivray SM, Mellon PL. Nuclear factor-Y and steroidogenic factor-1 physically and functionally interact to contribute to cell-specific expression of the mouse follicle-stimulating hormone-β gene. Mol Endocrinol. 2003;17:1470–1483. doi: 10.1210/me.2002-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham KE, Nusser KD, Low MJ. LβT2 gonadotroph cells secrete follicle stimulating hormone (FSH) in response to activin A. J Endocrinol. 1999;162:R1–R5. doi: 10.1677/joe.0.162r001. [DOI] [PubMed] [Google Scholar]
- 38.Schreihofer DA, Stoler MH, Shupnik MA. Differential expression and regulation of estrogen receptors (ERs) in rat pituitary and cell lines: estrogen decreases ERβ protein and estrogen responsiveness. Endocrinology. 2000;141:2174–2184. doi: 10.1210/endo.141.6.7505. [DOI] [PubMed] [Google Scholar]
- 39.Khurshid S, Weinbauer GF, Nieschlag E. Effect of administration of testosterone and gonadotrophin-releasing hormone (GnRH) antagonist on basal and GnRH-stimulated gonadotrophin secretion in orchidectomized monkeys. J Endocrinol. 1991;129:363–370. doi: 10.1677/joe.0.1290363. [DOI] [PubMed] [Google Scholar]
- 40.Kumar TR, Low MJ. Hormonal regulation of human follicle-stimulating hormone-β subunit gene expression: GnRH stimulation and GnRH-independent androgen inhibition. Neuroendocrinology. 1995;61:628–637. doi: 10.1159/000126889. [DOI] [PubMed] [Google Scholar]
- 41.Burger LL, Haisenleder DJ, Aylor KW, Dalkin AC, Pren-dergast KA, Marshall JC. Regulation of LHβ and FSHβ gene transcription by androgens: testosterone directly stimulates FSHβ transcription independent from its role on follistatin gene expression. Endocrinology. 2003;145:71–78. doi: 10.1210/en.2003-1047. [DOI] [PubMed] [Google Scholar]
- 42.Ibrahim SN, Moussa SM, Childs GV. Morphometric studies of rat anterior pituitary cells after gonadectomy: correlation of changes in gonadotropes with the serum levels of gonadotropins. Endocrinology. 1986;119:629–637. doi: 10.1210/endo-119-2-629. [DOI] [PubMed] [Google Scholar]
- 43.Stefaneanu L. Pituitary sex steroid receptors: localization and function. Endocrinol Pathol. 1997;8:91–108. doi: 10.1007/BF02739938. [DOI] [PubMed] [Google Scholar]
- 44.Kimura N, Mizokami A, Oonuma T, Sasano H, Nagura H. Immunocytochemical localization of androgen receptor with polyclonal antibody in paraffin-embedded human tissues. J Histochem Cytochem. 1993;41:671–678. doi: 10.1177/41.5.8468448. [DOI] [PubMed] [Google Scholar]
- 45.Pelletier G, Labrie C, Labrie F. Localization of oestrogen receptor β, oestrogen receptor β and androgen receptors in the rat reproductive organs. J Endocrinol. 2000;165:359–370. doi: 10.1677/joe.0.1650359. [DOI] [PubMed] [Google Scholar]
- 46.Renner U, Pagotto U, Arzt E, Stalla GK. Autocrine and paracrine roles of polypeptide growth factors, cytokines and vasogenic substances in normal and tumorous pituitary function and growth: a review. Eur J Endocrinol. 1996;135:515–532. doi: 10.1530/eje.0.1350515. [DOI] [PubMed] [Google Scholar]
- 47.Lerchl A, Nieschlag E. Diurnal variations of serum and testicular testosterone and dihydrotestosterone (DHT) in Djungarian hamsters (Phodopus sungorus): testes are the main source for circulating DHT. Gen Comp Endocrinol. 1995;98:129–136. doi: 10.1006/gcen.1995.1053. [DOI] [PubMed] [Google Scholar]
- 48.Dohler KD, Wuttke W. Circadian fluctuations of serum hormone levels in prepubertal male and female rats. Acta Endocrinol (Copenh) 1976;83:269–279. doi: 10.1530/acta.0.0830269. [DOI] [PubMed] [Google Scholar]
- 49.Chipuk JE, Cornelius SC, Pultz NJ, Jorgensen JS, Bonham MJ, Kim SJ, Danielpour D. The androgen receptor represses transforming growth factor-β signaling through interaction with Smad3. J Biol Chem. 2002;277:1240–1248. doi: 10.1074/jbc.M108855200. [DOI] [PubMed] [Google Scholar]
- 50.Hayes SA, Zarnegar M, Sharma M, Yang F, Peehl DM, ten Dijke P, Sun Z. SMAD3 represses androgen receptor-mediated transcription. Cancer Res. 2001;61:2112–2118. [PubMed] [Google Scholar]
- 51.Kang HY, Lin HK, Hu YC, Yeh S, Huang KE, Chang C. From transforming growth factor-β signaling to androgen action: identification of Smad3 as an androgen receptor coregulator in prostate cancer cells. Proc Natl Acad Sci USA. 2001;98:3018–3023. doi: 10.1073/pnas.061305498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mather JP, Moore A, Li R-H. Activin, inhibin, and follistatin: further thoughts on a growing family of regulators. Proc Soc Exp Biol Med. 1997;215:209–222. doi: 10.3181/00379727-215-44130. [DOI] [PubMed] [Google Scholar]
- 53.Naor Z, Clayton RN, Catt KJ. Characterization of gonadotropin-releasing hormone receptors in cultured rat pituitary cells. Endocrinology. 1980;107:1144–1152. doi: 10.1210/endo-107-4-1144. [DOI] [PubMed] [Google Scholar]
- 54.Young LS, Naik SI, Clayton RN. Increased gonadotrophin releasing hormone receptors on pituitary gonadotrophs: effect on subsequent LH secretion. Mol Cell Endocrinol. 1985;41:69–78. doi: 10.1016/0303-7207(85)90143-1. [DOI] [PubMed] [Google Scholar]
- 55.Bedecarrats GY, Kaiser UB. Differential regulation of gonadotropin subunit gene promoter activity by pulsatile gonadotropin-releasing hormone (GnRH) in perifused L β T2 cells: role of GnRH receptor concentration. Endocrinology. 2003;144:1802–1811. doi: 10.1210/en.2002-221140. [DOI] [PubMed] [Google Scholar]
- 56.Dalkin AC, Haisenleder DJ, Gilrain JT, Aylor K, Yasin M, Marshall JC. Regulation of pituitary follistatin and inhibin/activin subunit messenger ribonucleic acids (mRNAs) in male and female rats: evidence for inhibin regulation of follistatin mRNA in females. Endocrinology. 1998;139:2818–2823. doi: 10.1210/endo.139.6.6057. [DOI] [PubMed] [Google Scholar]
- 57.Bauer-Dantoin AC, Weiss J, Jameson JL. Gonadotropin-releasing hormone regulation of pituitary follistatin gene expression during the primary follicle-stimulating hormone surge. Endocrinology. 1996;137:1634–1639. doi: 10.1210/endo.137.5.8612495. [DOI] [PubMed] [Google Scholar]
- 58.Moeller H, Goecke B, Herter F. Seasonal and diurnal changes of prostatic androgen receptor and circulating testosterone in young mature rats. Res Exp Med (Berl) 1988;188:451–462. doi: 10.1007/BF01852003. [DOI] [PubMed] [Google Scholar]
- 59.Miller AE, Riegle GD. Temporal patterns of serum luteinizing hormone and testosterone and endocrine response to luteinizing hormone releasing hormone in aging male rats. J Gerontol. 1982;37:522–528. doi: 10.1093/geronj/37.5.522. [DOI] [PubMed] [Google Scholar]
- 60.Bronson F, Heideman P. Seasonal regulation of reproduction in mammals. In: Knobil E, Neil JD, editors. The physiology of reproduction. New York: Raven Press Ltd; 1994. pp. 570–572. [Google Scholar]
- 61.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 62.Vasilyev VV, Lawson MA, DiPaolo D, Webster NJG, Mellon PL. Different signaling pathways control acute induction versus long-term repression of LHβ transcription by GnRH. Endocrinology. 2002;143:3414–3426. doi: 10.1210/en.2001-211215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenberg SB, Mellon PL. An Otx-related homeo-domain protein binds an LHβ promoter element important for activation during gonadotrope maturation. Mol Endocrinol. 2002;16:1280–1298. doi: 10.1210/mend.16.6.0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao M, Zhou Z, Wilson EM. Redox-dependent DNA binding of the purified androgen receptor: evidence for disulfide-linked androgen receptor dimers. Biochemistry. 1999;38:9718–9727. doi: 10.1021/bi990589i. [DOI] [PubMed] [Google Scholar]