

Abstract
GnRH regulates pituitary gonadotropin gene expression through GnRH receptor activation of the protein kinase C (PKC) and calcium signaling cascades. The pulsatile pattern of GnRH release is crucial for induction of LHβ-subunit (LHβ) gene expression; however, continuous prolonged GnRH exposure leads to repression of LHβ gene transcription. Although in part, long-term repression may be due to receptor down-regulation, the molecular mechanisms of this differential regulation of LHβ transcription are unknown. Using transfection into the LH-secreting immortalized mouse gonadotrope cell line (LβT4), we have demonstrated that LHβ gene transcription is increased by acute activation (6 h) of GnRH receptor or PKC but not calcium influx; in contrast long-term activation (24 h) of GnRH receptor, PKC, or calcium influx each repress LHβ transcription. Whereas blockade of PKC prevented the acute action of GnRH and unmasked an acute repression of LHβ transcription by calcium, it did not prevent long-term repression by GnRH or calcium. Removal of calcium resulted in potentiation of acute GnRH and PKC induction of LHβ gene expression but prevented long-term repression by GnRH and reduced long-term repression by either calcium or 12-O-tetradecanoyl-phorbol-13-acetate (TPA). We conclude that GnRH uses PKC for acute induction, and calcium signaling is responsible for long-term repression of LHβ gene expression by GnRH. Furthermore, analysis of the responsiveness of truncated and mutated LHβ promoter regions demonstrated that not only do acute induction and long-term repression use different signaling systems, but they also use different target sequences for regulating the LHβ gene.
LH is a heterodimeric glycoprotein consisting of a unique β-subunit and a common α-subunit that is also shared by FSH, thyroid-stimulating hormone, and chorionic gonadotropin (1–4). LH is synthesized in anterior pituitary gonadotrope cells in which it is regulated by GnRH. Because the LH β-subunit is present at lower levels than the α-subunit, the concentration of LH β-subunit is the limiting factor in LH synthesis and secretion (5). Thus, understanding the regulation of the LHβ gene by GnRH is key to understanding the synthesis and secretion of LH.
GnRH is synthesized by hypothalamic neurons and delivered through the hypophyseal portal system to the pituitary in a pulsatile fashion. The intermittent pattern of release is critical for normal sexual development and gametogenesis because interruption of GnRH pulses or administration of long-acting GnRH analogs and antagonists result in suppression of both gonadotropin and gonadal steroid production, resulting in infertility (6). Specifically, GnRH pulsatility is essential for induction of LHβ gene expression because continuous incubation for 24 h with GnRH leads to desensitization of GnRH receptors (GnRH-Rs) and down-regulation of LH β-subunit mRNA, although not α-subunit mRNA (7, 8).
GnRH acts on gonadotropin gene expression through the GnRH-R, a G protein-coupled receptor that activates several signal transduction pathways (9, 10). This receptor activates L-type calcium channels, causing an influx of extracellular calcium (11, 12), and also activates phospholipase C (PLC). PLC cleaves phosphatidylinositoldiphosphate located in the cell membrane into inositol triphosphate, which mediates the calcium release from intracellular stores, and produces diacylglycerol (DAG) (13). Increased concentrations of intra-cellular calcium together with DAG, lead to activation of protein kinase C (PKC), which, in turn, activates other proteins by phosphorylation. This generally results in activation of downstream protein kinases such as those of the MAPK pathway (13, 14). The increase in calcium concentration in the cytoplasm can also activate other protein kinases such as the c-Jun N-terminal kinase (15, 16) independently of PKC or other members of the MAPK family. In addition, there are examples in other G protein-coupled receptor signaling systems in which nuclear calcium may change gene expression independently from cytoplasmic calcium (17, 18).
Incoming GnRH signals acting through this single receptor differentially regulate the gonadotropin subunit genes during the estrous cycle and in a variety of pharmacologic and pathophysiologic conditions. Differences in GnRH pulse frequency and/or receptor density allow gonadotrope cells to differentially activate LH vs. FSH synthesis (19–22), likely caused by differential activation of downstream signaling pathways. It is thought that signaling pathways regulating the α-subunit gene are distinct from those activating the LH β-subunit gene (23). Moreover, it is possible that GnRH-R activation of different signal transduction pathways is involved in acute induction vs. chronic repression of LHβ gene expression. There are several reports regarding regulation of α and LHβ mRNAs by GnRH, but the results of these studies differ as to the effects of various GnRH time courses, concentrations, and the signal transduction pathways involved. Some studies report that GnRH or other GnRH-R agonists are capable of decreasing LHβ mRNA after 24-h incubation (7, 8, 24, 25). Other studies show either increased or unchanged levels of LHβ mRNA by GnRH-R agonists (26, 27). All of these studies, however, agree that there is no down-regulation of the α-subunit after 24-h incubation with GnRH.
Until recently, there was no ideal cell model for studying the regulation of LHβ gene expression by GnRH. In primary pituitary cultures and in vivo systems, the influence of other cell types and paracrine interactions can interfere with the effect of GnRH on pituitary gonadotropes (28, 29). In addition, these complex cultures do not allow direct quantification of signal cascades in response to GnRH in the gonadotropes because these cells compose only 5–10% of the cells in the pituitary (5, 30). Cell lines such as GGH3 and αT3-1 have also been used as models for GnRH action (31–34). The GGH3 cell line consists of rat somatomammotropic tumor cells (GH3) stably transfected with an expression vector for GnRH-R (33, 35). It is likely that the heterologous cellular environment causes coupling to signaling components not normally used or even present in gonadotropes (35). For example, GnRH-R couples to cAMP in GGH3 cells (35), a signaling pathway not activated in the αT3-1 gonadotrope-lineage cell line (34). Furthermore, GGH3 cells do not express steroidogenic factor-1 (SF-1), an important activator of gonadotrope-specific genes that is thought to interact with EGR-1 (NGFI-A), an early response gene that is induced by GnRH and is also important for LHβ gene expression (36–41). In the GGH3 system, transcription from the LHβ promoter is activated by PKC, and transcription from the α-subunit promoter is activated by calcium (32, 42).
αT3-1 cells are immortalized mouse pituitary tumor cells belonging to the gonadotrope lineage. These cells express the glycoprotein hormone α-subunit, GnRH-R and SF-1, but they are derived from an early stage of pituitary development when LHβ is not yet expressed, and these cells therefore do not naturally express LHβ (43, 44). To circumvent this deficiency, Weck et al. (23) employed a chimeric reporter gene consisting of −617 to −245 of the LHβ gene placed upstream of the thymidine kinase (TK) promoter in transfections of αT3-1 cells. Because SF-1 and EGR-1 are bound to a more proximal region of the LHβ promoter (36, 45) and EGR-1 can be regulated by signaling pathways, this reporter gene does not fully address the regulation of LHβ by GnRH. Nevertheless, it was shown that transcriptional activation through this LHβ promoter fragment is induced by calcium, and the transcription of the α-subunit gene is activated by PKC. These data directly oppose those obtained using GGH3 cells, described above. Thus, a more homologous gonadotrope cell model was needed to address these contradictions.
We have developed an immortalized LH-secreting gonadotrope, the LβT4 cell line, by the method of targeted tumorigenesis in transgenic mice (46). These cells express GnRH-R, SF-1, and the α and β-subunits of LH (43). A second cell line, LβT2, cloned in the same manner from the same line of transgenic mice, was also shown to release LH in response to pulsatile GnRH (47, 48) and express FSHβ (49, 50). Therefore, the LβT cell lines are valuable gonadotrope cell models for the study of LH regulation by GnRH.
In the current study, we have addressed the mechanisms by which the LH β-subunit gene is regulated by GnRH. We show that short-term incubation (6 h) of LβT4 immortalized pituitary gonadotrope cells with GnRH leads to induction of LHβ transcription, whereas continuous long-term incubation (24 h) leads to repression of LHβ transcription. We also show that acute induction is mediated by the PKC signal transduction pathway, and long-term repression is mediated by the calcium signal transduction pathway. Not only do short- vs. long-term GnRH treatments act through different signaling systems; they also act through different elements on the rat LHβ gene. Activation localizes to the upstream region (−451 to −384) that contains SP-1 and CArG elements (31, 51, 52), and repression localizes to an evolutionarily conserved element found between −153 and −143. We conclude that the interaction of these two signal transduction pathways may function to balance the level of LHβ mRNA synthesis and perhaps modulate levels throughout the estrous cycle.
Materials and Methods
Cells, media, and transfection protocols
LβT4 cells were cultured in 80-cm2 flasks and passed weekly by trypsin dispersion. The cells were maintained in DMEM (Life Technologies, Inc., Grand Island, NY) with 4.5 mg/ml glucose, 10% fetal bovine serum, and penicillin/streptomycin and maintained at 37 C with 5% CO2. All of the transfections were performed using the calcium phosphate precipitation method unless noted otherwise. For the calcium phosphate precipitation method, on the first day of the experiment, confluent flasks were split into 100-mm2 tissue culture plates (one flask to eight plates). On the next day, the cells were transfected for 16 h with 15 μg/plate reporter gene and 5 μg/plate internal control TK-chloramphenicol acetyl transferase (CAT) plasmid. On the morning of the next day, the cells were subjected to glycerol shock (5 ml 10% glycerol in 1× PBS per plate for 80–90 sec) and then washed two times with 1× PBS. For the transfections performed using FuGENE 6 transfection reagent (Roche Molecular Biochemicals Corp., Indianapolis, IN), 3 μg reporter plasmid, and 1 μg internal control were used, following the manufacturer’s protocol. One day following the transfection, appropriate compounds were added in fresh medium. Cells were harvested either 6 h or 24 h later, as indicated. Protein extracts were prepared by freeze thawing as described (53). CAT and luciferase assays were performed as previously described (54, 55), and the luciferase activity of each sample was normalized to the internal control CAT activity.
Reagents
Ionomycin (0.5 μM), 12-O-tetradecanoyl-phorbol-13-acetate (TPA) (100 nM), EGTA (2 mM), and GnRH agonist (des-Glu10,[d-Ala6]-LHRH ethylamide, 0.1, 1, 10, or 100 nM) were purchased from Sigma (St. Louis, MO). Bisindolylmaleimide I hydrochloride (BMM; 100 nM) and U0126 (750 nM) were purchased from Calbiochem (La Jolla, CA).
Plasmids
The LHβ-luciferase plasmid was prepared by fusing 1.8 kb of the rat LHβ gene 5′ flanking sequence into the HindIII restriction site of the pUC18 plasmid containing the luciferase gene (49). The TK-CAT plasmid contains −109 to +55 of the TK promoter (derived from pBL-CATII) driving the CAT reporter gene (56).
The −451 and −384 truncations of the LH β-subunit promoter were created by amplifying fragments from −451 or −384 to −216 by PCR, digesting the PCR products with HindIII and NheI, and then subcloning the 190- or 130-bp fragment into –1800LHβLuc plasmid digested with HindIII and NheI. The forward primers for the PCR were 5′-CCGTACAAGCTTACCACACCCATTTTTGGACCCAAT-3′ and 5′-CCGTGCAAGCTTCTCTGGTTGTATTTAAAGCAAATT-3′ for the −451 and −384 truncations, respectively. The reverse primer corresponds to the reverse DNA strand of the LHβ promoter from −240 to −216.
The truncations containing 146, 179 nucleotides of LHβ promoter or 179 nucleotides of LHβ promoter with the mutation in the putative activator protein-1 (AP-1) site were created by subcloning the synthetic oligonucleotides corresponding to the regions of the LHβ promoter from −146 to −121 or −179 to −121 (with added HindIII and native Tth111I half-sites) into the –1800LHβLuc plasmid digested with HindIII and Tth111I.
EMSA assays
Annealed oligonucleotides (20 ng) containing sequences of rat, human, human variant, or equine LHβ promoter were radiolabeled with γ [32P]dATP (3000 Ci/mmol, NEN Life Science Products, Boston, MA) using the polynucleotide kinase method (57). Probes were purified by passing through the G-50 microcolumns (Pharmacia Biotech, Piscataway, NJ), counted in a scintillation counter, and then diluted to 1 fmol/μl in water. Binding reactions were carried out in 5 mM HEPES (pH 7.8), 30 mM KCl, 1 mM EDTA, 5 mM spermidine, 5 mM dithiothreitol, 0.2 mg/ml bovine serum albumin, 5 mM polydeoxyinosinic-deoxycytidylic acid, 10% (vol/vol) glycerol, and 20 mg/ml Ficoll; 3 fmol of each probe were incubated with 2 μg crude nuclear extract in 40-μl reactions. Reactions were incubated at room temperature for 1 h and loaded into a 5% polyacrylamide gel (30:1 acrylamide/bisacrylamide, 0.25× modified Tris-borate EDTA) and electrophoresed for 2 h at 225 V. Gels were prerun for 15–30 min in 0.25× Tris-borate EDTA. After electrophoresis, gels were dried and subjected to autoradiography. Competition reactions were performed by mixing of radiolabeled probe and the specified amount of unlabeled oligonucleotide, and then adding the nuclear extract. Sequences are shown (see Figs. 9A and 10A) with the exception of the AP-1 consensus oligonucleotide, which has the sequence 5′-CTAGTGATGAGTCAGCCGGATC-3′. Oligonucleotides used in the EMSAs were obtained from Operon Technologies Inc. (Alameda, CA).
Fig. 9.

Competition assays localize the binding site of the protein bound to the −163 LHβ probe. A, The oligonucleotides used for the competition analysis. Mutated bases are shown in bold and the mutation used for the transfection experiment is underlined. B, Radioactive labeled probe (3 fm) and 100-fold molar excess of unlabeled competitor were added simultaneously to the nuclear extract from LβT4 cells obtained as described in Materials and Methods. The competition oligonucleotides were added in the order shown in A.
Fig. 10.

Evolutionary conservation of the GnRH-repression element in the LHβ promoter. A, The oligonucleotides from the different species used for the EMSA assays. The rLH, mouse LH, hLH, variant (64) VLH, and eLH LHβ 5′ flanking regions show that the ACTG(A/G)NNCT sequence is conserved among these four species. Identical bases in the putative binding site are shown in bold. The boxed nucleotides are those mutated in the VLH vs. hLH genes. B, Radioactive-labeled probe (3 fm) and 100-fold molar excess of unlabeled competitor were added simultaneously to either nuclear extract from untreated LβT4 cells or nuclear extract from LβT4 cells treated with GnRH for 24 h. In lanes 1–5, the rLH oligonucleotide was used as probe; in lanes 6–10, the hLH oligonucleotide was used as probe; in lanes 11–15, the VLH oligonucleotide was used as probe; and in lanes 16–20, the eLH oligonucleotide was used as probe. Lanes 1, 6, 11, and 16 had no competitor; lanes 2, 7, 12, and 17 had the −163 oligonucleotide as a competitor; lanes 3, 8, 13, and 18 had the hLH oligonucleotide as a competitor; lanes 4, 9, 14, and 19 had the VLH oligonucleotide as a competitor; lanes 5, 10, 15, and 20 had the eLH oligonucleotide as a competitor.
Western blotting
LβT4 cells were grown to confluence in six-well plates, washed once with PBS, and incubated in serum-free DMEM overnight. For inhibition experiments, the cells were pretreated with U0126 (720 nM) for 30 min at 37 C. Cells were stimulated with increasing concentrations of GnRH (1, 10, 100 nM) for 5 min at 37 C. Thereafter cells were washed with ice-cold PBS and then lysed on ice in sodium dodecyl sulfate sample buffer (50 mM Tris, 5% glycerol, 2% sodium dodecyl sulfate, 0.005% bromophenol blue, 84 mM dithiothreitol, 100 mM sodium fluoride, 10 mM sodium pyrophosphate, and 2 mM sodium orthovanadate, pH 6.8), boiled for 5 min to denature proteins, and sonicated for 5 min to shear the chromosomal DNA. Equal volumes (30–40 μl) of these lysates were separated by SDS-PAGE on 10% gels and electrotransferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore Corp., Bedford, MA). The membranes were blocked with 5% BSA in Tris-buffered saline-Tween [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% Tween-20]. Blots were incubated with primary anti-ACTIVE MAPK antibodies (Promega Corp., Madison, WI) at a dilution of 1:2500 in blocking buffer for 60 min at room temperature and then incubated with horseradish peroxidase-linked secondary antibodies followed by chemiluminescent detection. The polyvinylidene difluoride membranes were immediately stripped by placing the membrane in stripping buffer (0.5 M NaCl and 0.5 M acetic acid) for 10 min at room temperature. The membrane was then washed once for 10 min in Tris-buffered saline-Tween, reblocked, and blotted with antibodies to the unphosphorylated form of the ERK2 enzyme to control for equal protein loading.
Statistical analysis
All values are expressed as ratios of luciferase activity to CAT activity. Differences between groups were examined by ANOVA and post hoc testing using Fisher’s protected least significant difference using the Statview program (SAS Institute, Inc., Cary, NC). Significant differences were declared for group comparisons returning an α ≤ 0.05.
Results
Acute GnRH treatment induced, but long-term GnRH treatment repressed, LHβ transcription
Previous studies of the effects of GnRH on gene expression have generally used 6-h incubations with 10 nM GnRH agonist (23, 32). Our first goal was to find the optimal GnRH dose for regulation of the −1800-bp rat LHβ promoter on luciferase transfected into LβT4 cells. As shown in Fig. 1A, 1 nM GnRH increased luciferase activity to 150%, and 10 nM GnRH increased luciferase activity to 160%, compared with untreated cells. To determine the optimal GnRH concentration for long-term repression of LHβ gene expression, transfected LβT4 cells were incubated with increasing concentrations of a GnRH agonist for 24 h. LHβ-luciferase is repressed in a pattern inversely correlated with dose. Addition of 100 nM GnRH agonist does not lead to changes in luciferase activity, compared with control. Incubation with 10 nM GnRH agonist resulted in a small decrease in luciferase activity to 78% of control. Incubation with 1 nM GnRH resulted in decreased luciferase activity to approximately 50% of control (Fig. 1B), although 0.1 nM has no statistically significant effect. All further studies used 10 nM GnRH agonist for acute treatments (6 h) and 1 nM GnRH agonist for long-term treatments (24 h).
Fig. 1.

Dose response of the GnRH agonist for induction and repression of LHβ gene expression. LβT2 cells were transiently transfected with 15 μg –1800LHβ-Luc reporter plasmid and 5.0 μg TK-CAT plasmid as an internal control. Sixteen hours after transfection, the cells were incubated with 1 nM and 10 nM GnRH agonist for 6 h (A) or with 0.1, 1, 10, or 100 nM GnRH analog for 24 h (B). The value of the untreated sample for each time point was set to 100 to allow direct comparison of the magnitude of the GnRH induction. Results are the mean ± SEM of three independent experiments, each performed in duplicate. Asterisks (*) indicate a significant difference, compared with untreated cells (P < 0.05).
Of note, a receptor for a related hormone, GnRH II, has been identified in primate gonadotropes (58, 59). It is unlikely that this novel receptor is activated by our treatments though because it exhibits a 40-fold preference for GnRH II, a hormone related to GnRH but not identical. The IC50 for GnRH on the primate GnRH II receptor is 42 nM as opposed to that for GnRH II, which is 1 nM (59), and our studies use 1–10 nM GnRH.
Acute GnRH induction of LHβ gene expression was reproduced by activation of PKC, and long-term GnRH repression was reproduced by inducing calcium influx
To determine whether induction of LHβ gene expression by GnRH could be mimicked by an influx of calcium or activation of PKC, we incubated transfected LβT4 cells with 0.5 μM ionomycin or 100 nM of the phorbol ester TPA, an activator of PKC, for 6 h (Fig. 2A). Incubation with TPA resulted in an induction of the LHβ promoter similar to that caused by 1 nM GnRH agonist. Incubation with ionomycin did not cause any significant change from control values (Fig. 2A) nor did treatment with 1 μM thapsigargin, an agent that releases intracellular stores of calcium (data not shown).
Fig. 2.
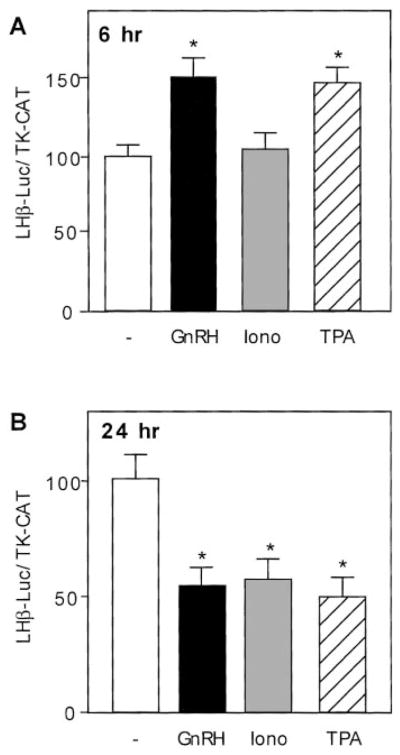
Treatment with a GnRH agonist, ionomycin, or TPA for 6 h or 24 h. LβT4 cells were transiently transfected with 15 μg –1800LHβ-Luc reporter plasmid and 5 μg TK-CAT plasmid as an internal control. Sixteen h after transfection, cells received GnRH treatment at 1 nM, or ionomycin at 500 nM, or TPA at 100 nM final concentrations for 6 h (A) or 24 h (B) before harvest. The value of the untreated sample for each time point was set to 100 to allow direct comparison of the magnitude of the GnRH induction. Results are the mean ± SEM of three independent experiments, each performed in duplicate. Asterisks (*) indicate a significant difference, compared with untreated cells (P < 0.05).
If the long-term repression caused by GnRH is mediated through calcium or PKC, then agents that increase the intracellular calcium concentration or activate PKC should also repress LHβ-luciferase in long-term treatments. Incubation with TPA or ionomycin (or thapsigargin, data not shown) for 24 h at the doses used in the previous experiment also produced repression of LHβ-luciferase (Fig. 2B). The magnitude of the repression was similar to that produced by 1 nM GnRH agonist. Thus, TPA mimics the biphasic regulation of the LHβ gene produced by GnRH, and calcium influx reproduces only the later repressive phase.
Acute GnRH induction of LHβ gene expression was prevented by blockade of PKC, and long-term repression was prevented by removal of extracellular calcium
To further assess the importance of the signal transduction pathway activated by calcium after 6-h incubation with GnRH, we used the calcium chelator EGTA. Treatment with EGTA alone did not result in significant changes, compared with the untreated cells or ionomycin-treated cells. As shown above (Fig. 2A), treatments with GnRH agonist or TPA (but not with ionomycin) led to induction of LHβ-luciferase activity. However, rather than blocking the induction, inclusion of EGTA with GnRH agonist or TPA produced statistically significant increases in luciferase activity, compared with untreated cells, cells treated with GnRH agonist alone, or TPA alone, respectively (Fig. 3A). This result may indicate that GnRH-R activation of calcium influx is exerting downward pressure on LHβ gene expression at the early time point (6 h) but that this repression is overcome by the stronger induction because of activation of PKC. This interpretation is supported by the observation that in the absence of calcium, induction by GnRH or TPA is more pronounced.
Fig. 3.
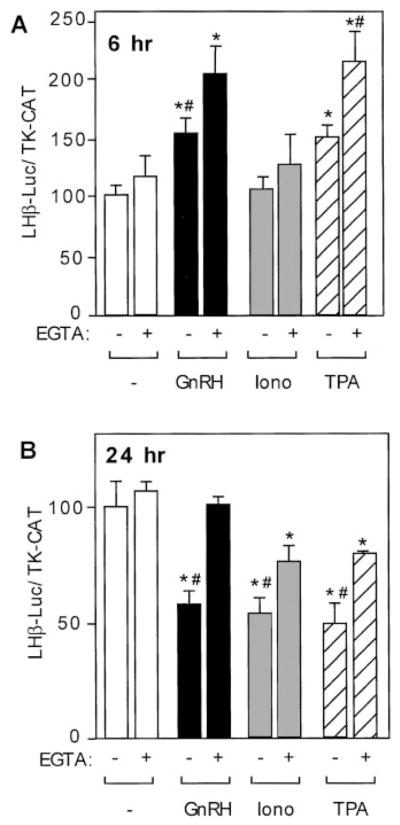
Repression of LHβ gene expression by GnRH is mediated by calcium. LβT4 cells were transiently transfected with 15 μg –1800LHβ-Luc reporter plasmid and 5 μg TK-CAT plasmid as an internal control. Sixteen hours after transfection, cells were pre-treated with EGTA (2 mM) for 30 min and then cotreated with or without GnRH agonist at 10 nM (A) or 1 nM (B), ionomycin (500 nM), or TPA (100 nM) for 6 h (A) or 24 h (B) before harvest. The value of the untreated sample for each time point was set to 100 to allow direct comparison of the magnitude of the GnRH induction. Results are the mean ± SEM of three independent experiments, each performed in duplicate. Asterisks (*) indicate a significant difference, compared with the untreated cells. Groups marked with # are significantly different between the groups treated with the given reagent alone (GnRH, ionomycin, or TPA only) vs. treated with the combination of a reagent and EGTA.
If the repression of LHβ transcription by GnRH after the 24-h treatment is indeed mediated by calcium, removal of the calcium source should abolish the repression of LHβ caused by the GnRH agonist. As in the previous experiment, cells were pretreated with EGTA and then cotreated with or without GnRH agonist, ionomycin, or TPA, but the time was increased to 24 h. Treatment with the GnRH agonist, ionomycin, or TPA resulted in statistically significant repression of luciferase activity, compared with control (Fig. 2B). Pre-treatment of transfected LβT4 cells with EGTA reversed the effects of the GnRH agonist under these conditions without causing changes in LHβ expression alone (Fig. 3B), indicating a key role for calcium influx in GnRH repression of the LHβ gene. Pretreatment with EGTA also partially reversed the repression because of ionomycin or TPA, indicating participation of the calcium signal cascade in repression by these treatments as well. The partial EGTA blockade may have been caused by utilization of insufficient concentrations to prevent the effect of ionomycin (although this level of EGTA is sufficient to block GnRH action). However, the cells do not tolerate higher levels of EGTA for 24 h.
If the induction of LHβ gene expression by acute GnRH treatment is indeed mediated through the PKC signal transduction system, then inhibition of this system should block the effect of GnRH. As before (Fig. 2A), 6-h treatment with GnRH or TPA, but not with ionomycin, produced significant induction in LHβ gene expression. As shown in Fig. 4A, the PKC inhibitor BMM blocked activation by GnRH or TPA, demonstrating a requirement for PKC action in GnRH induction of LHβ gene expression. Moreover, although BMM alone did not affect LHβ gene expression, it reduced LHβ gene expression in combination with ionomycin, indicating again that the calcium-activated signal transduction pathway was capable of repressing expression at 6 h of incubation with GnRH. However, PKC activity apparently masked the repression by the calcium system, thus preventing observation of repression without blockade of PKC activity.
Fig. 4.
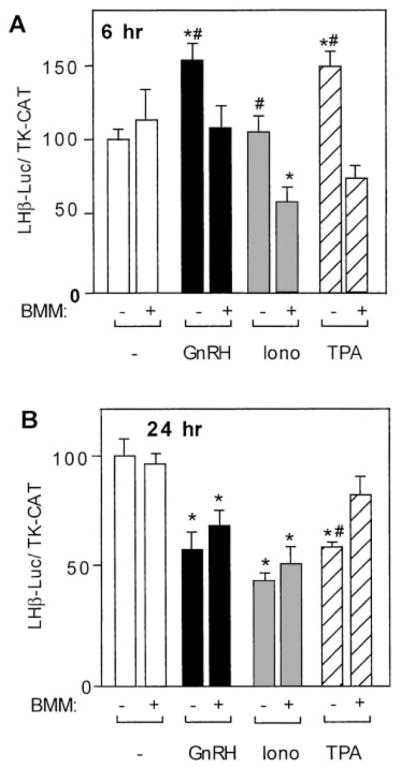
Induction of LHβ gene expression by GnRH is mediated by the PKC signaling system. LβT4 cells were transiently transfected with 15 μg –1800LHβ-Luc reporter plasmid and 5 μg TK-CAT plasmid as an internal control. Sixteen hours after transfection, cells were treated with BMM I (100 nM) with or without GnRH agonist at 10 nM (A) or 1 nM (B), ionomycin (500 nM), or TPA (100 nM) for 6 h (A) or 24 h (B) before harvest. The value of the untreated sample for each time point was set to 100 to allow direct comparison of the magnitude of the GnRH induction. Results are the mean ± SEM of three independent experiments, each performed in duplicate. Asterisks (*) indicate a significant difference compared with the untreated cells. Groups marked with # are significantly different between the groups treated with the given reagent alone (GnRH, ionomycin, or TPA only) vs. treated with the combination of a reagent and BMM I.
In Fig. 4B, cells incubated with GnRH agonist, ionomycin, or TPA in the absence of BMM for 24 h showed decreased luciferase activity as was observed in Fig. 2B. Treatment with BMM alone for 24 h did not lead to significant changes in luciferase activity, compared with untreated cells. However, although BMM prevented repression by TPA as expected, it did not prevent repression by GnRH agonist or ionomycin, indicating that long-term repression of LHβ gene expression by GnRH is independent of the PKC signaling system.
The MAPK pathway is not involved in either short-term induction or long-term repression of the LHβ promoter
One of the possible candidates acting downstream of the PKC or calcium systems to induce or repress the LHβ promoter is the MAPK pathway. This pathway is active in gonadotropes and can be regulated by both the PKC and calcium systems independently from each other (11–13). In our experiments, coincubation of GnRH agonist or TPA with the MEK inhibitor U0126 (final concentration 750 nM) for 6 h or coincubation of GnRH agonist or ionomycin with U0126 for 24 h did not lead to any significant changes in luciferase activity, compared with the groups treated by GnRH agonist, ionomycin, or TPA without U0126 (Fig. 5, A and B). Under similar conditions, U0126 did block GnRH induction of control genes (data not shown). Therefore, we conclude that although the MAPK pathway is activated by GnRH and U0126 blocks this activation (Fig. 5C), the MAPK pathway is not required for either short-term induction or long-term repression of LHβ promoter by GnRH.
Fig. 5.
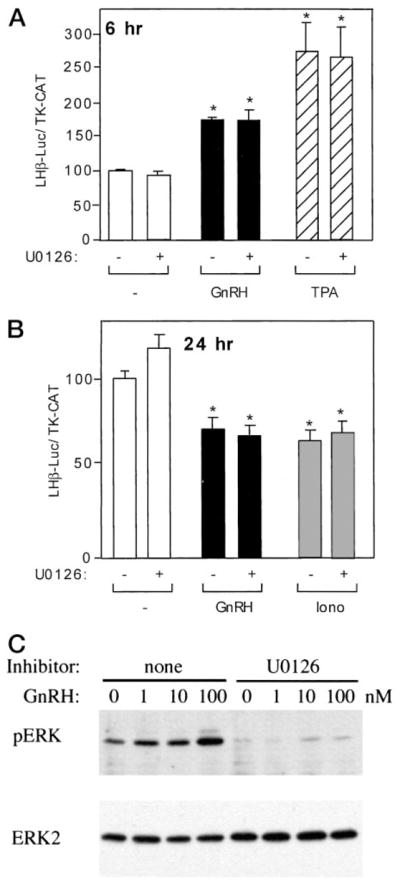
Inhibition of the MAPK system does not abolish either the induction or the repression of LHβ gene expression. A and B, LβT4 cells were transiently transfected with 3 μg –1800LHβ-Luc reporter plasmid and 1 μg TK-CAT plasmid as an internal control, using FuGENE 6 transfection reagent. Sixteen hours after transfection, cells were treated with U0126 (750 nM) with or without GnRH agonist at 10 nM (A) or 1 nM (B), ionomycin (500 nM), or TPA (100 nM) for 6 h (A) or 24 h (B) before harvest. The value of the untreated sample for each time point was set to 100 to allow direct comparison of the magnitude of the GnRH induction. Results are the mean ± SEM of three independent experiments, each performed in duplicate. Asterisks (*) indicate a significant difference, compared with the untreated cells. C, LβT4 cells were serum starved overnight and then pretreated with 720 nM U0126 or DMSO vehicle for 30 min and stimulated with 0, 1, 10, or 100 nM GnRH for 5 min at 37 C. Whole-cell extracts were separated by SDS-PAGE and immunoblotted with an antibody to phospho-ERK1/2 (top panel). The blots were stripped and reblotted for ERK1/2 protein, demonstrating equivalent loading (bottom panel).
Acute induction and long-term repression use different regions of the LHβ promoter
To further support our finding that calcium and PKC act through different, though interacting, pathways, we performed analysis of 5′ truncations of the LHβ promoter. If calcium and PKC indeed act through different pathways, they might exert their effects through different regions of the LHβ promoter. Truncation of the LHβ promoter sequence to −451 upstream of the transcription site did not disrupt responsiveness to incubation with GnRH agonist or TPA for 6 h (−1800 and −451), but truncation to −384 eliminated induction (Fig. 6A). In contrast, a promoter truncated to only the proximal 179 bp of LHβ promoter sequence but not induced after 6 h of GnRH agonist or TPA treatment (data not shown) was still repressed after 24 h of GnRH agonist or ionomycin (Fig. 6B). The LHβ promoter region from −146 bp to the mRNA start site was unable to mediate induction or repression of luciferase activity by GnRH (Fig. 6B and data not shown) despite containing the EGR-1, SF-1, and Ptx-1 binding sites previously reported to participate in GnRH responses (39, 41, 52, 60).
Fig. 6.

Acute induction and long-term repression by GnRH occur through different regions of the LHβ promoter. A, For the short-term treatment, LβT4 cells were transiently transfected with either 15 μg –1800LHβ-Luc or the equimolar amount of the one of the following −451 or −384 truncated LHβ reporter plasmid and 5 μg TK-CAT plasmid as an internal control. Sixteen hours after transfection, cells were treated with ethanol vehicle (−), 10 nM GnRH agonist (G), or 100 nM TPA (T) for 6 h before harvest. B, For the long-term treatment, LβT4 cells were transiently transfected with either 15 μg –1800LHβ-Luc or the equimolar amount of the one of the following: −179, −179M, or −146 truncated LHβ reporter plasmids and 5 μg TK-CAT as an internal control. Sixteen hours after transfection, cells were treated with ethanol vehicle (−), 1 nM GnRH agonist (G), or 500 nM ionomycin (I) for 24 h before harvest. The value of the untreated sample for each time point was set to 100 to allow direct comparison of the magnitude of the GnRH induction. Results are the mean ± SEM of three independent experiments, each performed in duplicate. Values marked with an asterisk are statistically different from the value of untreated cells within the reporter transfected (P < 0.05).
Although the sequence between −179 and −146 of the rat LHβ 5′ flanking region has not been shown to bind specific proteins, Kaiser et al. (51) have noted the presence of a putative AP-1 binding site (TGAGACA, a six of seven match to the AP-1 consensus of TGAC/GTCA). To investigate whether this site is involved in the repression of LHβ by GnRH or ionomycin after 24 h, we created a mutation in the putative AP-1 site of the −179 truncated LHβ promoter (−179M), as described in Materials and Methods. It has been previously shown by EMSA that the equivalent CA to TG 2-bp mutation completely eliminates the binding of AP-1 complexes to an AP-1 consensus site (61). As shown in Fig. 6B, this mutation eliminated repression of luciferase activity caused by GnRH or ionomycin, suggesting the importance of the mutated bases for repression of LHβ by GnRH or ionomycin.
Long-term repression of LHβ transcription is not mediated by AP-1 binding
To determine whether AP-1 mediates LHβ repression by GnRH through this binding site, we performed EMSA using an oligonucleotide probe representing the sequence from −163 to −142 of the LHβ promoter, which contains the putative AP-1 element (51). We performed competition assays with 10-, 50-, 100-, or 200-fold excess of nonradioactive oligonucleotides: self (−163), a mutated oligonucleotide (−163M) altered in the same two base pairs as mutated in the transfections in Fig. 6 (see −179M), and with an AP-1 consensus oligonucleotide described in Materials and Methods (Fig. 7). As expected, addition of the unlabeled self-competitor at 100- and 200-fold excess completely abolished the major band binding to the radiolabeled probe (arrow, Fig. 7), but the faster migrating doublet was not well self-competed and is likely to be nonspecific binding. The addition of the −163M competitor resulted in a decrease of the major band, but substantial binding to the probe remained. Interestingly, the oligonucleotide containing an AP-1 consensus-binding site did not compete with the binding of the complex to the radiolabeled −163 probe. Therefore, although this binding complex uses at least part of the putative AP-1 site (the 2 bp mutated in −163M), it is not likely to be AP-1.
Fig. 7.

Competition assays with an AP-1 consensus oligonucleotide. Radioactive labeled probe (3 fm) and 10-, 50-, 100-, or 200-fold molar excess of unlabeled competitor were added simultaneously to 2 μ g nuclear extract from LβT4 cells obtained as described in Materials and Methods. The competitors were the unlabeled probe (−163), unlabeled probe with a mutation in the putative AP-1 site (−163M), and an AP-1 consensus oligonucleotide.
To further establish whether the protein complex that binds to the −163 to −142 region of the LHβ promoter is AP-1, we performed EMSA using a labeled probe containing the AP-1 consensus site and incubated with a c-Fos antibody (Santa Cruz Biotechnology, Santa Cruz, CA) to detect the c-Fos protein, which is a component of the AP-1 complex. We saw an increase of binding intensity of one of the complexes (the upper band of the doublet, arrow, Fig. 8A) after cells were treated with GnRH for 6 h, and formation of this complex was blocked by incubation with c-Fos antibody but not with IgG (Fig. 8A). On the other hand, when the −163 to −142 oligonucleotide was used as a probe, we did not observe any changes in the binding intensity in the extracts from cells treated with GnRH for 6 h, compared with extracts from untreated cells (data not shown), and the c-Fos antibody did not have any effect on the protein complex that binds to the −163 to −142 oligonucleotide (Fig. 8B). Finally, the AP-1 complex did not comigrate with the complex bound to the −163 probe (data not shown). These data further show that AP-1 does not bind to the element mediating repression of the LHβ promoter by GnRH agonist or ionomycin.
Fig. 8.

The nuclear protein complex bound to the LHβ promoter element responsible for GnRH-repression is not supershifted by the c-fos antibody. LβT4 cells were treated for 6 h without (C) or with GnRH (G) or ionomycin (I), and nuclear extracts were harvested as described in Materials and Methods. Two micrograms nuclear extract were used for each EMSA reaction. A, AP-1 consensus oligonucleotide used as the probe. Lanes 1–3, Six-hour treatments; lanes 4–6, 24-h treatments; lane 7, cells were treated for 6 h with GnRH and then the c-fos antibody was added; lane 8, cells were treated for 6 h with GnRH and then IgG was added. B, The region of the LHβ promoter from −163 to −142 was used as a probe. Lane 1, Extracts from cells treated with GnRH for 6 h. Lanes 2 and 3, Extracts from cells treated with GnRH for 6 h with c-fos antibody (lane 2) or IgG (lane 3) added to the reaction mix 15 min before the labeled probe.
If this complex is involved in repression by chronic GnRH or calcium, nuclear extracts isolated from cells treated with GnRH agonist or ionomycin might reveal changes in the intensity or migration of the complex. However, no changes in the binding pattern were observed between the untreated cells and cells treated with either GnRH or ionomycin after 6 h or 24 h (data not shown). The absence of differences in binding intensity after 6- or 24-h treatment suggests that the protein or protein complex bound to the −163 probe may mediate the repression, not by direct changes in its binding to the DNA, but rather by modification of the protein that is already bound to the DNA or, alternatively, by recruitment of other cofactors or corepressors.
To further characterize the complex binding to the −163 probe, we performed competition assays with an excess of the unlabeled −163 oligonucleotide containing a variety of mutations. The oligonucleotides used for the competition analysis are shown in Fig. 9A. The mutated bases are shown in bold and the 2-bp mutation used in transfection analysis and in Fig. 7 is underlined. As shown in Fig. 9B, addition of 100-fold excess of the oligonucleotide containing a mutation eliminating the AP-1 binding site (M1) still completely abolished binding to the radiolabeled probe, again supporting the conclusion that the major complex does not contain AP-1. However, the mutations downstream of M1 either partially decreased or did not change, the intensity of the protein binding to the radiolabeled probe indicating that these competitors do not bind the complex. From these data, it seems likely that the binding site for this protein is downstream of the putative AP-1 site in the sequence ACACTGGAGCT from −153 to −143.
Next, we determined the level of conservation of this site across species. The comparison of rat LH (rLH), mouse LH, human LH (hLH), and equine LH (eLH) LHβ 5′ flanking regions shows that the ACTG(A/G)NNCT sequence is conserved among these four species (Fig. 10A). Searches in the TRANSFAC transcription factor database (http://transfac.gbf.de/TRANSFAC/) did not produce any likely candidates for this DNA-binding protein. An equivalent EMSA complex was present using extracts from the LβT4, αT3-1, αT1-1 (62), HeLa, and NIH3T3 cell lines, showing that it is not specific to the gonadotrope lineage (data not shown).
It has been previously shown that naturally occurring mutations (63) in the human LHβ promoter sequence (variant LH, VLH) result in higher expression of the human LHβ reporter gene in both LβT2 and human embryonic kidney 293 cell lines, and one of these mutations is in the putative AP-1 site of the human LHβ promoter in a nucleotide (equivalent to −147 in the rat) that would also affect the binding of the complex bound to the downstream repression element (64). In the VLH gene, this nucleotide is the same as the rat and mouse (G), but in human and equine it is different (A; boxed in Fig. 10A). When we performed EMSA using hLH, VLH, or eLH oligonucleotides as the radiolabeled probes, we found that all of these probes bound a complex that comigrated with the complex bound to the rLH (−163) probe (Fig. 10B). Moreover, cross-competition assays showed that all of these oligonucleotides fully cross-compete with each other, indicating that they bind the same protein.
Discussion
Long-term treatment with GnRH agonists in patients with gonadal-steroid sensitive cancers produces a profound repression of the pituitary gonadotropins resulting in suppression of gonadal steroid production (65). The power of GnRH to induce LH gene expression in the short term then repress it below basal levels after chronic treatment is particularly intriguing and provides an opportunity for understanding the signaling mechanisms used by GnRH in temporal control of gonadotropin gene expression.
In this article, we have addressed the difference between acute and chronic treatment with GnRH on the regulation of LHβ gene transcription in LβT4 cells, an immortalized mouse gonadotrope cell line. Our results suggest that there are at least two signaling systems downstream of GnRH-R in the LβT4 cells. One system is activated through PKC and is responsible for acute induction of LHβ transcription. Another system is activated through calcium-signaling pathways independent of PKC and is responsible for intense down-regulation of LHβ transcription. At early time points, the PKC system is more potent, and therefore transcription of the LHβ gene is induced. After long-term treatment, when the PKC system exhausts, the calcium system predominates (or the combination of these two effects occurs), resulting in down-regulation of the LHβ gene.
We observed that repression of LHβ gene transcription after 24-h incubation with GnRH was sensitive to the dose of GnRH agonist used. The reversal of repression at the higher doses may be due to desensitization and/or down-regulation of the GnRH-R, preventing chronic signaling. Cheng et al. (66) have shown that in αT3-1 cells, incubation with 100 nM GnRH agonist for 24 h leads to repression of GnRH receptor promoter activity. In GGH3 cells, in which transcription of the GnRH-R is driven by a heterologous promoter and thus likely to be refractory to GnRH, high doses of Buserelin down-regulate the Gq/11α protein involved in GnRH signaling (67). A higher dose (10 nM), although failing to repress, also failed to induce the LHβ gene at 24 h, indicating that the induction produced by this dose at 6 h is lost by 24 h.
To delineate the signaling cascades downstream of the GnRH-R that are used in induction vs. repression, we studied the effects of direct activation or inhibition of the calcium, kinase C, and MAPK pathways. Activation of LHβ transcription after 6-h incubation with the PKC activator TPA (Fig. 2A) and the loss of induction by GnRH in the 6 h coincubation with the PKC inhibitor BMM (Fig. 4A) show that acute induction of LHβ transcription occurs through the PKC system. The absence of any induction at 6 h with the calcium ionophore, ionomycin (Fig. 2A), supports the conclusion that calcium is not involved in acute transcriptional induction of the LHβ gene by GnRH. Moreover, LHβ is still induced by GnRH or TPA in the presence of the calcium chelator, EGTA (Fig. 3B).
Previous reports on the role of the MAPK pathway in the induction of LHβ by GnRH are controversial. Haisenleder et al. (68), using primary pituitary cultures, have shown that the inhibition of MEK abolished the GnRH induction of α-subunit, GnRH-R, and FSHβ but not of LHβ. Furthermore, overexpression of dominant negative MAPKs was sufficient to repress GnRH induction of mouse α-subunit (69, 70). On the other hand, Wolfe et al. (71) and Weck et al. (23), using the αT3-1 cell model, showed that MAPK is involved in the induction of LHβ gene transcription by GnRH in this non-LHβ-expressing cell type. In our experiments, inhibition of the MAPK system with U0126 does not change either induction by GnRH or TPA after 6 h of treatment or the repression by GnRH or ionomycin after 24 h of treatment. These results demonstrate that in the LβT4 cell model, the MAPK system is not involved in either the acute induction or the chronic repression of the LHβ 5′ regulatory region.
GnRH-R activation leads to an increased concentration of intracellular calcium. It has been shown that this increase is due to two events: calcium channel opening and calcium release from intracellular stores (11). We have found that chronic GnRH represses LHβ transcription through the calcium system. Although GnRH, TPA, and ionomycin can all repress LHβ gene expression at 24 h, only the calcium chelator, EGTA (but not the MAPK or kinase C inhibitors), blocks the effect of GnRH after the 24-h incubation.
Two observations support a role for calcium as a negative regulator of LHβ at the earlier 6-h treatment time point as well. Blockade of calcium by EGTA augments both GnRH and TPA induction at 6 h (Fig. 3A), indicating an existing downward pressure on LHβ transcription at 6 h by calcium. Furthermore, when ionomycin is coadministered with the kinase C inhibitor, BMM, at the 6-h time point (Fig. 4A), repression of the LHβ gene below basal levels becomes evident. These data suggest that the calcium system is responsible for repression at earlier time points but that it is not sufficient to overcome the acute activation of LHβ gene expression by the PKC system. However, GnRH does not repress LHβ at the 6-h time point when given in combination with BMM. GnRH may not be as potent an activator of the calcium system as ionomycin. However, later, after the 24-h treatment, PKC activity is exhausted, so that repression of LHβ transcription can be observed after 24 h of incubation with a GnRH agonist or TPA.
Induction of the kinase C system, although not required for chronic GnRH repression, can also cause repression of LHβ gene expression in that 24 h of TPA treatment down-regulates the LHβ gene. TPA is known to cause degradation of some kinase C isoforms after 24 h and this may contribute to the decrease in LHβ gene expression. Cross-talk between these two systems makes it possible that PKC activates the calcium system before the PKC system is down-regulated, resulting in the same outcome (repression of LHβ) as chronic GnRH or ionomycin treatment. This possibility of cross-talk may also explain why the 24-h treatment with EGTA partially blocks the repression caused by TPA (Fig. 3B).
GnRH stimulation plays a crucial role in the regulation of the molecular markers of the gonadotrope lineage, such as α-subunit, LHβ, FSHβ, and GnRH-R. Regulation of α-subunit transcription is relatively well studied. It has been shown that GnRH regulation of the transcription of α-subunit is species specific and is gradual, requiring longer incubation with tonic GnRH. In rodents, this regulation occurs through activation of ETS elements (52, 72), and in humans, cAMP response elements are involved (73). Studies using αT3-1 cells show that GnRH up-regulates the mouse GnRH-R gene through the PKC system at an AP-1 site (74). Another study that also used αT3-1 cells identified two elements, called SURGE-1 and SURGE-2, that mediate the induction of mouse GnRH-R gene transcription by GnRH. SURGE-2 was identified as an AP-1 site (75).
Several transcription factors, tissue specific as well as ubiquitous, bind to the LHβ promoter. In GGH3 cells, two regions of the rat LHβ promoter are important for induction by GnRH (51). The distal region, termed region A (− 451 to − 386 bp), contains several SP-1 binding sites and is capable of binding SP-1 (31). In addition to SP-1 sites, this region has been shown to have CArG elements important for GnRH induction in LβT2 cells (52). GnRH action through the proximal region, spanning from − 207 bp downstream, was shown to require EGR-1 binding sites in both GGH3 (51) and LβT2 cells (52, 76). Other tissue-specific proteins, such as SF-1 and Ptx-1, also have binding sites in the proximal region. In cotransfections into CV-1 or JEG-3 cells, it has been shown that SF-1, Ptx-1, and EGR-1 interact to increase the activity of the LHβ promoter (39, 41, 60) and mutation of an EGR site inhibits GnRH induction in LβT2 cells (41, 52, 60). This region also contains putative sites for AP-1 (seven of eight of consensus site) and cAMP response element-binding protein (six of eight of consensus) (51).
Our experiments (Fig. 6, A and B) show not only that the PKC and calcium systems act on different regions of the LHβ promoter, but they also provide insight into the protein(s) that may be responsible for induction vs. repression of LHβ by GnRH. It is likely that GnRH induces activity of the LHβ promoter in LβT4 cells through CArG-1 and SP-1 sites, located in region A. Despite the fact that SP-1 is a ubiquitous protein, the localization of GnRH-R to the gonadotrope may be sufficient to provide cell-specific regulation of LHβ by GnRH. On the other hand, the possibility cannot be excluded that an EGR-1/SF-1/Ptx-1 complex is also necessary for LHβ induction by GnRH and that it interacts with SP-1. Weck et al. (52) showed that mutation of the CArG box or the proximal SP-1 site completely abolishes the induction of the LHβ promoter by GnRH in LβT2 cells, thus supporting the hypothesis of the interaction between the transcription factors binding to region A and region B. Interestingly, however, the authors of that study were not able to show induction of LHβ by continuous stimulation with GnRH after 8 h, but in our study GnRH is capable of LHβ induction after 6 h of tonic treatment. It is likely that GnRH induces LHβ gene expression after 6 h of tonic treatment, but in their study this induction had disappeared by 8 h.
We have also shown that the LHβ promoter region from −179 to −146 is necessary for repression of the LHβ gene by GnRH. The SF-1, EGR-1, and Ptx-1 sites are all located more proximal within −127 of the rat LHβ promoter. EMSAs show that the region involved in repression of the LHβ promoter binds a protein that overlaps a nonconsensus AP-1 site but is not AP-1. A similar site is present in the LHβ promoters of other species, and the fact that the eLH, hLH, and VLH probes all bind the same complex suggests that this protein may play a conserved role in the regulation of LHβ gene expression.
The human GnRH-R gene has been shown to be down-regulated after transfection into αT3-1 cells by a wide range of GnRH doses as early as 6 h but maximal at 24 h with 100 nM (66). In contrast to repression of the LHβ gene by GnRH, this repression is mediated by the PKC system in that TPA treatment reproduces the repression and inhibition of the PKC system prevents GnRH repression. Mutation of an AP-1 site at −1000 abolishes the repression and AP-1 was found to bind this element and be induced by 100 nM GnRH at 24 h. This AP-1 site is not the same as the one found in the proximal promoter of the mouse GnRH-R gene responsible for induction (74, 75). Thus, although AP-1 may be involved in repression of the human GnRH-R gene, it is not responsible for repression of the rat LHβ gene.
It is clear that the signaling cascades activated by GnRH are diverse, employing a variety of different transcription factors to differentially induce or repress the expression of key genes expressed in the gonadotrope. In the case of the LHβ gene, GnRH activation of the PKC cascade is required for acute induction, but this cascade acts independently of downstream MAPK activation and does not require calcium influx. In contrast, repression of the LHβ gene by GnRH occurs through the calcium signaling system, again independently of MAPK but also independently of PKC. Not only does GnRH use different signaling systems to induce, as opposed to repress, LHβ gene expression, but it also uses different elements within the gene for induction vs. repression of LHβ gene expression.
Acknowledgments
We thank Brian Powl, Rachel White, Scott Anderson, and Melinda Merrill for technical assistance; Bernardo Yusta and members of the Mellon laboratory for helpful discussions; and Djurdjica Coss, Tom Spady, and Suzanne Rosenberg for careful reading of the manuscript.
This work was supported by National Institute of Child Health and Human Development/NIH through cooperative agreement (U54-HD-12303) as part of the Specialized Cooperative Centers Program in Reproduction Research (to P.L.M., M.A.L., and N.J.G.W.). This work was also supported by NIH Grant R37-HD-20377 (to P.L.M.). V.V.V. was supported by an NIH Training Grant (T32-DK-07541).
Abbreviations
- AP-1
Activator protein-1
- BMM
bisindolylmaleimide I hydrochloride
- CAT
chloramphenicol acetyl transferase
- DAG
diacylglycerol
- eLH
equine LH
- GnRH-R
GnRH receptor
- hLH
human LH
- PKC
protein kinase C
- PLC
phospholipase C
- rLH
rat LH
- SF-1
steroidogenic factor-1
- TK
thymidine kinase
- TPA
12-O-tetradecanoyl-phorbol-13-acetate
- VLH
variant LH
References
- 1.Albanese C, Colin IM, Crowley WF, Ito M, Pestell RG, Weiss J, Jameson JL. The gonadotropin genes: evolution of distinct mechanisms for hormonal control. Recent Prog Horm Res. 1996;51:23–58. [PubMed] [Google Scholar]
- 2.Chin WW, Gharib SD. Organization and expression of gonadotropin genes. Adv Med Biol. 1986;205:245–265. doi: 10.1007/978-1-4684-5209-9_11. [DOI] [PubMed] [Google Scholar]
- 3.Chin W, Boime I. Glycoprotein hormones. Norwell, MA: Serono Symposia USA; 1990. [Google Scholar]
- 4.Gharib SD, Wierman ME, Shupnik MA, Chin WW. Molecular biology of the pituitary gonadotropins. Endocr Rev. 1990;11:177–199. doi: 10.1210/edrv-11-1-177. [DOI] [PubMed] [Google Scholar]
- 5.Bhasin S, Swerdloff RS. Follicle-stimulating hormone and luteinizing hormone. In: Melmed S, editor. The pituitary. Cambridge, MA: Blackwell Science, Inc; 1995. pp. 230–276. [Google Scholar]
- 6.Fallest PC, Trader GL, Darrow JM, Shupnik MA. Regulation of rat luteinizing hormone β gene expression in transgenic mice by steroids and a gonadotropin-releasing hormone antagonist. Biol Reprod. 1995;53:103–109. doi: 10.1095/biolreprod53.1.103. [DOI] [PubMed] [Google Scholar]
- 7.Shupnik MA, Fallest PC. Pulsatile GnRH regulation of gonadotropin subunit gene transcription. Neurosci Biobehav Rev. 1994;18:597–599. doi: 10.1016/0149-7634(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 8.Chedrese PJ, Kay TW, Jameson JL. Gonadotropin-releasing hormone stimulates glycoprotein hormone α-subunit messenger ribonucleic acid (mRNA) levels in α T3 cells by increasing transcription and mRNA stability. Endocrinology. 1994;134:2475–2481. doi: 10.1210/endo.134.6.7515001. [DOI] [PubMed] [Google Scholar]
- 9.Stojilkovic SS, Reinhart J, Catt KJ. Gonadotropin-releasing hormone receptors: structure and signal transduction pathways. Endocr Rev. 1994;15:462–499. doi: 10.1210/edrv-15-4-462. [DOI] [PubMed] [Google Scholar]
- 10.Stojilkovic SS, Catt KJ. Expression and signal transduction pathways of gonadotropin-releasing hormone receptors. Recent Prog Horm Res. 1995;50:161–205. doi: 10.1016/b978-0-12-571150-0.50012-3. [DOI] [PubMed] [Google Scholar]
- 11.Merelli F, Stojilkovic SS, Iida T, Krsmanovic LZ, Zheng L, Mellon PL, Catt KJ. Gonadotropin-releasing hormone-induced calcium signaling in clonal pituitary gonadotrophs. Endocrinology. 1992;131:925–932. doi: 10.1210/endo.131.2.1379169. [DOI] [PubMed] [Google Scholar]
- 12.Stutzin A, Stojilkovic SS, Catt KJ, Rojas E. Characteristics of two types of calcium channels in rat pituitary gonadotrophs. Am J Physiol. 1989;257:C865–C874. doi: 10.1152/ajpcell.1989.257.5.C865. [DOI] [PubMed] [Google Scholar]
- 13.Naor Z, Harris D, Shacham S. Mechanism of GnRH receptor signaling: combinatorial cross-talk of Ca2+ and protein kinase C. Front Neuroendocrinol. 1998;19:1–19. doi: 10.1006/frne.1997.0162. [DOI] [PubMed] [Google Scholar]
- 14.Reiss N, Llevi LN, Shacham S, Harris D, Seger R, Naor Z. Mechanism of mitogen-activated protein kinase activation by gonadotropin-releasing hormone in the pituitary of αT3-1 cell line: differential roles of calcium and protein kinase C. Endocrinology. 1997;138:1673–1682. doi: 10.1210/endo.138.4.5057. [DOI] [PubMed] [Google Scholar]
- 15.Mulvaney JM, Roberson MS. Divergent signaling pathways requiring discrete calcium signals mediate concurrent activation of two mitogen-activated protein kinases by gonadotropin-releasing hormone. J Biol Chem. 2000;275:14182–14189. doi: 10.1074/jbc.275.19.14182. [DOI] [PubMed] [Google Scholar]
- 16.Johnson CM, Hill CS, Chawla S, Treisman R, Bading H. Calcium controls gene expression via three distinct pathways that can function independently of the Ras/mitogen-activated protein kinases (ERKs) signaling cascade. J Neurosci. 1997;17:6189–6202. doi: 10.1523/JNEUROSCI.17-16-06189.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 18.Malviya AN, Rogue PJ. “Tell me where is calcium bred”: clarifying the roles of nuclear calcium. Cell. 1998;92:17–23. doi: 10.1016/s0092-8674(00)80895-8. [DOI] [PubMed] [Google Scholar]
- 19.Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Differential effects of gonadotropin-releasing hormone (GnRH) pulse frequency on gonadotropin subunit and GnRH receptor messenger ribonucleic acid levels in vitro. Endocrinology. 1997;138:1224–1231. doi: 10.1210/endo.138.3.4968. [DOI] [PubMed] [Google Scholar]
- 20.Haisenleder D, Dalkin A, Ortolano G, Marshall J, Shupnik M. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology. 1991;128:509–517. doi: 10.1210/endo-128-1-509. [DOI] [PubMed] [Google Scholar]
- 21.Haisenleder DJ, Khoury S, Zmeili SM, Papavasiliou S, Ortolano GA, Dee C, Duncan JA, Marshall JC. The frequency of gonadotropin-releasing hormone secretion regulates expression of α and luteinizing hormone β-subunit messenger ribonucleic acids in male rats. Mol Endocrinol. 1987;1:834–838. doi: 10.1210/mend-1-11-834. [DOI] [PubMed] [Google Scholar]
- 22.Kaiser UB, Sabbagh E, Katzenellenbogen RA, Conn PM, Chin WW. A mechanism for the differential regulation of gonadotropin subunit gene expression by gonadotropin-releasing hormone. Proc Natl Acad Sci USA. 1995;92:12280–12284. doi: 10.1073/pnas.92.26.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weck J, Fallest PC, Pitt LK, Shupnik MA. Differential gonadotropin-releasing hormone stimulation of rat luteinizing hormone subunit gene transcription by calcium influx and mitogen-activated protein kinase-signaling pathways. Mol Endocrinol. 1998;12:451–457. doi: 10.1210/mend.12.3.0070. [DOI] [PubMed] [Google Scholar]
- 24.Kim WH, Yuan QX, Swerdloff RS, Bhasin S. Regulation of α and luteinizing hormone β subunit messenger ribonucleic acids during stimulatory and downregulatory phases of gonadotropin-releasing hormone action. Biol Reprod. 1988;39:847–853. doi: 10.1095/biolreprod39.4.847. [DOI] [PubMed] [Google Scholar]
- 25.Lerrant Y, Kottler ML, Bergametti F, Moumni M, Blumberg-Tick J, Counis R. Expression of gonadotropin-releasing hormone (GnRH) receptor gene is altered by GnRH agonist desensitization in a manner similar to that of gonadotropin β-subunit genes in normal and castrated rat pituitary. Endocrinology. 1995;136:2803–2808. doi: 10.1210/endo.136.7.7789305. [DOI] [PubMed] [Google Scholar]
- 26.Ben-Menahem D, Shraga-Levine Z, Limor R, Naor Z. Arachidonic acid and lipoxygenase products stimulate gonadotropin α-subunit mRNA levels in pituitary α T3-1 cell line: role in gonadotropin releasing hormone action. Biochemistry. 1994;33:12795–12799. doi: 10.1021/bi00209a010. [DOI] [PubMed] [Google Scholar]
- 27.Andrews WV, Maurer RA, Conn PM. Stimulation of rat luteinizing hormone-β messenger RNA levels by gonadotropin releasing hormone. Apparent role for protein kinase C. J Biol Chem. 1988;263:13755–13761. [PubMed] [Google Scholar]
- 28.Schwartz J, Cherny R. Intercellular communication within the anterior pituitary influencing the secretion of hypophysial hormones. Endocr Rev. 1992;13:453–475. doi: 10.1210/edrv-13-3-453. [DOI] [PubMed] [Google Scholar]
- 29.Farquhar MG, Skutelsky EH, Hopkins CR. Structure and function of pituitary cells. In: Tixier-Vidal A, Farquhar MG, editors. The anterior pituitary. New York: Academic Press; 1975. pp. 83–135. [Google Scholar]
- 30.Childs GV, Unabia G, Lee BL, Lloyd J. Maturation of follicle-stimulating hormone gonadotropes during the rat estrous cycle. Endocrinology. 1992;131:29–36. doi: 10.1210/endo.131.1.1612007. [DOI] [PubMed] [Google Scholar]
- 31.Kaiser UB, Sabbagh E, Chen MT, Chin WW, Saunders BD. Sp1 binds to the rat luteinizing hormone β (LHβ) gene promoter and mediates gonadotropin-releasing hormone-stimulated expression of the LHβ subunit gene. J Biol Chem. 1998;273:12943–12951. doi: 10.1074/jbc.273.21.12943. [DOI] [PubMed] [Google Scholar]
- 32.Saunders BD, Sabbagh E, Chin WW, Kaiser UB. Differential use of signal transduction pathways in the gonadotropin-releasing hormone-mediated regulation of gonadotropin subunit gene expression. Endocrinology. 1998;139:1835–1843. doi: 10.1210/endo.139.4.5972. [DOI] [PubMed] [Google Scholar]
- 33.Kaiser UB, Conn PM, Chin WW. Studies of gonadotropin-releasing hormone (GnRH) action using GnRH receptor-expressing pituitary cell lines. Endocr Rev. 1997;18:46–70. doi: 10.1210/edrv.18.1.0289. [DOI] [PubMed] [Google Scholar]
- 34.Horn F, Bilezikjian LM, Perrin MH, Bosma MM, Windle JJ, Huber KS, Bount AL, Hille B, Vale W, Mellon PL. Intracellular responses to GnRH in a clonal cell line of the gonadotrope lineage. Mol Endocrinol. 1991;5:347–355. doi: 10.1210/mend-5-3-347. [DOI] [PubMed] [Google Scholar]
- 35.Kuphal D, Janovick JA, Kaiser UB, Chin WW, Conn PM. Stable transfection of GH3 cells with rat gonadotropin-releasing hormone receptor complementary deoxyribonucleic acid results in expression of a receptor coupled to cyclic adenosine 3′, 5′-monophosphate-dependent prolactin release via a G-protein. Endocrinology. 1994;135:315–320. doi: 10.1210/endo.135.1.8013367. [DOI] [PubMed] [Google Scholar]
- 36.Barnhart KM, Mellon PL. The orphan nuclear receptor, steroidogenic factor-1, regulates the glycoprotein hormone α-subunit gene in pituitary gonadotropes. Mol Endocrinol. 1994;8:878–885. doi: 10.1210/mend.8.7.7527122. [DOI] [PubMed] [Google Scholar]
- 37.Keri RA, Nilson JH. A steroidogenic factor-1 binding site is required for activity of the luteinizing hormone β subunit promoter in gonadotropes of transgenic mice. J Biol Chem. 1996;271:10782–10785. doi: 10.1074/jbc.271.18.10782. [DOI] [PubMed] [Google Scholar]
- 38.Halvorson LM, Kaiser UB, Chin WW. Stimulation of luteinizing hormone β gene promoter activity by the orphan nuclear receptor, steroidogenic factor-1. J Biol Chem. 1996;271:6645–6650. doi: 10.1074/jbc.271.12.6645. [DOI] [PubMed] [Google Scholar]
- 39.Lee SL, Sadovsky Y, Swirnoff AH, Polish JA, Goda P, Gavrilina G, Milbrandt J. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1) Science. 1996;273:1219–1221. doi: 10.1126/science.273.5279.1219. [DOI] [PubMed] [Google Scholar]
- 40.Topilko P, Schneider-Maunoury S, Levi G, Trembleau A, Gourdji D, Driancourt MA, Rao CV, Charnay P. Multiple pituitary and ovarian defects in Krox-24 (NGFI-A, Egr-1)-targeted mice. Mol Endocrinol. 1998;12:107–122. doi: 10.1210/mend.12.1.0049. [DOI] [PubMed] [Google Scholar]
- 41.Tremblay JJ, Drouin J. Egr-1 is a downstream effector of GnRH and synergizes by direct interaction with Ptx1 and SF-1 to enhance luteinizing hormone β gene transcription. Mol Cell Biol. 1999;19:2567–2576. doi: 10.1128/mcb.19.4.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halvorson LM, Kaiser UB, Chin WW. The protein kinase C system acts through the early growth response protein 1 to increase LHβ gene expression in synergy with steroidogenic factor-1. Mol Endocrinol. 1999;13:106–116. doi: 10.1210/mend.13.1.0216. [DOI] [PubMed] [Google Scholar]
- 43.Alarid ET, Windle JJ, Whyte DB, Mellon PL. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development. 1996;122:3319–3329. doi: 10.1242/dev.122.10.3319. [DOI] [PubMed] [Google Scholar]
- 44.Windle JJ, Weiner RI, Mellon PL. Cell lines of the pituitary gonadotrope lineage derived by targeted oncogenesis in transgenic mice. Mol Endocrinol. 1990;4:597–603. doi: 10.1210/mend-4-4-597. [DOI] [PubMed] [Google Scholar]
- 45.Halvorson LM, Ito M, Jameson JL, Chin WW. Steroidogenic factor-1 and early growth response protein 1 act through two composite DNA binding sites to regulate luteinizing hormone β-subunit gene expression. J Biol Chem. 1998;273:14712–14720. doi: 10.1074/jbc.273.24.14712. [DOI] [PubMed] [Google Scholar]
- 46.Mellon PL, Windle JJ, Goldsmith P, Pedula C, Roberts J, Weiner RI. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-e. [DOI] [PubMed] [Google Scholar]
- 47.Turgeon JL, Kimura Y, Waring DW, Mellon PL. Steroid and pulsatile gonadotropin-releasing hormone (GnRH) regulation of luteinizing hormone and GnRH receptor in a novel gonadotrope cell line. Mol Endocrinol. 1996;10:439–450. doi: 10.1210/mend.10.4.8721988. [DOI] [PubMed] [Google Scholar]
- 48.Thomas P, Mellon PL, Turgeon JL, Waring DW. The LβT2 clonal gonadotrope: A model for single cell studies of endocrine cell secretion. Endocrinology. 1996;137:2979–2989. doi: 10.1210/endo.137.7.8770922. [DOI] [PubMed] [Google Scholar]
- 49.Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang H-J, Miller WL, Mellon PL. Cell-specific transcriptional regulation of FSHβ by activin and GnRH in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- 50.Graham KE, Nusser KD, Low MJ. LβT2 gonadotroph cells secrete follicle stimulating hormone (FSH) in response to activin A. J Endocrinol. 1999;162:R1–R5. doi: 10.1677/joe.0.162r001. [DOI] [PubMed] [Google Scholar]
- 51.Kaiser UB, Sabbagh E, Saunders BD, Chin WW. Identification of cis-acting deoxyribonucleic acid elements that mediate gonadotropin-releasing hormone stimulation of the rat luteinizing hormone β-subunit gene. Endocrinology. 1998;139:2443–2451. doi: 10.1210/endo.139.5.6003. [DOI] [PubMed] [Google Scholar]
- 52.Weck J, Anderson AC, Jenkins S, Fallest PC, Shupnik MA. Divergent and composite gonadotropin-releasing hormone-responsive elements in the rat luteinizing hormone subunit genes. Mol Endocrinol. 2000;14:472–485. doi: 10.1210/mend.14.4.0453. [DOI] [PubMed] [Google Scholar]
- 53.Gorman C. High efficiency gene transfer into mammalian cells. In: Glover DM, editor. DNA cloning: a practical approach. II. Oxford: IRL Press; 1985. pp. 143–190. [Google Scholar]
- 54.Seed B, Sheen JY. A simple phase-extraction assay for chloramphenicol acyltransferase activity. Gene. 1988;67:271–277. doi: 10.1016/0378-1119(88)90403-9. [DOI] [PubMed] [Google Scholar]
- 55.Mellon PL, Clegg CH, Correll LA, McKnight GS. Regulation of transcription by cyclic AMP-dependent protein kinase. Proc Natl Acad Sci USA. 1989;86:4887–4891. doi: 10.1073/pnas.86.13.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luckow B, Schütz G. CAT constructions with multiple unique restriction sites for the analysis of eukaryotic promoters and regulatory elements. Nucleic Acids Res. 1987;15:5490. doi: 10.1093/nar/15.13.5490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lawson MA, Whyte DB, Mellon PL. GATA factors are essential for activity of the neuron-specific enhancer of the gonadotropin-releasing hormone gene. Mol Cell Biol. 1996;16:3596–3605. doi: 10.1128/mcb.16.7.3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neill JD, Duck LW, Sellers JC, Musgrove LC. A gonadotropin-releasing hormone (GnRH) receptor specific for GnRH II in primates. Biochem Biophys Res Commun. 2001;282:1012–1018. doi: 10.1006/bbrc.2001.4678. [DOI] [PubMed] [Google Scholar]
- 59.Millar R, Lowe S, Conklin D, Pawson A, Maudsley S, Troskie B, Ott T, Millar M, Lincoln G, Sellar R, Faurholm B, Scobie G, Kuestner R, Terasawa E, Katz A. A novel mammalian receptor for the evolutionarily conserved type II GnRH. Proc Natl Acad Sci USA. 2001;98:9636–9641. doi: 10.1073/pnas.141048498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dorn C, Ou Q, Svaren J, Crawford PA, Sadovsky Y. Activation of luteinizing hormone β gene by gonadotropin-releasing hormone requires the synergy of early growth response-1 and steroidogenic factor-1. J Biol Chem. 1999;274:13870–13876. doi: 10.1074/jbc.274.20.13870. [DOI] [PubMed] [Google Scholar]
- 61.Madireddi MT, Dent P, Fisher PB. AP-1 and C/EBP transcription factors contribute to mda-7 gene promoter activity during human melanoma differentiation. J Cell Physiol. 2000;185:36–46. doi: 10.1002/1097-4652(200010)185:1<36::AID-JCP3>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 62.Alarid ET, Holley S, Hayakawa M, Mellon PL. Discrete stages of anterior pituitary differentiation recapitulated in immortalized cell lines. Mol Cell Endocrinol. 1998;140:25–30. doi: 10.1016/s0303-7207(98)00025-2. [DOI] [PubMed] [Google Scholar]
- 63.Huhtaniemi I, Jiang M, Nilsson C, Pettersson K. Mutations and polymorphisms in gonadotropin genes. Mol Cell Endocrinol. 1999;151:89–94. doi: 10.1016/s0303-7207(99)00015-5. [DOI] [PubMed] [Google Scholar]
- 64.Jiang M, Pakarinen P, Zhang FP, El-Hefnawy T, Koskimies P, Pettersson K, Huhtaniemi I. A common polymorphic allele of the human luteinizing hormone β-subunit gene: additional mutations and differential function of the promoter sequence. Hum Mol Genet. 1999;8:2037–2046. doi: 10.1093/hmg/8.11.2037. [DOI] [PubMed] [Google Scholar]
- 65.Yen SSC, Jaffe RB. Reproductive endocrinology: physiology, pathophysiology, and clinical management. Philadelphia: W. B. Saunders; 1999. [Google Scholar]
- 66.Cheng KW, Ngan ES, Kang SK, Chow BK, Leung PC. Transcriptional down-regulation of human gonadotropin-releasing hormone (GnRH) receptor gene by GnRH: role of protein kinase C and activating protein 1. Endocrinology. 2000;141:3611–3622. doi: 10.1210/endo.141.10.7730. [DOI] [PubMed] [Google Scholar]
- 67.Stanislaus D, Janovick JA, Brothers S, Conn PM. Regulation of G(q/11)α by the gonadotropin-releasing hormone receptor. Mol Endocrinol. 1997;11:738–746. doi: 10.1210/mend.11.6.0005. [DOI] [PubMed] [Google Scholar]
- 68.Haisenleder DJ, Cox ME, Parsons SJ, Marshall JC. Gonadotropin-releasing hormone pulses are required to maintain activation of mitogen-activated protein kinase: role in stimulation of gonadotrope gene expression. Endocrinology. 1998;139:3104–3111. doi: 10.1210/endo.139.7.6091. [DOI] [PubMed] [Google Scholar]
- 69.Hill CS, Treisman R. Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 1995;80:199–211. doi: 10.1016/0092-8674(95)90403-4. [DOI] [PubMed] [Google Scholar]
- 70.Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- 71.Wolfe AM, Wray S, Westphal H, Radovick S. Cell-specific expression of the human gonadotropin-releasing hormone gene in transgenic animals. J Biol Chem. 1996;271:20018–20023. doi: 10.1074/jbc.271.33.20018. [DOI] [PubMed] [Google Scholar]
- 72.Roberson MS, Misra-Press A, Laurance ME, Stork PJ, Maurer RA. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol. 1995;15:3531–3539. doi: 10.1128/mcb.15.7.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kay TW, Jameson JL. Identification of a gonadotropin-releasing hormone-responsive region in the glycoprotein hormone α-subunit promoter. Mol Endocrinol. 1992;6:1767–1773. doi: 10.1210/mend.6.11.1282668. [DOI] [PubMed] [Google Scholar]
- 74.White BR, Duval DL, Mulvaney JM, Roberson MS, Clay CM. Homologous regulation of the gonadotropin-releasing hormone receptor gene is partially mediated by protein kinase C activation of an activator protein-1 element. Mol Endocrinol. 1999;13:566–577. doi: 10.1210/mend.13.4.0262. [DOI] [PubMed] [Google Scholar]
- 75.Norwitz ER, Cardona GR, Jeong KH, Chin WW. Identification and characterization of the gonadotropin-releasing hormone response elements in the mouse gonadotropin-releasing hormone receptor gene. J Biol Chem. 1999;274:867–880. doi: 10.1074/jbc.274.2.867. [DOI] [PubMed] [Google Scholar]
- 76.Kaiser UB, Halvorson LM, Chen MT. Sp1, steroidogenic factor 1 (SF-1), and early growth response protein 1 (egr-1) binding sites form a tripartite gonadotropin-releasing hormone response element in the rat luteinizing hormone-β gene promoter: an integral role for SF-1. Mol Endocrinol. 2000;14:1235–1245. doi: 10.1210/mend.14.8.0507. [DOI] [PubMed] [Google Scholar]