

Abstract
LH β-subunit (LHβ), which is essential for ovulation and reproductive fitness, is synthesized specifically in pituitary gonadotropes. In this study, we show that LHβ gene expression is induced by activin in mouse primary pituitary cells if the cells are treated within 24 h after dispersion in culture. Furthermore, male mice deficient in Smad3, and therefore in activin signaling, have lower expression of both LHβ and FSHβ mRNAs compared with their wild-type littermates. Using the LβT2 immortalized mouse gonadotrope cell line that endogenously expresses LH, we identify specific elements in the regulatory region of the rat LHβ gene necessary for its induction by activin. Activin responsiveness is conferred by a promoter-proximal region located −121/−86 from the transcriptional start site. Maximal LHβ induction by activin requires a homeobox element (HB) and a 5′-early growth response (Egr) site found in this region of the promoter. Juxtaposed to the HB are three Smad-binding elements (SBEs), which are essential for LHβ induction. Interestingly, two of the SBEs are also critical for basal expression of the LHβ gene. We demonstrate that Smad proteins are necessary and sufficient for activin induction of the LHβ gene. Furthermore, Smad proteins can bind one of the identified SBEs. In addition to binding this SBE, Smad proteins interact with pituitary homeobox 1 (Ptx-1) and orthodenticle homeobox 1 (Otx-1), which can bind the HB located close to the Smad-binding site. Thus, activin induction of LHβ gene expression requires a combination of several transcription factors, both basal and activin induced, as well as cooperation between multiple DNA elements.
LH IS ESSENTIAL for steroidogenesis and reproductive function in both males and females, because a lack of this hormone leads to hypogonadism and infertility in both sexes (1). LH synthesis is restricted exclusively to the anterior pituitary gonadotropes (2). It is a heterodimeric glycoprotein, composed of an α-subunit, in common with FSH and TSH, and a unique β-subunit, which confers biological specificity. Transcription of the β-subunit is the rate-limiting step for LH production (3, 4). LH β-subunit (LHβ) gene transcription is induced by GnRH and repressed by gonadal steroids (5).
Activin and inhibin, members of the TGFβ family of growth factors, also play important roles in the modulation of gonadotropins. These glycoproteins were initially described as functioning in gonadal feedback on gonadotropin synthesis. Activin increases release of FSH (6) from the pituitary and induces FSHβ expression in gonadotrope cells (7), whereas inhibin antagonizes activin action. Follistatin, a potent activin-binding protein (8), can inhibit the biosynthesis and secretion of FSH (9). Interestingly, activin, follistatin, and inhibin are expressed in the mature pituitary gonadotrope and can function in an autocrine manner (10–12). Follistatin is also synthesized by folliculostellate cells in the pituitary and regulates activin availability in a paracrine manner (13, 14).
TGFβ family members, including activin, activate signaling molecules known as receptor-associated Smads, which, in the case of activin, are Smad2 and/or Smad3 (15). Smad2 or Smad3 then associate with a common Smad, Smad4 (DPC4). The activated heteromeric Smad complex translocates into the nucleus, where it binds a Smad-binding element GTCTAGAC, or either half of this palindrome, within DNA to regulate the expression of target genes (16).
As mentioned above, there is considerable evidence that activin regulates FSH synthesis and secretion. However, the initial reports regarding the role of activin in gonadotropes failed to detect an effect of activin on LH secretion (6, 17). In contrast, a number of subsequent studies suggest that activin can influence LH synthesis, both in vivo and in cell culture. Stouffer et al. (18) have shown an acute and sustained increase in LH secretion in response to activin in female rhesus monkeys. McLachlan et al. (19) reported that 2-d activin infusion in adult male monkeys significantly increased both LH and FSH release in response to GnRH injection. Attardi and Miklos (20) demonstrated that treatment of primary rat pituitary cell cultures with activin results in significant increases in both LH secretion and protein content in the cells, as well as LHβ mRNA levels. More recently, this observation was confirmed in human fetal primary cell culture, where recombinant human activin caused a significant increase in LH secretion into the medium. In addition, inhibin decreased FSH and LH secretion, but the LH response to inhibin was less prominent than that of FSH (21). Furthermore, our laboratory has previously shown that an LHβ reporter gene is induced by activin in transiently transfected LβT2 cells (22), and, recently, this finding was extended in a study in which the endogenous LHβ gene was induced by activin in this mature gonadotrope cell model (23). Despite these reports, activin is still generally regarded as a selective regulator of FSH.
In this report, we used primary mouse pituitary cells enzymatically dispersed in culture to address this discrepancy in the literature by showing that the LHβ gene can be induced by activin treatment. Moreover, using Smad3-deficient mice, we confirmed a role for activin in regulation of LHβ expression in vivo, when we found that these mice have lower expression of LHβ mRNA, in addition to lower levels of FSHβ, compared with their wild-type littermates. We then sought to identify the molecular mechanism of activin induction of LHβ gene expression using LβT2 cells, the mature gonadotrope cell model derived from a transgenic mouse pituitary tumor, which endogenously expresses LHβ. We found that in LβT2 cells, LHβ responds to activin in a time- and dose-dependent manner, and the response maps between −86 and −121 bp from the transcriptional start site. This is an active region of the promoter that contains a homeobox element (HB) at −100 that is crucial for basal and cell-specific expression of the LHβ gene (24). In close proximity, on either side of the HB, are tandem steroidogenic factor 1 (SF-1) and early growth response (Egr) sites (25). The SF-1 sites are located at −127 and −59 and are outside the −121/−86 region where activin response maps. The Egr sites are located at −112 and −50 and convey GnRH responsiveness, upon induction of Egr proteins by GnRH (26, 27). GnRH regulation of the LHβ gene occurs through the combinatorial action of several transcription factors that bind the HB element, the SF-1 and Egr sites, because they show synergistic interaction on the induction of the LHβ gene (26–28). The 5−-Egr site is found in the −121/−86 region of the promoter where activin response is located. We found that mutation of either the HB or the 5′-Egr site dramatically decreases LHβ induction by activin, in addition to decreasing basal expression. In this region, we also identified three Smad-binding elements (SBEs), and mutation of any one of these sites completely abolished activin response. Interestingly, two of these SBEs are also important for basal expression of LHβ. Finally, we demonstrate that overexpressed Smad proteins activate LHβ, bind a SBE in gel-shift assay, and interact with orthodenticle homeobox (Otx) and pituitary homeobox (Ptx) proteins, which can bind the HB. Therefore, activin regulation of LHβ gene expression, similar to LHβ regulation by GnRH, requires multiple, composite DNA elements and involves a combinatorial action of several DNA binding proteins.
RESULTS
Activin Induces LHβ in Primary Mouse Pituitary Cell Culture
There is little doubt that activin is a major regulator of FSH synthesis; however, examination of the effects of activin on LH has resulted in conflicting reports. Some studies have shown that activin regulates LHβ induction in animals and cell culture, both in rat primary pituitary cells and a gonadotrope cell model derived from the mouse; however, others did not detect this response. In this study, we sought to provide a possible explanation for these differing results regarding activin regulation of LH synthesis as well as determine whether activin regulates LHβ induction in mouse primary cells. Using mouse cells instead of rat primary cells allowed us to directly correlate our results with experiments in genetically modified mice and more detailed mechanistic studies using the mouse-derived gonadotrope cell line, LβT2. We enzymatically dispersed mouse pituitary cells and treated them with activin or GnRH for 5 h, either 1 d or 3 d after the dispersion. When cells were treated within 24 h after dispersion, both GnRH and activin induced robust expression of LHβ compared with the vehicle-treated cells (Fig. 1). Therefore, activin, as well as GnRH, induces LHβ gene expression. However, when cells were allowed to recover for 66 h before treatment with GnRH or activin, GnRH treatment still caused a significant induction of LHβ, although to a lesser extent than at 24 h, whereas activin induction failed to reach a significant level. Because the level of LHβ [normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH)], did not differ dramatically in the control cells between d 1 and d 3, the cells’ responsiveness to GnRH and activin seems to have decreased from d 1 to d 3. Thus, it is possible that the length of time in culture before treatment provides an explanation for the contradictory results obtained in other studies of LHβ induction by activin.
Fig. 1.

LHβ Is Induced by Activin in Primary Mouse Pituitary Cells
Primary pituitary cells were dispersed enzymatically from 8-wk-old male mice and plated in culture. They were treated with vehicle (white bars), 30 nM GnRH (hatched bars), or 200 ng/ml activin (black bars) for 5 h, on the first day or on the third day after dispersion. Total RNA was purified and reverse transcribed, and the level of LHβ expression was assayed by real-time PCR. In each sample the amount of LHβ, calculated from the standard curve, was compared with the amount of GAPDH. Asterisk indicates significant difference from the same day control as determined by one-way ANOVA and Tukey’s post hoc test.
Activin Induces LHβ Gene Expression through Proximal Regulatory Sequences
Activin is critical for gonadotrope cell function, and LHβ is crucial for both male and female reproduction (1). Thus, it was important to determine the molecular mechanisms by which activin induces LHβ gene expression. The proximal 1800 bp of the rat LHβ 5′-regulatory region linked to a luciferase reporter gene (LHβ-luc) was transiently transfected into LβT2 cells. This region of the LHβ promoter is sufficient for gonadotrope-specific expression and for GnRH regulation (24, 29). LβT2 cells transiently transfected with LHβ-luc were treated with various concentrations of activin to determine whether this region of the LHβ gene is also sufficient for activin responsiveness. Treatment with activin over a range of doses and at 3, 6, or 24 h revealed that this regulatory region contains elements that allow response to activin in a time- and dose-dependent manner. Maximal induction is achieved after 6 h of treatment and the induction is maintained for at least 24 h of activin treatment (Fig. 2A). There appears to be a decreasing trend at 24 h as compared with the induction at6hof treatment; however, the difference is not statistically significant.
Fig. 2.
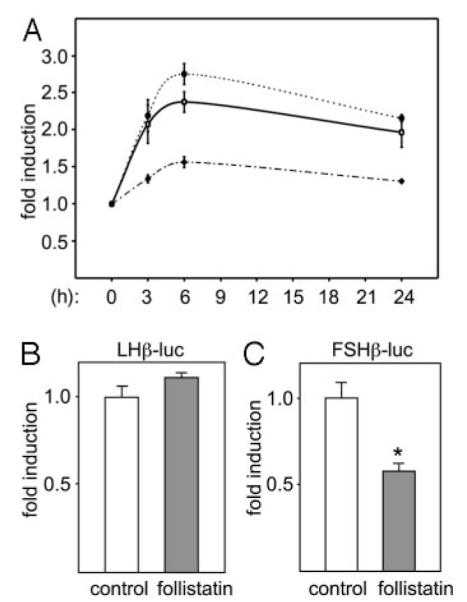
The LHβ Promoter Responds to Activin Treatment
A, The 1800-bp rat LHβ promoter was linked to a luciferase reporter gene (LHβ-luc) and transiently transfected into gonadotrope-derived LβT2 cells. The thymidine kinase β-galactosidase reporter was cotransfected as an internal control. After overnight starvation in serum-free DMEM, the cells were treated with 1 ng/ml activin (dashed line, solid triangles), 10 ng/ml activin (solid line, open squares), or 100 ng/ml activin (dashed line, solid circles) for different lengths of time (indicated on the abscissa). Luciferase activity in the lysates was measured and normalized to β-galactosidase. Results represent the mean ± SEM of at least three independent experiments, each performed in triplicate, and are presented as fold induction from vehicle control. Statistical significance was determined by one-way ANOVA followed by Tukey’s post hoc test. B, LβT2 cells were transiently transfected with either −1800 bp rat LHβ or a −398 bp mouse FSHβ promoter linked to a luciferase reporter and treated with vehicle (white bars) or with 100 ng/ml of follistatin (gray bars) for 24 h, after which the luciferase levels were measured and normalized to the internal control. Results represent the mean ± SEM of at least three independent experiments, each performed in triplicate, and are presented as fold induction from vehicle control. *, Statistically significant difference from vehicle control, as calculated by Student’s t test.
Because LβT2 cells secrete activin and express the activin receptors, thus having a fully functional autocrine loop (22), we assessed the contribution of endogenous activin on LHβ expression. We treated the cells with follistatin for 24 h and measured the expression of LHβ-luc (Fig. 2B). Consistent with our previous report, follistatin treatment does not diminish LHβ levels in these cells (22). This is in contrast to FSHβ in which follistatin treatment decreases expression of FSHβ by about 50%. The concentration of follistatin used in this experiment was sufficient to inhibit induction of either LHβ (data not shown) or FSHβ expression by exogenous activin (30). It is possible that the response of LHβ expression differs with endogenous vs. exogenous activin. It is also possible that there is a threshold level of activin needed to achieve induction that is higher for LHβ as compared with FSHβ and that this threshold is not achieved by endogenous activin produced by LβT2 cells under our conditions.
To identify which promoter elements convey activin responsiveness, we mapped the regions of the LHβ gene promoter that confer activin response using truncation/deletion analysis. LβT2 cells were transiently transfected with a series of truncations of the LHβ gene 5′-flanking region, ranging in length from 1800 bp to 86 bp upstream of the transcription start site, and the ability of activin to induce LHβ gene transcription was tested (Fig. 3). Samples treated with activin were normalized to the vehicle-treated samples for each truncation, and results are represented as fold induction because the basal levels of expression of the truncations vary (24). The response of LHβ to activin mapped between −86 and −121. The 121 most proximal bp of the rat LHβ promoter linked to luciferase was induced about 2-fold, as was the longer 1800 bp of the promoter, whereas truncation to −86 reduced the induction to the level of the vector control.
Fig. 3.

Activin Response Localizes to a 35-bp Region in the LHβ Proximal Promoter
Different lengths of the LHβ regulatory region were transiently transfected into LβT2 cells and, after overnight starvation, the cells were treated with 10 ng/ml activin for 6 h. The results are represented as fold induction from vehicle-treated cells for each truncation. *, Significantly different induction in the treated cells vs. the control cells for each truncation, as determined by Student’s t test. Results represent the mean ± SEM of three independent experiments each performed in triplicate.
Homeobox and Smad-Binding Elements Are Critical for LHβ Induction by Activin
Because the activin response maps directly to this region of 35 bp, we then determined what promoter elements in this region are necessary for this response. The proximal region of the LHβ promoter contains a homeobox element, GATTA, with tandem SF-1 and Egr sites on either side. The Egr-1 sites are important for GnRH induction of LHβ because Egr-1 is induced by GnRH. SF-1 is expressed specifically in the gonadotropes of the pituitary and is important for expression of genes in these cells. The truncations of 86 and 121 bp from the transcription start site of LHβ gene were originally created to narrow this region and limit the number of transcription factor-binding sites. Only the 5′-Egr site and the HB are located in this small region, as illustrated in Fig. 4A with uppercase letters. We created mutations in the Egr and the HB sites and named them mEgr and mHB, respectively (Fig. 4B). As has been shown previously, mutations in these sites decreased basal expression of the LHβ gene. In addition, they decreased the induction by activin (Fig. 4C). The HB site mutation significantly decreased the fold induction by activin by 57%, whereas the mutation of the 5′-Egr site decreased fold induction by activin by 33%. Thus, the 5′-Egr site and especially the HB site are involved in the activin regulation of the LHβ gene. The homeodomain protein, which binds the HB in the gonadotrope cell, was determined to be either an Otx-1-like protein the identity of which is not known, or potentially a posttranslationally modified Otx-1. This protein is expressed at a high basal level in LβT2 cells (24). The involvement of the HB site in activin regulation of LHβ expression may, therefore, involve this Otx-1-like protein. The involvement of the 5′-Egr in activin induction is not clear, because Egr proteins are not expressed in the gonadotrope cell until they are induced by GnRH. Therefore, it was important to assess whether activin can also induce Egr-1 (Fig. 4D). Egr-1 is the major Egr family member induced by GnRH to bind the Egr site in the LHβ promoter. We performed a Western blot to detect Egr-1 protein following GnRH (G) or activin (A) treatment of LβT2 cells. Because Egr-1 is an immediate early gene, induced very rapidly after GnRH treatment (25), we used the same duration of activin treatment to assess the activin affect on Egr-1 protein levels in LβT2 cells. We determined that activin, contrary to GnRH, did not induce Egr-1 protein under these conditions.
Fig. 4.

The Homeobox and 5′-Egr Sites Are Required for Maximal Induction of LHβ by Activin
A, Rat LHβ promoter sequence from −130 to −86 bp from the start site of transcription is shown. The truncations between which the activin response maps are marked with uppercase letters; however, the larger sequence was presented to identify the SF-1 site. The 5′-Egr and the homeobox element (HB) are indicated with an oval and a rectangle, respectively. B, Mutations in the homeobox element (mHB) and Egr (mEgr) were introduced into the LHβ-luc vector, and transfections were performed in LβT2 cells. Cells were treated with vehicle (white bars) or with 10 ng/ml activin (black bars) for 6 h, after which the luciferase activity was measured and normalized to β-galactosidase. The results are normalized to the luciferase/β-galactosidase ratio in the cells transfected with the wild-type reporter and treated with vehicle control. Statistical significance was determined by one-way ANOVA followed by Tukey’s post hoc test. *, Significant induction by activin in the treated cells vs. the control cells for each reporter; #, a significant difference in expression from the wild-type reporter in control-treated cells. Results represent the mean ± SEM of four independent experiments, each performed in triplicate. C, The same results from panel B, presented as percent change in fold induction of the mutated reporters from the induction of the wild-type reporter. #, Statistically significant change in fold induction by one-way ANOVA and Tukey’s test. D, Western blot of LβT2 whole-cell extract after 2-h treatment with vehicle control (C), 10 nM GnRH (G), or 10 ng/ml activin (A). Equal amounts of protein were run on the gel and, after transfer, membranes were probed with antibodies specific for Egr-1 protein. After the secondary antibody, enhanced chemiluminescence was performed and the blots were exposed to film. The experiment was repeated three times with the same result and a representative gel was shown. WT, wild type.
To identify promoter elements that may further contribute to LHβ induction by activin, we examined the DNA sequence in this region more closely. As mentioned, activin signals primarily through Smad2 and/or Smad3, which, together with Smad4, bind SBE, GTCTAGAC, or its half-sites. Sequence analysis revealed that the −121/−86 region contains three SBEs, which may play roles in activin induction of LHβ gene expression (Fig. 5A). Starting from the 5′-end, we termed them S1, S2, and S3 and mutated each of these SBEs to investigate their roles in activin responsiveness (Fig. 5B). Each mutation abolished activin responsiveness of the LHβ gene. Surprisingly, two of these SBEs, S2 and S3, also had a strong influence on LHβ basal expression. The basal expression was reduced by 50% by either mutation as compared with the wild-type sequence.
Fig. 5.

The SBE Sites Are Crucial to the LHβ Response to Activin
A, Rat LHβ promoter sequence from −121 to −86 bp from the start site of transcription is shown. In addition to the indicated homeobox and Egr sites, SBEs (S1, S2 and S3 numbering from 5′ to 3′) are circled for clear observation. B, Point mutations (2 bp) in each of the SBEs were separately introduced into the reporter (mS1, mS2, and mS3), and their induction by activin treatment was compared with the wild-type reporter. *, Significantly different fold induction in the treated cells vs. the control cells for each reporter; #, a significant difference in the level of luciferase expression from the control treated cells transfected with wild-type reporter.
Smad Proteins Are Essential for LHβ Induction by Activin
To determine whether activin signals to the LHβ promoter through its classical pathway by phosphorylation of the Alk4 receptor and activation of the Smadsignaling pathway, we overexpressed Smad7 in LβT2 cells and, after activin treatment, assessed the induction of LHβ. Smad7 serves as a dominant-negative Smad because it prevents phosphorylation of receptor Smads and formation of an active Smad heterodimer of Smad2/3 and Smad4 (31, 32). Complete inhibition of activin signaling by overexpression of Smad7 would indicate that activin uses the classical signaling pathway to induce LHβ gene expression. Indeed, overexpression of Smad7 completely abolishes activin induction of the LHβ gene, indicating that an active Smad pathway is necessary for induction (Fig. 6A). Again, consistent with our results with follistatin treatment, Smad7 does not affect basal LHβ levels, indicating that endogenous activin does not play a role in the regulation of LHβ under these culture conditions.
Fig. 6.

Smad Proteins Are Necessary and Sufficient for LHβ Induction by Activin
A, Cells transfected with wild-type LHβ-luc were treated for 6 h with vehicle (white bars) or 10 ng/ml activin (black bars). Cells were cotransfected with an expression vector for Smad7 or its empty vector control, to assess the necessity for Smad protein activation in the induction by activin. Results represent the means of three independent experiments each performed in triplicate. *, Statistically significant difference from the control cells transfected with the wild-type vector. B, Cells transfected with wild-type LHβ-luc, or each SBE mutation introduced into LHβ-luc, were cotransfected with Smad3 or empty vector control (control). Activin was added to the cells 24 hr after transfection (filled bars are activin treated and white bars are control) and after6hof treatment, luciferase activity was measured and normalized to β-galactosidase. Results represent the means of three independent experiments each performed in triplicate. *, Statistically significant induction by activin; #, statistically significant induction by Smad3 overexpression as analyzed by ANOVA and Tukey’s post hoc test. WT, Wild type.
To further assess the role of Smad proteins in induction of LHβ gene expression, we cotransfected a Smad3 expression vector with LHβ-luc and assessed luciferase levels with and without activin treatment. A recent report has shown that Smad2 cannot be overexpressed in LβT2 cells and that Smad3 alone is sufficient for FSHβ induction, presumably because Smad4 levels are not the limiting factor for activin effect in these cells (33). Similar to FSHβ regulation, LHβ is induced by 3.2-fold with overexpression of Smad3 (Fig. 6B). Mutation of any one of the three SBE sites diminished LHβ-luciferase induction by overexpression of Smad3. Specifically, mutation of the SBE 1 or 2 sites completely abolished induction, not only by activin, as we have shown in the previous figure, but also by Smad3 overexpression. The SBE 3 site mutation considerably reduced the level of induction. Promoter induction was reduced from 3.2-fold for the wild type to 1.65-fold. None of the mutant promoters showed an additional increase with treatment with activin, which is similar to what was observed in Fig. 5B. This result indicates that all three sites play roles in induction by activin and by Smad3 protein. As in previous reports studying FSHβ induction (33, 34), overexpression of Smad3 alone is sufficient to reproduce the effect of activin on LHβ induction.
Smad Proteins Bind an SBE in the LHβ Promoter and Can Interact with Ptx-1 and Otx-1 Proteins
To assess whether Smad proteins can bind the SBE sequences in the −121/−86 region of the promoter, we used EMSA. The whole −121/−86 region was used as a probe (Fig. 7A) with nuclear extracts from Cos 1 cells transfected with the expression vector for Smad4 or empty vector control. Smad proteins have relatively low affinity in binding DNA, and overexpression is one way to overcome this limitation. Comparing complexes that bind this region from cells transfected with empty vector control (first lane) with cells transfected with the Smad4 expression vector (second lane), we detected two strong bands not present in the control lane (marked with arrows) (Fig. 7B). The same pattern was observed when nuclear extract from Cos 1 cells transfected with both Smad3 and Smad4 was used (data not shown). These bands are supershifted (labeled “ss”) with antibodies to Smad4 protein (third and last lane), indicating that Smad proteins are indeed a part of this complex. Thus, Smad proteins can bind this region in vitro. To determine which nucleotides are needed for Smad protein binding, competitions with 200-fold excess of unlabeled mutant oligonucleotides were used. Scanning mutations (3 bp) were introduced into the −121/−86 oligonucleotide and were termed mutations A–K (Fig. 7A). Oligonucleotides with mutations in the second SBE (S2) element (Fig. 7, B and C) could not bind Smads and did not compete with the labeled wild-type probe. Therefore, SBE2at −116 from the start site of the transcription is the site that can be bound by Smad proteins with high enough affinity to be detected in EMSA.
Fig. 7.

Smad Proteins Bind a SBE at −116 in the LHβ Promoter
A, An alignment of wild-type sequence (WT) from the rat LHβ promoter −121/−86, and the oligonucleotides used as competitors, labeled A–K, is shown. Scanning mutations were introduced into oligonucleotides A–K, and these changes are underlined. These unlabeled oligonucleotides were used in 200-fold excess in EMSA experiments in panel B, whereas wild-type sequence (WT) was used as a probe. The SBE sites are indicated with a circle in the wild-type sequence, and the HB site is indicated with a square. B, Nuclear extracts from transfected Cos 1 cells were subjected to EMSA with radiolabeled wild-type probe. Extracts from cells transfected with the control plasmid were used in the lane labeled C, whereas extracts from Cos 1 cells transfected with Smad4 expression vector were used in the remaining lanes. Complexes that change in Smad4-containing lysates are indicated with arrows. Mutated oligonucleotides were used as competitors in 200-fold excess in the corresponding lanes. In the lanes labeled 1 and 2, the antibody to Smad4 was included to induce the supershift, marked with “ss.” The antibody in the lane labeled 1 was obtained from Upstate USA, whereas the antibody in the lane labeled 2 was purchased from Santa Cruz Biotechnology. NE, Nuclear extract; ab, antibody.
In the next EMSA, we used nuclear extracts from LβT2 cells treated with vehicle control or activin, to assess whether activin treatment causes changes in complexes that bind this promoter region. Activin treatment for 30 min, 2 h, or 6 h increased the intensity of a complex indicated with an asterisk in Fig. 8A. The same complex is supershifted with antibodies to Smad4 (indicated with “ss” in the figure), confirming the identity of the band as a Smad4-containing complex. Next, we introduced mutations into the wild-type sequence to assess the ability of each of the SBEs to bind Smad proteins. Subsequently, these mutants were used as probes in comparison with the wild-type sequence. Smad4 antibody was included with nuclear extracts to allow clearer observation of the Smad complex due to the supershift caused by Smad4 antibody (Fig. 8B). A supershift was observed when the wild-type sequence was used as a probe, as demonstrated in Fig. 8A. When the mutated sequence that contains only SBE 2 was used as a probe (2), a supershift with Smad4 antibody was retained, although SBE 1 and SBE 3 were not present. On the other hand, the probes containing only SBE 1 (1) or SBE 3 (3) sites, but lacking SBE 2, did not bind Smad4 in this assay. Thus, again, as with Cos-1 extract, the SBE 2 site is critical for Smad protein binding of LHβ promoter. The SBE 1 and 3 may contribute to the affinity of binding because the intensity of the complex binding probe 2 was lower than that with the wild-type probe. As expected, when all three SBE sites were mutated (Fig. 8A), Smad4 antibody did not cause a supershift, indicating that Smad proteins cannot be tethered to the promoter solely through interaction with other promoter-binding proteins. Furthermore, when we used a mutant probe that contains only the three SBE sites, with the Egr and HB sites mutated (Fig. 8B), Smad4 antibody caused a supershift, indicating that Smad proteins do not need the Egr or HB sites to bind the LHβ promoter.
Fig. 8.

Smad Proteins from Activin-Treated LβT2 Cells Bind the LHβ Promoter
A, EMSA was performed with nuclear extracts from activin-treated LβT2 cells for the times indicated above each lane, using the −121/−86 region of LHβ promoter as a probe. In the IgG lane, nonspecific antibody was included, whereas in the S4 lane, antibody to Smad4 from Santa Cruz Biotechnology was included. “ss” indicates a supershift, whereas * marks a complex that changed after activin treatment. B, Wild-type and mutant probes used in EMSA are illustrated. In the bottom panel, each lane represents a corresponding probe used to assess binding in EMSA with activin-treated nuclear extract and Smad4 antibodies. Ab, Antibody; WT, wild type.
Herein, we have determined in transfection assays that all of the three SBEs found in the −121/−86 region of the LHβ promoter are required for activin responsiveness and that the HB element at −100 is necessary for maximal induction of LHβ by activin. We also determined by EMSA that Smad proteins can bind one of these SBEs found in this region, whereas previously, it has been shown that Ptx-1 or Otx-1 proteins can bind the HB element (24, 35). Further, it has also been shown that proteins that bind this region of the promoter can interact synergistically (27). Consequently, it was important to assess whether Smad proteins can interact with Ptx or Otx proteins. We used a glutathione-S-transferase (GST) pull-down assay with in vitro transcribed and translated Ptx-1 and Otx-1 proteins to test whether they can interact with Smad2-, 3-, or 4-GST fusion proteins. GST pull-down assays indicate that both Ptx-1 and Otx-1 interact with the Smad2, 3, and 4 proteins (Fig. 9). 35S-labeled Ptx-1 and Otx-1 precipitate with glutathione beads through an interaction with GST-Smad2, 3, or 4. No interactions were observed using GST alone, nor did labeled green fluorescent protein (GFP), which serves as a control, interact with any of the GST-fusion proteins. Thus, Ptx-1 and Otx-1 can form heteromeric complexes with Smad proteins in vitro, through protein-protein interaction.
Fig. 9.

Smads2, 3, and 4 Can Interact Directly with Ptx-1 and Otx-1
35S-labeled proteins, indicated on the left of the panels, were used in the binding assay with GST-fusion proteins, labeled above each lane. GST-fusion proteins were induced with isopropyl-β-D-thiogalactosidase overnight, and the bacterial pellets were sonicated. These proteins were bound to Glutathione Sepharose beads and incubated with in vitro transcribed and translated labeled proteins. After extensive washing, the precipitates were run on a gel and subjected to autoradiography. The experiment was repeated three times with the same results and a representative experiment is shown.
Activin Is Critical for LHβ Expression in Vivo
To validate our results obtained using the LβT2 cells and assess the role of activin in LHβ gene expression in vivo, we determined the level of LHβ and FSHβ gene expression in Smad3-deficient mice. These mice lack exon 8 of the Smad3 gene, which prevents synthesis of the functional Smad3 protein (36). Females are infertile due to an ovarian phenotype (37) and, therefore, pituitary effects are impossible to assess. For that reason, we used only male animals in our studies at 8 wk of age. To determine the levels of LHβ and FSHβ mRNA in the pituitary, quantitative PCR was used, and values were normalized to the levels of GAPDH mRNA expression for each animal (Fig. 10). We determined that both LHβ and FSHβ are expressed at a lower level in Smad3-deficient animals than in their wild-type littermates. LHβ was decreased by 38%, whereas FSHβ was reduced by 32%, in comparison with gonadotropin mRNA levels in wild-type mice. Thus, activin plays a role in both LHβ and FSHβ expression in vivo.
Fig. 10.
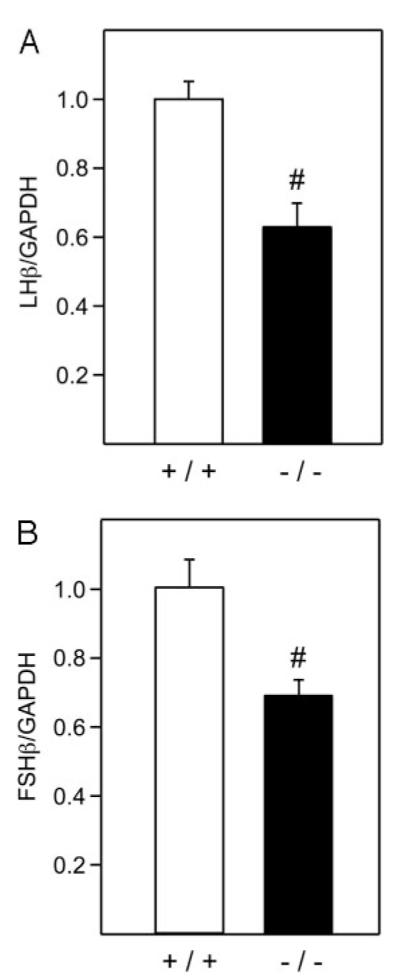
Smad3-Deficient Male Mice Have Lower Levels of LHβ and FSHβ mRNA
Total RNA from individual pituitary was reverse transcribed and LHβ, FSHβ, and GAPDH cDNA were quantified using real-time PCR. For each animal, the amount of LHβ or FSHβ mRNA was calculated from a standard curve performed simultaneously. The amount was compared with the amount of GAPDH also calculated from its standard curve. n = 9 animals per group; the level of LHβ/GAPDH ratio in +/+ animals was set to 1, to allow comparison of the change from wild type. #, Significant difference from the wild-type littermates by Student’s t test: P = 0.0007 for LHβ and P = 0.008 for FSHβ.
DISCUSSION
Recently, a number of studies have shown that activin regulates both LH synthesis and secretion in vivo and in primary pituitary cell culture, both in primates and in rodents. However, due to initial reports that did not detect increased LH synthesis after activin treatment, activin is still generally regarded as a selective regulator of FSH. In gonadotrope-derived LβT2 cells, both endogenous LHβ gene and a reporter gene under LHβ promoter regulation were significantly induced by activin. Herein, we show that activin induces LHβ mRNA, if the primary pituitary culture is treated within 24 h after enzymatic cell dispersion. Several possibilities can be proposed to explain the differences in these various studies. First, it is possible that higher activin doses are necessary for stimulation of LH synthesis as opposed to FSH, and the initial studies did not detect the smaller, but significant, changes in LH due to the lower activin concentrations used. In our studies, activin consistently induced LHβ gene in primary cells with the higher concentration (200 ng/ml), as presented in Fig. 1, whereas lower concentrations of activin (20 ng/ml) significantly increased LHβ gene expression in about 50% of experiments. Second, the difference may be due to the physiological state of the animals from which the pituitaries were extracted or to the stress level the animals experienced during transport or procedure. For example, the animals used could have been from a stage of the estrous cycle when ovarian follistatin is at its highest. On the other hand, circulating activin may have been at a different level. As demonstrated in Fig. 2B, autocrine, endogenous activin does not seem to play a role in LHβ expression, whereas it contributes significantly to FSHβ expression. Circulating activin, however, may have a different effect. It is also credible that different levels of steroid hormones due to cycling of the female animals may have contributed to the differential regulation of FSH and LH by activin. FSH synthesis has been shown to be stimulated by progesterone or testosterone, which also synergize with activin to induce FSH, whereas LH can be suppressed by progesterone or testosterone in LβT2 cells (Thackray, V. G., S. M. McGillivray, and P. L. Mellon, manuscript submitted). To eliminate fluctuation of hormones due to the estrous cycle, we used male animals for our studies. Third, the studies that failed to detect activin induction of LHβ may have used less sensitive techniques to analyze changes in the hormone or gene expression levels. Lastly, the studies that did not observe activin regulation of LH may have had a higher level of folliculostellate cells in their primary pituitary cultures due to having been in culture longer before the experiment. Traditionally, primary cells were left to recover for 48 h after the pituitary dispersion and then were treated with the stimulus of interest, because that was the optimal time for studying secretory response to the releasing hormones. Some studies in the literature still follow this protocol, despite more recent studies with primary pituitary cultures that have reported that secretory cells are rapidly overgrown by folliculostellate cells, which secrete follistatin, a potent activin inhibitor (14). Our results appear to correlate with the rationale of overgrowth of folliculostellate cells, because the LHβ induction by activin was more robust when the treatment was conducted 1 d after dispersion. When the cells were treated on the third day after dispersion in culture, induction of LHβ was far less prominent, perhaps due to high levels of follistatin secreted by the predominant folliculostellate cells.
Smad3-deficient mice have lower levels of both LHβ and FSHβ mRNA when compared with their wild-type littermates, confirming a role for activin in gonadotropin regulation in vivo. Female animals have a severe ovarian phenotype, and any effect on the gonadotropes may be due to the lack of gonadal feedback. To circumvent these problems, we have used only male animals. Although fertile, Smad3-deficient males are considerably smaller and we determined that they also have lower gonadotropin levels than their littermates. Because of the importance of activin in appropriate gonadotropin gene expression and the necessity for LHβ in both male and female reproductive function, we proceeded to identify the molecular mechanism of activin regulation of LHβ gene expression.
Activin response maps to a very active region of the promoter that contains elements for basal, cell-specific, and GnRH regulation of the LHβ gene. Mutation of the HB site, GATTA, in this region, dramatically reduces activin responsiveness. The HB is critical for basal regulation, and it is found to bind the Ptx proteins overexpressed in CV-1 cells (27) or an Otx-1-related protein, expressed endogenously in the LβT2 cells (24). Therefore, this site is also involved in cell specificity of LHβ expression, because LβT2 cells, which represent the most mature gonadotrope cells and endogenously express LHβ, are the only gonadotrope-derived cell type that expresses the protein that binds this site. A less mature gonadotrope cell model, such as the αT3–1 cell, which does not express LHβ, does not express this protein. An SF-1 site is found in close proximity to the HB, and it may also contribute to cell-specific expression, because SF-1 is limited to the gonadotrope population within the pituitary. Ptx proteins were found to synergize with SF-1 to induce LHβ gene expression in CV-1 cells, which do not express endogenous SF-1 (28). LβT2 cells express high levels of endogenous SF-1, and thus it is difficult to observe this regulation in these cells. Regardless, the SF-1 site was not present in the −121/−86 region of the promoter that remains activin responsive, and therefore we concentrated on the HB and Egr sites. Here we find that both HB and the 5′-Egr site have roles in activin regulation of LHβ gene expression, because mutation of these reduces the magnitude of activin induction.
GnRH regulation of LHβ occurs through the induction of Egr proteins, which bind their two sites also found in this region of the promoter. Interestingly, there is a functional difference between the 5′- and 3′-Egr sites. Mutation of the 3′-Egr site diminishes GnRH induction, whereas the 5′-site does not seem to play a role in GnRH regulation of LHβ gene (38). Also, in vitro binding to these sites by EMSA showed that Egr-1 binds with high affinity to the 3′-Egr site, whereas the intensity of binding to the 5′-site is orders of magnitude lower (25, 39). This raises the question whether the 5′-Egr site is bound in vivo by a different transcription factor. Because the 5′-Egr site does not seem to bind Egr proteins with high affinity nor is it involved in GnRH induction of LHβ, this site may bind a different member of the Egr/SP-1 superfamily. It is interesting to speculate that this protein may be induced by activin and may play a role in activin regulation of LHβ. On the other hand, it has been demonstrated that Ptx can also synergize with Egr to induce LHβ (27). Interestingly, mutation of the Ptx element has a more profound effect, and it is possible that the Egr site contributes to the activin induction only because of its synergistic interaction with Ptx or Otx.
Mutation of any of the three SBE sites completely abolishes induction by activin and dramatically diminishes induction by Smad3 cotransfection. This requirement for multiple sites has been found in other TGFβ/ activin-responsive promoters (40). Multiple SBEs in close proximity to each other allow for cooperative binding of Smad proteins and allow them to overcome their low binding affinity. Furthermore, the requirements for other promoter elements for full activin response have also been reported (41). It is likely that a similar situation occurs with the LHβ promoter, in which the Ptx/Otx-binding homeobox site is required for full activin response. Interaction of Smad proteins with other transcription factors has been reported (42). Most notably, Smad2 is known to interact with the homeobox proteins Milk and Mixer (43). Herein we detect that Smads 2, 3, and 4 can interact with homeobox proteins Ptx-1 and Otx-1. It is likely that, because Smads can interact with Ptx-1 in addition to Otx-1, they can also interact with a related family member expressed endogenously in LβT2 cell, the Otx-1-like protein, the identity of which is not known. Possibly, that interaction with other proteins stabilizes Smad interaction with DNA and, in the case of LHβ, may play a role in activin induction. Additionally, SBE site 2, which has a dramatic effect both on basal and activin-induced gene expression, and is bound by Smad4 in EMSA, is juxtaposed to the 5′-Egr site. Thus, it is possible that Smad proteins are interacting with this putative Egr family member to regulate both basal and activin-regulated LHβ gene expression.
We determined that Smad proteins bind the DNA in the −121/−86 region of the promoter using EMSA both from Cos-1 cells with overexpressed Smad proteins and from activin-treated LβT2 cells. Specifically, we established that the middle SBE is needed for this binding. This SBE is located 5′ to the HB and to the Egr site. It is possible that the binding affinity for the other SBEs is too low to detect in a gel shift, because our transient transfection experiments indicate that all of the SBEs are required for activin response. SBE 1 and 3 may contribute to the stability of Smad protein binding, because, when only SBE 2 is present, the intensity of binding is lower than when all three sites are present.
Surprisingly, two of the three SBEs identified are critical for LHβ basal expression as well. These two SBEs are located around the HB, which has been shown to be crucial for the basal expression of LHβ gene. However, these SBE sites are sufficiently separate such that the sequences do not overlap the HB, and neither site is needed for binding of the homeodomain protein (24). Because neither follistatin nor overexpression of Smad7 can reduce the basal expression of LHβ, it appears that endogenous activin does not play a role in supporting basal expression under these culture conditions, and therefore what we are measuring are changes in “true” basal expression due to these cis mutations. It would be interesting to determine whether Smad proteins have a role in basal expression and the manner in which they accomplish it. Smad4 has been shown to shuttle in and out of the nucleus independent of activin treatment (44). Given that Smad4 can bind the SBE 2 that is critical for basal regulation, whereas Ptx or Otx bind the homeobox element also involved in basal expression, Smad4 interaction with Ptx and Otx may contribute to basal expression of LHβ gene though interaction with these homeodomain proteins. Alternatively, whether some other proteins, which are involved in both basal and activin induction, can bind these sites remains to be determined.
In this report, we determined that activin plays a role in LHβ expression in vivo and induces LHβ mRNA in primary mouse pituitary cells before overgrowth of secretory cells by folliculostellate cells, which may diminish bioavailability of activin by secreting follistatin. Activin acts through three SBE sites, the mutation of which abolishes activin induction, and, of which, two are critical for basal expression of the LHβ gene as well. Interestingly, the region of the LHβ promoter where these SBE sites are found is also critical for induction by GnRH and for basal expression. Transcription factors that bind elements in this region of the promoter have been shown to physically interact, forming a complex. Because Smads bind and induce LHβ and they can interact with homeodomain proteins, this suggests that Smad proteins are a part of a large, transcriptionally active complex that is critical for the regulation of the LHβ gene transcription.
MATERIALS AND METHODS
Primary Mouse Pituitary Cell Culture and Treatment
Male C57 Black 6 mice (6 wk of age) were purchased from Harlan Sprague Dawley (Indianapolis, IN), and housed for 2 wk in UCSD animal facility under standard conditions. At 8 wk of age, mice were killed by decapitation and pituitaries were collected in ice-cold Dulbecco’s A PBS (PBS). After a PBS rinse, pituitaries were placed in Hank’s balanced salt solution with 25 mM HEPES, pH 7.2, containing 0.25% collagenase and 0.25% trypsin. After cell dispersion for 30 min at 37 C in a shaking waterbath, the same volume of DMEM with 10% fetal bovine serum was added to stop the reaction and then deoxyribonuclease was added to a final concentration of 20 μg/ml and incubated for another 15 min at 37 C. Subsequent to removal of tissue debris, cells were pelleted by centrifugation and plated at 106 cells/2-cm2 well density. The medium was changed to serum-free DMEM with 0.1% bovine serum albumin (BSA) 16 h before treatment. Cells were treated with GnRH (Sigma Chemical Co., St. Louis, MO) or activin (Calbiochem, La Jolla, CA) for 5 h, after which the cells were lysed to obtain total RNA.
Smad 3-deficient mice and their littermates were housed in our facility in accordance with approved animal protocols. At 8 wk of age, mice were killed and pituitaries were removed for total RNA extraction.
Quantitative Real-Time PCR
RNA was obtained with Trizol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Contaminating DNA was removed with DNA-free reagent (Ambion, Inc., Austin, TX) and 2 μg RNA was reverse transcribed using Superscript III First-strand Synthesis System (Invitrogen, Carlsbad, CA). Quantitative real-time PCR was performed in iCycler from Bio-Rad Laboratories, Inc. (Hercules, CA), using QuantiTect SYBR Green PCR Kit (QIAGEN, Valencia, CA) and the following primers:
LH forward: CTGTCAACGCAACTCTGG
LH reverse: ACAGGAGGCAAAGCAGC
GAPDH forward: TGCACCACCAACTGCTTAG
GAPDH reverse: GGATGCAGGGATGATGTTC
FSH forward: GCCGTTTCTGCATAAGC
FSH reverse: CAATCTTACGGTCTCGTATACC
under the following conditions: 95 C for 15 min, followed by 40 cycles at 95 C for 15 sec, 54 C for 30 sec, and 72 C for 30 sec. For LHβ and FSHβ measurements, equivalent to 50 ng of starting RNA (as quantified before reverse transcription) was used in each reaction, whereas for GAPDH 10 ng was sufficient. Each sample was assayed in triplicate, and the experiment was repeated four times. A standard curve with dilutions of 10 pg/well, 1 pg/well, 100 fg/well, and 10 fg/well of a plasmid containing LHβ cDNA, FSHβ cDNA, or GAPDH cDNA was generated in each run with the samples. In each experiment, the amount of LHβ was calculated by comparing threshold cycle obtained for each sample with the standard curve generated in the same run. Replicates were averaged and divided by the mean value of GAPDH in the same sample. After each run, a melting curve analysis was performed to confirm that a single amplicon was generated in each reaction.
Cell Culture and Transient Transfection
LβT2 cells were plated in 12-well plates 1 d before transfection. Transfection was performed in DMEM with 10% FBS using Fugene 6 reagent (Roche Molecular Biochemicals, Indianapolis, IN) following the manufacturer’s instructions. Each well was transfected with 0.5 μg rat LHβ-luciferase or mouse FSHβ-luciferase plasmid (45). Plasmid construction and preparation were described previously (24). The sequences of all of the promoter fragments were confirmed by dideoxynucleotide sequencing. An expression plasmid containing β-galactosidase driven by the Herpes virus thymidine kinase promoter was co-transfected with LHβ-luc and used as an internal control. Sixteen hours after transfection, the cells were switched to serum-free DMEM supplemented with 0.1% BSA, 5 mg/liter transferrin, and 50 nM sodium selenite. The following day, the cells were treated with 10 ng/ml activin for 6 h, unless otherwise indicated, or follistatin (R&D Systems, Minneapolis, MN) for 24 h. The cells were then lysed with 0.1 M K-phosphate buffer (pH 7.8) with 0.2% Triton X-100. Equal volumes of each lysate were placed in 96-well plates, and luciferase activity was measured on a luminometer (EG&G Microplate, Berthold Technologies, Oak Ridge, TN) by injecting 100 μl of a buffer containing 100 mM Tris-HCl (pH 7.8), 15 mM MgSO4,10mM ATP, and 65 μM luciferin per well. Galactosidase activity was measured using the Galactolight assay (Tropix, Bedford, MA) following the manufacturer’s instructions. All transfection experiments were performed in triplicate and repeated at least three times. Luciferase values from reporter gene-transfected cells were consistently at least 100 times higher than values from mock transfected cells. Results represent the mean ± SEM of all samples analyzed. Asterisk marks a statistically significant difference from the control-treated cells, determined by one-way ANOVA analysis followed by Tukey’s post hoc multiple range test for individual comparison with P ≤ 0.05 or Student’s t test as indicated.
Western Blot
After overnight starvation and 2-h treatment with vehicle control, 10 nM GnRH, or 10 ng/ml activin, LβT2 cells were rinsed with PBS and lysed with lysis buffer [20 mM Tris-HCl (pH 7.4), 140 mM NaCl, 0.5% Nonidet P-40 (NP-40), 0.5 mM EDTA], with protease inhibitors (aprotinin, pepstatin, leupeptin) at 10 μg/ml each, and 1 mM phenyl-methyl-sulfonyl-fluoride (PMSF). Protein concentration was determined with Bradford reagent (Bio-Rad Laboratories), and an equal amount of protein per sample was loaded on an SDS-PAGE gel. After proteins had been resolved by electrophoresis and transferred to a polyvinylidene fluoride membrane, they were probed with specific antibodies for Egr-1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The bands were detected with secondary antibodies linked to horseradish peroxidase and enhanced chemiluminescence reagent (Amersham Pharmacia Biotech, Piscataway, NJ).
EMSA
LβT2 cells, after vehicle control or 10 ng/ml activin treatment for the times indicated, or Cos 1 cells, transfected for 24 h with expression vectors containing Smad 3 and/or Smad 4 cDNAs (kindly provided by Rik Derynck), or vector control, were scraped in hypotonic buffer (20 mM Tris-HCl, pH 7.4; 10 mM NaCl, 1 mM MgCl2) with protease inhibitors (aprotinin, pepstatin, leupeptin) at 10 μg/ml each, and 1 mM PMSF and allowed to swell on ice. Cells were broken by passing through a 25-gauge needle, and the nuclei were pelleted by centrifugation. Nuclear proteins were extracted in hypertonic buffer (20 mM HEPES, pH 7.8; 420 mM KCl; 1.5 mM MgCl2 with protease inhibitors; and 20% glycerol). Nuclear protein (2 μg per sample) was used in the binding reaction (10 mM HEPES, pH 7.8; 50 mM KCl; 5 mM MgCl2;5mM dithiothreitol; 0.1% BSA; 0.1% NP-40 with 0.5 μg/ml polydeoxyinosinic deoxycytidylic acid; and 2 fmol per reaction of end-labeled probe). Oligonucleotides were labeled with T4 kinase using γ-32P-labeled ATP. In the competition experiments, competitor oligonucleotide was added 10 min before addition of the probe, as were the antibodies in the supershift assays. The Smad4 antibody and nonspecific IgG were obtained from Santa Cruz Biotechnology and from Upstate USA, Inc. (Charlottesville, VA). The reaction was loaded on a 5% acrylamide gel in 0.25× TBE and run on 0.5 V/ cm2 constant voltage. After drying, gels were exposed to autoradiography.
Mutagenesis
Mutagenesis of the LHβ-luc plasmid was performed using the QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. The HB mutation and Egr site mutation were described in a previous report (24). From the wild-type sequence 5′-TGTCTGTCTCGCCCCCAAAGAGATTAGTGTCTAGG-3′, three SBEs (S1, S2, and S3; GTCT, underlined) were individually mutated to GTAG and named mutant SBE 1 (mS1, most 5′), mutant SBE 2 (mS2, middle SBE), and mutant SBE 3 (mS3, most 3′). Mutations were confirmed by dideoxyribonucleotide sequencing performed by the DNA Sequencing Shared Resource, UCSD Cancer Center.
GST Interaction Assay
The GST-Smad2, 3, and 4 in the pGEX vector were kindly provided by Dr. Rik Derynck. The Otx-1 and Ptx-1 expression vectors were obtained from Dr. Antonio Simeone and Dr. Jacques Drouin, respectively. The GFP expression vector was provided by Dr. Douglass Forbes. 35S-labeled proteins were produced using the TnT T7 Coupled Reticulocyte Lysate System (Promega Corp., Madison, WI). Bacteria transformed with the pGEX vectors were grown to an OD of 0.6, upon which protein expression was induced by addition of 0.25 mM isopropyl-β-D-thiogalactosidase. Bacterial pellets were sonicated in PBS with 5 mM EDTA and 0.1% Triton X-100 and centrifuged, and the supernatant was bound to glutathione sepharose beads (Amersham Pharmacia Biotech). Beads were washed four times with sonication buffer followed by equilibration in the binding buffer (below) and split equally between different samples and the control. 35S-labeled proteins were added to the beads and bound for 1 h at 4 C in 20 mM HEPES (pH 7.8), with 50 mM NaCl, 10 mg/ml BSA, 0.1% NP-40, and 5 mM dithiothreitol. After extensive washing, samples were eluted from the beads by boiling in Laemmli sample buffer and subjected to SDS-PAGE. After-ward, the gels were dried and autoradiographed.
Acknowledgments
We thank Rik Derynck who kindly provided the GST-Smad2, 3, and 4 in the pGEX vector and Smad3 and 4 expression vectors, and Suzanne Rosenberg Jacobs for many LHβ reporter plasmids. We are grateful to Antonio Simeone and Jacques Drouin from whom we obtained the Otx-1 and Ptx-1 expression vectors, respectively, and to Douglass Forbes for the GFP expression vector. We appreciate the time and insight of the Mellon laboratory members and Janice Sue Bailey for helpful discussions.
This work was supported by National Institute of Child Health and Human Development/National Institutes of Health (NIH) through a cooperative agreement (U54 HD12303) as part of the Specialized Cooperative Centers Program in Reproduction Research (to P.L.M.). This work was also supported by NIH Grant R37 HD20377 (to P.L.M.). D.C. was supported by NIH National Research Service Award F32 HD41301 and the Lalor Foundation. V.G.T. was supported by NIH National Research Service Award F32 DK065437. University of California San Diego Cancer Center was funded in part by National Cancer Institute Cancer Center Support Grant P30 CA23100.
Abbreviations
- BSA
Bovine serum albumin
- Egr
early growth response
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- GST
glutathione-S-transferase
- HB
homeobox element
- LHβ
LH β-subunit
- NP-40
Nonidet P-40
- Otx
orthodenticle homeobox
- Ptx
pituitary homeobox
- SBE
Smad-binding element
- SF-1
steroidogenic factor 1
REFERENCES
- 1.Ma X, Dong Y, Matzuk MM, Kumar TR. Targeted disruption of luteinizing hormone β-subunit leads to hypogonadism, defects in gonadal steroidogenesis, and infertility. Proc Natl Acad Sci USA. 2004;101:17294–17299. doi: 10.1073/pnas.0404743101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Annu Rev Biochem. 1981;50:465–495. doi: 10.1146/annurev.bi.50.070181.002341. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Differential effects of gonadotropin-releasing hormone (GnRH) pulse frequency on gonadotropin subunit and GnRH receptor messenger ribonucleic acid levels in vitro. Endocrinology. 1997;138:1224–1231. doi: 10.1210/endo.138.3.4968. [DOI] [PubMed] [Google Scholar]
- 4.Papavasiliou SS, Zmeili S, Khoury S, Landefeld TD, Chin WW, Marshall JC. Gonadotropin-releasing hormone differentially regulates expression of the genes for luteinizing hormone α and β subunits in male rats. Proc Natl Acad Sci USA. 1986;83:4026–4029. doi: 10.1073/pnas.83.11.4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jorgensen JS, Quirk CC, Nilson JH. Multiple and overlapping combinatorial codes orchestrate hormonal responsiveness and dictate cell-specific expression of the genes encoding luteinizing hormone. Endocr Rev. 2004;25:521–542. doi: 10.1210/er.2003-0029. [DOI] [PubMed] [Google Scholar]
- 6.Ling N, Ying S-Y, Ueno N, Shimasaki S, Esch F, Hotta O, Guillemin R. Pituitary FSH is released by a heterodimer of the β-subunits from the two forms of inhibin. Nature. 1986;321:779–782. doi: 10.1038/321779a0. [DOI] [PubMed] [Google Scholar]
- 7.Weiss J, Guendner MJ, Halvorson LM, Jameson JL. Transcriptional activation of the follicle-stimulating hormone β-subunit gene by activin. Endocrinology. 1995;136:1885–1891. doi: 10.1210/endo.136.5.7720634. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura T, Takio K, Eto Y, Shibai H, Titani K, Sugino H. Activin-binding protein from rat ovary is follistatin. Science. 1990;247:836–838. doi: 10.1126/science.2106159. [DOI] [PubMed] [Google Scholar]
- 9.Shimasaki S, Koga M, Esch F, Cooksey K, Mercado M, Koba A, Ueno N, Ying S-Y, Ling N, Guillemin R. Primary structure of the human follistatin precursor and its genomic organization. Proc Natl Acad Sci USA. 1988;85:4218–4222. doi: 10.1073/pnas.85.12.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaiser UB, Lee BL, Carroll RS, Unabia G, Chin WW, Childs GV. Follistatin gene expression in the pituitary: localization in the gonadotropes and folliculostellate cells in diestrous rats. Endocrinology. 1992;130:3048–3056. doi: 10.1210/endo.130.5.1572312. [DOI] [PubMed] [Google Scholar]
- 11.Roberts V, Meunier H, Vaughan J, Rivier J, Rivier C, Vale W, Sawchenko P. Production and regulation of inhibin subunits in pituitary gonadotropes. Endocrinology. 1989;124:552–554. doi: 10.1210/endo-124-1-552. [DOI] [PubMed] [Google Scholar]
- 12.Kogawa K, Nakamura T, Sugino K, Takio K, Titani K, Sugino H. Activin-binding protein is present in pituitary. Endocrinology. 1991;128:1434–1440. doi: 10.1210/endo-128-3-1434. [DOI] [PubMed] [Google Scholar]
- 13.Gospodarowicz D, Lau K. Pituitary follicular cells secrete both vascular endothelial growth factor and follistatin. Biochem Biophys Res Commun. 1989;165:292–298. doi: 10.1016/0006-291x(89)91068-1. [DOI] [PubMed] [Google Scholar]
- 14.Kawakami S, Fujii Y, Okada Y, Winters SJ. Paracrine regulation of FSH by follistatin in folliculostellate cell-enriched primate pituitary cell cultures. Endocrinology. 2002;143:2250–2258. doi: 10.1210/endo.143.6.8857. [DOI] [PubMed] [Google Scholar]
- 15.Attisano L, Wrana JL. Signal transduction by the TGF-β superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 16.Massague J. TGF-β signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 17.Katayama T, Shiota K, Takahashi M. Activin A increases the number of follicle-stimulating hormone cells in anterior pituitary cultures. Mol Cell Endocrinol. 1990;69:179–185. doi: 10.1016/0303-7207(90)90011-v. [DOI] [PubMed] [Google Scholar]
- 18.Stouffer RL, Woodruff TK, Dahl KD, Hess DL, Mather JP, Molskness TA. Human recombinant activin-A alters pituitary luteinizing hormone and follicle-stimulating hormone secretion, follicular development, and steroidogenesis, during the menstrual cycle in rhesus monkeys. J Clin Endocrinol Metab. 1993;77:241–248. doi: 10.1210/jcem.77.1.8325947. [DOI] [PubMed] [Google Scholar]
- 19.McLachlan RI, Dahl KD, Bremner WJ, Schwall R, Schmelzer CH, Mason AJ, Steiner RA. Recombinant human activin-A stimulates basal FSH and GnRH-stimulated FSH and LH release in the adult male macaque, Macaca fascicularis. Endocrinology. 1989;125:2787–2789. doi: 10.1210/endo-125-5-2787. [DOI] [PubMed] [Google Scholar]
- 20.Attardi B, Miklos J. Rapid stimulatory effect of activin-A on messenger RNA encoding the follicle-stimulating hormone β-subunit in rat pituitary cell cultures. Mol Endocrinol. 1990;4:721–726. doi: 10.1210/mend-4-5-721. [DOI] [PubMed] [Google Scholar]
- 21.Blumenfeld Z, Ritter M. Inhibin, activin, and follistatin in human fetal pituitary and gonadal physiology. Ann NY Acad Sci. 2001;943:34–48. doi: 10.1111/j.1749-6632.2001.tb03788.x. [DOI] [PubMed] [Google Scholar]
- 22.Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang H-J, Miller WL, Mellon PL. Cell-specific transcriptional regulation of FSHβ by activin and GnRH in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- 23.Yamada Y, Yamamoto H, Yonehara T, Kanasaki H, Nakanishi H, Miyamoto E, Miyazaki K. Differential activation of the luteinizing hormone β-subunit promoter by activin and gonadotropin-releasing hormone: a role for the mitogen-activated protein kinase signaling pathway in LβT2 gonadotrophs. Biol Reprod. 2004;70:236–243. doi: 10.1095/biolreprod.103.019588. [DOI] [PubMed] [Google Scholar]
- 24.Rosenberg SB, Mellon PL. An Otx-related homeodomain protein binds an LHβ promoter element important for activation during gonadotrope maturation. Mol Endocrinol. 2002;16:1280–1298. doi: 10.1210/mend.16.6.0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halvorson LM, Ito M, Jameson JL, Chin WW. Steroidogenic factor-1 and early growth response protein 1 act through two composite DNA binding sites to regulate luteinizing hormone β-subunit gene expression. J Biol Chem. 1998;273:14712–14720. doi: 10.1074/jbc.273.24.14712. [DOI] [PubMed] [Google Scholar]
- 26.Dorn C, Ou Q, Svaren J, Crawford PA, Sadovsky Y. Activation of luteinizing hormone β gene by gonadotropin-releasing hormone requires the synergy of early growth response-1 and steroidogenic factor-1. J Biol Chem. 1999;274:13870–13876. doi: 10.1074/jbc.274.20.13870. [DOI] [PubMed] [Google Scholar]
- 27.Tremblay JJ, Drouin J. Egr-1 is a downstream effector of GnRH and synergizes by direct interaction with Ptx1 and SF-1 to enhance luteinizing hormone β gene transcription. Mol Cell Biol. 1999;19:2567–2576. doi: 10.1128/mcb.19.4.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tremblay JJ, Lanctôt C, Drouin J. The pan-pituitary activator of transcription, Ptx1 (pituitary homeobox 1), acts in synergy with SF-1 and Pit1 and is an upstream regulator of the Lim-homeodomain gene Lim3/Lhx3. Mol Endocrinol. 1998;12:428–441. doi: 10.1210/mend.12.3.0073. [DOI] [PubMed] [Google Scholar]
- 29.Vasilyev VV, Lawson MA, DiPaolo D, Webster NJG, Mellon PL. Different signaling pathways control acute induction versus long-term repression of LHβ transcription by GnRH. Endocrinology. 2002;143:3414–3426. doi: 10.1210/en.2001-211215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobs SBR, Coss D, McGillivray SM, Mellon PL. Nuclear factor-Y and steroidogenic factor-1 physically and functionally interact to contribute to cell-specific expression of the mouse follicle-stimulating hormone-β gene. Mol Endocrinol. 2003;17:1470–1483. doi: 10.1210/me.2002-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 32.Whitman M. Signal transduction. Feedback from inhibitory SMADs. Nature. 1997;389:549–551. doi: 10.1038/39202. [DOI] [PubMed] [Google Scholar]
- 33.Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone β subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623. doi: 10.1210/me.2003-0264. [DOI] [PubMed] [Google Scholar]
- 34.Suszko MI, Lo DJ, Suh H, Camper SA, Woodruff TK. Regulation of the rat follicle-stimulating hormone β-subunit promoter by activin. Mol Endocrinol. 2003;17:318–332. doi: 10.1210/me.2002-0081. [DOI] [PubMed] [Google Scholar]
- 35.Tremblay JJ, Marcil A, Gauthier Y, Drouin J. Ptx1 regulates SF-1 activity by an interaction that mimics the role of the ligand-binding domain. EMBO J. 1999;18:3431–3441. doi: 10.1093/emboj/18.12.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomic D, Brodie SG, Deng C, Hickey RJ, Babus JK, Malkas LH, Flaws JA. Smad 3 may regulate follicular growth in the mouse ovary. Biol Reprod. 2002;66:917–923. doi: 10.1095/biolreprod66.4.917. [DOI] [PubMed] [Google Scholar]
- 38.Weck J, Anderson AC, Jenkins S, Fallest PC, Shupnik MA. Divergent and composite gonadotropin-releasing hormone-responsive elements in the rat luteinizing hormone subunit genes. Mol Endocrinol. 2000;14:472–485. doi: 10.1210/mend.14.4.0453. [DOI] [PubMed] [Google Scholar]
- 39.Wolfe MW, Call GB. Early growth response protein 1 binds to the luteinizing hormone-promoter and mediates gonadotropin-releasing hormone-stimulated gene expression. Mol Endocrinol. 1999;13:752–763. doi: 10.1210/mend.13.5.0276. [DOI] [PubMed] [Google Scholar]
- 40.Lopez-Rovira T, Chalaux E, Massague J, Rosa JL, Ventura F. Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic proteinspecific transcriptional activation of Id1 gene. J Biol Chem. 2002;277:3176–3185. doi: 10.1074/jbc.M106826200. [DOI] [PubMed] [Google Scholar]
- 41.Datta PK, Blake MC, Moses HL. Regulation of plasminogen activator inhibitor-1 expression by transforming growth factor-β-induced physical and functional interactions between smads and Sp1. J Biol Chem. 2000;275:40014–40019. doi: 10.1074/jbc.C000508200. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Feng XH, Derynck R. Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-β-induced transcription. Nature. 1998;394:909–913. doi: 10.1038/29814. [DOI] [PubMed] [Google Scholar]
- 43.Germain S, Howell M, Esslemont GM, Hill CS. Homeodomain and winged-helix transcription factors recruit activated Smads to distinct promoter elements via a common Smad interaction motif. Genes Dev. 2000;14:435–451. [PMC free article] [PubMed] [Google Scholar]
- 44.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 45.Coss D, Jacobs SB, Bender CE, Mellon PL. A novel AP-1 site is critical for maximal induction of the follicle-stimulating hormone β gene by gonadotropin-releasing hormone. J Biol Chem. 2004;279:152–162. doi: 10.1074/jbc.M304697200. [DOI] [PMC free article] [PubMed] [Google Scholar]