

Abstract
Gonadotropin-releasing hormone (GnRH) is the central regulator of the hypothalamic-pituitary-gonadal axis, controlling sexual maturation and fertility in diverse species from fish to humans. GnRH gene expression is limited to a discrete population of neurons that migrate through the nasal region into the hypothalamus during embryonic development. The GnRH regulatory region contains four conserved homeodomain binding sites (ATTA) that are essential for basal promoter activity and cell-specific expression of the GnRH gene. MSX and DLX are members of the Antennapedia class of non-Hox homeodomain transcription factors that regulate gene expression and influence development of the craniofacial structures and anterior forebrain. Here, we report that expression patterns of the Msx and Dlx families of homeodomain transcription factors largely coincide with the migratory route of GnRH neurons and co-express with GnRH in neurons during embryonic development. In addition, MSX and DLX family members bind directly to the ATTA consensus sequences and regulate transcriptional activity of the GnRH promoter. Finally, mice lacking MSX1 or DLX1 and 2 show altered numbers of GnRH-expressing cells in regions where these factors likely function. These findings strongly support a role for MSX and DLX in contributing to spatiotemporal regulation of GnRH transcription during development.
Proper sexual maturation and fertility are dependent upon the correct function of the hypothalamic-pituitary-gonadal axis, initiated by a small, yet critical population of gonadotropin-releasing hormone (GnRH)1 neurons. The GnRH gene is expressed in a complex spatiotemporal manner during embryonic development and into postnatal life with several populations of GnRH-expressing neurons originating at different developmental stages and locations. These populations include the classical, septohypothalamic neurons, as well as populations in the lateral septum, posterior bed nucleus stria terminalis (pBNST), and tectum (1, 2). Although the role of each of these populations of GnRH-producing neurons remains to be elucidated, the contribution of the septohypothalamic population is required for maturation of the hypothalamic-pituitary-gonadal axis and fertility (3).
The precursor cells of the septohypothalamic GnRH neurons have been reported to originate within the olfactory placode (4–6) or the neural crest (7) and begin to express the GnRH transcript in a discrete population of cells located in close proximity to the olfactory placode of the embryonic mouse by 11.5-days postcoitum (11.5 dpc). By 12.5 dpc, the full complement of septohypothalamic GnRH neurons (~800) in the adult population is established (6), and over the course of the next several days, these neurons migrate toward the CNS, closely associated with the established position of the peripherin-positive nerve bundle of the olfactory nerve (8, 9). By 16.5 dpc the majority of the septohypothalamic neurons have reached their destination, scattered throughout the preoptic area, the diagonal band of Broca and the medial septum (5, 10). Expression of GnRH during prenatal development actively contributes to the identity and migratory ability of the septohypothalamic population. Indeed, GnRH itself, is involved in furthering the commitment and migratory state of the GnRH neuronal precursor in an ultrashort, positive feedback loop by upregulating expression of the GnRH gene, stimulating axon growth, cytoskeletal remodeling and increasing the migration of neuronal precursors (11). Thus, expression of GnRH during embryonic development likely promotes the characteristics of the maturing GnRH neuron that are essential for reproductive ability.
Because of the limited number and dispersed nature of GnRH neurons, the difficulty in examining GnRH promoter expression in vivo is evident. The generation of immortalized GnRH neuronal cell lines, the GT1-7, GN11, and NLT cells, has provided cellular model systems in which to study GnRH (12, 13). Whereas the murine hypothalamic cell line, GT1-7, exhibits key characteristics of a mature, fully differentiated GnRH-expressing neuron, the GN11 and NLT cell lines are migratory in culture and suggested to represent immature GnRH neurons (12–15). Studies with these two models have been invaluable for identifying the protein transcription factor complexes that facilitate expression of GnRH.
Both in vitro and in vivo studies have demonstrated that well defined promoter and enhancer regions of the GnRH gene, containing ATTA homeodomain-binding sites, are both necessary and sufficient for proper GnRH gene expression during embryonic development and in the adult (12, 16). Moreover, these homeodomain-binding sites are essential for appropriate basal, as well as cell-specific expression of the GnRH promoter (17, 18). A role for homeoproteins, such as MSX and DLX, in regulation of gene expression during development is well documented in vertebrate and invertebrate species (19–23). Interestingly, the MSX and DLX homeodomain transcription factor families have the appropriate expression patterns in the developing olfactory region and fore-brain of the mouse to be considered as potential regulators of the GnRH gene (24, 25). These factors can also occupy homeodomain binding sites in a variety of upstream promoter regions (26–28). Here, we report the contribution of DLX and MSX family members to regulation of GnRH promoter expression in vivo and in vitro. We show that spatiotemporal expression of Msx and Dlx family members coincides with the migratory, septohypothalamic GnRH neuronal population. MSX and DLX family members are also expressed in the GnRH model cell lines, bind to upstream regulatory regions of the GnRH gene, and regulate promoter activity in vitro. In addition, analyses of mouse mutants revealed that DLX1 and -2 as well as MSX1 contribute to proper GnRH expression in the prenatal septohypothalamic areas. Therefore, we propose that MSX and DLX family members participate in developmental regulation of GnRH gene expression.
MATERIALS AND METHODS
Nuclear Extract Preparation and Electromobility Shift Assays (EMSA)
Nuclear extracts for EMSA were prepared according to Clark and Mellon (29). Oligonucleotide probes were annealed and labeled with [γ-32P]ATP (6000 Ci/mmol; PerkinElmer Life Science Products) in a polynucleotide kinase reaction. Radiolabeled probes were then cleaned over MicroSpin G-25 columns (Amersham Biosciences). 1 fmol of probe was incubated with 2 μg from GT1-7 cells or 6 μg from NLT or GN11 cells of nuclear protein extract in binding buffer (50 mM KCl, 10 mM HEPES (pH 7.8), 1 mM EDTA, 5 mM spermidine, 2% Ficoll, 1.25 mM dithiothreitol, 6% glycerol, and 4 mM phenylmethylsulfonyl fluoride) for 5 min before loading on a 5% polyacrylamide nondenaturing gel. Gels were run at 250 V for 2 h, then dried under a vacuum and exposed to film for 1–3 days. For supershift assays, 1 or 2 μl of anti-DLX1 (Chemicon AB5724), DLX2 (Chemicon AB5726), DLX5 (Chemicon AB5728), MSX1 (Covance MMS-261R), or MSX2 (Santa Cruz SC-15396) antibody was incubated in the reaction mix for 5 min before loading.
Cell Culture and Transfection
GT1-7, GN11, and NLT cells were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, penicillin (100 units/ml), streptomycin (0.1 mg/ml), and 4.5% glucose in a 5% CO2 atmosphere at 37 °C. Cells were split into 24-well plates for transfection. 400 ng of pGL3-GnRHe/p reporter or pGL3-mutant GnRHe/p reporter were co-transfected with 100 or 200 ng of pCB6+-Msx1, pCMV-Msx2, pCDNA3-Dlx5, pCAGGS-Dlx1, or pCB6+-Dlx2 expression vectors in addition to 200 ng of thymidine kinase-β-galactosidase reporter vector as an internal control using FuGENE 6 reagent (Roche Applied Science). The mutant GnRHe/p reporter contained a linker-scanner mutation deleting the two homeodomain enhancer sites (−1636 to −1631 and −1623 to −1618) (17) and two point mutations in each of the promoter sites, −55 to −50 and −42 to −38 (CAATTA to CAgTTg and CATTA to CgTTg). Transfections were harvested 24 h later in lysis buffer (100 mM potassium phosphate, pH 7.8; 0.2% Triton X-100) to yield cellular proteins. β-Galactosidase assays were performed as advised by the manufacturer (Tropix, Bedford, MA), and luciferase assays were performed as previously described (17). To control for transfection efficiency, luciferase values were normalized to β-galactosidase activity. Values of the normalized expression vectors were compared with empty vector control or an RSVe/RSVp control. All experiments were performed in triplicate and repeated a minimum of three times.
In Situ Hybridization and Immunohistochemistry
Mouse colonies were maintained in agreement with protocols approved by the Institutional Animal Care and Use Committee at the University of California, San Diego. The Dlx1&2-null, or Msx1-null embryos were produced by heterozygous intercrosses of Dlx1&2 (30) or Msx1-nLacZ (31), and maintained in a C57BL/6J background. Fertilization was assumed to occur at midnight, and pregnant females were sacrificed at 12–16 dpc accordingly to harvest embryos (noon on the day after finding the plug is considered 0.5 dpc). Genotypes were confirmed by PCR as previously described (30, 31).
Mouse embryos were fixed (10% acetic acid, 30% formaldehyde, 60% ethanol) overnight at 4 °C and then dehydrated in a series of ethanol/water washes before embedding in paraffin. Sections were cut ~7-μm thick and then placed onto Superfrost Plus Slides (Fisher) and left at room temperature to dry overnight. Sections were then deparaffinized with xylene washes, hydrated in ethanol/water solutions and digested with 1 μg/ml proteinase K for 7 min at 37 °C, followed by postfixation in 10% neutral buffered formalin for 20 min at room temperature. The sections were then washed with 1× phosphate-buffered saline and 2× SSC for 5 min.
Antisense probes corresponded to sequences as previously reported for Dlx2 (25), Msx1 and Msx2 (32), Dlx1 and Dlx5 (33, 34). The GnRH antisense sequence corresponds to 325 bp of the cDNA from +1 to the endogenous BamHI site. Probes corresponding to the sense sequence were used as controls. Briefly, 1 μg of linearized plasmid DNA was incubated with 10× digoxigenin (DIG) RNA labeling mix (Roche Applied Science) as well as 5× transcription buffer (Promega) and an RNA polymerase T7, T3, or SP6 for 2 h at 37 °C. The reaction was stopped by adding 0.2 M EDTA, pH 8.0, and probes were then cleaned over G-50 MicroSpin columns (Amersham Biosciences).
The DIG-labeled probes, diluted in hybridization buffer (1× NaCl, 50% formamide, 10% dextran sulfate, 1 mg/ml yeast RNA, 1× Denhardt’s solution) were added to slides and incubated overnight at 65 °C. Sections were washed three times at 65 °C in 50% formamide, 1× SSC, with 0.1% Tween-20 and then twice at room temperature in MABT (100 mM maleic acid, pH 7.5, 150 mM NaCl, 0.1% Tween-20). The slides were blocked with MABT plus 5% normal goat serum for 3–4 h. The hybridized DIG-labeled probe was detected using alkaline phosphatase-conjugated anti-DIG antibody (Roche Applied Science) at a dilution of 1:2000 and visualized with the chromogen combination 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (BCIP/NBT). The single probe in situ hybridization sections were counterstained with nuclear fast red (Vector).
Immunohistochemical analysis was performed after in situ hybridization. These slides were soaked in immunogen stabilizing buffer (1 mM citric acid, 8 mM sodium citrate) for 20 min at 65 °C to retrieve the antigen. Immunohistochemistry was then carried out as previously described (16). The GnRH peptide was recognized with the LR-1 antibody at a dilution of 1:2000 (gift of R. Benoit) and visualized using the horseradish peroxidase-conjugated ABC kit (Vector).
Counting of the GnRH-positive neurons was carried out by sectioning whole embryo heads (starting from the inner ear upon appearance of the cochlear partition) in sagittal orientation at ~10-μm thickness and performing in situ hybridization on all sections as described above to identify GnRH-expressing cells. All sections were tallied to obtain the number of GnRH-positive cells. Approximately 150 sections were analyzed per embryo head for the earlier developmental stages, and 250 sections were analyzed per embryo head for the later developmental stage. Three or more embryos were examined at each time point with the exception of the Dlx1&2 embryos analyzed at 16.5 dpc, which represent two whole embryo heads.
Reverse Transcriptase PCR
Total RNA extraction was carried out as described by the method of Chomczynski and Sacchi (35). The reverse transcriptase reaction was performed according to the methods of Pernasetti et al. (36). PCR analysis was done with 30 cycles of: 45 s at 95 °C, 45 s at the proper annealing temperature for the primers, and 1 min at 72 °C, followed by an elongation step for 10 min at 72 °C. Specific primers for Msx1-3, Dlx1-6, and β-actin were used in the PCR reaction. PCR products were separated by electrophoresis on a 2% agarose gel.
RESULTS
Msx and Dlx Are Candidates for Regulating GnRH during Embryonic Development
Although ongoing research has examined the human and mouse GnRH gene promoters, the rat gene has been investigated most extensively. The two well characterized regulatory regions of the rat GnRH gene are a 300-bp enhancer located at −1863 to −1571 relative to the start site and an evolutionarily conserved 173-bp promoter just 5′ of the transcriptional start site. Within these regulatory regions, four conserved, consensus homeodomain sites (ATTA) have been identified (Fig. 1, panel A) (17, 18). These sites were extensively characterized and shown to be essential for appropriate basal and cell-specific expression of the GnRH promoter (17, 18). Thus, identifying the transcriptional regulators that occupy and function through these homeodomain sites is important for understanding the regulation of GnRH transcriptional activity.
Fig. 1. Conserved homeodomain binding sites in the GnRH upstream sequence and expression of candidate homeodomain regulators.

Panel A, four consensus Q50 homeodomain (CAATTA) sites in the rat GnRH gene are located at −1636/−1631, −1623/−1618, −55/−50, and an additional CATTA site in the GnRH promoter at −42/−38 (underlined in the rat sequence). The homologous human and mouse GnRH sequences are shown with lowercase letters indicating mismatches from the rat sequence. B, expression of Msx and Dlx in relation to the migratory GnRH neuron in vivo. In situ hybridization was carried out on parasagittal sections from mouse embryos at 13.5 dpc and probed in series. Sense probes served as controls. Three separate embryos were used for the sections in series, grouped horizontally. Adjacent sections were hybridized using probes for GnRH (panel B), Dlx1 (panel C), and Dlx5 (panel D), or GnRH (panel E), Dlx2 (panel F), and Msx1 (panel G), or GnRH (panel H), and Msx2 (panel I). Abbreviations: olfactory epithelium (OE), cribriform plate (CP), olfactory bulb (OB), primordium of the septum (PS), olfactory placode (OP). Black scale bars represent 100 μm in the lower left of each panel.
Several candidate homeoproteins were considered for their binding properties, developmental expression patterns, and transcriptional activity, among them the MSX and DLX families of transcriptional regulators. Given the previously described similarities in expression patterns of these transcription factors and the septohypothalamic population of GnRH neurons (24, 25), we sought to compare the expression patterns of Dlx1, Dlx2, Dlx5, Msx1, and Msx2 to that of GnRH in the embryonic forebrain and nasal region during development. Adjacent series of parasagittal sections were taken from mouse embryos at age 13.5 dpc, when the septohypothalamic population of GnRH neurons has been established. By RNA in situ hybridization, we observed a trail of GnRH-expressing neurons localizing from the olfactory placode, along the olfactory nerve and crossing the cribriform plate into the anterior forebrain, caudal to the olfactory bulb, in a region termed the primordium of the septum (Fig. 1, panels B, E, and H). Adjacent sections show strong expression of Msx1 and 2 (panels G and I, respectively) as well as Dlx1, 2, and 5 (panels C, D, and F, respectively) in the nasal region and anterior forebrain, consistent with their previously reported expression patterns. These results indicate that the spatial expression of these Msx and Dlx family members correlates with the developing septohypothalamic GnRH neuronal population.
We then examined the expression of Msx and Dlx family members in the model GnRH neuronal lines, GT1-7 and NLT/GN11. Reverse transcriptase PCR was performed on RNA from the GT1-7, GN11, NLT, and NIH3T3 cell lines, as well as total olfactory bulb RNA to assess the Msx and Dlx family members being expressed. Msx1, 2, and 3, as well as Dlx2, 4, 5, and 6, were expressed in the GT1-7 cells (Fig. 2A). Interestingly, NLT cells expressed Msx2 and 3 but not Msx1, and Dlx1 but not Dlx2 through 6. This same expression profile was seen in GN11 cells (data not shown). As expected, based on previous observations in vivo, Dlx family members were expressed in the olfactory bulb (37). In addition, the NIH3T3 cells served as a negative control for expression of Msx1 or Dlx2 (17). Thus, distinct members of the Dlx and Msx families are expressed in the different GnRH model cell lines.
Fig. 2. Expression of Msx and Dlx family members in model GnRH cell lines and co-expression with GnRH in vivo.

A, reverse transcriptase PCR analysis was used to identify the Msx and Dlx genes expressed in the GT1-7 and NLT cells. Olfactory bulb (OB) RNA was used as a positive control for the Dlx family members. NIH3T3 RNA is a mouse fibroblast control. β-Actin was used as a control for equal RNA loading. Ethidium-stained, 2% agarose gels are shown here. B–E, Msx and Dlx family members co-express with GnRH. Double in situ hybridization/immunohistochemistry was performed on parasagittal sections of 13.5-dpc embryos with an antibody recognizing GnRH (brown) and RNA probes (blue) for Msx1 (B), Msx2 (C), Dlx1 (D), and Dlx2 (E). The expanded panels to the right indicate higher magnification images of the boxed region to the left. Abbreviations: olfactory epithelium (OE), cribriform plate (CP), primordium of the septum (PS), olfactory placode (OP). Black scale bars represent 100 μm in the lower right of each panel.
Consistent with the RT-PCR studies in vitro, we found that both Msx and Dlx family members co-expressed with GnRH-positive neurons at embryonic stages. Using RNA probes for the Msx and Dlx family members and an antibody recognizing the GnRH peptide, we performed double in situ hybridization/immunohistochemistry on sagittal sections of 13.5-dpc embryos. Fig. 2B shows expression of Msx1 in the olfactory epithelium and along the olfactory sensory neuron tracts in the nasal region and GnRH-immunoreactive neurons expressing Msx1 in this region (Fig. 2B, expanded panel). Msx2 exhibited a similar expression pattern to that of Msx1 and neurons co-expressing Msx2 and GnRH along the olfactory sensory neuron tracts in the nasal region were observed (Fig. 2C, expanded panel). Fig. 2D indicates GnRH neurons located along the cribriform plate and in the forebrain primordium of the septum that expressed Dlx1 (expanded panel). Dlx2 exhibited a similar expression pattern to that of Dlx1 and also was co-expressed with GnRH-positive neurons (Fig. 2E, expanded panel). Like Dlx1 and 2, Dlx5 was expressed in GnRH-positive cells in the forebrain. However, we also observed GnRH neurons in the nasal region expressing Dlx5 (data not shown). These results show that Msx and Dlx are expressed in the GnRH-positive neurons during embryonic development and are plausible candidates for regulating GnRH promoter activity through the conserved homeodomain binding sites.
DLX and MSX Bind to Conserved ATTA Sequences in the GnRH Enhancer and Promoter
Previously, detailed analyses of the protein complexes binding to the ATTA homeodomain sites in the upstream region of the GnRH gene suggested that they are composed of Q50 homeodomain proteins (17, 18). MSX and DLX are members of the Antennapedia class of non-Hox homeodomain proteins that contain a glutamine residue at amino acid position 50 (Gln-50) of the homeodomain (38). Furthermore, MSX and DLX have been shown to mediate transcription by binding consensus ATTA sites in upstream regulatory regions (28). Thus, we hypothesized that one or more members of the MSX and DLX families may regulate GnRH transcription by binding to ATTA sequences within the promoter and enhancer.
To determine whether these proteins could directly bind to the ATTA sequences in the GnRH enhancer and promoter, we performed EMSA with antibody supershifts (Fig. 3). Radiolabeled oligonucleotides containing the homeodomain sites of the GnRH enhancer and promoter were incubated with nuclear extracts from GT1-7 cells. As described previously (17, 18), a specific complex was formed in GT1-7 nuclear extracts incubated with probes containing the ATTA sequences of the GnRH enhancer or promoter. Interestingly, a faint complex of faster mobility than the major complex also formed. This minor complex had been observed previously but not characterized (17, 18). The addition of an antibody specific to MSX1 inhibited protein binding of the major complex (thick arrow). In contrast, an antibody against DLX2 supershifted the minor complex (thin arrow) on the probe representing the 5′-enhancer site, −1642/−1623 (Fig. 3A, lanes 3 and 4) as well as the complex formed on the promoter element (data not shown). These results indicate that the complexes formed in GT1-7 cells on the ATTA sites within the GnRH enhancer and promoter contain DLX2 and MSX1.
Fig. 3. MSX and DLX family members bind to the homeodo-main sites.

A, EMSA analysis and antibody supershifts were performed using GT1-7 nuclear extract as well as antibodies recognizing MSX1, and DLX2 (lanes 3 and 4) and an IgG control (lane 5). The oligonucleotide probe corresponds to −1642/−1623 of the rat GnRH gene and contains the 5′ most homeodomain-binding site in the enhancer. The thick arrow marks the major complex and the thin arrow marks the minor complex. An arrowhead indicates the supershift of the minor complex. B, EMSA analysis and antibody supershifts were performed using the same oligonucleotide probe, NLT nuclear extract and an antibody recognizing DLX1 or MSX2 (lanes 2 and 3) or an IgG control (lane 4). The thick arrow represents the major complex and the thin arrow represents the minor complex. N.S. represents nonspecific binding, and F.P. indicates excess, free probe.
The EMSA binding pattern with the GT1-7 nuclear proteins on the DNA probe corresponding to the GnRH enhancer homeodomain binding site (−1642/−1623) is unique in comparison to many other patterns observed from non-GnRH producing cell lines (17, 18). Interestingly, the binding pattern of GN11 and NLT cell nuclear extracts is similar to the GT1-7 cell nuclear extracts (17, 18). Therefore, we explored the possibility that these similar complexes formed in GN11 and NLT cells might also contain DLX and MSX. Fig. 3B represents EMSA analysis of the homeodomain site in the GnRH enhancer incubated with NLT nuclear extracts. Five specific complexes formed on the GnRH enhancer probe with NLT extracts. Addition of an antibody recognizing DLX1 specifically inhibited binding of complex 4 (thin arrow) in NLT cells (Fig. 3B, lane 2), while an anti-MSX2 antibody inhibited binding of complex 3 (thick arrow, lane 3). EMSA performed with GN11 extracts showed the same binding pattern as that observed with the NLT extracts including results utilizing the anti-DLX1, or MSX2 antibodies (data not shown). Antibodies recognizing additional MSX and DLX family members did not supershift complexes formed on the −1642 probe (anti-MSX2 in the GT1-7 extract and anti-DLX5 in the GT1-7 and GN11/NLT extracts; data not shown). These results suggest that despite the molecular differences between the GT1-7 and GN11/NLT cells, the complexes bound to the homeodomain sites in the GnRH gene contain MSX and DLX family members in both model GnRH neuron cell lines.
DLX and MSX Regulate GnRH Gene Expression
Transcriptional assays have revealed that the MSX and DLX proteins interact with each other and recognize the same DNA consensus sequence (28). In addition, members of the MSX family have been characterized as transcriptional repressors, whereas the DLX family activates transcription (28, 34, 39–42). To characterize the effect of these transcription factors on the expression of the GnRH gene, we expressed MSX or DLX family members in the GT1-7 cells by transient co-transfections. The GnRH reporter plasmid utilized consists of the well characterized regulatory regions of the GnRH gene, the evolutionarily conserved enhancer and promoter (GnRHe/p), driving the luciferase gene. These DNA sequences are sufficient to target appropriate expression to GnRH neurons in vivo (16). MSX1 and MSX2 repressed GnRHe/p reporter activity to 47 and 36%, respectively, as compared with an empty vector control (Fig. 4). In contrast, expression of DLX2 or DLX5 in the GT1-7 cells increased activity of the GnRHe/p reporter 169 and 254%, respectively, while introduction of DLX1 increased reporter activity 227%. Moreover, co-transfection of expression vectors for DLX1, 2, and 5 together significantly increased reporter activity (417%) over the empty vector control. These results show that MSX expression represses, while DLX expression activates GnRH transcription in GT1-7 cells.
Fig. 4. Transcriptional activity of MSX and DLX on the GnRH gene in GT1-7 cells.

A, the GnRHe/p-luciferase reporter was co-transfected with expression vectors for MSX1, MSX2, DLX1, DLX2, or DLX5 in transient transfection of GT1-7 cells. B, wild-type and mutant Gn-RHe/p luciferase reporters were co-transfected into GT1-7 cells with expression vectors for these same MSX and DLX family members. In both panels, a TK-β-galactosidase reporter was used to control for transfection efficiency. Percent activity is relative to empty vector control. Asterisks indicate statistical difference from the control and the pound sign (#) signifies difference between activity on the wild-type and mutant reporter by analysis of variance with Tukey-Kramer HSD (p < 0.05).
Having identified four homeodomain sites in the GnRH promoter that are bound by MSX and DLX in vitro, we next asked whether the activities of the MSX and DLX family members were dependent upon these ATTA sequences. Mutations previously shown to inhibit protein binding to the homeodomain element (17, 18) were introduced in the GnRHe/p-luciferase vector at the two enhancer sites (−1636 to −1631 and −1623 to −1618) and the two promoter sites (−55 to −50 and −42 to −38). Interestingly, transient transfection of the reporter plasmid containing these four mutated sites (mutant GnRHe/p) in GT1-7 cells revealed 70% reduced reporter activity compared with a wild-type GnRHe/p reporter (Fig. 4B), demonstrating that these sites are important for proper basal activity of the GnRH promoter in GT1-7 cells. Although the mutant GnRHe/p reporter showed greatly reduced basal reporter activity compared with the wild-type plasmid, activity was still significantly above background. While expression of MSX1 or MSX2 significantly reduced activity of the GnRHe/p reporter, it did not significantly affect the mutant GnRHe/p reporter. Further, expression of DLX2 or DLX5 activated the GnRHe/p reporter but showed no statistically significant increase in activity on the mutant GnRHe/p reporter. However, co-expression of DLX1 in concert with either the wild-type or mutant GnRHe/p reporter significantly increased transcriptional activity, although activation was reduced with the mutant reporter. These results suggest that MSX and DLX family members function through the homeodomain binding sites and these sites are important for GnRH promoter activity.
MSX and DLX Compete for Occupation of the Q50 Homeodomain Binding Sites and Show Functional Antagonism on the GnRH Gene
Given the opposing transcriptional activity of MSX and DLX family members on the GnRH promoter (Fig. 4), we asked whether the major (MSX) and minor (DLX) complexes compete for binding to the ATTA elements of the GnRH regulatory region. Competition between MSX1 and DLX2 for DNA binding has been previously described and indicates that while MSX1 and DLX2 can form heterodimers, this interaction is mutually exclusive of DNA binding (28). To address this question, we generated nuclear extracts from GT1-7 cells overexpressing the DLX2 protein for EMSA analysis. As seen in Fig. 5A, the intensity of the minor complex increased while the major complex had reduced intensity compared with the binding pattern of nontransfected GT1-7 nuclear extract (Fig. 5A, lanes 2 and 1, respectively). These same results were seen in the extracts from NLT cells overexpressing the DLX1 protein (Fig. 5B). Furthermore, in transient transfection of GT1-7 cells, expression of increasing amounts of DLX2 overcame MSX1-repressed GnRH promoter activity (Fig. 5C). These results indicate that MSX1 and DLX2 compete for occupation of the ATTA repeats in the GnRH enhancer and exhibit functionally antagonistic activity.
Fig. 5. Functional antagonism of MSX1 and DLX2 on the GnRH gene in GT1-7 cells.
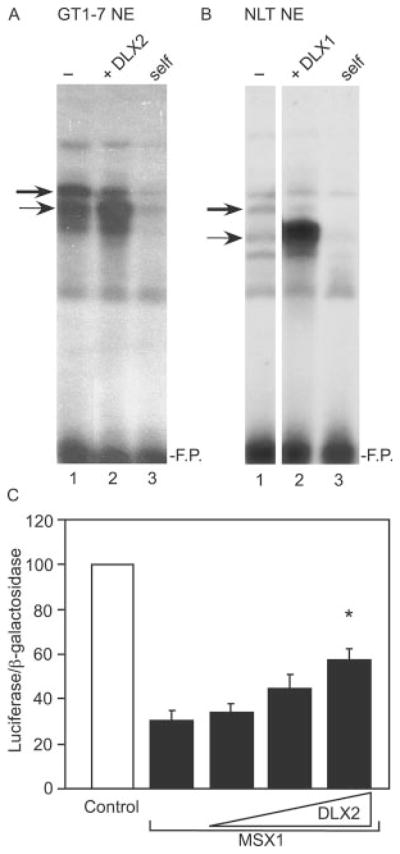
Competitive binding between MSX and DLX on the rat GnRH gene is shown. EMSA was performed with A, GT1-7 nuclear extract or B, NLT nuclear extract. The thick arrow represents the major complex (MSX) and the thin arrow represents the minor complex (DLX). Lane 1 represents control nuclear extract, and lane 2 shows extracts overexpressing DLX protein. Lane 3 represents self-competition with 100× unlabeled oligonucleotide. The probe corresponds to the 5′-enhancer homeodomain binding site, −1642/−1623. F.P. represents excess, free probe. C, transient co-transfection of the GT1-7 cells was performed with the GnRHe/p-luciferase reporter and a plasmid vector overexpressing MSX1 in addition to 1×, 2×, and 3× the amount of an expression vector for DLX2. Percent activity is relative to empty vector control. The asterisk indicates statistical difference from MSX1 alone by analysis of variance with Tukey-Kramer HSD (p < 0.05).
Differential GnRH Promoter Expression in Model GnRH Neuronal Cell Lines
Characterization of the GT1-7 and GN11 or NLT cell lines suggests that the GT1-7 cells represent a differentiated, mature GnRH neuron while the GN11 or NLT cells mimic the immature, migratory stages of GnRH neuron development (12–15). Previous findings suggest that GnRH expression itself is involved in furthering the commitment and migratory state of the GnRH neurons (11). We therefore hypothesized that GnRH transcriptional activity would differ between the migratory and mature GnRH neuronal cell lines. We examined the level of GnRH promoter activity in these cells by transient transfection with the GnRHe/p reporter. Because the GN11 cell line has higher transfection efficiency than the NLT line, these cells were used to investigate reporter activity. GnRH promoter activity in the GT1-7 cells was more than 11-fold higher than a pGL3-luciferase control (Fig. 6A). Interestingly, GN11 cells transiently transfected with the GnRHe/p reporter showed a greater than 20-fold reduction in promoter activity compared with the GT1-7 cells despite the lack of a statistical difference in expression of the control plasmid. Finally, the GnRH regulatory regions showed extremely low levels of activity in the NIH3T3 cells (Fig. 6A), as the activity of the GnRHe/p reporter in these cells was significantly lower than that of an empty pGL3-luciferase vector, suggesting that the promoter may be actively repressed in these cells.
Fig. 6. Relative GnRH expression in model cell lines.

A, transient transfection of the GnRHe/p-luciferase reporter was performed in GT1-7, GN11, and NIH3T3 cells. In addition, an RSVe/RSVp-β-galactosidase reporter was included to normalize for transfection efficiency. All values are normalized to the RSVe/RSVp-luciferase to control for metabolic differences in the cell lines. The asterisk indicates statistical difference from the pGL3-luc control while the pound sign indicates statistically significant difference from the GnRHe/p across cell lines by analysis of variance with Tukey-Kramer HSD (p < 0.05). B, overexpression of DLX family members in GN11 cells enhances promoter activity. Transient transfection of GN11 cells was performed with the GnRHe/p or mutant GnRHe/p in concert with expression vectors for DLX1, -2, and -5 (represented as + DLX). The asterisk indicates statistical difference from the empty vector control.
One explanation for the significantly reduced activity of the GnRHe/p in GN11 cells may be the absence of key transcriptional activators in the GN11 cells that are present in the GT1-7 cell line. Indeed, using RT-PCR (Fig. 2A), we did not detect expression of Dlx2-6 in the GN11/NLT cells. Thus, we explored the possibility that expression of the DLX family members in GN11 cells would restore the level of GnRH transcription in these cells. Transient co-transfection of DLX1, 2 and 5 with the GnRHe/p reporter in the GN11 cells significantly increased GnRH promoter expression by 13-fold (Fig. 6B). Furthermore, DLX activation of GnRH promoter expression required the presence of the homeodomain binding sites within the GnRH enhancer and promoter. This suggests that the reduced promoter activity of the GnRH gene in the GN11 cells may be caused by the absence of the DLX family of activators.
MSX1 and DLX1 and 2 Contribute to the Development of the GnRH Neuronal Population in Vivo
The data we have presented herein indicate that DLX1 and 2 function as key regulatory activators and MSX1 as a repressor of GnRH promoter expression through binding to homeodomain elements within the GnRH regulatory region (Figs. 3–5). In addition, double in situ hybridization/immunohistochemical analysis revealed co-expression of Msx and Dlx in the developing GnRH-positive neurons (Fig. 2). To determine whether MSX1 or DLX1 and DLX2 play a role in maintaining proper spatial and temporal GnRH expression in vivo, we performed RNA in situ hybridization to compare the number of GnRH-positive neurons between wild-type and Dlx1&2-null or Msx1-null mice at developmental stages when the complete population of septohypothalamic GnRH neurons has been established.
In the Dlx1&2 background on 13.5 dpc, wild-type embryos had ~945 ± 62 GnRH-positive cells within the olfactory epithelium as well as along the cribriform plate and into the CNS, consistent with previous findings (6). In contrast, Dlx1&2-null littermates showed significantly reduced numbers of GnRH-positive neurons, ~612 ± 177 (Table I). Interestingly, the number of GnRH-positive neurons that have not yet crossed into the central nervous system was similar in the Dlx1&2 wild-type (668 ± 74) and null (515 ± 125) mice. In contrast, the number of GnRH-positive neurons in the CNS of Dlx1&2-null mice (97 ± 60) was significantly reduced compared with Dlx1&2 wild-type mice (277 ± 92) (Table I and Fig. 7, panel A). By 16.5 dpc, Dlx1&2-null embryos had a similar number of septohypothalamic, GnRH-positive neurons overall. However, the position of these neurons had changed such that the majority of GnRH-positive cells were in the CNS. In the Dlx1&2-null mice there were 681 ± 81, whereas in the wild-type littermates there were 985 ± 16 total GnRH-positive neurons (Table I and Fig. 7, panel B). Thus, embryos lacking DLX1 and 2 have a reduced number of GnRH-positive cells in the septohypothalamic population, suggesting that DLX1 and DLX2 promote expression of GnRH in vivo.
Table I. GnRH-positive cells counted by in situ hybridization of mouse embryos.
In situ hybridization was performed using an antisense RNA probe for GnRH to assess the number of GnRH-positive cells. Whole heads of mouse embryo littermates were sectioned, hybridized, and then counted for GnRH-positive cells.
| Genotype | Embryo age | Nose | Primordium of the septum | Tectum | Total (septohypothalamic) | |
|---|---|---|---|---|---|---|
| dpc | ||||||
| Dlx1&2 | +/+ | 13.5 | 668 ± 74 | 277 ± 92 | 0 | 945 ± 62 |
| Dlx1&2 | −/− | 13.5 | 515 ± 125 | 97 ± 60 | 0 | 612 ± 177 |
| Dlx1&2 | +/+ | 16.5 | 153 ± 40 | 833 ± 34 | 203 ± 29 | 985 ± 16 |
| Dlx1&2 | −/− | 16.5 | 72 ± 36 | 610 ± 45 | 204 ± 18 | 681 ± 81 |
| Msx1 | +/+ | 12.5 | 733 ± 15 | 125 ± 3 | 0 | 858 ± 18 |
| Msx1 | −/− | 12.5 | 896 ± 52 | 137 ± 14 | 0 | 1,032 ± 54 |
| Msx1 | +/+ | 13.5 | 710 ± 44 | 229 ± 31 | 0 | 939 ± 20 |
| Msx1 | −/− | 13.5 | 1,021 ± 99 | 267 ± 25 | 19 ± 6 | 1,300 ± 116 |
| Msx1 | +/+ | 16.5 | 309 ± 58 | 728 ± 63 | 238 ± 26 | 1,037 ± 103 |
| Msx1 | −/− | 16.5 | 227 ± 49 | 643 ± 64 | 215 ± 31 | 871 ± 53 |
Fig. 7. GnRH-positive cells in the Dlx1&2 or Msx1-null mouse embryos.

In situ hybridization of Dlx1&2 or Msx1-positive and null embryonic mice ages 12.5, 13.5, and 16.5 dpc was carried out as described using a DIG-GnRH probe. Images represent parasagittal sections of the wild-type (left panel) and null (right panel) embryos from 13.5-dpc Dlx1&2 (panel A), 16.5-dpc Dlx1&2 (panel B), 12.5-dpc Msx1 (panel C), 13.5-dpc Msx1 (panel D), and 16.5-dpc Msx1 (panel E). Panel F, 13.5-dpc Msx1-null embryo with GnRH-positive cells in the olfactory epithelium (arrow) at low (left panel) and higher (right panel) magnification are indicated. Panel G, immunohistochemical analysis of GnRH-positive cells located in the olfactory epithelium of Msx1-null embryos at 13.5 dpc. GnRH-positive cells located in the tectum of 13.5-dpc Msx1-null (panel H), 16.5-dpc Dlx1&2-null (panel I), 16.5-dpc wild-type (panel J), and 16.5-dpc Msx1-null (panel K) embryo. Arrows indicate GnRH-positive cells located outside of the characterized migratory route for the septohypothalamic population. Black scale bars represent 100 μm in the lower left of each panel. The schematic represents a parasagittal view of a mouse embryo head in the region where GnRH-positive neurons are found.
In addition to the septohypothalamic population, another population of GnRH-positive cells was present at 16.5 dpc in the tectal region of the developing midbrain. Recent studies suggest that the tectal population is regulated by the same promoter elements as the septohypothalamic population but different transcription factor complexes because of the unique spatiotemporal expression of GnRH in the midbrain (2). We examined this population of GnRH-positive cells in the Dlx1&2-null embryos in order to assess GnRH promoter activity that is not directly regulated by DLX, as the Dlx genes are not expressed in this region of the brain. Interestingly, by in situ hybridization, we observed no differences between wild-type and null embryos in GnRH-positive cells in the tectal population (Table I and Fig. 7, panels I and J). This suggests that the reduced population of septohypothalamic, GnRH neurons in the Dlx1&2-null may be because of a loss of DLX1 and 2 and not an indirect, global, developmental misregulation.
Examination of the Msx1 wild-type and null littermates at 12.5 and 13.5 dpc also revealed significant differences in the population of GnRH-positive cells. By 12.5 dpc, ~858 ± 18 GnRH-positive cells were present in the wild-type embryos while 1,032 ± 54 positive cells were observed in the Msx1-null littermates (Table I, Fig. 7, panel C). Although the difference did not reach statistical significance, the higher number of GnRH-positive cells seen in the null embryos could be attributed to a larger population of positive cells in the nasal region in addition to a few GnRH-positive cells in the olfactory epithelium (Fig. 7, panel C, arrow). Wild-type Msx1 embryos on 13.5 dpc had ~939 ± 20 GnRH-positive cells scattered throughout the nasal region, extending into the primordium of the septum, while the Msx1-null embryos had significantly more (1,300 ± 116 and Table I). Interestingly, on 13.5 dpc, the nasal populations of GnRH-positive cells in Msx1-null embryos were significantly greater than those in wild-type littermates (1,021 ± 99 compared with 710 ± 44) (Table I and Fig. 7, panel D). Moreover, the GnRH-positive cells in the Msx1-null embryos were scattered throughout the nasal region but also located in the olfactory epithelium, outside of the typical location of the septohypothalamic population (Fig. 7, panel F, arrows). In addition, these cells were identified by an antibody recognizing the GnRH peptide, confirming that they indeed expressed GnRH (Fig. 7, panel G). There was also a small population of GnRH-positive cells (~20) located in the developing tectum at 13.5 dpc (Table I and Fig. 7, panel H), several days before these cells begin expressing GnRH in the wild-type embryos.
By 16.5 dpc, no significant difference in the number of GnRH-positive cells between wild-type and Msx1-null was observed. Approximately 871 ± 53 GnRH-positive cells were present in the Msx1-null embryos compared with 1,037 ± 103 in the wild-type embryos (Table I and Fig. 7, panel E). Interestingly, the nasal and CNS population of GnRH-positive cells did not differ between the wild-type and null embryos. Furthermore, the population of GnRH-positive cells located in the olfactory epithelium was still present at 16.5 dpc (Fig. 7, panel E, arrows). These findings suggest that Msx1, as an essential factor in the development of craniofacial morphology, also disrupts the development of the septohypothalamic population of GnRH neurons and likely regulates GnRH promoter expression during early stages of embryonic development.
In addition to the septohypothalamic population of GnRH-positive cells on 16.5 dpc, the tectal population of GnRH-expressing cells was present in the wild-type and Msx1-null embryos. Tectal GnRH-positive cells could be seen in the Msx1-nulls as early as 13.5 dpc, though this population did not appear to differ in number or morphology in the 16.5-dpc embryos as compared with the wild-type litter mates (Table I and Fig. 7, panel K). Although the functional role for these tectal GnRH-expressing cells has not yet been elucidated, tectal GnRH expression is driven by the same promoter that regulates the septohypothalamic population (2) and, intriguingly, is also affected by an absence of MSX1 but not DLX1 and 2. The absence of MSX1, which is normally expressed in this region (43, 44), may have resulted in premature differentiation or improper regulation of the GnRH promoter in these cells.
DISCUSSION
Regulated transcriptional activity forms the foundation for promoting the molecular identity and maturation of the septohypothalamic population of GnRH neurons during embryonic development. Investigation of immature, migratory and adult GnRH-expressing neurons has revealed differences in the expression pattern of transcriptional factors that regulate spatiotemporal transcriptional control of the GnRH gene (45, 46). Thus, GnRH neurons are a heterogeneous population that differentially expresses specific genes at distinct stages of embryonic development, which may, in turn, result in dynamic regulation of GnRH promoter expression. Indeed, the transcriptional activity of the GnRH gene appears to be highly dynamic during the developmental, migratory stage of the septohypothalamic neurons. A recent study reported that the GnRH promoter expresses only at very low levels in neurons located in the olfactory region and becomes increasingly active in neurons in close proximity to the cribriform plate and in those that have entered the CNS (47). Thus, it has been reported that GnRH promoter activity is dependent upon the position of the neuron (47), which in turn is determined by the stage of embryonic development.
The findings presented herein implicate members of the DLX and MSX Q50 homeodomain families as important transcriptional regulators of GnRH gene expression. Repression and activation of the GnRH regulatory region by MSX and DLX, respectively, are dependent upon the conserved ATTA homeodomain sites (Fig. 4). Moreover, MSX and DLX bind identical homeodomain elements within the GnRH gene enhancer and promoter (Figs. 1 and 4A) and have functionally antagonistic activities (Fig. 5B). Interestingly, similar observations have been made in other contexts, such as the reciprocal activity of MSX2 and DLX5 on the osteocalcin (OC) promoter (48) and between PAX3 and MSX1 on the MyoD promoter (49).
In addition, we found that expression of Msx and Dlx family members coincides with the migratory route of septohypothalamic GnRH neurons during embryonic development (Fig. 2, B–E). The repressors, Msx1 and 2 are expressed in the nasal region where GnRH promoter activity is low, while the transcriptional activators Dlx1 and 2 are expressed at later times along the migratory route of GnRH neurons into the hypothalamus when GnRH promoter activity is high (47). In support of this, two model cell lines with characteristics of GnRH neurons at distinct developmental stages differ in their expression of several Msx and Dlx family members. The more mature GnRH neuronal cell model, GT1-7, expresses nearly all of the Dlx family of transcriptional activators as well as the Msx repressors (Fig. 2A). In contrast, the GN11/NLT cell lines, which represent the immature stage of the GnRH neuron, predominantly express the Msx repressors. Furthermore, GnRH promoter activity was 20-fold lower in GN11 versus GT1-7 cells, which may be due in part to absent expression of some DLX family members. Indeed, exogenous expression of the DLX family members in GN11 cells resulted in significant increases in GnRH promoter expression (Fig. 6B). Thus, we hypothesize that the coordinated spatial and temporal expression of Msx and Dlx family members in the embryo contributes to dynamic regulation of the GnRH promoter.
In support of our hypothesis, DLX and MSX family members appear to be required to maintain normal spatiotemporal populations of GnRH neurons in mice. Analysis of Dlx1&2 and Msx1-null mutant mice revealed significant differences in the number of GnRH-positive neurons during development compared with wild-type mice (Table I). The reduced number of GnRH-positive cells in these Dlx1&2-null mutants may be directly due to misregulation of GnRH promoter activity. Whereas our in vitro and in vivo data strongly support the roles for DLX1 and DLX2 in GnRH expression, we cannot rule out an additional phenotypic contribution from decreased Dlx5 expression, which has been observed in the Dlx1&2 mutants (50, 51). DLX5 can activate the GnRH promoter in vitro (Fig. 4) and is expressed in the appropriate spatiotemporal pattern relevant for the developing GnRH neuron (Fig. 1). Another potential explanation for the decreased numbers of GnRH-positive cells in the CNS of the Dlx1&2-null is improper migration. In fact, we observed a small portion of mis-migrating GnRH-positive cells as early as 13.5 dpc in these null embryos. Upon mis-migration, it is possible that these few neurons down-regulate expression of GnRH or apoptose in the absence of appropriate environmental cues.
The number of GnRH-positive cells in Msx1-null embryos was significantly greater than the number present in wild-type littermates on 13.5 dpc (Fig. 7, panel D and Table I). Intriguingly, the abnormality in the number and location of the GnRH population observed in the Msx1-null embryo is similar to that of mouse embryos with null mutations in AP-2α, which plays an important role in development of craniofacial features including the nasal environment of the GnRH neuron (52, 53). Like MSX1, AP-2α co-localizes with GnRH-positive neurons as early as e13.5, but not in the adult (54) and AP-2α-null embryos show a considerable increase in the nasal population of GnRH-positive cells between 13.5 and 15.5 dpc, developmental stages when AP-2α co-expresses with GnRH in the wild-type embryo (55). In that study, expansion of the GnRH-positive nasal population was attributed to proliferation and misregulation of mitotic signals in the absence of AP-2α (55). Thus, an alternative hypothesis is that an increase in the population of GnRH-positive cells in the Msx1 mutant represents a proliferation of the GnRH neuronal population, as MSX has also been implicated in cell cycle control (56). However, Msx1 wild-type and null embryos showed no difference in the number of GnRH-positive cells also positive for the nuclear proliferation antigen, Ki67 (data not shown). Thus, the specific increase in GnRH-positive cells in the Msx1-null, rather than an actual increase in overall cell number, might also represent cells that express the GnRH transcript at levels below detection in a wild-type embryo and would be consistent with the role of Msx1 as a transcriptional repressor of GnRH promoter activity.
Herein, we have found that the MSX and DLX transcription factor families are important for proper regulation of the GnRH promoter during development. Understanding the molecular identity of the GnRH neuronal population throughout development will facilitate a better understanding of the key factors involved in cell-specific as well as temporal expression of the GnRH gene. Such knowledge will also provide insight into the development of the hypothalamic-pituitary-gonadal axis and reproductive function.
Acknowledgments
We thank Dr. R. Benoit for providing the LR-1 GnRH antibody, Dr. C. Abate-Shen for expression vectors for MSX1 and DLX2, Dr. D. Sassoon for Msx1 and Msx2 in situ plasmids, Dr. D. A. Towler for the Msx2 expression vector, and Dr. S. Radovick for the GN11 and NLT cell lines. We also appreciate the efforts of M. A. Lawson, D. Coss, J. S. Bailey, N. L. G. Miller, K. Pinson, M. J. Barsoum, and T. K. Sato for discussions and critical reading, as well as K. Pinson and R. S. White for technical assistance.
Footnotes
This work was supported by National Institutes of Health Grant R01 DK44838 (to P. L. M.).
The abbreviations used are: GnRH, gonadotropin-releasing hormone; dpc, days postcoitum; EMSA, electromobility shift assay; DIG, digoxigenin; CNS, central nervous system.
References
- 1.Wu TJ, Gibson MJ, Silverman AJ. J Neuroendocrinol. 1995;7:899–902. doi: 10.1111/j.1365-2826.1995.tb00733.x. [DOI] [PubMed] [Google Scholar]
- 2.Skynner MJ, Slater R, Sim JA, Allen ND, Herbison AE. J Neurosci. 1999;19:5955–5966. doi: 10.1523/JNEUROSCI.19-14-05955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mason AJ, Hayflick JS, Zoeller RT, Young WS, Phillips HS, Nikolics K, Seeburg PH. Science. 1986;234:1366–1371. doi: 10.1126/science.3024317. [DOI] [PubMed] [Google Scholar]
- 4.Dubois EA, Zandbergen MA, Peute J, Goos HJ. Brain Res Bull. 2002;57:413–418. doi: 10.1016/s0361-9230(01)00676-1. [DOI] [PubMed] [Google Scholar]
- 5.Schwanzel-Fukuda M, Pfaff DW. Nature. 1989;338:161–164. doi: 10.1038/338161a0. [DOI] [PubMed] [Google Scholar]
- 6.Wray S, Grant P, Gainer H. Proc Natl Acad Sci U S A. 1989;86:8132–8136. doi: 10.1073/pnas.86.20.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitlock KE, Wolf CD, Boyce ML. Dev Biol. 2003;257:140–152. doi: 10.1016/s0012-1606(03)00039-3. [DOI] [PubMed] [Google Scholar]
- 8.Wray S, Key S, Qualls R, Fueshko SM. Dev Biol. 1994;166:349–354. doi: 10.1006/dbio.1994.1320. [DOI] [PubMed] [Google Scholar]
- 9.Livne I, Gibson MJ, Silverman AJ. Dev Biol. 1993;159:643–656. doi: 10.1006/dbio.1993.1271. [DOI] [PubMed] [Google Scholar]
- 10.Schwanzel-Fukuda M, Jorgenson KL, Bergen HT, Weesner GD, Pfaff DW. Endocr Rev. 1992;13:623–634. doi: 10.1210/edrv-13-4-623. [DOI] [PubMed] [Google Scholar]
- 11.Romanelli RG, Barni T, Maggi M, Luconi M, Failli P, Pezzatini A, Pelo E, Torricelli F, Crescioli C, Ferruzzi P, Salerno R, Marini M, Rotella CM, Vannelli GB. J Biol Chem. 2004;279:117–126. doi: 10.1074/jbc.M307955200. [DOI] [PubMed] [Google Scholar]
- 12.Mellon PL, Windle JJ, Goldsmith P, Pedula C, Roberts J, Weiner RI. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-e. [DOI] [PubMed] [Google Scholar]
- 13.Radovick S, Wray S, Lee E, Nicols DK, Nakayama Y, Weintraub BD, Westphal H, Cutler J, GB, Wondisford FE. Proc Natl Acad Sci U S A. 1991;88:3402–3406. doi: 10.1073/pnas.88.8.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prioni S, Loberto N, Prinetti A, Chigorno V, Guzzi F, Maggi R, Parenti M, Sonnino S. Neurochem Res. 2002;27:831–840. doi: 10.1023/a:1020217309987. [DOI] [PubMed] [Google Scholar]
- 15.Pimpinelli F, Redaelli E, Restano-Cassulini R, Curia G, Giacobini P, Cariboni A, Wanke E, Bondiolotti GP, Piva F, Maggi R. Eur J Neurosci. 2003;18:1410–1418. doi: 10.1046/j.1460-9568.2003.02866.x. [DOI] [PubMed] [Google Scholar]
- 16.Lawson MA, MacConell LA, Kim J, Powl BT, Nelson SB, Mellon PL. Endocrinology. 2002;143:1404–1412. doi: 10.1210/endo.143.4.8751. [DOI] [PubMed] [Google Scholar]
- 17.Kelley CG, Givens ML, Rave-Harel N, Nelson SB, Anderson S, Mellon PL. Mol Endocrinol. 2002;16:2413–2425. doi: 10.1210/me.2002-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson SB, Lawson MA, Kelley CG, Mellon PL. Mol Endocrinol. 2000;14:1509–1522. doi: 10.1210/mend.14.9.0521. [DOI] [PubMed] [Google Scholar]
- 19.Alappat S, Zhang ZY, Chen YP. Cell Res. 2003;13:429–442. doi: 10.1038/sj.cr.7290185. [DOI] [PubMed] [Google Scholar]
- 20.Kraus P, Lufkin T. J Cell Biochem. 1999;(Suppl):32–33. 133–140. doi: 10.1002/(sici)1097-4644(1999)75:32+<133::aid-jcb16>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 21.Zerucha T, Ekker M. Biochem Cell Biol. 2000;78:593–601. [PubMed] [Google Scholar]
- 22.Panganiban G, Rubenstein JL. Development. 2002;129:4371–4386. doi: 10.1242/dev.129.19.4371. [DOI] [PubMed] [Google Scholar]
- 23.Cornell RA, Ohlen TV. Curr Opin Neurobiol. 2000;10:63–71. doi: 10.1016/s0959-4388(99)00049-5. [DOI] [PubMed] [Google Scholar]
- 24.Porteus MH, Bulfone A, Liu JK, Puelles L, Lo LC, Rubenstein JL. J Neurosci. 1994;14:6370–6383. doi: 10.1523/JNEUROSCI.14-11-06370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bendall AJ, Abate-Shen C. Gene (Amst) 2000;247:17–31. doi: 10.1016/s0378-1119(00)00081-0. [DOI] [PubMed] [Google Scholar]
- 26.Iler N, Rowitch DH, Echelard Y, McMahon AP, Abate-Shen C. Mech Dev. 1995;53:87–96. doi: 10.1016/0925-4773(95)00427-0. [DOI] [PubMed] [Google Scholar]
- 27.Iler N, Abate-Shen C. Biochem Biophys Res Commun. 1996;227:257–265. doi: 10.1006/bbrc.1996.1498. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Hu G, Wang H, Sciavolino P, Iler N, Shen MM, Abate-Shen C. Mol Cell Biol. 1997;17:2920–2932. doi: 10.1128/mcb.17.5.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clark ME, Mellon PL. Mol Cell Biol. 1995;15:6169–6177. doi: 10.1128/mcb.15.11.6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu M, Bulfone A, Ghattas I, Meneses JJ, Christensen L, Sharpe PT, Presley R, Pedersen RA, Rubenstein JL. Dev Biol. 1997;185:165–184. doi: 10.1006/dbio.1997.8556. [DOI] [PubMed] [Google Scholar]
- 31.Houzelstein D, Cohen A, Buckingham ME, Robert B. Mech Dev. 1997;65:123–133. doi: 10.1016/s0925-4773(97)00065-8. [DOI] [PubMed] [Google Scholar]
- 32.Reginelli AD, Wang YQ, Sassoon D, Muneoka K. Development. 1995;121:1065–1076. doi: 10.1242/dev.121.4.1065. [DOI] [PubMed] [Google Scholar]
- 33.Bulfone A, Puelles L, Porteus MH, Frohman MA, Martin GR, Rubenstein JL. J Neurosci. 1993;13:3155–3172. doi: 10.1523/JNEUROSCI.13-07-03155.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zerucha T, Stuhmer T, Hatch G, Park BK, Long Q, Yu G, Gambarotta A, Schultz JR, Rubenstein JL, Ekker M. J Neurosci. 2000;20:709–721. doi: 10.1523/JNEUROSCI.20-02-00709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 36.Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang HJ, Miller WL, Mellon PL. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- 37.Eisenstat DD, Liu JK, Mione M, Zhong W, Yu G, Anderson SA, Ghattas I, Puelles L, Rubenstein JL. J Comp Neurol. 1999;414:217–237. doi: 10.1002/(sici)1096-9861(19991115)414:2<217::aid-cne6>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 38.Gauchat D, Mazet F, Berney C, Schummer M, Kreger S, Pawlowski J, Galliot B. Proc Natl Acad Sci U S A. 2000;97:4493–4498. doi: 10.1073/pnas.97.9.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Catron KM, Wang H, Hu G, Shen MM, Abate-Shen C. Mech Dev. 1996;55:185–199. doi: 10.1016/0925-4773(96)00503-5. [DOI] [PubMed] [Google Scholar]
- 40.Catron KM, Zhang H, Marshall SC, Inostroza JA, Wilson JM, Abate C. Mol Cell Biol. 1995;15:861–871. doi: 10.1128/mcb.15.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Catron KM, Abate-Shen C. Proc Natl Acad Sci U S A. 1996;93:1764–1769. doi: 10.1073/pnas.93.5.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu G, Zerucha T, Ekker M, Rubenstein JL. Brain Res Dev Brain Res. 2001;130:217–230. doi: 10.1016/s0165-3806(01)00239-5. [DOI] [PubMed] [Google Scholar]
- 43.Grove EA, Tole S, Limon J, Yip L, Ragsdale CW. Development. 1998;125:2315–2325. doi: 10.1242/dev.125.12.2315. [DOI] [PubMed] [Google Scholar]
- 44.Bach A, Lallemand Y, Nicola MA, Ramos C, Mathis L, Maufras M, Robert B. Development. 2003;130:4025–4036. doi: 10.1242/dev.00609. [DOI] [PubMed] [Google Scholar]
- 45.Lawson MA, Mellon PL. Mol Cell Endocrinol. 1998;140:157–161. doi: 10.1016/s0303-7207(98)00044-6. [DOI] [PubMed] [Google Scholar]
- 46.Rave-Harel N, Givens ML, Nelson SB, Duong HA, Coss D, Clark ME, Hall SB, Kamps MP, Mellon PL. J Biol Chem. 2004;279:30287–30297. doi: 10.1074/jbc.M402960200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simonian SX, Herbison AE. Neuroendocrinology. 2001;73:149–156. doi: 10.1159/000054631. [DOI] [PubMed] [Google Scholar]
- 48.Newberry EP, Latifi T, Towler DA. Biochemistry. 1998;37:16360–16368. doi: 10.1021/bi981878u. [DOI] [PubMed] [Google Scholar]
- 49.Bendall AJ, Ding J, Hu G, Shen MM, Abate-Shen C. Development. 1999;126:4965–4976. doi: 10.1242/dev.126.22.4965. [DOI] [PubMed] [Google Scholar]
- 50.Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JL. Neuron. 1997;19:27–37. doi: 10.1016/s0896-6273(00)80345-1. [DOI] [PubMed] [Google Scholar]
- 51.Yun K, Fischman S, Johnson J, Hrabe de Angelis M, Weinmaster G, Rubenstein JL. Development. 2002;129:5029–5040. doi: 10.1242/dev.129.21.5029. [DOI] [PubMed] [Google Scholar]
- 52.Schorle H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Nature. 1996;381:235–238. doi: 10.1038/381235a0. [DOI] [PubMed] [Google Scholar]
- 53.Zhang J, Hagopian-Donaldson S, Serbedzija G, Elsemore J, Plehn-Dujowich D, McMahon AP, Flavell RA, Williams T. Nature. 1996;381:238–241. doi: 10.1038/381238a0. [DOI] [PubMed] [Google Scholar]
- 54.Kramer PR, Krishnamurthy R, Mitchell PJ, Wray S. Endocrinology. 2000;141:1823–1838. doi: 10.1210/endo.141.5.7452. [DOI] [PubMed] [Google Scholar]
- 55.Kramer PR, Guerrero G, Krishnamurthy R, Mitchell PJ, Wray S. Mech Dev. 2000;94:79–94. doi: 10.1016/s0925-4773(00)00316-6. [DOI] [PubMed] [Google Scholar]
- 56.Hu G, Lee H, Price SM, Shen MM, Abate-Shen C. Development. 2001;128:2373–2384. doi: 10.1242/dev.128.12.2373. [DOI] [PubMed] [Google Scholar]