

Abstract
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) play important roles in regulation of cell survival. In general, moderate levels of ROS/RNS may function as signals to promote cell proliferation and survival, whereas severe increase of ROS/RNS can induce cell death. Under physiologic conditions, the balance between generation and elimination of ROS/RNS maintains the proper function of redox-sensitive signaling proteins. Normally, the redox homeostasis ensures that the cells respond properly to endogenous and exogenous stimuli. However, when the redox homeostasis is disturbed, oxidative stress may lead to aberrant cell death and contribute to disease development. This review focuses on the roles of key transcription factors, signal-transduction pathways, and cell-death regulators in affecting cell survival, and how the redox systems regulate the functions of these molecules. The current understanding of how disturbance in redox homeostasis may affect cell death and contribute to the development of diseases such as cancer and degenerative disorders is reviewed. We also discuss how the basic knowledge on redox regulation of cell survival can be used to develop strategies for the treatment or prevention of those diseases. Antioxid. Redox Signal. 10, 1343–1374.
-
Redox Regulation of Signaling Proteins Affecting Cell Death and Survival
-
Role of Redox Regulation of Cell Survival in Pathogenesis of Diseases
-
Therapeutic Strategies Based on Redox Regulation of Cell Survival
I. Redox Biology and Regulatory Mechanisms
A. Redox homeostasis: ROS production and elimination
The redox system is essential in maintaining cellular homeostasis. Under physiologic conditions, cells maintain redox balance through generation and elimination of reactive oxygen/nitrogen species (ROS/RNS). ROS include radical species such as superoxide (O2−) and hydroxyl radical (HO·), along with nonradical species such as hydrogen peroxide (H2O2). RNS include nitric oxide (NO·) and peroxynitrite (ONOO−) (143). ROS are derived from oxygen, an obligate component of eukaryotic organisms. Reduction of molecular oxygen is the principal mechanism for ROS formation. The initial product, superoxide, results from the addition of a single electron to molecular oxygen. Superoxide can be rapidly dismutated by superoxide dismutase (SOD), yielding H2O2 and O2, which can be reused to generate superoxide radical. In the presence of reduced transition metals, H2O2 can be converted into the highly reactive hydroxyl radical HO· (73).
Both exogenous and endogenous sources contribute to the formation of intracellular ROS/RNS. Exogenous sources include irradiation (i.e., UV irradiation, x-ray, gamma-ray), atmospheric pollutants, and chemicals. For example, exposure to metabolites of polychlorinated biphenyls (PCBs) has been shown to increase ROS production in HL-60 cells (286). As illustrated in Fig. 1, a major endogenous source of cellular ROS is from the mitochondria, where O2− is generated by electron leakage from complex I and III of the electron-transport chain (177, 286). Microsomes and peroxisomes are also sources of ROS, primarily H2O2, whereas immune cells such as neutrophils and macrophages possess oxygen-dependent mechanisms to fight against invading microorganisms. Other endogenous sources of ROS include the membrane-associated NAD(P)H oxidase, cytochrome c oxidase, and xanthine oxidase. The presence of redox-active metals such as Fe and Cu also contributes to ROS generation. In the presence of Fe(II) and Fe(III), HO· can be generated through the Fenton reaction or Haber–Weiss reaction (151). Similarly, NO· generation occurs through specific nitric oxide synthase isozymes, including mitochondrial nitric oxide synthase (mtNOS), neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS) (93). NO can react with O2− to generate ONOO− (293).
FIG. 1.
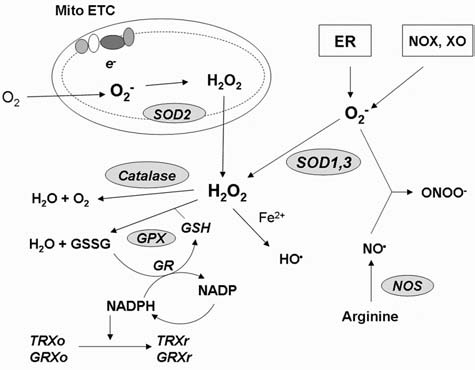
Redox homeostasis. Major sites of cellular ROS generation include the mitochondrial electron transport chain (Mito ETC), the endoplasmic reticulum (ER) system, and the NAD(P)H oxidase (NOX) complex. Nitric oxide synthases (NOS) are key enzymes for production of NO. Major ROS-scavenging enzymes are highlighted in grey. GSH and NAPDH play roles in maintaining the reduced cellular redox state. GPX, glutathione peroxidase; GR, glutathione reductase; TRXo, thioredoxin (oxidized); TRXr, thioredoxin (reduced); GRXo, glutaredoxin (oxidized); GRXr, glutaredoxin (reduced); HO·, hydroxyl radical; NO·, nitric oxide; ONOO−, peroxynitrite; SOD, superoxide dismutase; GSH, reduced glutathione; GSSG, oxidized glutathione; NADPH, reduced nicotinamide adenine dinucleotide phosphate; XO, xanthine oxidase.
Cells are equipped with enzymatic and nonenzymatic antioxidant systems to eliminate ROS/RNS and maintain redox homeostasis. A major class of enzymatic antioxidants, which catalyze the dismutation of O2− to H2O2, is known as superoxide dismutase (SOD). Multiple isoforms of SOD exist in different cellular compartments. SOD1 (CuZnSOD) is the major superoxide scavenger found in the cytoplasm, mitochondrial intermembrane space, nucleus, and lysosomes, whereas SOD2 (MnSOD) and SOD3 are found in the mitochondria and extracellular matrix, respectively (83). Further conversion of H2O2 to H2O + O2 occurs through the action of catalase, a heme-based enzyme that is normally localized in the peroxisome. Interestingly, catalase has extremely high substrate-turnover rates, scavenging ∼6 million molecules of H2O2 per minute (309). H2O2 also can be converted to O2 through coupled reactions with the conversion of reduced glutathione (GSH) to oxidized glutathione (GSSG), catalyzed by glutathione peroxidase (GPX). Five isoforms of selenium (Se)-dependent GPXs are found in humans [for review, see (33)]. The reduction of hydroperoxides by wild-type GPXs is nearly 1,000-fold the rate found in mutated GPXs, which have a cysteine replacing selenocysteine at the active site (197). Glutathione peroxidase 1 (GPX1) is ubiquitously expressed and a major scavenger for H2O2 and lipid hydroperoxides. GPX2 is epithelium-specific and highly expressed in the gastrointestinal tract, whereas GPX3 is an extracellular glycosylated enzyme found in plasma. Interestingly, GPX3 can use thioredoxin and glutaredoxin in addition to GSH as electron donors to reduce a broad range of hydroperoxides. GPX4 is present in cytosolic, mitochondrial, and nuclear forms by alternative splicing, and is a major enzyme preventing oxidation of membrane phospholipids. A newly discovered GPX6 is localized preferentially in olfactory mucosa and embryonic tissue. Furthermore, enzymes such as glutathione S-transferases (GSTs) are known to have Se-independent peroxidase activity (279).
Nonenzymatic antioxidants, recognized to execute thiol–disulfide exchange reactions, also play a major role in maintaining cellular redox balance. In addition to being a cofactor of various antioxidant enzymes, GSH, which is the most abundant peptide in cells, possesses a plethora of functions. These include direct scavenging of HO·, singlet oxygen, and regeneration of other antioxidants such as vitamin C and E to their active forms (222). The thioredoxin system is another important thiol antioxidant consisting of thioredoxin (Trx) and thioredoxin reductase. Thioredoxin is a multifunctional selenoprotein containing two redox-active cysteines and a conserved active site (Cys-Gly-Pro-Cys) (27). Although many ROS are quenched by GSH through reaction with its thiol group, other thiol-containing proteins are also attacked by ROS, leading to their oxidation (184). Therefore, it is essential for cells to change these oxidized proteins to their reduced forms to maintain proper function. The thioredoxin system, in collaboration with the GSH system, plays an important role in reducing oxidized thiol-containing proteins. Similarly, the glutaredoxin (Grx) system, also with the CXXC conserved active site, functions to reduce protein disulfides. Grx1, Grx2, and Grx3 obtain their protein-reducing capacity from the GSH/glutathione reductase system, which is maintained by NADPH (123). Peroxiredoxins (Prxs) are a large family of proteins with cysteine-containing redox active centers (260). The six mammalian isoforms of Prxs are classified into two groups: the two-cysteine peroxiredoxins (I–IV, V) and one-cysteine class (Prx VI). Peroxiredoxins use the peroxidatic cysteine (reactive center) to reduce hydroperoxides in a two-step reaction [for review, see (332)].
B. Oxidative stress and its consequences
The delicate balance between ROS generation and elimination is maintained by many complex mechanisms, and a dysfunction of any of these mechanisms could lead to alterations in cellular redox status. An increase in ROS production or a decrease in ROS-scavenging capacity due to exogenous stimuli or endogenous metabolic alterations can disrupt redox homeostasis, leading to an overall increase of intracellular ROS levels, or oxidative stress. Increased oxidative stress plays a crucial role in a variety of pathologic conditions including cancer, neurodegenerative diseases, and aging (310). Under normal physiologic conditions, the reactive nature of ROS/RNS at moderate levels allows their incorporation into the structure of macromolecules in a reversible fashion. Such reversible oxidative modifications play a critical role in regulating cellular function. However, under oxidative stress, excessive ROS/RNS constantly attack lipids, proteins, and DNA, leading to severe and irreversible oxidative damage.
Lipids are most susceptible to oxidative modification. Lipid peroxidation generates lipid radicals, which can further attack the subsequent lipid molecules and propagate as a chain reaction. The chain-reaction process consists of three stages: initiation, propagation, and termination [for review, see (98)]. Polyunsaturated fatty acid residues of phospholipids are attacked by a radical either at an internal position or near the end of the conjugated system, generating a peroxyl radical (309). Attack at an internal position allows the peroxyl radical to further undergo either a cyclization or metal-catalyzed reaction and produce reactive alkoxyl radicals. After cyclization, the fatty acid may form a hydroperoxide or undergo another cyclization, which produces aldehydes, including malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE) (243). While MDA can further react with DNA bases, resulting in gene mutations, HNE reacts mostly with proteins, leading to significant functional alterations affecting signaling pathways. Oxidative damage of phospholipids can lead to cell death, not only through membrane damage but also through the lipid peroxidation product HNE. Attack of various proteins such as c-Jun N-terminal kinase (JNK) and caspase-3 activation was found to be a mechanism of cell death induced by lipid peroxidation (8).
Protein oxidation can be reversible or irreversible, depending on the target and the form of oxidative damage. The highly reactive OH·, generated through ionizing radiation or the Fenton reaction, and ONOO− are common reactive species that target proteins. Although all amino acid residues could be oxidized by ROS/RNS, certain side chains are particularly susceptible to oxidation. For instance, lysine, arginine, histidine, proline, and threonine are highly sensitive to metal-catalyzed oxidation (309). Oxidation of these side chains results in carbonyl derivatives, which can also be generated through glycation/glycoxidation pathways, lipid peroxidation, α-amidation, and glutamic acid pathways (257). Because a variety of mechanisms of protein oxidation can lead to formation of protein carbonyls, which are easily detectable, the level of protein carbonyls has been used as a quantitative marker of protein oxidation and oxidative stress (62). Sulfur-containing amino acids such as cysteine and methionine are also susceptible to either reversible or irreversible oxidation. Reversible oxidation of the sulfhydryl group includes intramolecular or intermolecular protein cross-linkages and glutathionylation (287). Irreversible protein oxidation includes nitrosylation of cysteine sulfhydryl groups, tyrosine, methionine, and tryptophan by ONOO−. Nitration of tyrosine residues may inhibit its phosphorylation or adenylation, important for protein function (249). Severe oxidative stress can induce disulfide bond–mediated protein cross-linkage or secondary oxidative modifications such as adduct formation between oxidized proteins and lipid peroxides or glycation products. These products can generate aggregation of bulky protein complexes, which may inactivate both 26S and 20S proteosome, leading to accumulation of damaged proteins and cell death (247).
Compared with lipids and proteins, nuclear DNA may seem less susceptible to oxidative modifications because of its double-helix structure and the protective shield from histone and other coating proteins. However, oxidative nuclear DNA damage is detectable under various conditions. Thus, oxidized products of DNA bases such as 8-OHdG have been used as a marker for damage caused by oxidative stress (109). The correlation between oxidative DNA damage and various stages of carcinogenesis has been studied (305). DNA is subject to damage in nearly all of its components. Both purine and pyrimidine bases and the sugar backbone contain N and O as nucleophilic centers, which are highly susceptible to react with electrophiles, especially OH·. Furthermore, the double bonds within the bases are prime targets for OH·. Reactions are primarily centered at the C-5 and C-6 of pyrimidines and C-4 and C-8 of purines (31). NO· and ONOO− have been found to react with DNA bases and induce single-strand breaks (305). Oxidative damage to the sugar backbone, through H abstraction, has been known to cause single-strand breaks and double-strand breaks (31). Unlike nuclear DNA, mitochondrial DNA (mtDNA) is more susceptible to oxidative damage, not only because of its close proximity to the major site of ROS generation (electron-transport chain), but also because of the limited capacity of mtDNA repair (129).
Under physiologic conditions, cellular DNA is constantly attacked by ROS. It has been estimated that in mammalian cells, ∼1.5 × 105 oxidative adducts in DNA per cell are found (15). As such, those hits may induce mutations and play a role in the evolution process. Moderate levels of DNA damage can trigger cell-cycle arrest and initiate DNA-repair processes that ensure DNA integrity. In contrast, excessive damage or failure in DNA repair can induce apoptosis (60). It is worth noting that oxidative modifications of lipids, proteins, or DNA play a crucial role in physiologic processes such as differentiation, maturation, and trafficking of intracellular vesicles (73). However, when the ROS/RNS levels are in excess, the biologic consequences are often deleterious. Therefore, regulation of ROS/RNS levels is critical in maintaining cellular homeostasis.
C. Redox-mediated mechanisms in regulation of cellular processes
Redox balance plays a critical role in maintaining the biologic process under normal conditions. However, disruption of the balance due to an increase in ROS/RNS production or decrease in ROS-scavenging capacity may alter cellular functions. Alterations in ROS/RNS levels can modulate biologic activity through aberrant stimulation/suppression of certain signaling pathways and through direct modifications of biomolecules, especially proteins. The redox system can modify functions of proteins through regulating their expression, posttranslational modifications, and stabilities, as depicted in Fig 2.
FIG. 2.
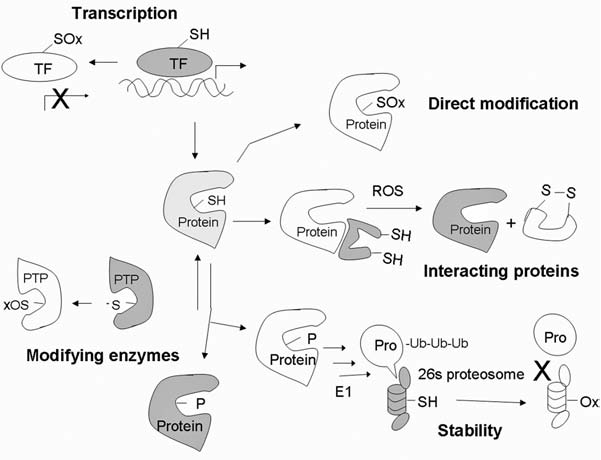
Redox-mediated mechanisms that regulate protein functions. Protein expression can be regulated through redox modification of transcription factors. Oxidation of Cys at or near the DNA-binding site may disrupt the transactivation activity. Newly synthesized protein can be directly modified by oxidation of amino acids such as Cys, Tyr, and Met, resulting in alteration of the protein functions. Certain proteins are stabilized by their redox-sensitive interacting proteins. Modification of the interacting proteins can dissociate the complex and allow activation of the functional proteins. Posttranslational modifications such as phosphorylation can either activate or inhibit protein functions. Phosphatases, which are responsible for dephosphorylation, can be oxidatively inactivated, promoting phosphorylation of proteins. Stability of signaling proteins determines both the level and duration of their functional effects. Most proteins can be degraded through the ubiquitin–proteosome system. Ubiquitin-activating enzyme E1 and proteosome 26S and 20S can be inactivated under oxidative stress. TF, transcription factor; -SH, reduced thiol; SOx, oxidized thiol; PTP, protein tyrosine phosphatase; ub, ubiquitin. X, inhibition; white, inactive state; light grey, partially activated; dark grey, fully activated molecules.
1. Transcriptional regulation
At the synthesis level, expression of signaling proteins can be tightly controlled through the rate of gene transcription. A number of transcription factors contain redox-sensitive cysteine residues at their DNA-binding sites (107). Examples of the factors are NF-κB, AP-1, HIF-1α and P53. In most cases, thiol oxidation of these proteins would inhibit their DNA-binding activities (306). Under physiologic conditions, nuclear GSH plays a critical role in maintaining the reducing environment to prevent excessive oxidative modifications of nuclear DNA and to ensure proper gene transactivation (103). Furthermore, transcriptional co-activators such as CBP/p300 are equipped with histone acetylation activity, which is required to uncoil DNA structure, allowing accessibility of transcription factors to promoter regions of target genes (229). Interestingly, the enzyme histone deacetylase (HDAC), which reverses the acetylation process, was recently found to be redox sensitive (252). Thus, in addition to direct modification of the transcription factors, alteration in ROS/RNS level may regulate gene expression through modulation of chromatin remodeling.
2. Direct oxidative modification
At the posttranslational level, oxidative modification was found to be a major mechanism for redox regulation of protein functions (81). Multiple types of amino acids can be oxidatively modified, with various susceptibilities (27). Direct oxidation is mostly mediated by HO· and NO·. Among those amino acids, sulfur-containing ones such as methionine and cysteine are preferential targets. Oxidative modifications of amino acid residues in a peptide may lead to structural and functional changes, ranging from a slight conformational change to a severe denaturation accompanied by fragmentation. The functional outcome of the oxidation depends on the types of modifications and the criticality of the modified amino acid in the protein function. It may lead to either activation or inhibition of the protein activities. Examples of common oxidative modification of proteins are illustrated in Fig. 3. Mild oxidative stress can induce modifications of Cys such as reversible glutathionylation (94), disulfide formation (4), and S-nitrosylation (292). These modifications are known to have regulatory roles in the function of many proteins such as TRX, p53, IκB, RAS, Akt, and protein tyrosine phosphatase. Conversely, a severe increase in oxidative stress likely promotes more-damaging types of modifications, such as sulfenic acid, sulfinic acid, and sulfonic acid formation (246). Protein carbonylation can occur through either direct oxidative attack (of Lys, Arg, Pro, Thr) or interaction between amino acids (such as Lys, Cys, His) and oxidation products of lipids and sugars. Protein carbonylation is often used as an indicator for protein oxidations, as it accumulates in vivo at high levels relative to other oxidative modifications and is readily detectable (59). Examples of proteins modified by carbonylation, which can impair their functions, include ANT, Hsp, and BCL2 (81). Besides Cys, Tyr is another attractive target for redox modification. Nitration of tyrosine by RNS yields nitrotyrosine, which causes the protein to lose its ability as a substrate for phosphorylation. Kinases such as JNK, p38MAPK, and PKC are targets for tyrosine nitration, which inhibits their activations (272). In contrast, reversible oxidation of a methionine residue in calmodulin (CaM) is essential for its function as a calcium-regulatory protein (23).
FIG. 3.
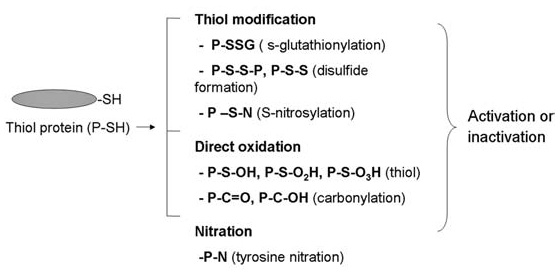
Oxidative modification of proteins. Protein can be oxidatively modified by multiple types of modifications, and the consequence can be either activation or inactivation of protein functions. Cys in thiol proteins is a major target, which could be modified by reversible S-glutathionylation, disulfide formation, S-nitrosylation, or formation of sulfinic, sulfenic, and sulfonic acid derivatives. Nitration of Tyr is known to modulate the function of multiple kinases. P, protein.
3. Regulation of redox-sensitive interacting proteins
Many proteins are stabilized by contact with others. Such protein-protein interactions may also modulate their functions, mostly being negative regulations of each other. Oxidative modification of the interacting partners can lead to dissociation of the protein complex, allowing activation of the free functional proteins (55) (see Fig. 2 for illustration). Examples of proteins whose function can be altered by such redox-sensitive mechanism include ASK1-TRX, JNK-GST, p53-JNK, and Nrf2-Keap1.
4. Regulation of redox-sensitive modifying enzymes
Posttranslational modification of proteins, especially by phosphorylation, has been known to be a critical regulatory mechanism for protein function. Phosphorylation of proteins may either lead to their activation or flag for degradation in a site-specific manner (137). Phosphorylation status is the outcome of the balance between kinases and phosphatases. Interestingly, whereas thiol oxidation of phosphotyrosine kinase (PTKs) leads to their activation, transient oxidation of protein tyrosine phosphatases (PTPases) inhibits their functions (48). Under physiologic conditions, because of its low pKa, the catalytic cysteine of active PTPases is in the thiolate anion form and thus susceptible to oxidative modification. Thiol modifications such as direct oxidation, inter- and intramolecular disulfide bridges, S-glutathionylation, and S-nitrosylation can all lead to inactivation of PTPases (119). The oxidative inhibition of PTPases consequently shifts the balance toward a phosphorylated state in target proteins. Besides PTPases, the lipid phosphatase PTEN and the low-molecular-weight phosphatase cdc25 can also be modified in a similar fashion (50).
5. Regulation of protein turnover
Stability of proteins can determine the extent of their functional effects. The rates of protein turnover can also be regulated by redox-mediated mechanisms. Many proteins are degraded by the proteo-some system, whereas certain proteins are substrates of other proteases such as caspases. Under nonstressed conditions, ubiquitin and 26S proteosome play a crucial role in the degradation of misfolded/damaged proteins (256). Redox-mediated phosphorylation of IκB, Bcl-2, and p53 seems to increase the binding to their specific ubiquitin ligase E3 and to promote the proteosome-mediated degradation of these proteins. However, under oxidative stress, although the ubiquitin-activating enzymes (E1) and 26S proteosome are oxidatively inactivated, oxidized proteins may no longer be ubiquitinated and degraded (22). Instead, such oxidized products can be eradicated by the 20S proteosome in a ubiquitin-independent manner (285).
II. Redox Regulation of Signaling Proteins Affeecting Cell Death and Survival
The roles of ROS and antioxidant systems in regulation of cell survival are bifurcated. In general, ROS at low levels act as signaling molecules that promote cell proliferation and cell survival. In contrast, a severe increase in ROS can induce cell death. Previous studies suggest that regulation of signaling pathways by the redox system relies mostly on direct oxidative modifications of the redox-sensitive signaling proteins (143). However, recent evidence shed new light on the novel role of redox regulation in chromatin remodeling, which affects death/survival signals at the transcriptional level (252). Furthermore, posttranslational modifications of signaling proteins such as phosphorylation have recently been shown to be regulated in part through a redox-mediated mechanism (119). Oxidative modification of ubiquitin-proteosome or other proteases can also affect the turnover of signaling proteins (247). The following sections summarize redox regulation of cell survival through modulation of those factors at the transcription, signal transduction, and death-execution levels. Figure 4 illustrates an overview of the redox regulation at these levels and their crosstalk.
FIG. 4.
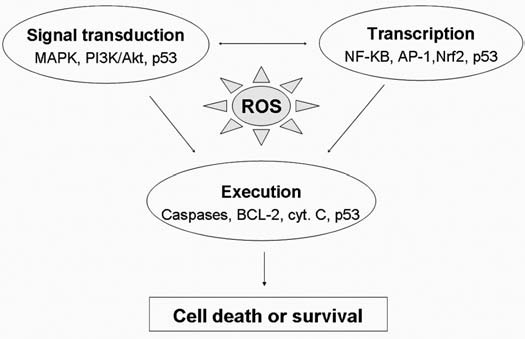
Redox-sensitive signaling pathways for regulation of cell survival. The redox system can regulate the cell-fate decision through regulations of many functional proteins involving cell life-or-death decisions. Many of those signaling proteins are redox sensitive, which controls survival at the levels of signal transduction, transcriptional regulation, or execution. Examples of the key redox-sensitive molecules involved at each level are indicated. The possible crosstalk among these regulators/executors is indicated by arrows. Signal transduction may involve in transcriptional regulation, and p53 is a redox-sensitive molecule that affects cell survival at all three levels. Therefore, oxidative stress not only serves as a type of stimulus to trigger stress-response signal-transduction pathways, but also can modulate cell death/survival through direct oxidative modification of those signal molecules.
A. Redox regulation of cell survival at the transcription level
Intracellular redox homeostasis regulates the expression of multiple gene-encoded proteins affecting cell death and survival. In response to alterations in oxidative status, the transcription of those genes can be modulated in part through a redox control of transcription factors. Here, we focus on the roles of transcription factors NF-κB, AP-1, Nrf2, and HIF in cell survival and how the redox system regulates the functions of these factors.
1. NF-κB
Nuclear factor kappa B (NF-κB) is a redox-sensitive transcription factor that coordinates regulators of immunity, inflammation, development, cell proliferation, and survival. In mammals, the NF-κB family consists of NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), c-Rel, and RelB. All members are characterized by the presence of the Rel homology domain (RHD). The RHD mediates DNA binding, dimerization between the family members, and the association of NF-κB dimers with the inhibitors of kappa B (IκB) (117). Normally, the NF-κB components are sequestered and inactivated by IκBs in the cytosol. A wide range of stimuli, including cytokines and ROS stress, are capable of activating NF-κB through activation of IκB kinase (IKK). Active IKK phosphorylates IκB, leading to dissociation of NF-κB from the inhibitor and the attraction of IκB to degradation by ubiquitin/proteasome system (96). Free NF-κB translocates to the nucleus, binds to DNA at the promoter region, and activates the transcription of target genes.
a. Role of NF-κB in cell survival
Active NF-κB controls cell survival through altering transcription of multiple genes as illustrated in Fig. 5. In response to oxidative stress, activation of NF-κB leads to elevated expression of (a) antiapoptotic Bcl-2 family members such as Bcl-xL and A1/Bfl-1; (b) the inactive homologue of caspase-8 (FLIPL); (c) caspase inhibitors such as IAPs that directly prevent activation of caspases; (d) TNF receptor–associated factor TRAF1; (e) Gadd45, which inhibits JNK-mediated cell death; and (f) antioxidants such as Mn-SOD and ferritin heavy chain (FHC) (147).
FIG. 5.
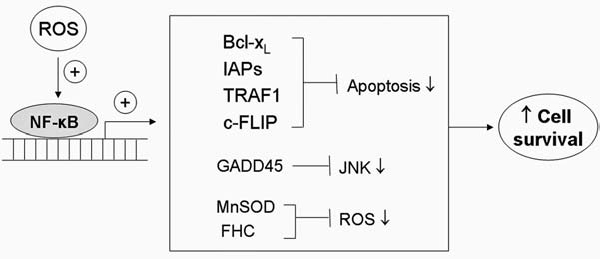
Role of NF-κB in cell survival. NF-κB functions as a transcription factor regulating the expression of multiple genes. Activation of NF-κB by stimuli such as oxidative stress or cytokines promotes increased expression of antiapoptotic proteins such as Bcl-xL and XIAP, which suppress the execution phase of cell death. Induction of GADD45 leads to inhibition of JNK and prevents JNK-induced apoptosis. NF-κB also promotes the expression of antioxidant genes such as MnSOD, which plays a major role in scavenging mitochondria superoxide and in maintaining redox homeostasis. Overall, the activation of NF-κB by ROS leads to inhibition of apoptosis, redox rebalance, and enhanced cell survival.
b. Redox regulation of NF-κB
NF-κB has long been recognized as a redox-sensitive transcription factor. Experimental evidence suggests that ROS seem to have paradoxic effects on NF-κB regulation. ROS can either activate or inhibit NF-κB activity, depending on the level of ROS, types of stimuli, and cell types (140, 236). Moderate increase of ROS often leads to NF-κB activation, which requires sequential steps in the cytosol and nucleus. Conversely, severe increase of ROS could inactivate NF-κB, leading to cell death. As depicted in Fig. 6, multiple redox-mediated mechanisms can regulate NF-κB activity at various stages.
FIG. 6.
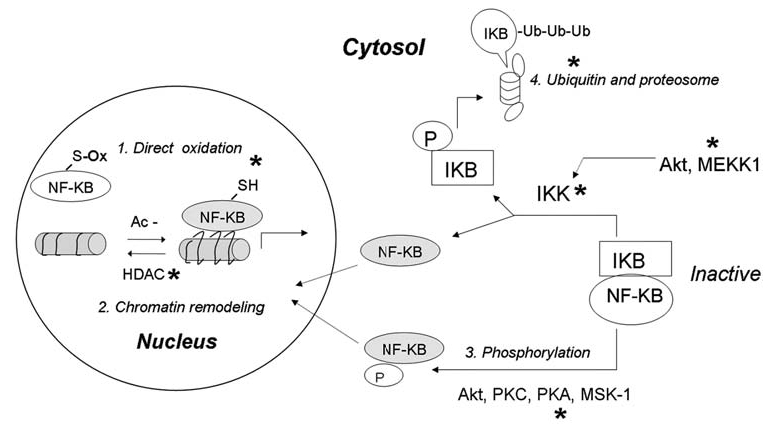
Redox regulation of NF-κB. The function of NF-κB can be activated or inhibited through various redox-mediated mechanisms at multiple levels of the activation pathways. In the nucleus, direct oxidation of Cys in the DNA-binding domain can inhibit NF-κB-DNA-binding activity. In contrast, enzyme histone deacetylase (HDAC), which catalyzes the removal of an acetyl (Ac-) group from histone, can be inactivated by oxidative stress, allowing histone acetylation, chromatin uncoiling, and increased accessibility for NF-κB. In cytosol, activation of NF-κB can be regulated through phosphorylation of NF-κB itself or phosphorylation of its inhibitor IκB. Normally, NF-κB and IκB form a complex, which is sequestered in cytosol. Increased ROS can activate IκB-kinase (IκK) either directly through redox modification of IκK, or indirectly through activation of Akt and/or MEKK1, which then phosphorylates and activates IκK. Active IκK phosphorylates IκB and liberates active NF-κB from the complex to translocate to the nucleus. Phosphorylated IκB undergoes ubiquitination and degradation by proteosomes. Because the proteosome system is also redox sensitive, ROS can also regulate NF-κB activity by affecting the stability of IκB. Furthermore, phosphorylation of NF-κB by certain kinases may dissociate NF-κB from IκB and promote its nuclear translocation. Grey, Active forms of the proteins. *Major target molecules of redox regulation.
c. Redox regulation of nuclear NF-κB
Studies since the early 1990s demonstrated that the reduced form of nuclear NF-κB is required for its DNA binding. Oxidation of the redox-sensitive site Cys62 of the p50 subunit inhibits its ability to bind DNA (210, 298). Oxidation of p50 is reversible, and DNA binding can be restored through reduction by thioredoxin (204). Detailed studies revealed that nuclear NF-κB can be inactivated by several redox modifications including glutathionylation (244) and S-nitrosylation (200). Besides direct structural modification, DNA binding activity of NF-κB can be modulated by chromatin remodeling (252). Histone acetylation, which is catalyzed by histone acetylase (HAT), uncoils the helical structure of DNA and exposes the binding sequence at the promoter regions, thus promoting NF-κB–DNA binding activity. Conversely, removal of the acetyl group from histone by histone deacetylase (HDAC) renders recoiling of DNA structure and prevents DNA binding by NF-κB. Acetylation of the RelA (p65) subunit of NF-κB was shown to increase DNA-binding activity, which can be reversed by HDAC (252). Interestingly, recent evidence suggests that HDAC activity can be inhibited by ROS. Oxidative inactivation of HDAC results in a shift of balance in favor of histone acetylation, which unwinds the DNA and promotes NF-κB activity (251). Further work suggested that inhibition of HDAC prevents apoptosis in leukemia cells by activation of NF-κB and downregulation of c-Jun kinase (57).
d. Cytoplasmic regulation of NF-κB
A key step in activation of NF-κB is the dissociation of NF-κB from IκB. Phosphorylation of either IκB or NF-κB subunits promotes such dissociations (236). Under certain conditions, hydrogen peroxide is able to activate NF-κB activity directly through phosphorylation/activation of IKK (144), or indirectly via MEKK-1 or Akt (196, 226). The transactivation of NF-κB induced by MEKK-1 or Akt was shown to be critical in mediating its antiapoptotic effect (225, 314). Conversely, in other system oxidants such as H2O2, arsenic, and lipid peroxidation product 4-HNE were found to inhibit IKK activity through direct oxidation, S-glutathionylation, or S-nitrosylation at Cys-179 of the IKK β-subunit (258, 259). Furthermore, MEKK-1 and Akt are redox sensitive. S-glutathionylation can inhibit their kinase activities (54, 219). It is worth noting that the oxidative activation or inhibition of IKK activity may or may not translate into corresponding changes in NF-κB activity. The overall consequences may be dependent on the different redox states of the cell types, the levels of the oxidants, and the durations of ROS exposure. For example, in human bronchial epithelial cells treated with H2O2, whereas the IKK activity was increased, leading to IκB phosphorylation, NF-κB–DNA-binding activity was inhibited. The unexpected result was attributed to an inhibitory effect on proteosomal degradation of IκB under oxidative stress (135). Besides phosphorylation of IκB by IKK, emerging evidence demonstrated that the NF-κB subunit may also be directly phosphorylated. Akt, protein kinase A (PKA), PKC ζ, mitogen and stress-activated kinase-1 (MSK-1), the 90-kDa ribosomal S6 kinase-1 (RSK-1), and casein kinase 2 (CK-2), can all phosphorylate RelA subunit. The phosphorylation of RelA can affect its binding affinity to IκB, to DNA and to the recruitment of essential cofactors (76, 236, 319, 344,). Interestingly, these kinases were all redox sensitive, thus providing another layer for redox regulation of NF-κB.
2. AP-1
The AP-1 family of proteins represents an example of transcription factors whose functions involve control of both cell growth and apoptosis. Under certain conditions, AP-1 activation could lead to cell death, whereas under other circumstances, AP-1 may promote cell proliferation and survival. The AP-1 family consists of several groups of basic leucine zipper domain (bZIP) proteins, including Jun (c-Jun, JunB, JunD), Fos (c-Fos, FosB, Fra-1, and Fra2), Maf (c-Maf, MafB, MafA, MafG/F/K, and Nrl), and ATF (ATF2, LRF1/ATF3, B-ATF, JDP1, JDP2) subfamilies (3). AP-1 proteins form heterodimers and bind to the target DNA sequence. Activation of AP-1 is regulated at both transcript and protein levels. The intracellular levels of c-jun and c-fos are controlled mainly by their transcription rates, which are tightly regulated by a variety of stimuli (278). The mitogen-activated protein kinase (MAPK) plays a major role in controlling activation of AP-1 proteins through phosphorylation. All three classes of MAPKs are involved in regulation of AP-1 activity (i.e., c-jun is regulated mainly by JNK and ERK in some cell types). c-Fos is a substrate of ERK, and ATF2 is regulated by JNK and p38 kinases (146). JNK and p38 are both activated by stress stimuli.
a. Role of AP-1 in cell survival
AP-1 transcription factors are involved in both the induction and prevention of apoptosis, and the exact outcomes are highly tissue and developmental-stage specific (278). The pivotal role of c-Jun in cell survival was evidenced by embryonic lethality of mice lacking c-Jun, which is associated with prominent apoptosis of liver cells, leading to liver failure (121). Furthermore, by using sorbitol as an osmotic stressor, a recent study showed that c-Jun–deficient fibroblasts were more sensitive to osmotic stress-induced cell death, and downregulation of c-jun promoted cell death in c-Jun +/+ cells (334). These findings suggested a protective role of c-Jun against cell death. In contrast, studies in PC12 cells revealed a dual effect of c-Jun on cell death, depending on the stage of differentiation. In differentiated cells, c-Jun mediated induction of apoptosis, whereas in cells that were not yet differentiated, c-Jun exerted its cytoprotective effect (181). The role of c-Jun as an inducer of apoptosis was also seen in other systems. For example, overexpression of c-Jun was found to induce apoptosis in 3T3 fibroblast (26). Inhibition of c-Jun by antisense oligonucleotides is known to increase survival of lymphoid cells deprived of growth factor (53). Some studies suggested that the apoptosis-inducing effect of c-Jun may be triggered by JNK (71, 338). The role of the JNK signaling pathway in cell survival is discussed later in this section.
The mechanism by which c-Jun mediates cell survival or death seems to depend on the balance between the proapoptotic and antiapoptotic target-gene transcriptions and may be further regulated by p53 and p21 through their cell-cycle regulatory activity. FasL, Bim, and Bcl3 are target genes of c-Jun. Induction of FasL and Bim may promote apoptosis, whereas upregulation of BCL3 by c-jun may potentiate its antiapoptotic function (278). It is the equilibrium between the positive and negative regulators of apoptosis that determines overall cell fate. This balance may be cell-type and stimulus dependent, as well as the integration of the effects from other transcription factors. Furthermore, c-Jun was shown to regulate the decision between p53-mediated cell-cycle arrest and apoptosis. A high level of c-Jun, which repressed p53-mediated p21 induction, was shown to prevent UV-induced growth arrest and shift most of p53 activity toward the induction of apoptosis (277). Interestingly, a recent report showed that Jun proteins (c-Jun, JunD, and JunB) upregulate antioxidant-responsive element (ARE)-mediated expression of antioxidant genes, such as thioredoxin, by associating with Nrf2 and Nrf1 and binding with ARE (318). This function may be important in the adaptive response to survive under oxidative stress.
b. Redox regulation of AP-1
Oxidative stress can activate c-Jun and ATF2 through phosphorylation by JNK and p38, respectively. Redox regulation of the JNK and p38 pathway is discussed later in this review. Like NF-кB, transcriptional activation of AP-1 can be regulated by chromatin remodeling (10, 211). Oxidative stress is known to promote AP1 activity through histone acetylation by inhibition of HDAC (251). Likewise, nitric oxide may suppress the DNA-binding activity of AP-1 through S-glutathionylation (158). Furthermore, the intracellular level of AP-1 can be regulated by redox-mediated mechanisms at the levels of transcription and protein turnover. Recent reports show that expression of c-Jun can be transcriptionally repressed by HDAC or proteosomally degraded through MEKK1-induced ubiquitination in response to osmotic stress. This downregulation of c-Jun plays an important role in apoptosis induction by oxidative stress (333, 334).
3. Nrf2
NF-E2–related factor 2 (Nrf2) is a member of p45 NF-E2–related proteins (p45 NF-E2, Nrf1, Nrf2, and Nrf3 (162, 163). The proteins in this family require a heterodimeric formation with small Maf proteins for DNA binding (215). Under normal conditions, Nrf2 localizes in the cytoplasm, where it interacts with the actin-binding protein, Kelch-like ECH-associating protein 1 (Keap1) (133). Keap1 functions as an adaptor of Cul3-based E3 ubiquitin ligase and targets Nrf2 for rapid degradation by the ubiquitin-proteasome (161). Oxidative stress and electrophiles are major activators of Nrf2 pathway. Dissociation of Nrf2 from Keap1 is a key step in activating Nrf2. The free Nrf2 translocates to the nucleus, heteromerizes with Maf(s), and binds to a cis-acting element known as antioxidant responsive element (ARE) or electrophile responsive element (EpRE) within the regulatory regions of many genes. Studies using Nrf2-deficient mice and microarray-based assays suggest that Nrf2 modulates transcription of almost 200 genes whose protein products function as antioxidants, phase II detoxification enzymes, proteosomes, heat-shock proteins, and glutathione-synthesis enzymes. These proteins all play a critical role in cellular defense against oxidative stress (132, 162).
a. Role of Nrf2 in cell survival
Nrf2 plays a critical role in protection against oxidative damage induced by acute injury, hyperoxia, nitrosative stress, ER stress, and exogenous prooxidative agents (43, 178, 189, 295). Nrf2 activation promotes cell survival under oxidative stress through multiple mechanisms. One major function is the transactivation of many antioxidant proteins, including heme oxygenase-1, ubiquitin/PKC-interacting protein A170, peroxiredoxin 1, the heavy and light chains of ferritin, catalase, glutathione peroxidase, superoxide dismutase, and thioredoxin (131). These proteins directly or indirectly scavenge free radicals and decrease the dose-dependent toxicity of ROS. Furthermore, Nrf2 regulates the synthesis of glutathione by controlling both the basal and inducible expression of genes encoding the heavy and light chains of glutamylcysteine synthetase (14). Because glutathione is not only the most abundant scavenger of ROS, but also the key controller of redox status of proteins affecting cell survival and death, the regulatory effect of Nrf2 on glutathione synthesis plays an important role in cell survival. Furthermore, Nrf2 was shown to modulate elimination of prooxidative electrophilic compounds through regulating expression of phase II detoxification enzymes such as glutathione-S-transferase (GST) and transporters such as multidrug resistance–associated protein 1/ATP-binding cassette transporter C. Direct roles of Nrf2 on cell survival and the death pathway are also evident. Nrf2 has been identified as an inhibitor of Fas-induced apoptosis (166, 213). In the absence of Nrf2, death-receptor–induced apoptosis was found to be enhanced. The cell death could be suppressed by supplementation of glutathione, suggesting that the antiapoptotic effect of Nrf2 was through elevating intracellular glutathione levels (166, 213).
Accumulation of unfolded polypeptides after oxidative stress could also trigger apoptosis. In response to unfolded protein stress, Nrf2 is a direct substrate of phosphorylation by PERK and acts as an effector of PERK-dependent cell survival (56). PERK is an ER transmembrane protein kinase that phosphorylates the subunit of translation-initiation factor 2 (eIF2a) in response to ER stress. Phosphorylation of eIF2a reduces the global translation, allowing cells sufficient time to correct the impaired protein folding (329). Induction of 26S proteosome and heat-shock proteins by Nrf2 facilitates the repair or elimination of the damaged proteins and thus protects cells from apoptosis (171).
b. Redox regulation of Nrf2
Association and dissociation of the Nrf2–Keap1 complex is considered as a key step in regulating Nrf2 activity. Multiple reactive Cys residues in Keap1 are targets of modifications by ROS and electrophiles. As illustrated in Fig. 7, sulfhydryl modifications dissociate Keap1 from Nrf2, allowing the translocation of Nrf2 to the nucleus, where it transactivates target-gene expression (70). Among the possible targeted residues for oxidation, Cys273 and Cys288 of Keap1 seem crucial for the ubiquitination-promoting activity. Therefore, the oxidative modification of Keap1 may also inhibit Keap1-mediated proteosomal degradation of Nrf2, allowing stabilization and nuclear accumulation of Nrf2 (163). In addition to targeting Keap1, oxidants and electrophiles can activate Nrf2 through phosphorylation by PKC and PERK. Phosphorylation of Nrf2 promotes its dissociation from Keap1, allowing the free Nrf2 to translocate to nucleus (56, 126). In the nucleus, Nrf2 can also be regulated at the step of DNA binding. Nrf2 cannot bind to the ARE without forming a heterodimer with one of the small Maf proteins (149, 215); therefore, the expression level of Maf protein likely regulates the Nrf2–DNA-binding capacity. Interestingly, expression of Maf can also be transcriptionally regulated by Nrf2/ARE itself, thus serving as an autoregulatory feedback mechanism (150). In addition to Maf, c-Jun and ATF-4 can heteromerize with Nrf2 and enhance Nrf2-DNA–binding activity (118). In contrast, Bach1, a transcriptional repressor of ARE/EpRE, can compete with Nrf2 to bind to the same DNA sequence, thus preventing Nrf2/ARE transcriptional activation. Recent study showed that oxidative stress can trigger nuclear accumulation of Bach1, leading to transcriptional suppression of ARE target genes (220).
FIG. 7.
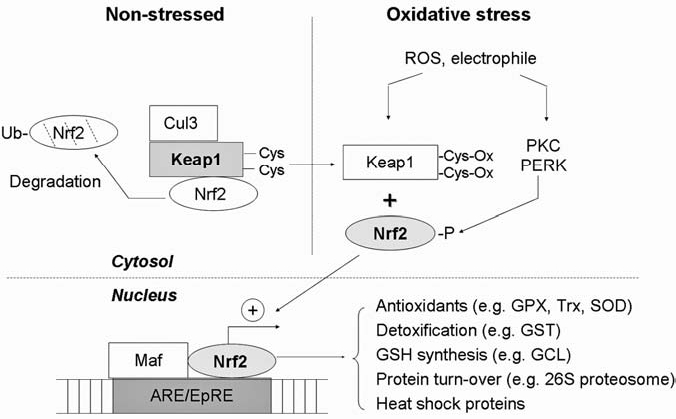
Redox regulation of Nrf2. In unstressed cells, Nrf2 is sequestered in cytosol by Keap1, which functions as an adaptor for Cul3 (a ubiquitin E3 ligase) to target Nrf2 for ubiquitination and degradation. On oxidative stress or electrophilic stimuli, Nrf2 is activated via two mechanisms: (a) thiol oxidation of Keap1 and (b) phosphorylation of Nrf2 by kinases such as PKC or PERK. These cause release of Nrf2 from the inactive complex. The free Nrf2 is translocated to the nucleus, where it forms a heterodimer with Maf proteins and then binds to the antioxidant-responsive element or electrophile-responsive element (ARE/EpRE). The active binding triggers transcription of multiple target genes that encode antioxidants, glutathione synthesis enzymes, proteosomes, and heat-shock proteins. Grey, Active forms of the molecules. GPX, glutathione peroxidase; Trx, thioredoxin; SOD, superoxide dismutase; GCL, glutamylcysteine ligase, GST; glutathione-S-transferase.
4. HIF
Hypoxia-inducible factor (HIF) is generally known as an important transcription factor that regulates cellular metabolism and cell survival under hypoxic stress (29). However, HIF can also be activated by nonhypoxic stimuli such as thrombin and CoCl2 under normoxia (248). HIF is composed of an alpha and beta subunit (HIF-α and HIF-β). Three α isoforms have been described (HIF-1α, HIF-2α, HIF-3α), with HIF-1 α being most intensively studied (30). Active HIF requires heterodimeric formation of the two subunits, which then translocates to nucleus, binds to the hypoxia-response element (HRE), and associates with coactivators such as CBP/p300. The binding results in activation or suppression of many genes involved in metabolism, angiogenesis, invasion/metastasis, and cell survival/death (29). The stability and activity of HIF1 can be regulated by oxygen-requiring hydroxylases [for review, see (270)]. Although HIF-β is constantly expressed regardless of oxygen levels, the stability of HIF-α is highly dependent on oxygen. Under atmospheric levels of oxygen (21%), the dioxygenase PHD (prolyl hydroxylase domain) hydroxylates HIF-1α on two proline residues. The hydroxylated HIF is then recognized by the von Hippel–Lindau (VHL) ubiquitin E3 ligase complex, which promotes the degradation of HIF-1α by the 26S proteosome (134). The hydroxylation of HIF requires iron in ferrous form (Fe2+), oxygen, and 2-oxoglutarate as cofactors for the PHD catalytic activity (30). Under hypoxia, ferrous iron is converted to ferric form (Fe3+), resulting in a decrease in HIF-α hydroxylation by PHD and subsequent stabilization of HIF-α (18). When the free HIF-1α binds to HIF-β and translocates to nucleus, another oxygen-dependent hydroxylase enzyme called factor-inhibiting HIF-1 (FIH-1) can regulate the DNA binding and transcriptional activity of HIF. Under normoxia, FIH-1 hydroxylates an N-terminal asparagines residue and renders HIF inactive by preventing the binding between HIF and its coactivators (175). Interestingly, genes encoding two isoforms of PHD proteins (phd 2 & 3) are HIF targets. Therefore, HIF can also be autoregulated under hypoxia by increased expression of its regulators. This response ensures a rapid and optimal degradation of HIF-α when the cells return to normoxia (30).
a. Role of HIF in cell survival
HIFs can act as both prosurvival and prodeath factors, depending on the stress conditions. Under most circumstances, HIF-1 actively contributes to adoptive responses to promote cell survival under hypoxia through transcriptional regulation of angiogenic factors and glycolytic enzymes (90). In tumor cells, HIF plays a major role in the metabolic switch that shunts glucose metabolites from mitochondria respiration to cytosolic glycolysis (Warburg effect) (194). HIF activation not only increases anaerobic glycolysis, but also attenuates mitochondrial respiration. The former occurs through upregulation of genes encoding glucose transporter (GLUT), glucokinases, aldolase A, and lactate dehydrogenase A (LDH-A), the enzymes that convert pyruvate to lactate. The latter occurs through the induction of pyruvate dehydrogenase kinase 1 (PDK1), which inhibits pyruvate dehydrogenase, the enzyme that converts pyruvate into acetyl-CoA in the mitochondria. These two phenomena were known to prevent the entry of pyruvate to the TCA cycle and shunt pyruvate toward lactate formation through glycolysis (29, 155). Recent study showed that silencing LDH-A resulted in a metabolic switch from glycolysis to the mitochondrial pathway and reduced tumor growth (82). These suggest that formation of lactate through glycolysis is important for tumor cell metabolism. Furthermore, it has been proposed that the increase in glycolysis mediated by HIF facilitates cell survival through maintaining ATP production and preventing the deleterious effect of ROS generated from mitochondrial respiration (155). Conversely, recent evidence suggests that HIF1 can also promote hypoxic cell death under certain conditions. It is proposed that active HIF may induce apoptosis though increased expression of several proapoptotic factors, including mitochondrial HGTD-P, Noxa, BNIP3, NIX, and IGFBP-3 (104). Thus, in response to hypoxia, HIF may promote or prevent cell death in a cell-type and stimulus-dependent manner.
b. Redox regulation of HIF
Under normoxia, a variety of stimuli [including growth factors such as IGF-1, hormones, vasoactive peptides such as thrombin, metal ions such as CoCl2, H2O2, and certain NO donors such as S-nitrosoglutathione (GSNO) and NOC-18] are known to stabilize HIF, in part through increase ROS/RNS production (17, 100, 208). Interestingly, attenuation of ROS/RNS levels by antioxidants, such as NAC, ascorbate, and catalase, or genetic downmodulation of ROS-producing enzymes, such as NADPH oxidase 4 (NOX4), were found to decrease HIF1 expression under certain conditions (17, 153, 160, 276). At least three mechanisms have been proposed to explain how ROS/RNS stabilize HIF under normoxia (18). The first possibility is that the increased generation of OH· from H2O2 through Fenton reactions, likely promotes the conversion of Fe2+ to Fe3+. Because Fe2+ but not Fe3+ is required for active PHD activity, accumulation of ROS would lead to inactivation of PHD and consequently stabilization of HIF1 (248). This idea is supported by the findings that ROS accumulation by genetic loss of JunD-dependent antioxidant pathways leads to increased HIF1 activation through decreased availability of Fe2+ and attenuated activity of PHD (92). Furthermore, direct oxidative modifications such as S-nitrosylation of HIF or pVHL have been shown to cause stabilization of HIF (183, 234). Another putative mechanism of oxidative stabilization of HIF1 is that ROS/RNS activate multiple signaling pathways, such as PI3K/Akt and p38 MAPK, which may render PHD catalytically inactive (80, 148, 216).
Under hypoxic conditions, both ROS and RNS were found to inhibit HIF-1 DNA-binding activity and HIF-1 accumulation (34, 127). A study in HEK293 cells demonstrated that NO can destabilize HIF under hypoxic conditions through an increase in PHD-dependent degradation of HIF-1α (108). The author proposed that under hypoxia, NO inhibited mitochondrial respiration; thus, oxygen may be redistributed to other oxygen-dependent targets, such as PHD, and consequently promotes prolyl hydroxylation of HIF (108). Another study suggests that NO mediates destabilization of HIF under hypoxia through increased ROS production (40). In vitro HIF-1α -pVHL interaction assays demonstrated that a low level of ROS formation increased PHD activity and promoted ubiquitination and degradation of HIF (40). In addition to redox modulation of PHD, several lines of evidence suggest that a reducing condition is required for HIF-DNA–binding activity (152, 187, 325), and oxidative agents such as H2O2 can decrease HIF-DNA–binding activity and the expression of its target genes, such as EPO, aldolase A, and glucokinase (152). Taken together, these studies suggest the important role of the redox system in regulating HIF under both normoxia and hypoxia. However, further investigation is needed to provide a clear understanding of how ROS/RNS modulate HIF stability and activity under hypoxia.
B. Redox regulation of cell survival at the signal-transduction level
1. Mitogen-activated protein kinase
MAPK operates in a cascade fashion with a MAP kinase kinase kinase (MAPKKK) phosphorylating and activating a MAP kinase kinase (MAPKK), which then phosphorylates and activates a MAP kinase (MAPK). The MAPK family consists of extracellular regulated kinases (ERK1/2), Jun N-terminal kinase (JNK), p38 kinase, ERK3/4, and the big mitogen-activated protein kinase 1 (BMK1/ERK5) pathways. The JNK and p38 kinase pathways are sometimes grouped together and referred to as the stress-activated protein kinases (SAPKs) (172).
a. Role of MAPK for cell survival under oxidative stress
Generally, ERK1/2 signaling promotes cell survival under mild oxidative stress, whereas SAPKs seem to induce cell death as a response to oxidative injuries (203). In response to oxidative stress, JNK and p38 activation can induce both intrinsic and extrinsic pathways of apoptosis and necrosis (206, 280). Induction of apoptosis by activated JNK involves direct phosphorylation of pro/anti-apoptotic BCL2 family members, trans-activation of AP-1 (c-Jun and ATF2), and stabilization of p53. Phosphorylation of Bcl-2, Bcl-xL, and Mcl-1 by JNK is known to inhibit their antiapoptotic activities, whereas phosphorylation of Bim, Bmf, and Bad by JNK results in activation of those BH3-only proteins. The inhibition of antiapoptotic proteins and the activation of BH3-only proteins may promote translocation and activation of Bax/Bak, leading to mitochondria-mediated apoptosis (331). Furthermore, activation of JNK was shown to promote its dissociation from p53, leading to stabilization of p53. Active p53 in combination with AP-1 leads to Bid cleavage followed by the translocation of Bax protein to mitochondria and initiation of apoptosis (291). Although JNK and p38 activation can lead to cell death, the requirement of kinases seems to be cell-type and stimulus specific. An example is the critical role of p38 MAPK but not JNK in induction of apoptosis in keratinocytes by UVB irradiation (311). A recent study showed that the SAPK pathways can also induce apoptosis through the death-receptor pathway (280, 282). Conversely, JNK or p38 MAPK activation has been reported to have an antiapoptotic effect in malignant B and T lymphocytes, respectively (41, 191). The bifunctional role of the SAPK pathways in cell-fate decision may be dependent on different cell types and stimuli. Furthermore, the duration of the SAPK activation may dictate its consequences; that is, a transient activation may promote cell survival, whereas a sustained activation tends to induce apoptosis (203).
b. Redox regulation of MAPK
Multiple evidence shows that oxidative stress can activate the ERK pathway (301). This activation involves the stimulation of growth-factor receptor (tyrosine kinase receptor), which activates Ras, recruits Raf-1MAPKKK to the plasma membrane, and sequentially phosphorylates and activates MEK1/2 and ERK1/2. Redox modification can regulate ERK activation at the level of the tyrosine kinase receptor and the Ras activation. Autophosphorylation and activation of the tyrosine kinase receptor can be promoted by direct thiol-modification of the receptor (46). Furthermore, sustained activation of the receptor by oxidative stress could be obtained through oxidative inactivation of the phosphatases, the enzymes that dephosphorylate and inactivate the receptors (159). At the level of Ras activation, oxidative stress can modulate the function of Ras through thiol modifications. S-nitrosylation or glutathionylation of Ras is known to activate the protein directly and initiate the Ras-Raf-MEK/ERK cascade (174, 199).
The SAPK pathways are major transducers that signal cell death or survival in response to oxidative stress. In most circumstances, activation of SAPK pathways by ROS stress results in induction of apoptosis. The stress-response pathways are regulated at multiple levels, as illustrated in Fig. 8. The apoptosis-regulating signal kinase 1 (ASK1) is an important redox sensor for initiation of the SAPK signaling cascade. ASK1/MEKK5 is a ubiquitously expressed MAP kinase kinase kinase, which activates JNK and p38 MAP kinase pathways by Ser/Thr phosphorylation of their respective MKKs: MKK4/MKK7 for JNK and MKK3/MKK6 for p38 MAP kinases α/γ (291). Under nonstressed condition, ASK1 is sequestered by Trx in an inactive form (264). The association of thioredoxin with ASK1 via Cys32 or Cys35 of Trx appears to be necessary and sufficient to promote ASK1 ubiquitination and degradation, leading to abrogation of the ASK1 apoptotic activity. Because the binding between Trx and ASK1 requires a reduced form of Trx, the ASK-1/Trx complex can be dissociated through oxidative modifications. Apoptotic stimuli such as ROS and TNF induce oxidation of the critical cysteine residues in Trx and cause its dissociation from ASK1. The free ASK1 can then form a multimeric complex with active kinase activity (101, 186). A study using genetic deletion of ASK1 has confirmed that ASK1 activation is required for sustained activation of JNK/p38 MAPK and oxidative stress-induced apoptosis (297). Interestingly, recent work suggested that in response to H2O2, the active ASK1 seemed to undergo further oxidation via interchain disulfide bond formation, which maintained a sustained activation of JNK and induction of apoptosis (221). Besides Trx, glutaredoxin (Grx) has been reported to function as another negative regulator of ASK1 in a redox-sensitive and glutathione-dependent fashion (283). As a negative regulatory mechanism, activation of ASK1 can be inhibited through its binding with Ser/Thr phosphatase 5 (PP5). In response to oxidative stress, PP5 was found to dephosphorylate and inactivate ASK1 (212). At the level of JNK, recent studies showed that under nonstressed conditions, the π and μ classes of glutathione-S-transferase (GST) are negative regulators of JNK (1, 326). Under oxidative stress, suppression of JNK activity by GST can be reversed through dissociation of the GST/JNK complex and oligomerization of GSTπ (1). The oxidative liberation of JNK from GST resulted in induction of JNK-mediated apoptosis. In contrast, others reported that oxidative stress can inhibit JNK activity and apoptosis through induction of Hsp72 (238). RNS can oxidatively modify various SAPK pathways and provide differential effects. For example, S-nitrosylation of JNK1 and JNK2 was found to inhibit their activities (237), whereas tyrosine nitration of p38MAPK by peroxynitrite was shown to induce immediate activation of the kinase (269). The fairly complex effect of oxidative stress on SAPK pathways may be owing to the differential dose and duration of the stimuli and types of oxidative modifications.
FIG. 8.
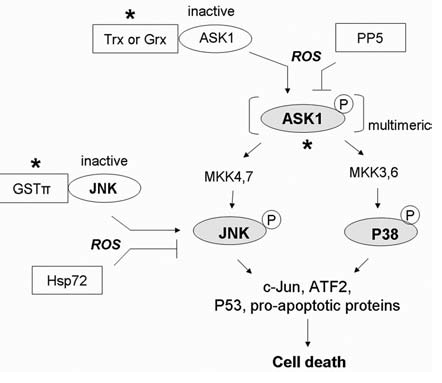
Redox regulation of stress-responsive kinase (SAPK) signaling pathways. In most cases, activation of the SAPK pathway transduces an oxidative stress signal to cell death. Under nonstressed conditions, apoptosis-regulating signal kinase 1(ASK1) is inhibited by the reduced form of thioredoxin (Trx) or glutaredoxin (Grx). Increased oxidative stress causes oxidation of Trx and Grx and releases ASK1 to form an active multimeric complex with proper trans- or autophosphorylation. The activation of ASK1 subsequently leads to activation of c-Jun N-terminal kinase (JNK) and p38-MAPK, resulting in induction of cell death. JNK can also be inhibited by complex formation with glutathione S-transferase-π (GST-π) under nonstressed conditions, and can be activated by ROS in a similar fashion as ASK1. Negative regulatory molecules include the Ser/Thr phosphatase 5 (PP5), which inhibits ASK1 kinase activity by causing its dephosphorylation, and heat-shock protein 72 (Hsp72), which inhibits JNK activity. *Site of redox regulation. Grey, Active forms of the proteins.
2. PI3K/Akt pathway
Signal transduction via PI3-kinases plays an important role in regulating cell growth, proliferation, survival, and motility. A moderate level of ROS activates PI3K signaling and promotes cell survival, whereas sustained oxidative stress may inhibit this pathway, allowing apoptosis to occur (182). The PI3K cascade is stimulated by phosphorylation of growth-factor receptor (tyrosine kinase receptor), which promotes its direct binding with PI3K or indirect binding through adapter proteins such as IRS docking protein for IGF-1 signaling. Activated PI3K converts the membrane phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] to the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3 or PIP3]. PIP3 recruits Akt and 3′-phospho-inositide-dependent kinase-1 (PDK1) to the plasma membrane through its binding with pleckstrin homology (PH) domains of these proteins (315). Then the PDK1 can phosphorylate and activate Akt (22, 315). Akt, which can also be phosphorylated by the mTOR complex 2 (TORC2), regulates the functions of downstream targets through its Ser/Thr kinase activity (266). The activation of the PI3K/Akt pathway is tightly regulated by phosphatases, especially the reversion of PIP3 back to PI(4,5)P2 by phosphatase and tensin homologue (PTEN) and the inactivation of receptor tyrosine kinases by protein tyrosine phosphatases (PTPases) (68).
a. Role of PI3K/Akt in cell survival
Compelling evidence suggests that oxidative stress–induced activation of the PI3K/Akt pathway is crucial for cell survival (327). Paradoxically, under certain circumstances, phosphorylation of Akt seemed to play a proapoptotic role through induction of ROS production (105) or activation of the Fas-mediated death pathway (193). The dual effects of PI3K/Akt are likely the results of crosstalk with other signaling pathways, such as JNK and PKC (105). Survival signals from PI3K/Akt pathways were transduced mainly through the phosphorylation and inactivation of proapoptotic proteins such as BAD, caspase-9, P53, and forkhead transcription factor (FKHRL1), which targets FasL, Bim, IGFBP1, and Puma. Akt phosphorylates and activates IKK and cyclic AMP response element–binding protein CREB, resulting in elevated transcription of genes encoding antiapoptotic proteins such as Bcl-2, Bcl-Xl, and Mcl-1 (203). Furthermore, Akt also exerts its antiapoptotic function by phosphorylation and inhibition of ASK1 activity, which prevent stress-induced apoptosis (154). This is an example of crosstalk between PI3K/Akt and SAPK pathways in the regulation of cell survival.
b. Redox regulation of PI3K/Akt
As depicted in Fig. 9, several components of the PI3K/Akt signaling pathway are redox sensitive. Activation or inhibition of this pathway by the redox system is mainly through oxidative modification of Cys-dependent phosphatases (CDPs) and protein kinases. Although the oxidative inactivation of CDPs is known to be critical in activation of the pathways, redox modifications of protein kinases seems to inhibit the survival signaling. CDPs comprises of a large family of enzymes that share a conserved catalytic domain containing a highly reactive Cys residue. At physiologic pH, the Cys exists as a thiolate anion, which is required for its phosphatase activity. Oxidative modification of the Cys significantly inhibits the enzyme activities (265). CDPs that regulate PI3K/Akt signaling include PI3-phosphatase, PTEN, and PTPases. Oxidative inactivation of PTEN through an intramolecular disulfide bond or S-nitrosylation of the active Cys is known as an important mechanism of PI3K/Akt activation by oxidative stress (179, 342). The reduction of oxidized PTEN in cells appears to be mediated predominantly by the Trx system (180). Redox modification of PTEN was recently shown to be a mechanism to promote survival of cancer cells with mitochondrial dysfunction (240). Normally, active PTEN is maintained in a reduced state by the NADPH/Trx system. Therefore, a defect in mitochondrial respiration, which causes an increase in NADH and a decrease in NADPH, can lead to oxidative inactivation of PTEN and activation of the PI3K/Akt survival pathway (240). Besides the lipid phosphatase, protein phosphatases such as PTP1B, SHP-2, and TC45 are targets of ROS-mediated oxidation (179, 207). These phosphatases are negative regulators of receptor tyrosine kinases; therefore, their oxidative inactivations result in sustained activation of the receptor-mediated PI3K/Akt signaling (328).
FIG. 9.
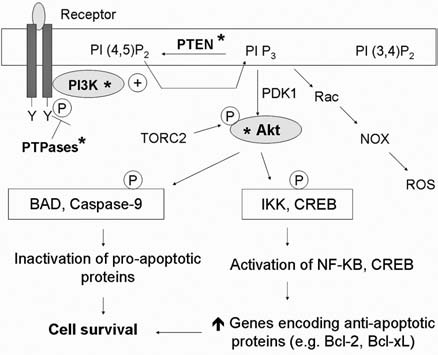
Redox regulation of the PI3K/Akt signaling pathway. PI3K/Akt transduces the signal for cell survival mainly through phosphorylation of target molecules by Akt. This results in inactivation of proapoptotic proteins and activation of transcription factors, which target antiapoptotic proteins. Under oxidative stress, this pathway is activated by oxidative inactivation of phosphatases [i.e., protein tyrosine phosphatases (PTPases) and PTEN], allowing constitutive activation of tyrosine kinase receptor and PI3K. However, direct oxidative modification of PI3K and Akt can result in their inactivation and compromise the survival signals. Furthermore, the PI3K/Akt pathway can also promote cellular production of ROS through activation of Rac and NADPH oxidase (NOX). *Site of redox regulation. Grey, Active forms of the proteins; TORC2, mTOR complex 2.
Besides redox regulation of phosphatases, protein kinases such as PI3K and Akt can be modified by oxidation. In contrast to phosphatases, redox modification of the kinases results in downregulation of PI3K/Akt signals and a decrease in survival capacity. In response to oxidative stress, a disulfide bridge is formed between Cys297 and Cys311 in the kinase domain of Akt (219). Interestingly, the oxidation of Akt does not directly affect the kinase activity in vitro. Instead, it promotes the binding of Akt to protein phosphatase PP2A, leading to dephosphorylation and inactivation of Akt (219). The oxidative inactivation of Akt was shown to be reversible by GRX, which appears to exert its antiapoptotic effect through this mechanism (219). Recently, Akt was also shown to be inhibited by S-nitrosylation (340). At the level of PI3K, the p85 subunit of PI3K was shown to be a direct target for tyrosine nitration, leading to inactivation of the Akt survival pathways (77).
It is worth noting that receptor-mediated activation of PI3K can stimulate Rac-NAD(P)H oxidase (NOX), leading to increased generation of ROS (330). Recent study demonstrated that ROS produced by NOX may contribute to monocyte/macrophage cell survival through activation of Akt and inhibition of p38 MAPK pathways (328).
C. Redox regulation of cell survival at the cell death–execution level
The redox regulation of transcription factors and signal-transduction pathways may affect cell survival through a series of molecular processes to activate or inhibit the cell-death execution molecules. Under certain conditions, ROS stress may directly modulate the activity of these cell-death effectors. As shown in Fig. 10, several apoptotic effectors, such as caspases, Bcl-2, and cytochrome c, are redox-sensitive, and their functions can be significantly affected by cellular ROS.
FIG. 10.
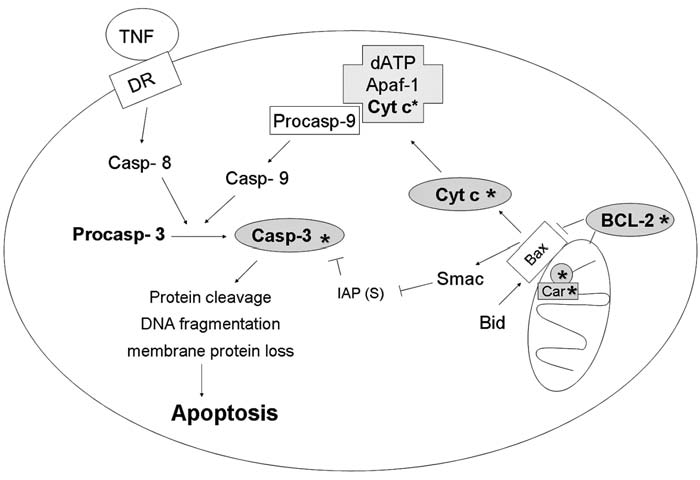
Redox regulation at the execution level. Apoptosis can be triggered through extrinsic or intrinsic pathways. External stimuli such as TNF-α or Fas ligand binds to death receptor and transduces the signal into activation of caspase-8, leading to initiation of the extrinsic pathway. Intrinsic signals, such as DNA damage and oxidative stress, can transduce the death signal by causing release of cytochrome c from mitochondria to cytosol, followed by activation of caspase-9 through formation of apoptosome (Apaf-1, cytochrome c, pro-caspase 9, and dATP). Active caspase 8 and caspase 9 can further cleave procaspase-3, producing an active fragment of caspase-3, which cleaves its protein substrates such as PARP, resulting in apoptosis. To keep apoptosis in check, Bcl-2 family proteins play an important role in regulation of mitochondria membrane permeability and cytochrome c release. Antiapoptotic proteins such as Bcl-2 prevent apoptosis through both direct interaction with proapoptotic proteins such as Bax and indirect control of oxidative stress via maintenance of a reducing environment. Grey*, Potential sites of redox regulation. Car, cardiolipin; Cyt c, cytochrome c; casp 3, caspase 3.
1. Caspases
Caspases are evolutionarily conserved aspartate-specific, cysteine-dependent proteases. Caspases include large prodomain caspases such as caspase-1, −2, −4, −5, −9, −8, −10, −11, and −12, and small prodomain caspases including caspase-3, −6, −7, and −14. Activation of the large-domain caspases (initiator caspases) occurs by forming multimeric complexes [i.e., apoptosome for caspase-9 and death-inducing signaling complex (DISC) for caspase-8]. Whereas the initiator caspases can be activated through proximity-induced autoactivation, effector caspases require proteolytic maturation (by the large-prodomain caspases) to be activated (281).
a. Role of caspases in cell death and survival
A hallmark of apoptosis is the activation of caspases, which requires sequential proteolysis of the initiator caspases and effector caspases. Apoptotic stimuli can trigger caspase activation either through the extrinsic (death receptor–mediated caspase-8 activation) or the intrinsic pathways (mitochondria-mediated caspase-9 activation). Activation of the caspase cascade ultimately leads to cleavage of a variety of substrates (e.g., PARP), DNA fragmentation, loss of membrane integrity, and cell death. Besides the critical role of caspases in apoptosis, emerging evidence suggests non-apoptotic functions of caspases, which includes an opposing role in promoting cell survival (173). An example is the requirement of caspase-8 and c-FLIP in NF-κB activation (288). Although oxidative stress is known to regulate the apoptotic activity of caspases, it is still unclear whether the prosurvival function of caspases can be regulated by redox modifications.
b. Redox regulation of caspases
As described earlier, oxidative stress can activate or inhibit caspases through signal-transduction pathways. Furthermore, the levels of oxidative stress can regulate the expression of FasL, which binds to its receptor and leads to caspase-8 activation (12). Increased endogenous levels of ROS, especially in the mitochondria, can stimulate the apoptotic machinery by promoting membrane permeabilization through mitochondrial permeability transition (MPT). The permeabilization of mitochondria may lead to a release of cytochrome c and an activation of caspase-9 through the formation of apoptosomes (6, 168, 177). In addition to the stimulation of the apoptotic apparatus, ROS can also directly affect the function of caspase proteins. The reduced state of Cys at the active site is required for the catalytic activity of caspases, and thus its function is redox sensitive. Caspases can be activated or inhibited by redox system, depending on the degree of the ROS stress (39). A study using various concentrations of H2O2 demonstrated that a low dose of H2O2 can activate caspases and induce apoptosis, whereas a high dose can cause oxidative inactivation of caspases, and the cells undergo necrosis (112). Several lines of evidence suggest that reducing environment is required for proper function of the Cys-containing active sites of caspases. Oxidative modifications of caspases, such as direct oxidation, glutathionylation, and S-nitrosylation, attenuated the proteolytic activities and inhibited apoptosis (112, 115, 156). Besides the regulation of its catalytic activity, a recent study with genetic silencing of Grx in endothelial cells showed that activation of caspase 3 and TNF-α–induced cell death can be suppressed by glutathionylation of procaspase-3 (235). This is because the glutathionylated procaspase 3 is less susceptible to cleavage by initiator caspase-8 (235). The important role of glutathione in modulating the activity of effector caspases may provide an explanation for the observation that the efflux of GSH is required for the execution of apoptosis (88, 95).
2. Bcl-2
Bcl-2 is the prototype of antiapoptotic BCL-2 family members (11). It contains BH1-BH4 domains and attaches to the outer mitochondrial membrane. Extensive studies highlight the importance of Bcl-2 in protecting cells against oxidative stress–induced apoptosis and its role in regulating redox signaling. These are evidenced by the suppression of lipid peroxidation and attenuated apoptosis observed in BCL-2–over-expressed cells, and the similar phenotypes between Bcl-2 knockout mice and mice exposed to chronic oxidative stress (122, 316). Furthermore, the increased amount of glutathione was observed in BCL-2–overexpressing cells, suggesting a role of BCL-2 in controlling the cellular redox status (79).
a. Role of Bcl-2 in cell survival
The antiapoptotic effects of Bcl-2 are observed at multiple levels. Bcl-2 can prevent mitochondrial membrane permeabilization through direct interaction with proapoptotic Bax/Bak or BH3-only proteins (6). Furthermore, Bcl-2 was shown to prevent ROS-induced mitochondrial permeability transition pore opening in certain experimental models (167). Besides its direct antiapoptotic role, Bcl-2 also functions to maintain redox homeostasis by regulating glutathione and NADPH levels (324). Interestingly, because the redox environment of the nucleus has an effect on the accessibility of transcription factors to their targets, nuclear GSH compartmentalization controlled by Bcl-2 is thought to play a role in regulating the transcription of genes encoding certain mitochondria proteins such as fatty acid–binding proteins (FABPs), VDAC, and UCP (324). These proteins may directly or indirectly modulate mitochondria-induced apoptosis. Interestingly, recent work showed that conformational change of Bcl-2 through its binding with Nur77 can convert Bcl-2 to a proapoptotic protein (185).
b. Redox regulation of Bcl-2
The function of Bcl-2 can be regulated through redox-sensitive signaling. Mild oxidative stress is shown to induce the expression of Bcl-2 through activation of transcription factors such as NF-κB, as an adaptive response to promote cell survival (42). In contrast, in response to oxidative stress, JNK can phosphorylate and inhibit Bcl-2 function, allowing apoptotic processes to occur (11, 65). Besides regulation at the signal-transduction level, recent evidence suggests that Bcl2 can be directly affected by oxidative stress. For example, oxidative carbonylation of Bcl-2 by NO has been shown to be an important mechanism of NO-induced apoptosis in insulin-secreting cells (38). Furthermore, inactivation of ERK1/2 was found to cause proteosomal degradation of Bcl-2 in a redox-dependent manner (32).
3. Cytochrome c
Cytochrome c is a small-molecular-weight heme-containing protein, which participates in an electron transfer from complex III to complex IV in mitochondrial electron-transport chain. Because of the nature of cytochrome c as a reversible electron donor/acceptor, this protein is highly redox-sensitive. Cytochrome c locates in inner mitochondria membrane and interacts with the membrane phospholipid cardiolipin (233). The release of cytochrome c is considered a hallmark of mitochondrial-mediated apoptosis (99).
a. Role of cytochrome c in cell survival
Under physiologic conditions, cytochrome c plays an essential role in oxidative phosphorylation and production of ATP, which is the major energy source for biologic reactions. Generation of superoxide in mitochondria occurs mainly through electron leakage from complex I and complex III. Therefore, cytochrome c, which accepts an electron from complex III and donates it to cytochrome c oxidase, also functions to prevent the electron outflow and superoxide generation. Because of this function, cytochrome c plays a critical role in keeping ROS generation at a low level, optimal for cell survival (343). Apoptotic stimuli trigger mitochondrial membrane permeabilization and promote the release of cytochrome c from mitochondria to cytosol, where it acts as an apoptotic inducer. As shown in Fig. 10, the released cytochrome c forms a complex (apoptosome) with procaspase-9, Apaf-1, and ATP or dATP, leading to activation of caspase-9 and the downstream caspase cascade (168). Furthermore, the loss of cytochrome c from the respiratory chain leads to electron leakage from complex III and increased mitochondrial generation of superoxide radicals (343). This may explain the increase of ROS level observed during apoptosis, even when the stimuli are not prooxidants. In addition to ROS generation from the electron leakage, direct oxidation of cytochrome c by p66Shc has recently been proposed as a novel mechanism of mitochondrial ROS generation during apoptosis (97). On proapoptotic signals, a mammalian adapter protein p66Shc can be liberated from its putative inhibitory complex in mitochondria. The active p66Shc then oxidizes cytochrome c and promotes the formation of ROS (97). The ROS increase mediated by cytochrome c may amplify the apoptotic signals and accelerate the death-execution process.
Besides its function as an electron carrier, cytochrome c may act as a peroxidase (13). During the cell-death process, the released cytochrome c in the cytosol binds and exerts a peroxidase activity on plasma membrane lipids, especially phosphatidyl serine, leading to lipid peroxidation (141). This structural modification of the plasma membrane can lead to exposure of the signals recognized by the macrophage to engulf the cell corpse (13).
b. Redox regulation of cytochrome c
Cytochrome c is tightly regulated by the redox system at multiple levels. In the mitochondria, the interaction between cytochrome c and cardiolipin plays a critical role in maintaining cytochrome c in its proper location and prevents its release to the cytosol (233). The possible role of cardiolipin in the release of cytochrome c from mitochondria during apoptosis has recently been discussed (233). Cardiolipin is sensitive to lipid peroxidation, and the oxidized cardiolipin will lose its binding affinity to cytochrome c. Therefore, under normal conditions, the cardiolipin-cytochrome c complex is stabilized by the ROS-scavenging effect of the mitochondrial glutathione and mitochondrial membrane glutathione peroxidase 4 (mtGPX4) (253). Interestingly, a novel role of cytochrome c as a cardiolipin peroxidase has recently been found (142). When ROS stress exceeds the capacity of the glutathione system or when the mitochondrial GSH pool is depleted, excessive ROS can activate the peroxidase activity of cytochrome c, leading to cardiolipin peroxidation (142). Oxidized cardiolipin loses its binding affinity to cytochrome c, allowing cytochrome c to be dissociated and released to cytosol. This interesting finding raises a novel concept that the redox status of cytochrome c can autoregulate the localization of cytochrome c. It is worth noting that the effect of ROS/RNS on the cardiolipin peroxidase activity of cytochrome c is species specific. For example, H2O2 is a very important cofactor for the peroxidase reaction, whereas the physiologic concentration of NO has been shown to effectively inhibit the peroxidase activity of cytochrome c (323).
The release of cytochrome c requires not only its dissociation from cardiolipin but also the permeabilization of the mitochondrial membrane. This provides another level of its redox regulation. The increased mitochondrial ROS and the disruption of the electron-transport chain can cause the collapse of the mitochondrial transmembrane potential (ΔΨm). This may result in permeabilization of the mitochondrial membrane, which allows cytochrome c to be released from the mitochondria. The mechanism of membrane permeabilization has been extensively reviewed (168). Although low levels of NO can inhibit the peroxidase activity of the cytochrome c/cardiolipin complex, a recent study showed that a direct nitrosylation of mitochondrial cytochrome c promotes its release to the cytosol (271).
Once cytochrome c is released to the cytosol, it can induce apoptosis only if it is in an oxidized form. Under physiologic conditions, the presence of high levels of cytoplasmic GSH keeps the released cytochrome c in an inactive (reduced) state, thus functioning as a fail-safe mechanism if cytochrome c is released from mitochondria. If the redox status of the cell is disturbed, however, perhaps in the presence of hydrogen peroxide or depletion of GSH or both, the cellular redox status will be shifted toward the oxidized state, and cytochrome c will be active, allowing caspase activation and apoptosis to proceed (114).
D. Integration of redox-sensitive signaling pathways in the regulation of cell survival
1. Crosstalk between signaling pathways
Redox regulation of the key factors affecting cell death/survival is often bifurcated (i.e., the same protein can either be activated or inhibited by redox alteration). Whether the consequence of oxidative stress will lead to cell survival or death is likely dependent on the integration of those redox-sensitive signals. This is exemplified by the crosstalk between the PI3K/Akt and SAPK pathways. As shown in Fig. 11, both pathways can be activated by oxidative stress. Activation of the PI3K/Akt pathway can activate nuclear translocation of NF-κB mainly through phosphorylation of the IKK or NF-κB subunit (314). Activated NF-κB induces transcription of multiple genes to promote cell survival (147). In contrast, activation of ASK1 may lead to phosphorylation of JNK and JNK-induced cell death (297). Under physiologic conditions, cell survival is a favorable process, and survival mechanisms are activated to inhibit the cell-death signals at multiple levels. For instance, Akt can phosphorylate and inhibit the ASK1 cascades (154), whereas NF-κB inhibits JNK-induced apoptosis through increasing the transcription of JNK inhibitor (GADD45, XIAP) (61, 147). Studies using knockout IKK or RelA cells showed that, in the absence of NF-κB activation, ROS stress induces JNK activation, leading to cell death (91, 145). Severe oxidative stress inhibited NF-κB signaling, which allowed not only sustained JNK activation but also amplification of ROS stress due to decreased transcription of antioxidants (35). These results suggest that the crosstalk between NF-κB and JNK signaling is important in determining the final cell fate in response to oxidative stress. In general, transient activation of JNK signaling by mild ROS stress leads to predominant antiapoptotic signals and cell survival, whereas sustained activation of JNK by severe ROS stress would be proapoptotic. The integration of NF-κB and JNK pathways in signaling apoptosis and cell survival in response to oxidative stress was recently reviewed (35, 224).
FIG. 11.

Integration of redox signaling. An increase of reactive oxygen species (ROS) can result in either cell survival or cell death, depending on the integration of the proapoptotic and antiapoptotic signals. The levels and durations of ROS stress determine the activation or inhibition of each signal-transduction pathway. The crosstalk between PI3K/Akt and stress-responsive MAPK pathway (SAPK) serves as an example. A low level or transient increase of ROS can activate PI3K/Akt, leading to cell survival through NF-κB. The predominant survival signals of the Akt and NF-κB pathways prevent cell death by inhibiting ASK1 and JNK, respectively. However, a high level of ROS causes a sustained activation of the ASK1-JNK cascade and inactivation of PI3K/Akt and NF-κB due to protein oxidation, leading to cell death.
2. Role of p53
Besides the crosstalk between multiple signaling pathways, certain factors such as p53, which is a redox-sensitive molecule, can regulate cell survival at multiple levels of signaling. Under normal physiologic conditions, p53 protein has a short half-life. It is maintained at a low level by MDM2-mediated inactivation and ubiquitin/proteasome degradation (116, 169, 198, 230). With certain stimuli, such as oxidative stress and DNA damage, p53 is stabilized by posttranslational modification and translocates to the nucleus. p53 serves as a master transcription factor to activate the expression of proteins involved in maintaining genomic stability and cellular homeostasis. As a tumor suppressor, p53 exerts its genome guardian effect by controlling various cell-cycle checkpoints and regulating DNA-damage repair, senescence, and apoptotic machineries (9). Recent studies suggested a novel function of p53 in maintaining redox homeostasis through regulating energy metabolism, mitochondrial biogenesis, and the expression of antioxidant enzymes (21). Loss of p53 function contributes to the development of many types of human cancer (284). A number of studies suggest that p53 plays an important role in controlling cell fate through regulation of cellular ROS level (28, 69, 300).
a. P53 serves as an antioxidant to maintain redox homeostasis and normal cell survival
Recent study revealed that under physiologic conditions or mild ROS stress, p53 is activated to induce transcriptions of multiple antioxidant molecules such as SESN1 and SESN2 and GPX1, which function to decrease ROS level and to promote cell survival (263). Suppression of p53 by siRNA caused an increase in cellular ROS, which can be completely reversed by the use of antioxidant N-acetylcysteine (NAC) (263). Splenocytes and thymocytes of Trp53−/− mice also exhibited increased ROS compared with that of the wild-type mice. Furthermore, a recent report showed that the p53 tumor suppressor also affects cellular redox homeostasis by inhibiting glycolysis and stimulating mitochondrial bioenergetics through transcriptional activation of TIGAR (TP53-induced glycolysis and apoptosis regulator) and the SCO2 (synthesis of cytochrome c oxidase 2), respectively (20, 202). TIGAR, a homologue of fructose-2,6-biphosphatase, functions to inhibit phospho-fructokinase activity and reduce fructose-2,6-bisphosphate levels, resulting in an inhibition of glycolysis. This causes a shift toward the pentose phosphate pathway, leading to the production of NADPH. The reducing equivalents of NADPH can be used to regenerate major cellular antioxidant GSH, which in turn promotes the scavenging of ROS, genomic stability, and prevention of cancer. Thus, TIGAR seems to function as a checkpoint to regulate glycolysis negatively (102). SCO2 is a nuclear gene that encodes a copper-binding protein required for the assembly of cytochrome c oxidase II (CO II) subunits of complex IV in the respiratory chain. Disruption of the SCO2 gene in wt-p53 cancer cells caused the metabolic switch toward glycolysis, as exhibited in p53-deficient cells (202). Interestingly, using new technology to increase an extra copy of p53 and Arf genes, Matheu and colleagues also confirm the redox modulating function of p53 in vivo (201). They found that the super Arf/p53 (s-Arf/p53) mice, which have higher stimulated expression of p53, exhibited an increase in glutathione and SESN1/SESN2 level, along with a decrease in ROS, lipid peroxidation, and oxidative damage to protein and DNA (201). Taken together, activation of p53 can decrease ROS stress, maintain redox homeostasis, and promote cell survival through transcriptional regulation of antioxidant enzymes, TIGAR, and SCO2, which regulate activities of glycolysis and mitochondrial electron-transport chain.
b. Role of p53 in cell death
P53 controls cell fate through several mechanisms, depending on the magnitude of the stress. High levels of ROS cause phosphorylation and stabilization of p53 protein, which often exhibits proapoptotic function under such conditions. Whereas a low level of stress induces p53 to upregulate the expression of genes encoding ROS-scavenging enzymes, high level of stress induces p53 to upregulate genes encoding prooxidant such as PIG3, p66Shc, and proline oxidase (97, 261, 263). Furthermore, p53 is also known to increase gene expression of proapoptotic proteins such as BAX, BBC3 (Puma) and decrease gene expression of antiapoptotic proteins such as Bcl-2 (45). As illustrated in Fig. 12, these observations suggest that the very same p53 protein can control ROS levels and cell fate by transcriptional regulation of different sets of genes in response to different intensities of stress. Interestingly, when cells are under severe stress, activated p53 can also directly interact with ARE-containing promoters and suppress Nrf2-dependent transcription of antioxidant response genes such as x-CT, NQO1, and GST-α1, resulting in an elevation of ROS and apoptosis (84). Reactivation of p53 in p53-deficient tumors can cause complete tumor regression, suggesting a proapoptotic role of p53 (336). Thus, p53 exerts antioxidant function in cells under moderate (physiologically relevant) levels of ROS stress, but exhibits prooxidant function in the severely damaged cells (263). Besides transcriptional regulation, p53 also modulates cell-survival pathways through direct interaction and inhibition of antiapoptotic protein Bcl-2 (299).
FIG. 12.
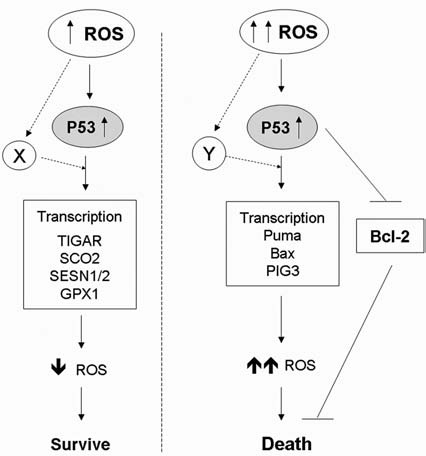
Regulation of cell fate by p53. Different levels of ROS stress activate p53, leading to various outcomes. Activation of p53 by a low level of ROS stress promotes cell survival, whereas severe ROS stress activates p53 and causes cell death. It is possible that the activated p53 might interact with different factors (indicated as “X” and “Y”) under different levels of ROS stress, leading to activation of distinct sets of target genes (see text for details).
c. Redox regulation of p53
Although p53 functions as a transcription factor to control the expression of several redox-regulating molecules, p53 itself is redox sensitive. p53 is a zinc-binding protein containing 10 cysteine residues susceptible to ROS oxidation. The mechanisms that regulate the redox status of p53 remain to be fully elucidated, but much evidence suggests that p53-DNA–binding ability can be strongly inhibited by oxidation and nitrosylation. In vitro experiments revealed that only the reduced form of p53 can specifically bind to the target DNA, whereas direct oxidation or S-glutathionylation of Cys can block its binding to DNA and transactivation activity (63, 110, 317). Further study suggested that the redox status of p53 can be controlled by redox-sensitive thioredoxin, thioredoxin reductase, and redox factor-1 (Ref-1) (111). Besides thiol oxidations, p53 protein can be oxidatively modified and inactivated through protein nitration by ONOO− in human glioblastoma cells (51, 52). The redox-sensitive nature of p53 protein suggests that this molecule may act as a redox sensor in mammalian cells, as OxyR in bacterial system. Therefore, it is possible that a loss of p53 function, as observed in many types of cancer cells, may disable this important redox homeostasis mechanism and allow cancer cells to escape senescence and cell death. It is worth noting that not only the redox system can regulate the function of p53 through direct oxidative modifications, but the indirect activation of p53 through its interacting partners such as ATM or MDM-2 also provides another layer of redox regulation of p53 (44, 120).
III. Role of Redox Regulation of Cell Survival in Pathogenesis of Diseases
As described in previous sections, the redox system plays a crucial role in regulating cell survival. Disruption of redox homeostasis will result in a deregulation of apoptosis associated with various diseases, including cancer, degenerative diseases, and aging. Increased oxidative stress can either promote cell survival or induce cell death, depending on the cellular context. Genetically unstable cells can adapt to live with the stress by adjusting the level of ROS to the extent that promotes cell survival, leading to development of cancer. In contrast, normal or aging cells that failed to maintain redox balance are prone to oxidative stress–induced cell death. This may act as a pathogenic mechanism of degenerative diseases. Figure 13 shows the overall roles of redox regulation in cell survival and the pathologic process of diseases.
FIG. 13.

Disturbance of redox homeostasis and pathogenesis of diseases. Alterations in redox homeostasis by exogenous stimuli or endogenous stress or both can result in increased oxidative stress with elevated cellular ROS. The ability to adapt to such ROS stress determines the overall fates of the cells. A successful adaptation to increased survival signals in combination with further ROS-mediated mutations and loss of critical regulatory mechanisms lead to defective cell death and aberrant proliferation. These may contribute to development of cancer. A failure in adaptation to the ROS stress while the cells accumulate oxidative DNA, protein, and lipid damage product may result in excessive cell death, leading to degenerative disorders and aging.
A. Aberrant prolonged cell survival leads to cancer
Cancer cells often exhibit increased generation of ROS compared with normal cells (294). Although the exact mechanisms responsible for the intrinsic ROS stress in cancer cells remain unclear, several lines of evidence suggest that ROS production is induced after the expression of genes associated with tumor transformation, such as Ras, Bcr-Abl, and c-Myc (16). Not only ROS production seems to be increased in cancer cells, but the levels of antioxidants also were shown to be profoundly altered in the malignant cells (228). These suggest that redox balance is impaired in cancer cells. The intrinsic oxidative stress in cancer cells is thought to play an important role not only in cell proliferation (125) and genetic instability (250), but also in evasion of cell death. Increased ROS stress in cancer cells may activate survival pathways (130), disrupt cell-death signaling (242), and evade senescence (47). Because high levels of oxidative stress can kill cells, the cells that are equipped with flexible machinery have a higher possibility of adapting and surviving the ROS stress. Genomic instability is likely the key mechanism that cancer cells use for that purpose. The loss of functional p53 is thought to play a pivotal role in the adaptation process. Prolonged cell survival, together with increased proliferation, metastasis, and angiogenesis, are known to be required for development of cancer (113). As illustrated in Fig. 14, oncogene activation, dysfunction of redox signaling, and loss of p53 can all lead to disruption of redox homeostasis, resulting in increased cell proliferation and survival, and can contribute to the development of cancer.
FIG. 14.

Redox alterations and cancer development. Disturbance of redox homeostasis by an increase in ROS production (due to oncogenic stimulation, mitochondrial dysfunction, etc.) or a decrease in ROS elimination (due to a deficit or dysfunction of the antioxidant system) can lead to elevated ROS. The increased ROS stress can induce DNA mutations and genetic instability, including a loss of tumor-suppressor genes such as p53. The loss of p53 function can in turn further contribute to mitochondrial dysfunction, ROS generation, and genomic instability, forming a vicious cycle. ROS stress may also induce the expression of prosurvival factors and certain ROS-scavenging proteins, which would enable the cells to adapt and survive under oxidative stress. The ROS-induced mutations and genetic instability further enhance the chance for selection of cells with malignant phenotypes (an increase in proliferation, survival capacity, cell motility, and angiogenesis), leading to development of cancer.
1. Oncogene activation
a. Ras
Ras is a guanine nucleotide triphosphatase (GTPase) that functions as a molecular switch in a large network of intracellular signaling pathways. Constitutive activation of Ras, either by overexpression or by mutation, is very common in various human cancers. Although sharing a high degree of sequence identity, the active mutation rates of three different Ras genes (H-Ras, K-Ras, and N-Ras) are tissue- and tumor-type specific. It has been shown that Ras expression promotes ROS production. In the H-Rasv12-transformed NIH3T3 fibroblasts cells; large amounts of superoxide were generated through pathways involving flavoprotein and Rac1, which activated NADPH oxidase–mediated ROS generation (130). Further study by conditional deletion of Rac1 confirmed that Rac1 function is required for Ras-mediated tumorigenesis, and loss of Rac1 causes a substantial reduction in cell proliferation (157). Besides the direct activation of ROS-production machinery, a recent study showed that Ras oncogenic signal also induced repression of the antioxidant gene SESN1 (165), resulting in a shift of redox balance toward increased ROS levels. Certain regulators of cell survival, such as PI3K/Akt, NF-kB, and c-Jun/AP-1, were shown to be influenced by the Ras oncogenic signal, which plays a major role in the transformation process (335). Although the increase in ROS production induced by Ras can transform some immortalized nontumorigenic cells to malignant cells, Ras transformation also provoked premature senescence in primary cells in the Em-N-Ras transgenic mouse model (275). These observations suggest that the increased ROS triggered by Ras can induce either cellular senescence or malignant transformation, depending on the cellular context. A study using doxycycline-inducible Ras transgenic mice showed that long-term low-level induction of K-Ras resulted in tumor formation after evasion of the senescence checkpoint, whereas high-levels of K-Ras activation led to upregulation of tumor suppressors and cellular senescence (267).
Besides the extent of ROS production induced by the oncogenic signal, the adaptation process for cell survival seems to be influenced by the ROS-scavenging systems. As shown in a genetically defined human ovarian cancer H-Rasv12 model, Ras-transformed cells, which had increased O2− and H2O2 levels, were shown to have an upregulation of multiple antioxidants such as SOD2 and peroxiredoxin 3, compared with their non-tumorigenic parental cells (341). This study also suggested that the enhanced antioxidant capability serves as an important mechanism to evade apoptosis induced by ROS stress. This was evidenced by the increased resistance to H2O2 induced cell death observed in the Ras-transformed cells (341). Furthermore, a recent study demonstrated that the Ras-transformed cells were more sensitive to depletion of glutathione, leading to massive ROS accumulation and preferential cell death (302). These suggested a critical role of antioxidants in cell survival. It is conceivable that maintaining redox homeostasis in a high dynamic state (active ROS scavenging to counteract increased ROS generation) may be an adaptation mechanism used by cancer cells to survive under Ras oncogenic stress.
b. c-Myc
Myc is a helix-loop-helix leucine zipper transcription factor that regulates the expression of many genes involved in normal cell growth, proliferation, apoptosis, and metabolism. Abnormal expression of the c-Myc protooncogene can lead to aberrant activation of its downstream pathways and deregulation of the chromatin state, subsequently causing genomic instability. Thus, it has been implicated in a wide spectrum of human cancers. Significant increases in H2O2 and double-strand breaks of DNA were detected after c-Myc activation (85, 308). Such ROS stress is reversible by antioxidant NAC (308) and vitamin C (139). The role of c-Myc in cell survival is rather complicated. Downregulation of c-Myc has been shown to sensitize cancer cells to cisplatin or radiation-induced apoptosis (24, 36), suggesting a prosurvival role of c-Myc against oxidative stress. In contrast, overexpression of c-Myc was shown to enhance serum-deprivation–induced apoptosis (296). This occurred through excessive accumulation of ROS, which inhibited NF-κB–mediated transactivation of SOD2 (296). Based on these observations, it is likely that the life/death decision under c-Myc expression is mediated through a differential dose effect of ROS. Studies using inducible c-Myc in melanoma cells revealed that apoptosis induced by downregulation of c-Myc was associated with cellular depletion of GSH. The change of GSH level after altered c-Myc expression occurred through the transcriptional control of the glutathione-synthesis enzyme (19). This indicates that the survival effect of c-Myc may be regulated by redox homeostasis. The upregulation of the glutathione antioxidant synthesis by c-Myc may represent an adaptive mechanism to survival under oxidative stress.
c. Bcr-Abl
Philadelphia (Ph)-positive leukemia cells contain a chromosomal translocation (t9/22) that results in a fusion of Bcr and c-Abl genes. The chimeric BCR-ABL tyrosine kinase is oncogenic and is responsible for the development of chronic myelogenous leukemia (CML), a relatively common adult leukemia. In experimental systems, the expression of Bcr-Abl completely abrogated growth factor dependence and transformed primary hematopoietic cells (58). Further study revealed that BCR-ABL exerts its antiapoptotic effects by activating multiple pathways, including RAS, PI3K/Akt, NF-κB, and STAT, leading to activation of antiapoptotic factors such as Bcl-xL (86). Compared with quiescent, nontransformed hematopoietic cells, Bcr-Abl transformed cells also have increased intracellular levels of ROS and decreased protein-tyrosine phosphatases (PTPases) (268). Treatment of Bcr-Abl–expressing cells with reducing agents such as NAC or PDTC decreased the ROS level and attenuated protein tyrosine phosphorylation, likely through activation of PTPases. This suggests a positive-feedback regulation between ROS generation and BCR-ABL activation (268). Furthermore, Bcr-Abl–induced ROS stress may induce DNA double-strand breaks. An inadequate repair of the damaged DNA may cause mutations and genetic instability, thus promoting the progression of CML (227).
2. Loss of functional p53
An activation of oncogenes and a defect in antioxidant systems can increase oxidative stress, which may contribute to tumorigenesis. However, the transformation process might not occur if the redox sensor p53 is still capable of maintaining redox homeostasis and protecting genome integrity. The role of p53 as a tumor suppressor and how oxidative stress modulates its function and contributes to tumor development were recently discussed (124). Cells and mice with defective p53 exhibited increased ROS stress, high mutagenesis, and increased tumor growth rate, which can be delayed by antioxidant NAC. Reactivation of p53 in p53-deficient tumors can cause a complete tumor regression (7). Furthermore, recent work demonstrated that s-Arf/s-p53 mice (which have increased levels of Arf and p53) show decreased ROS levels and reduced oxidative DNA damage (201). These mice seemed resistant to H-RasV12 and E1A oncogenic transformation (201). The incidences of both sporadic cancer and carcinogen-induced cancer (fibrosarcomas and papillomas) were significantly decreased in the s-Arf/s-p53 mice (201). Taken together, this evidence suggests that p53 plays a key role as tumor suppressor, and that its functions to maintain redox homeostasis and genomic stability plays an important role in suppressing tumor formation.
3. Aberrant expression of antioxidant enzymes
Maintaining redox homeostasis is essential for cell survival. Alterations in the antioxidant system could induce redox imbalance and promote the development of cancer. Most evidence linking a deficit in antioxidant capacity to cancer development came mainly from animal studies in which antioxidant molecules were either knocked out or overexpressed. Then, the tumor incidence in the genetically altered animals was compared with those in the wild-type animals.
a. Superoxide dismutase (SOD)
More than 30% of SOD1−/− mice developed liver tumors, and >70% of the mice developed tumor nodules (78). Further study showed that the mutation frequency of the SOD1-deficient mice was significantly increased. The mutation types of the mice were mainly GC to AT transversions and GC to AT transitions, which was consistent with mutations induced by oxidative stress (37). Heterozygous SOD2+/- mice, which exhibited a 50% reduction in SOD levels, appeared normal but had mitochondrial oxidative damage and decreased mitochondrial membrane potential (312). Furthermore, significantly elevated levels of 8-oxo-2-deoxyguanosine (8-oxo-dG) in nuclear and mitochondrial DNA and premature induction of apoptosis were observed (164). Tumor incidence, particularly for lymphoma and pituitary adenoma, increased 100% in old SOD+/- mice compared with the wild-type mice. Interestingly, transgenic mice (SOD3TG) with skin-specific overexpression of SOD3 exhibited a decrease in DNA damage and a 50% reduction in skin tumor formation in a chemically induced carcinogenesis model treated with DMBA/TPA.
b. Glutathione peroxidase (GPX) and peroxiredoxin (Prx)
Transgenic mice overexpressing GPX-1 or coexpressing GPX-1 and SOD1 were found to have an increased incidence of tumorigenesis in a DMBA/TPA two-stage skin carcinogenesis model (195). This suggests that a precise redox homeostasis is essential and that overexpression of GPX-1 might disturb the redox balance and contribute to cancer development. Prx1 knock-out mice revealed higher levels of ROS and an increased predisposition to cancer (217). Furthermore, Prx1 seems to abrogate c-Myc-mediated transformation through interaction with its transcriptional regulatory domain (217), suggesting the potential role of Prx1 as a tumor suppressor.
B. Diseases with excessive cell death: aging and degenerative disorders
One characteristic of aging is the loss of cellularity and the gradual decline of tissue function (304). This may be caused by progressive senescence of postmitotic tissues, likely due to chronic damage by ROS (218). Multiple evidence suggests the involvement of redox imbalance in the aging process. For example, increased NO level and nitrosative stress were shown to induce protein misfolding and neuronal cell death, which may play a role in development of brain aging and neurodegenerative conditions (223). Mutations of the antioxidant gene SOD1 were linked to 20% of cases of the familial amyotrophic lateral sclerosis (ALS), an age-dependent degenerative disorder of motor neurons in cortex, brainstem, and spinal cord (64, 262). Furthermore, mice lacking SOD1 exhibited premature aging and neuromuscular dysfunction. The aged mice have elevated oxidative damage to lipid, protein, and DNA, which are associated with increased apoptosis and reduced life span (78). In addition to the increase of ROS associated with aging, impairments in antioxidant capacity were often observed. For instance, the level of glutathione and the expression of glutathione-synthesis enzymes were shown to be decreased in aged mice and rats (188, 255). Recent study suggested that this phenomenon may be caused by attenuation of Nrf2/ARE transcriptional activity (289). Studies of normal aging, of genetic mutations that cause disease, and of environmental factors that affect disease risk have revealed multiple mechanisms underlying how excessive oxidative stress can cause neuronal cell death. For example, accumulation of oxidative DNA, protein, and lipid products; accumulation of self-aggregating proteins such as amyloid β-peptide, tau, α-synuclein, and huntingtin; oxidative perturbation of lipid metabolism and disruption of calcium homeostasis were shown to induce mitochondria-mediated apoptosis of neurons in several neurodegenerative disorders [for review, see (205)]. Furthermore, ROS-mediated activation of insulin-receptor signaling is known to cause impairment in autophagy, which has been associated with altered life span and decline in cognitive functions (75).
As mitochondria play a role in ROS production and apoptosis, the role of mitochondrial ROS in aging has been extensively investigated (192). In a study of transgenic mice with catalase targeted to peroxisome, nucleus, or mitochondria, the catalase transgene targeting mitochondria was designed by removing the peroxisomal localization signal along with the initial methionine and the addition of the mitochondrial localization signal to the amino terminal to target catalase expression to the mitochondria. Similarly, a nuclear-localization sequence was added to the amino terminus of peroxisomal catalase for nuclear expression. Only the mice overexpressing mitochondria-targeted catalase exhibited a 5-month increase in life span. These mice also exhibited a decrease in H2O2 level and in mtDNA damage (273). This study suggested the important role of mitochondrial H2O2 in aging. Another study found that the activity of aconitase, a redox-sensitive mitochondrial enzyme, was severely inhibited by excess superoxide and hydroxyl radicals in aging cells (339). Mice lacking p66Shc, an adapter protein that promotes ROS generation in mitochondria, were shown to have longer life span. This work supported the causative role of mitochondrial ROS in aging (209). In addition to ROS, mitochondrial dysfunction may also lead to development of aging in a redox-independent manner. In a study using homozygous mutation in mitochondrial DNA replication enzyme (DNA polymerase-γ), the mutator mice had a reduced life span of ∼46 weeks on average (303). The mice developed premature onset of the aging-related phenotypes, which included weight loss, reduced subcutaneous fat, alopecia, kyphosis, osteoporosis, reduced fertility, and heart enlargement (303). Nevertheless, accumulation of mtDNA mutations seemed not to be associated with increased oxidative stress in this model (170).
Surprisingly, a recent report showed that although an 11-fold increase in mitochondrial point mutations has been observed with age, a mitochondrial mutator mouse was able to sustain a 500-fold higher mutation burden than normal mice, without obvious features of accelerated aging (320). The author concluded, based on this work, that the mitochondrial mutations do not limit the life span of the mice. Apparently, further studies are needed in this area.
Recent evidence suggests a crucial role of p53 in prevention of aging. As described earlier, s-Arf/p53 mice not only showed a decrease in ROS level and increased resistance to oxidative DNA damage, but also showed a 9-month delay in aging. However, mice harboring s-Arf or s-p53 alone did not show an increase in life span. These observations suggest that p53 may require Arf to be stabilized and to exert its antiaging effect.
In another study, p53+/- mice lacking exon 1 to 6 of the p53 gene were generated through an aberrant gene-targeting event in embryonic stem cells (307). Despite only having one wild-type allele, the mice showed a premature aging phenotype, but seemed not prone to cancer development (307). It would be interesting to examine the cellular redox status in these mice.
IV. Therapeutic Strategies Based on Redox Regulation of Cell Survival
Alterations in redox homeostasis can promote cell death or cell survival, depending on the magnitude of the stimuli. Because the redox alteration may contribute to development of diseases, the dose-dependent effects of ROS may provide an opportunity for potential clinical applications in therapeutics and prevention of diseases. As described previously, the final outcomes of cell fates (survival or death) under oxidative stress depend largely on the levels and types of ROS. Also, the functional status of cellular antioxidant systems and the redox-sensitive survival-signaling pathways can significantly influence the cell-fate decision. Therefore, the redox-based therapeutic/preventive strategies should include manipulations of redox homeostasis and modulations of the redox-sensitive factors that regulate cell survival and apoptosis.
A. Manipulating redox homeostasis
The principle of redox homeostasis and the strategies to modulate redox dynamics are illustrated in Fig. 15. Under physiologic conditions, normal cells maintain redox homeostasis by controlling the proper balance between ROS generation and elimination. ROS are a double-edge sword. A moderate increase of ROS may promote cell proliferation and survival. However, when the increase of ROS reaches a certain level (the threshold), it may overwhelm the cellular antioxidant capacity and trigger the cell-death process. Therefore, cells with higher basal ROS generation (as in the case of cancer cells) would be more dependent on the antioxidant system and more vulnerable to further oxidative stress–inducing agents. A further increase of ROS stress by using exogenous ROS-generating agents or drugs that disable the endogenous antioxidant system may preferentially increase ROS above the threshold level in cancer cells, leading to cell death. In contrast, normal cells may be able to tolerate better such exogenous ROS stress because of their low basal ROS outputs and normal metabolic regulations. Prolonged accumulation of ROS-induced damage in neurons and other normal cells may result in cell death, leading to aging or degenerative disorders. Reducing ROS levels with proper antioxidants may be a useful strategy to prevent or delay these pathologic processes.
FIG. 15.
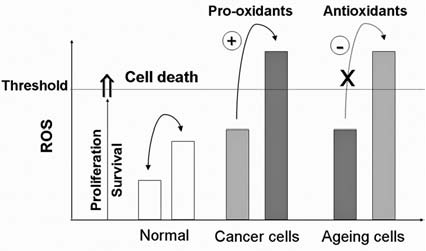
Redox homeostasis and strategies to modulate redox dynamics for potential therapeutic applications. Under physiologic conditions, normal cells maintain redox homeostasis by controlling the proper balance between ROS generation and elimination. The redox dynamics may fluctuate within a tolerable range. An increase of ROS may promote cell proliferation and survival, as in the case of many cancer cells. However, when the increase of ROS reaches a critical level (the threshold), it may overwhelm the cellular antioxidant capacity and trigger the cell-death process. Chronic ROS stress may cause accumulation of damage to a level that induces cell death. This is thought to be a mechanism contributing to neural degenerative diseases and aging. For therapeutic purposes, it is possible to use agents that promote ROS generation or inhibit the cellular antioxidant system to trigger cancer cell death by pushing the ROS above the threshold level. In contrast, antioxidants may be used to prevent cells from oxidative damage and delay aging and the neurodegenerative process.
1. Pro-oxidants as a therapeutic strategy for cancer
A defect in apoptosis plays a major role in development of most cancers. A moderate increase of ROS is known to activate survival pathways and inhibit apoptosis in cancer cells. Thus, increased ROS in cancer have been viewed as an adverse event. However, because severe increases of ROS can cause lethal damage and kill the cells, it is possible to use ROS-generating agents or compounds that abrogate the antioxidant system to further increase ROS in cancer cells to a level that triggers cell death (for review see refs. 89 and 239). Such ROS-mediated therapeutic strategies have been tested in various experimental systems with promising results (128). Because toxic side effects in normal tissue is a major problem in clinical treatment of cancer by using cytotoxic drugs, new agents with high therapeutic selectivity are urgently needed. Because cancer cells exhibit increased ROS compared with normal cells, this redox difference provides a biochemical basis for development of new therapeutics with high selectivity. In the Ras-transformed ovarian cancer model, it was recently shown that the oncogenic transformed cells exhibited increased ROS stress and were more dependent on the glutathione antioxidant system to maintain homeostasis and survive. Disruption of glutathione by a natural compound, β-phenylethyl isothiocyanate (PEITC), was shown to preferentially increase ROS stress and selectively kill the malignant cells in vitro with minimal toxicity to their nontumorigenic parental cells (302). In vivo, treatment with PEITC prolonged survival of mice bearing Ras-transformed ovarian cancer (302). These results demonstrated the critical role of redox homeostasis in regulation of cancer cell survival and cell death and suggest that it is possible to kill cancer cells preferentially through ROS-mediated mechanism. Because ROS stress is prevalent in cancer cells, the ROS-based approach may have broad therapeutic applications.
Defective apoptosis in cancer not only promotes disease progression but also confers resistance to many therapeutic agents (138). Because alterations of multiple redox regulators such as Trx, GST, GPX1, and Prx were observed in drug-resistant cancer cells (241), it was speculated that an altered redox homeostasis may play a role in the drug-resistance mechanism, and that manipulation of the redox system may be a useful strategy to overcome the problem of apoptosis resistance in tumor cells (242). Recent work showed that a compound known as TDZD-8 (4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione) can induce depletion of thiols and rapid accumulation of ROS and selectively kill leukemic cells expressing stem-cell marker, with minimal toxicity to normal hematopoietic stem cells (106). Because tumor stem cells are thought to be the subpopulation of cells highly resistant to chemotherapy and play a critical role in disease relapse after treatment, the potency of the prooxidative compound in removing those cells underscores a key role of the redox system in regulating survival of stem cells and highlights the promising therapeutic potential of using redox-based strategy in cancer treatment.
2. Antioxidants for prevention of degenerative diseases
Because accumulation of ROS was known to promote excessive or premature cell death, leading to aging and neural degenerative diseases, the use of antioxidants in prevention of aging has been established for decades. This involves the use of both supplementation of natural ROS scavengers and treatment with exogenous antioxidants [for review see (75)]. For example, cysteine supplementation in addition to the normal protein diet has shown significant beneficial effects on several parameters relevant to aging, including skeletal muscle functions (74). N-acetylcysteine (NAC), a glutathione precursor, has been shown to protect against oxidative stress–induced neuronal death and thus might delay neurogenerative diseases (5). Melatonin, a physiologic hormone and antioxidant, seems to have a protective effect against neurodegenerative diseases, especially Alzheimer's disease (245). Phenolic compounds such as resveratrol are potent antioxidants and have been reported to be protective against neuronal apoptosis associated with the pathogenesis of Alzheimer's disease (72). Experimental and clinical studies showed that ebselen (PZ51), a GPX mimetic that can inhibit lipid peroxidation and decrease iNOS expression, was shown to suppress cell death of cortical neurons and prevent cerebral ischemia (stroke) in clinical trials (290, 337). Mitochondria-targeted antioxidants such as MitoQ and MitoVitamin E were developed as pharmaceutical products for prevention of stroke and cardiovascular disease (321). GSNO, a physiologic metabolite of glutathione (GSH) and NO, was shown to be several-fold more potent than GSH in protecting cells against oxidative stress caused by peroxynitrite. In rats, GSNO therapy was proven to be neuroprotective in cerebral ischemia (254).
In addition to the pharmacologic interventions, epidemiologic studies have shown that regular consumption of diets rich in antioxidants and antiinflammatory agents, such as those found in fruits and vegetables, may reduce the risk of developing age-related neurodegenerative diseases such as Parkinson's disease and Alzheimer's disease (176). These suggest that maintaining redox homeostasis may prevent degenerative diseases.
It is worth noting that, although accumulation of ROS is thought to play a role in development of cancer, the benefit of using antioxidants as cancer chemopreventive agents is somewhat controversial. An early study showed that certain antioxidants may enhance the therapeutic efficacy of chemotherapy in a colorectal cancer model (49). Antioxidants may also mitigate the adverse effects of radiation therapy (214). However, compounds such as carotenoids, tocopherols, and ascorbate derivatives may act as antioxidants or prooxidants, depending on doses (274). The use of β-carotene and vitamin A in lung-cancer prevention trials showed no chemopreventive effect and might even increase the risk of lung cancer incidence and mortality in smokers (231, 232). Recent evidence suggests that antioxidants such as NAC may blunt the therapeutic activity of chemotherapeutic agents, including cisplatin and paclitaxel, against tumor cells (2, 67). Because ROS stress can either promote cell survival or induce cell death, depending on dosage and duration, cautions should be exercised in using antioxidants and prooxidants to modulate cellular redox, with full consideration given to the time- and dose-dependent nature of such modulations.
B. Modulating redox-sensitive signaling molecules
Besides manipulating ROS level by using prooxidants or antioxidants, cell death and survival can also be modulated by targeting redox-sensitive signaling molecules at the signal-transduction, transcription, or death-execution levels (241). For example, at the level of signal transduction, JNK activation was found to be associated with ROS-induced neuronal death in Parkinson's and Alzheimer's disease (25). Chemical inhibitors of this signaling pathway have proven to be effective in vivo to reduce brain damage and some of the symptoms of arthritis in animal models (25). At the level of execution, proapoptotic or antiapoptotic factors, such as caspases and Bcl-2, are attractive targets for cancer therapies. Pharmacologic inhibitors, mimetics, activators, and anti-sense oligonucleotides targeting these molecules are of potential therapeutic utility (87). Because molecular pathways of apoptosis are excessively activated in aging and neurodegenerative disorders, pharmacologic/genetic inhibitions of apoptotic players such as Fas, caspases, and p53 are emerging strategies to prevent or retard the degenerative diseases (322). At the level of transcription, redox-sensitive transcription factors regulating expression of multiple antioxidants, such as NF-κB and Nrf2, are of particular interest. Since activation of the prosurvival NF-κB pathway was shown to play a role in cancer development induced by carcinogens and oncogenic viruses, a variety of NF-κB inhibitors such as curcumin have been under clinical evaluation for use in cancer prevention and treatment. New agents targeting the proteasome, IKK, and other upstream molecules involved in NF-κB activation seem to show anticancer activity in clinical or preclinical studies (313). The fact that Nrf2 can be activated not only by oxidative stress, but also by electrophilic compounds such as isothiocyanates, provides therapeutic and preventive opportunities. For example, increased Nrf2 transactivation is considered a major mechanism for the cancer chemopreventive effects of isothiocyanates (136). Furthermore, a recent study suggests the essential roles of PI3K and PKC signaling in the activation of the Nrf2/ARE in the absence of general oxidative stress (190). Because ARE-targeted genes were known to play a protective role against apoptosis in cortical neuron (66), compounds activating Nrf2 via PI3K and PKC signaling would likely prevent neuronal cell death. This may provide a new basis for development of chemopreventive agents for degenerative diseases.
V. Concluding Remarks
Cellular redox systems control the functions of multiple signaling proteins affecting cell survival at the levels of signal transduction, transcriptional regulation, and cell-death execution. Oxidative stress can either enhance cell survival or promote cell death, depending on the magnitude and duration of the stress and the genetic background and redox states of the cells. Oxidative stress not only serves as a type of stimulus to trigger stress-response signal-transduction pathways but also can modulate cell death/survival through direct oxidative modifications of the execution molecules. Under physiologic conditions, the balance between production and elimination of ROS ensures the proper maintenance of cellular metabolism and other functions. The final decision, whether the cells will survive or die, is the overall outcome of the integration of signals from redox-sensitive factors and other regulatory mechanisms. However, when the redox homeostasis is disturbed by either oncogenic activation, mitochondrial dysfunction, or accumulation of oxidative stress, cells that acquire genetic instability may adapt to survive under the stress by acquiring mutations to attenuate programmed cell death. The defective cell death in conjunction with increased cell proliferation, angiogenesis, and metastatic potentials can promote the development of cancer. Conversely, accumulation of oxidative-damage products and failure to adapt to ROS stress may result in excessive cell death, leading to degenerative disorders and aging. Strategies to modulate cellular redox status, either by prooxidants and antioxidants or by affecting redox-sensitive signaling pathways, may have significant clinical applications in disease treatment and prevention. Logical combinations of ROS-modulating agents and compounds that affect redox-sensitive signaling pathways may further enhance therapeutic activity and selectivity. A comprehensive understanding of the redox biology underlying the disease processes and the mechanisms of action of pro-oxidants and antioxidants is essential for developing effective therapeutic strategies.
Footnotes
Reviewing Editors: Junichi Sadoshima, Chandan K. Sen, Bor-Luen Tang, and Keith Webster
Acknowledgments
This work is supported by NIH grants CA085563, CA100428, and CA109041. D.T. is a recipient of a scholarship from Ananda-Mahidol foundation under the Royal Patronage of His Majesty the King of Thailand. D.T. is also supported by a Lummis Family fellowship in Biomedical Sciences.
Abbreviations Used
ARE, antioxidant responsive element; ASK1, apoptosis-regulating signal kinase 1; EpRE, electrophile responsive element; ER, endoplasmic reticulum; ERK, extracellular regulated kinase; GPX, glutathione peroxidase; GRX, glutaredoxin; GSH, reduced glutathione; GSSG, oxidized glutathione; GST, glutathione-S-transferase; HAT, histone acetylase; HDAC, histone deacetylase; HIF, hypoxia-inducible factor; HNE, 4-hydroxy-2-nonenal; HO·, hydroxyl radical; H2O2, hydrogen peroxide; Hsp, heat-shock protein; IкB, inhibitor of NF-κB; JNK, c-Jun-N-terminal kinase; Keap1, Kelch-like ECH-associating protein 1; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; MPT, mitochondrial permeability transition; NAC, N-acetylcysteine; NADPH, nicotinamide adenine dinucleotide phosphate (reduced); NF-κB, nuclear factor kappa B; NO·, nitric oxide; NOS, nitric oxide synthase; NOX, NAD(P)H oxidase; Nrf2, NF-E2–related factor 2; O2−, superoxide; ONOO−, peroxynitrite; Prx, peroxyredoxins; PTEN, phosphatase and tensin homologue; PTK, phosphotyrosine kinase; PTPase, protein tyrosine phosphatase; RNS, reactive nitrogen species; ROS, reactive oxygen species; SAPK, stress-activated protein kinases; SCO2, synthesis of cytochrome c oxidase 2; SOD, superoxide dismutase; TIGAR, TP53-induced glycolysis and apoptosis regulator; TRX, thioredoxin; ub, ubiquitin; XO, xanthine oxidase.
References
- 1.Adler V. Yin Z. Fuchs SY. Benezra M. Rosario L. Tew KD. Pincus MR. Sardana M. Henderson CJ. Wolf CR. Davis RJ. Ronai Z. Regulation of JNK signaling by GSTp. EMBO J. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandre J. Nicco C. Chereau C. Laurent A. Weill B. Goldwasser F. Batteux F. Improvement of the therapeutic index of anticancer drugs by the superoxide dismutase mimic mangafodipir. J Natl Cancer Inst. 2006;98:236–244. doi: 10.1093/jnci/djj049. [DOI] [PubMed] [Google Scholar]
- 3.Angel P. Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 4.O'Brian CA. Chu F. Review: post-translational disulfide modifications in cell signaling: role of inter-protein, intra-protein, S-glutathionyl, and S-cysteaminyl disulfide modifications in signal transmission. Free Radic Res. 2005;39:471–480. doi: 10.1080/10715760500073931. [DOI] [PubMed] [Google Scholar]
- 5.Arakawa M. Ito Y. N-acetylcysteine and neurodegenerative diseases: basic and clinical pharmacology. Cerebellum. 2007;6:308–314. doi: 10.1080/14734220601142878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armstrong JS. Mitochondrial membrane permeabilization: the sine qua non for cell death. Bioessays. 2006;28:253–260. doi: 10.1002/bies.20370. [DOI] [PubMed] [Google Scholar]
- 7.Attardi LD. Donehower LA. Probing p53 biological functions through the use of genetically engineered mouse models. Mutat Res. 2005;576:4–21. doi: 10.1016/j.mrfmmm.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 8.Awasthi YC. Sharma R. Cheng JZ. Yang Y. Sharma A. Singhal SS. Awasthi S. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol Aspects Med. 2003;24:219–230. doi: 10.1016/s0098-2997(03)00017-7. [DOI] [PubMed] [Google Scholar]
- 9.Aylon Y. Oren M. Living with p53, dying of p53. Cell. 2007;130:597–600. doi: 10.1016/j.cell.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Bannister AJ. Miska EA. Regulation of gene expression by transcription factor acetylation. Cell Mol Life Sci (CMLS) 2000;57:1184–1192. doi: 10.1007/PL00000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basu A. DuBois G. Haldar S. Posttranslational modifications of Bcl2 family members: a potential therapeutic target for human malignancy. Frontiers Biosci. 2006;11:1508–1521. doi: 10.2741/1900. [DOI] [PubMed] [Google Scholar]
- 12.Bauer MK. Vogt M. Los M. Siegel J. Wesselborg S. Schulze-Osthoff K. Role of reactive oxygen intermediates in activation-induced CD95 (APO-1/Fas) ligand expression. J Biol Chem. 1998;273:8048–8055. doi: 10.1074/jbc.273.14.8048. [DOI] [PubMed] [Google Scholar]
- 13.Bayir H. Fadeel B. Palladino MJ. Witasp E. Kurnikov IV. Tyurina YY. Tyurin VA. Amoscato AA. Jiang J. Kochanek PM. DeKosky ST. Greenberger JS. Shvedova AA. Kagan VE. Apoptotic interactions of cytochrome c: redox flirting with anionic phospholipids within and outside of mitochondria. Biochim Biophys Acta. 2006;1757:648–659. doi: 10.1016/j.bbabio.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Bea F. Hudson FN. Chait A. Kavanagh TJ. Rosenfeld ME. Induction of glutathione synthesis in macrophages by oxidized low-density lipoproteins is mediated by consensus antioxidant response elements. Circ Res. 2003;92:386–393. doi: 10.1161/01.RES.0000059561.65545.16. [DOI] [PubMed] [Google Scholar]
- 15.Beckman KB. Ames BN. Oxidative decay of DNA. J Biol Chem. 1997;272:19633–19636. doi: 10.1074/jbc.272.32.19633. [DOI] [PubMed] [Google Scholar]
- 16.Behrend L. Henderson G. Zwacka RM. Reactive oxygen species in oncogenic transformation. Biochem Soc Trans. 2003;31:1441–1444. doi: 10.1042/bst0311441. [DOI] [PubMed] [Google Scholar]
- 17.BelAiba RS. Djordjevic T. Bonello S. Flugel D. Hess J. Kietzmann T. Gorlach A. Redox-sensitive regulation of the HIF pathway under non-hypoxic conditions in pulmonary artery smooth muscle cells. Biol Chem. 2004;385:249–257. doi: 10.1515/BC.2004.019. [DOI] [PubMed] [Google Scholar]
- 18.Bell EL. Chandel NS. Mitochondrial oxygen sensing: regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem. 2007;43:17–27. doi: 10.1042/BSE0430017. [DOI] [PubMed] [Google Scholar]
- 19.Benassi B. Fanciulli M. Fiorentino F. Porrello A. Chiorino G. Loda M. Zupi G. Biroccio A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol Cell. 2006;21:509–519. doi: 10.1016/j.molcel.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Bensaad K. Tsuruta A. Selak MA. Vidal MN. Nakano K. Bartrons R. Gottlieb E. Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 21.Bensaad K. Vousden KH. p53: New roles in metabolism. Trends Cell Biol. 2007;17:286–291. doi: 10.1016/j.tcb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Bhaskar PT. Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 23.Bigelow DJ. Squier TC. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim Biophys Acta. 2005;1703:121–134. doi: 10.1016/j.bbapap.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 24.Biroccio A. Benassi B. Amodei S. Gabellini C. Del Bufalo D. Zupi G. c-Myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in M14 human melanoma cells. Mol Pharmacol. 2001;60:174–182. doi: 10.1124/mol.60.1.174. [DOI] [PubMed] [Google Scholar]
- 25.Borsello T. Forloni G. JNK signalling: a possible target to prevent neurodegeneration. Curr Pharm Des. 2007;13:1875–1886. doi: 10.2174/138161207780858384. [DOI] [PubMed] [Google Scholar]
- 26.Bossy-Wetzel E. Bakiri L. Yaniv M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997;16:1695–1709. doi: 10.1093/emboj/16.7.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bourdon E. Blache D. The importance of proteins in defense against oxidation. Antioxid Redox Signal. 2001;3:293–311. doi: 10.1089/152308601300185241. [DOI] [PubMed] [Google Scholar]
- 28.Bragado P. Armesilla A. Silva A. Porras A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis. 2007;12:1733–1742. doi: 10.1007/s10495-007-0082-8. [DOI] [PubMed] [Google Scholar]
- 29.Brahimi-Horn MC. Pouyssegur J. Oxygen, a source of life and stress. FEBS Lett. 2007;581:3582–91. doi: 10.1016/j.febslet.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 30.Brahimi-Horn MC. Pouyssegur J. Hypoxia in cancer cell metabolism and pH regulation. Essays Biochem. 2007;43:165–178. doi: 10.1042/BSE0430165. [DOI] [PubMed] [Google Scholar]
- 31.Breen AP. Murphy JA. Reactions of oxyl radicals with DNA. Free Radic Biol Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- 32.Breitschopf K. Haendeler J. Malchow P. Zeiher AM. Dimmeler S. Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway. Mol Cell Biol. 2000;20:1886–1896. doi: 10.1128/mcb.20.5.1886-1896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brigelius-Flohe R. Glutathione peroxidases and redox-regulated transcription factors. Biol Chem. 2006;387:1329–1335. doi: 10.1515/BC.2006.166. [DOI] [PubMed] [Google Scholar]
- 34.Brune B. Zhou J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1alpha (HIF-1alpha) Curr Med Chem. 2003;10:845–855. doi: 10.2174/0929867033457746. [DOI] [PubMed] [Google Scholar]
- 35.Bubici C. Papa S. Dean K. Franzoso G. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene. 2006;25:6731–6748. doi: 10.1038/sj.onc.1209936. [DOI] [PubMed] [Google Scholar]
- 36.Bucci B. D'Agnano I. Amendola D. Citti A. Raza GH. Miceli R. De Paula U. Marchese R. Albini S. Felsani A. Brunetti E. Vecchione A. Myc down-regulation sensitizes melanoma cells to radiotherapy by inhibiting MLH1 and MSH2 mismatch repair proteins. Clin Cancer Res. 2005;11:2756–2767. doi: 10.1158/1078-0432.CCR-04-1582. [DOI] [PubMed] [Google Scholar]
- 37.Busuttil RA. Garcia AM. Cabrera C. Rodriguez A. Suh Y. Kim WH. Huang TT. Vijg J. Organ-specific increase in mutation accumulation and apoptosis rate in CuZn-superoxide dismutase-deficient mice. Cancer Res. 2005;65:11271–11275. doi: 10.1158/0008-5472.CAN-05-2980. [DOI] [PubMed] [Google Scholar]
- 38.Cahuana GM. Tejedo JR. Jimenez J. Ramirez R. Sobrino F. Bedoya FJ. Nitric oxide-induced carbonylation of Bcl-2, GAPDH and ANT precedes apoptotic events in insulin-secreting RINm5F cells. Exp Cell Res. 2004;293:22–30. doi: 10.1016/j.yexcr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Cai J. Wu M. Nelson KC. Sternberg P., Jr Jones DP. Oxidant-induced apoptosis in cultured human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1999;40:959–966. [PubMed] [Google Scholar]
- 40.Callapina M. Zhou J. Schmid T. Kohl R. Brune B. NO restores HIF-1alpha hydroxylation during hypoxia: role of reactive oxygen species. Free Radic Biol Med. 2005;39:925–936. doi: 10.1016/j.freeradbiomed.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 41.Cappellini A. Tazzari PL. Mantovani I. Billi AM. Tassi C. Ricci F. Conte R. Martelli AM. Antiapoptotic role of p38 mitogen activated protein kinase in Jurkat T cells and normal human T lymphocytes treated with 8-methoxypsoralen and ultraviolet-A radiation. Apoptosis. 2005;10:141–152. doi: 10.1007/s10495-005-6069-4. [DOI] [PubMed] [Google Scholar]
- 42.Catz SD. Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20:7342–7351. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 43.Chan K. Han X-D. Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen K. Albano A. Ho A. Keaney JF., Jr Activation of p53 by oxidative stress involves platelet-derived growth factor-beta receptor-mediated ataxia telangiectasia mutated (ATM) kinase activation. J Biol Chem. 2003;278:39527–39533. doi: 10.1074/jbc.M304423200. [DOI] [PubMed] [Google Scholar]
- 45.Chen K. Hu Z. Wang LE. Sturgis EM. El-Naggar AK. Zhang W. Wei Q. Single-nucleotide polymorphisms at the TP53-binding or responsive promoter regions of BAX and BCL2 genes and risk of squamous cell carcinoma of the head and neck. Carcinogenesis. 2007;28:2008–2012. doi: 10.1093/carcin/bgm172. [DOI] [PubMed] [Google Scholar]
- 46.Chen W. Martindale JL. Holbrook NJ. Liu Y. Tumor promoter arsenite activates extracellular signal-regulated kinase through a signaling pathway mediated by epidermal growth factor receptor and Shc. Mol Cell Biol. 1998;18:5178–5188. doi: 10.1128/mcb.18.9.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Z. Trotman LC. Shaffer D. Lin HK. Dotan ZA. Niki M. Koutcher JA. Scher HI. Ludwig T. Gerald W. Cordon-Cardo C. Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiarugi P. PTPs versus PTKs: the redox side of the coin. Free Radic Res. 2005;39:353–364. doi: 10.1080/10715760400027987. [DOI] [PubMed] [Google Scholar]
- 49.Chinery R. Brockman JA. Peeler MO. Shyr Y. Beauchamp RD. Coffey RJ. Antioxidants enhance the cytotoxicity of chemotherapeutic agents in colorectal cancer: a p53-independent induction of p21WAF1/CIP1 via C/EBPbeta. Nat Med. 1997;3:1233–1241. doi: 10.1038/nm1197-1233. [DOI] [PubMed] [Google Scholar]
- 50.Cho SH. Lee CH. Ahn Y. Kim H. Kim H. Ahn CY. Yang KS. Lee SR. Redox regulation of PTEN and protein tyrosine phosphatases in H(2)O(2) mediated cell signaling. FEBS Lett. 2004;560:7–13. doi: 10.1016/s0014-5793(04)00112-7. [DOI] [PubMed] [Google Scholar]
- 51.Cobbs CS. Samanta M. Harkins LE. Gillespie GY. Merrick BA. MacMillan-Crow LA. Evidence for peroxynitrite-mediated modifications to p53 in human gliomas: possible functional consequences. Arch Biochem Biophys. 2001;394:167–172. doi: 10.1006/abbi.2001.2540. [DOI] [PubMed] [Google Scholar]
- 52.Cobbs CS. Whisenhunt TR. Wesemann DR. Harkins LE. Van Meir EG. Samanta M. Inactivation of wild-type p53 protein function by reactive oxygen and nitrogen species in malignant glioma cells. Cancer Res. 2003;63:8670–8673. [PubMed] [Google Scholar]
- 53.Colotta F. Polentarutti N. Sironi M. Mantovani A. Expression and involvement of c-fos and c-jun protooncogenes in programmed cell death induced by growth factor deprivation in lymphoid cell lines. J Biol Chem. 1992;267:18278–18283. [PubMed] [Google Scholar]
- 54.Cross JV. Templeton DJ. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem J. 2004;381:675–683. doi: 10.1042/BJ20040591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cross JV. Templeton DJ. Regulation of signal transduction through protein cysteine oxidation. Antioxid Redox Signal. 2006;8:1819–1827. doi: 10.1089/ars.2006.8.1819. [DOI] [PubMed] [Google Scholar]
- 56.Cullinan SB. Zhang D. Hannink M. Arvisais E. Kaufman RJ. Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dai Y. Rahmani M. Dent P. Grant S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. 2005;25:5429–5444. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daley GQ. Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc Natl Acad Sci U S A. 1988;85:9312–9316. doi: 10.1073/pnas.85.23.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dalle-Donne I. Aldini G. Carini M. Colombo R. Rossi R. Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.David SS. O'Shea VL. Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Smaele E. Zazzeroni F. Papa S. Nguyen DU. Jin R. Jones J. Cong R. Franzoso G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 62.Dean RT. Fu S. Stocker R. Davies MJ. Biochemistry and pathology of radical-mediated protein oxidation. Biochem J. 1997;324:1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Delphin C. Cahen P. Lawrence JJ. Baudier J. Characterization of baculovirus recombinant wild-type p53: dimerization of p53 is required for high-affinity DNA binding and cysteine oxidation inhibits p53 DNA binding. Eur J Biochem. 1994;223:683–692. doi: 10.1111/j.1432-1033.1994.tb19041.x. [DOI] [PubMed] [Google Scholar]
- 64.Deng HX. Hentati A. Tainer JA. Iqbal Z. Cayabyab A. Hung WY. Getzoff ED. Hu P. Herzfeldt B. Roos RP. Warner C. Deng G. Soriano E. Smyth C. Parge HE. Ahmed A. Roses AD. Hallewell RA. Pericak-Vance MA. Siddique T. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 65.Deng X. Xiao L. Lang W. Gao F. Ruvolo P. May WS., Jr Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem. 2001;276:23681–23688. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- 66.Dhakshinamoorthy S. Porter AG. Nitric oxide-induced transcriptional up-regulation of protective genes by Nrf2 via the antioxidant response element counteracts apoptosis of neuroblastoma cells. J Biol Chem. 2004;279:20096–20107. doi: 10.1074/jbc.M312492200. [DOI] [PubMed] [Google Scholar]
- 67.Dickey DT. Wu YJ. Muldoon LL. Neuwelt EA. Protection against cisplatin-induced toxicities by N-acetylcysteine and sodium thiosulfate as assessed at the molecular, cellular, and in vivo levels. J Pharmacol Exp Ther. 2005;314:1052–1058. doi: 10.1124/jpet.105.087601. [DOI] [PubMed] [Google Scholar]
- 68.Dillon RL. White DE. Muller WJ. The phosphatidyl inositol 3-kinase signaling network: implications for human breast cancer. Oncogene. 2007;26:1338–1345. doi: 10.1038/sj.onc.1210202. [DOI] [PubMed] [Google Scholar]
- 69.Ding B. Chi SG. Kim SH. Kang S. Cho JH. Kim DS. Cho NH. Role of p53 in antioxidant defense of HPV-positive cervical carcinoma cells following H2O2 exposure. J Cell Sci. 2007;120:2284–2294. doi: 10.1242/jcs.002345. [DOI] [PubMed] [Google Scholar]
- 70.Dinkova-Kostova AT. Holtzclaw WD. Cole RN. Itoh K. Wakabayashi N. Katoh Y. Yamamoto M. Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dong C. Yang DD. Wysk M. Whitmarsh AJ. Davis RJ. Flavell RA. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 72.Draczynska-Lusiak B. Doung A. Sun AY. Oxidized lipoproteins may play a role in neuronal cell death in Alzheimer disease. Mol Chem Neuropathol. 1998;33:139–148. doi: 10.1007/BF02870187. [DOI] [PubMed] [Google Scholar]
- 73.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 74.Droge W. Kinscherf R. Hildebrandt W. Schmitt T. The deficit in low molecular weight thiols as a target for antiageing therapy. Curr Drug Targets. 2006;7:1505–1512. doi: 10.2174/1389450110607011505. [DOI] [PubMed] [Google Scholar]
- 75.Droge W. Schipper HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell. 2007;6:361–70. doi: 10.1111/j.1474-9726.2007.00294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Duran A. Diaz-Meco MT. Moscat J. Essential role of RelA Ser311 phosphorylation by zetaPKC in NF-kappaB transcriptional activation. EMBO J. 2003;22:3910–3918. doi: 10.1093/emboj/cdg370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.el-Remessy AB. Bartoli M. Platt DH. Fulton D. Caldwell RB. Oxidative stress inactivates VEGF survival signaling in retinal endothelial cells via PI 3-kinase tyrosine nitration. J Cell Sci. 2005;118:243–252. doi: 10.1242/jcs.01612. [DOI] [PubMed] [Google Scholar]
- 78.Elchuri S. Oberley TD. Qi W. Eisenstein RS. Jackson Roberts L. Van Remmen H. Epstein CJ. Huang TT. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 79.Ellerby LM. Ellerby HM. Park SM. Holleran AL. Murphy AN. Fiskum G. Kane DJ. Testa MP. Kayalar C. Bredesen DE. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J Neurochem. 1996;67:1259–1267. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- 80.Emerling BM. Platanias LC. Black E. Nebreda AR. Davis RJ. Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005;25:4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.England K. Cotter TG. Direct oxidative modifications of signalling proteins in mammalian cells and their effects on apoptosis. Redox Rep. 2005;10:237–245. doi: 10.1179/135100005X70224. [DOI] [PubMed] [Google Scholar]
- 82.Fantin VR. St-Pierre J. Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 83.Faraci FM. Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- 84.Faraonio R. Vergara P. Di Marzo D. Pierantoni MG. Napolitano M. Russo T. Cimino F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem. 2006;281:39776–39784. doi: 10.1074/jbc.M605707200. [DOI] [PubMed] [Google Scholar]
- 85.Felsher DW. Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fernandez-Luna JL. Bcr-Abl and inhibition of apoptosis in chronic myelogenous leukemia cells. Apoptosis. 2000;5:315–318. doi: 10.1023/a:1009623222534. [DOI] [PubMed] [Google Scholar]
- 87.Fischer U. Schulze-Osthoff K. Apoptosis-based therapies and drug targets. Cell Death Differ. 2005;12(suppl 1):942–961. doi: 10.1038/sj.cdd.4401556. [DOI] [PubMed] [Google Scholar]
- 88.Franco R. Cidlowski JA. SLCO/OATP-like transport of glutathione in fasL-induced Apoptosis: glutathione efflux is coupled to an organic anion exchange and is necessary for the progression of the execution phase of apoptosis. J Biol Chem. 2006;281:29542–29557. doi: 10.1074/jbc.M602500200. [DOI] [PubMed] [Google Scholar]
- 89.Fruehauf JP. Meyskens FL., Jr Reactive oxygen species: a breath of life or death? Clin Cancer Res. 2007;13:789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 90.Fulda S. Debatin KM. HIF-1-regulated glucose metabolism: a key to apoptosis resistance? Cell Cycle. 2007;6:790–792. doi: 10.4161/cc.6.7.4084. [DOI] [PubMed] [Google Scholar]
- 91.Geisler F. Algul H. Paxian S. Schmid RM. Genetic inactivation of RelA/p65 sensitizes adult mouse hepatocytes to TNF-induced apoptosis in vivo and in vitro. Gastroenterology. 2007;132:2489–2503. doi: 10.1053/j.gastro.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 92.Gerald D. Berra E. Frapart YM. Chan DA. Giaccia AJ. Mansuy D. Pouyssegur J. Yaniv M. Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 93.Ghafourifar P. Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26:190–195. doi: 10.1016/j.tips.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 94.Ghezzi P. Review: regulation of protein function by glutathionylation. Free Radic Res. 2005;39:573–580. doi: 10.1080/10715760500072172. [DOI] [PubMed] [Google Scholar]
- 95.Ghibelli L. Fanelli C. Rotilio G. Lafavia E. Coppola S. Colussi C. Civitareale P. Ciriolo MR. Rescue of cells from apoptosis by inhibition of active GSH extrusion. FASEB J. 1998;12:479–486. doi: 10.1096/fasebj.12.6.479. [DOI] [PubMed] [Google Scholar]
- 96.Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- 97.Giorgio M. Migliaccio E. Orsini F. Paolucci D. Moroni M. Contursi C. Pelliccia G. Luzi L. Minucci S. Marcaccio M. Pinton P. Rizzuto R. Bernardi P. Paolucci F. Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 98.Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res. 1998;39:1529–1542. [PubMed] [Google Scholar]
- 99.Gogvadze V. Orrenius S. Zhivotovsky B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim Biophys Acta. 2006;1757:639–647. doi: 10.1016/j.bbabio.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 100.Gorlach A. Diebold I. Schini-Kerth VB. Berchner-Pfannschmidt U. Roth U. Brandes RP. Kietzmann T. Busse R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: role of the p22(phox)-containing NADPH oxidase. Circ Res. 2001;89:47–54. doi: 10.1161/hh1301.092678. [DOI] [PubMed] [Google Scholar]
- 101.Gotoh Y. Cooper JA. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J Biol Chem. 1998;273:17477–17482. doi: 10.1074/jbc.273.28.17477. [DOI] [PubMed] [Google Scholar]
- 102.Green DR. Chipuk JE. p53 and metabolism: Inside the TIGAR. Cell. 2006;126:30–32. doi: 10.1016/j.cell.2006.06.032. [DOI] [PubMed] [Google Scholar]
- 103.Green RM. Graham M. R.O'Donovan M. Chipman JK. Hodges JN. Subcellular compartmentalization of glutathione: correlations with parameters of oxidative stress related to genotoxicity. Mutagenesis. 2006;21:383–390. doi: 10.1093/mutage/gel043. [DOI] [PubMed] [Google Scholar]
- 104.Greijer AE. van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57:1009–1014. doi: 10.1136/jcp.2003.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guo J. Zhu T. Xiao Z-XJ. Chen C-Y. Modulation of intracellular signaling pathways to induce apoptosis in prostate cancer cells. J Biol Chem. 2007;282:24364–24372. doi: 10.1074/jbc.M702938200. [DOI] [PubMed] [Google Scholar]
- 106.Guzman ML. Li X. Corbett CA. Rossi RM. Bushnell T. Liesveld JL. Hebert J. Young F. Jordan CT. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8) Blood. 2007;110:4436–4444. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haddad JJ. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal. 2002;14:879–897. doi: 10.1016/s0898-6568(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 108.Hagen T. Taylor CT. Lam F. Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- 109.Haghdoost S. Czene S. Naslund I. Skog S. Harms-Ringdahl M. Extracellular 8-oxo-dG as a sensitive parameter for oxidative stress in vivo and in vitro. Free Radic Res. 2005;39:153–162. doi: 10.1080/10715760500043132. [DOI] [PubMed] [Google Scholar]
- 110.Hainaut P. Milner J. Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Res. 1993;53:4469–4473. [PubMed] [Google Scholar]
- 111.Hainaut P. Mann K. Zinc binding and redox control of p53 structure and function. Antioxid Redox Signal. 2001;3:611–623. doi: 10.1089/15230860152542961. [DOI] [PubMed] [Google Scholar]
- 112.Hampton MB. Orrenius S. Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis. FEBS Lett. 1997;414:552–556. doi: 10.1016/s0014-5793(97)01068-5. [DOI] [PubMed] [Google Scholar]
- 113.Hanahan D. Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 114.Hancock JT. Desikan R. Neill SJ. Does the redox status of cytochrome c act as a fail-safe mechanism in the regulation of programmed cell death? Free Radic Biol Med. 2001;31:697–703. doi: 10.1016/s0891-5849(01)00646-3. [DOI] [PubMed] [Google Scholar]
- 115.Hentze H. Kunstle G. Volbracht C. Ertel W. Wendel A. CD95-Mediated murine hepatic apoptosis requires an intact glutathione status. Hepatology. 1999;30:177–185. doi: 10.1002/hep.510300111. [DOI] [PubMed] [Google Scholar]
- 116.Haupt Y. Maya R. Kazaz A. Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 117.Hayden MS. Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2204. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 118.Hayes JD. McMahon M. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 2001;174:103–113. doi: 10.1016/s0304-3835(01)00695-4. [DOI] [PubMed] [Google Scholar]
- 119.Heneberg P. Draber P. Regulation of cys-based protein tyrosine phosphatases via reactive oxygen and nitrogen species in mast cells and basophils. Curr Med Chem. 2005;12:1859–1871. doi: 10.2174/0929867054546636. [DOI] [PubMed] [Google Scholar]
- 120.Hess DT. Matsumoto A. Kim SO. Marshall HE. Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 121.Hilberg F. Aguzzi A. Howells N. Wagner EF. c-Jun is essential for normal mouse development and hepatogenesis. Nature. 1993;365:179–181. doi: 10.1038/365179a0. [DOI] [PubMed] [Google Scholar]
- 122.Hockenbery DM. Oltvai ZN. Yin XM. Milliman CL. Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 123.Holmgren A. Aslund F. Glutaredoxin. Methods Enzymol. 1995;252:283–292. doi: 10.1016/0076-6879(95)52031-7. [DOI] [PubMed] [Google Scholar]
- 124.Horn HF. Vousden KH. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–1316. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- 125.Hu Y. Rosen DG. Zhou Y. Feng L. Yang G. Liu J. Huang P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J Biol Chem. 2005;280:39485–39492. doi: 10.1074/jbc.M503296200. [DOI] [PubMed] [Google Scholar]
- 126.Huang HC. Nguyen T. Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 127.Huang LE. Arany Z. Livingston DM. Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem. 1996;271:32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- 128.Huang P. Feng L. Oldham EA. Keating MJ. Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- 129.Inoue M. Sato EF. Nishikawa M. Park AM. Kira Y. Imada I. Utsumi K. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr Med Chem. 2003;10:2495–2505. doi: 10.2174/0929867033456477. [DOI] [PubMed] [Google Scholar]
- 130.Irani K. Xia Y. Zweier JL. Sollott SJ. Der CJ. Fearon ER. Sundaresan M. Finkel T. Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 131.Ishii T. Itoh K. Takahashi S. Sato H. Yanagawa T. Katoh Y. Bannai S. Yamamoto M. Transcription factor nrf2 coordinately regulates a group of oxidative stress-inducible genes in macro-phages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 132.Itoh K. Chiba T. Takahashi S. Ishii T. Igarashi K. Katoh Y. Oyake T. Hayashi N. Satoh K. Hatayama I. Yamamoto M. Nabeshima Y-i. An Nrf2/Small maf heterodimer mediates the induction of phase ii detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 133.Itoh K. Wakabayashi N. Katoh Y. Ishii T. Igarashi K. Engel JD. Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the aminoterminal NeH2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jaakkola P. Mole DR. Tian YM. Wilson MI. Gielbert J. Gaskell SJ. Kriegsheim A. Hebestreit HF. Mukherji M. Schofield CJ. Maxwell PH. Pugh CW. Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 135.Jaspers I. Zhang W. Fraser A. Samet JM. Reed W. Hydrogen peroxide has opposing effects on IKK activity and Ikappa-Balpha breakdown in airway epithelial cells. Am J Respir Cell Mol Biol. 2001;24:769–777. doi: 10.1165/ajrcmb.24.6.4344. [DOI] [PubMed] [Google Scholar]
- 136.Jeong WS. Jun M. Kong AN. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxid Redox Signal. 2006;8:99–106. doi: 10.1089/ars.2006.8.99. [DOI] [PubMed] [Google Scholar]
- 137.Johnson L. Protein kinases and their therapeutic exploitation. Biochem Soc Trans. 2007;035:7–11. doi: 10.1042/BST0350007. [DOI] [PubMed] [Google Scholar]
- 138.Johnstone RW. Ruefli AA. Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 139.KC S. Carcamo JM. Golde DW. Antioxidants prevent oxidative DNA damage and cellular transformation elicited by the over-expression of c-MYC. Mutat Res. 2006;593:64–79. doi: 10.1016/j.mrfmmm.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 140.Kabe Y. Ando K. Hirao S. Yoshida M. Handa H. Redox regulation of NF-κB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 141.Kagan VE. Fabisiak JP. Shvedova AA. Tyurina YY. Tyurin VA. Schor NF. Kawai K. Oxidative signaling pathway for externalization of plasma membrane phosphatidylserine during apoptosis. FEBS Lett. 2000;477:1–7. doi: 10.1016/s0014-5793(00)01707-5. [DOI] [PubMed] [Google Scholar]
- 142.Kagan VE. Tyurin VA. Jiang J. Tyurina YY. Ritov VB. Amoscato AA. Osipov AN. Belikova NA. Kapralov AA. Kini V. Vlasova , II Zhao Q. Zou M. Di P. Svistunenko DA. Kurnikov IV. Borisenko GG. cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 143.Kamata H. Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 144.Kamata H. Manabe T. Oka S. Kamata K. Hirata H. Hydrogen peroxide activates IkappaB kinases through phosphorylation of serine residues in the activation loops. FEBS Lett. 2002;519:231–237. doi: 10.1016/s0014-5793(02)02712-6. [DOI] [PubMed] [Google Scholar]
- 145.Kamata H. Honda S. Maeda S. Chang L. Hirata H. Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 146.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 147.Karin M. Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 148.Kasuno K. Takabuchi S. Fukuda K. Kizaka-Kondoh S. Yodoi J. Adachi T. Semenza GL. Hirota K. Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J Biol Chem. 2004;279:2550–2558. doi: 10.1074/jbc.M308197200. [DOI] [PubMed] [Google Scholar]
- 149.Katsuoka F. Motohashi H. Engel JD. Yamamoto M. Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. J Biol Chem. 2005;280:4483–4490. doi: 10.1074/jbc.M411451200. [DOI] [PubMed] [Google Scholar]
- 150.Katsuoka F. Motohashi H. Ishii T. Aburatani H. Engel JD. Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol. 2005;25:8044–8051. doi: 10.1128/MCB.25.18.8044-8051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kehrer JP. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology. 2000;149:43–50. doi: 10.1016/s0300-483x(00)00231-6. [DOI] [PubMed] [Google Scholar]
- 152.Kietzmann T. Freimann S. Bratke J. Jungermann K. Regulation of the gluconeogenic phosphoenolpyruvate carboxykinase and glycolytic aldolase A gene expression by O2 in rat hepatocyte cultures: involvement of hydrogen peroxide as mediator in the response to O2. FEBS Lett. 1996;388:228–232. doi: 10.1016/0014-5793(96)00557-1. [DOI] [PubMed] [Google Scholar]
- 153.Kietzmann T. Gorlach A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol. 2005;16:474–486. doi: 10.1016/j.semcdb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 154.Kim AH. Khursigara G. Sun X. Franke TF. Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kim JW. Tchernyshyov I. Semenza GL. Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 156.Kim Y-M. Talanian RV. Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 157.Kissil JL. Walmsley MJ. Hanlon L. Haigis KM. Bender Kim CF. Sweet-Cordero A. Eckman MS. Tuveson DA. Capobianco AJ. Tybulewicz VL. Jacks T. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007;67:8089–8094. doi: 10.1158/0008-5472.CAN-07-2300. [DOI] [PubMed] [Google Scholar]
- 158.Klatt P. Molina EP. Lamas S. Nitric oxide inhibits c-jun DNA binding by specifically targeted S-glutathionylation. J Biol Chem. 1999;274:15857–15864. doi: 10.1074/jbc.274.22.15857. [DOI] [PubMed] [Google Scholar]
- 159.Knebel A. Rahmsdorf HJ. Ullrich A. Herrlich P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996;15:5314–5325. [PMC free article] [PubMed] [Google Scholar]
- 160.Knowles HJ. Raval RR. Harris AL. Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 2003;63:1764–1768. [PubMed] [Google Scholar]
- 161.Kobayashi A. Kang M-I. Okawa H. Ohtsuji M. Zenke Y. Chiba T. Igarashi K. Yamamoto M. Oxidative stress sensor keap1 functions as an adaptor for cul3-based e3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kobayashi M. Yamamoto M. Molecular mechanisms activating the nrf2-keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 163.Kobayashi M. Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 164.Kokoszka JE. Coskun P. Esposito LA. Wallace DC. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci U S A. 2001;98:2278–2283. doi: 10.1073/pnas.051627098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kopnin PB. Agapova LS. Kopnin BP. Chumakov PM. Repression of sestrin family genes contributes to oncogenic Ras-induced reactive oxygen species up-regulation and genetic instability. Cancer Res. 2007;67:4671–4678. doi: 10.1158/0008-5472.CAN-06-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kotlo KU. Yehiely F. Efimova E. Harasty H. Hesabi B. Shchors K. Einat P. Rozen A. Berent E. Deiss LP. Nrf2 is an inhibitor of the Fas pathway as identified by Achilles' heel method, a new function-based approach to gene identification in human cells. Oncogene. 2003;22:797–806. doi: 10.1038/sj.onc.1206077. [DOI] [PubMed] [Google Scholar]
- 167.Kowaltowski AJ. Vercesi AE. Fiskum G. Bcl-2 prevents mitochondrial permeability transition and cytochrome c release via maintenance of reduced pyridine nucleotides. Cell Death Differ. 2000;7:903–910. doi: 10.1038/sj.cdd.4400722. [DOI] [PubMed] [Google Scholar]
- 168.Kowaltowski AJ. Castilho RF. Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- 169.Kubbutat MH. Jones SN. Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 170.Kujoth GC. Hiona A. Pugh TD. Someya S. Panzer K. Wohlgemuth SE. Hofer T. Seo AY. Sullivan R. Jobling WA. Morrow JD. Van Remmen H. Sedivy JM. Yamasoba T. Tanokura M. Weindruch R. Leeuwenburgh C. Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 171.Kwak MK. Wakabayashi N. Itoh K. Motohashi H. Yamamoto M. Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 172.Kyriakis JM. Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 173.Lamkanfi M. Festjens N. Declercq W. Vanden Berghe T. Vandenabeele P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 2007;14:44–55. doi: 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]
- 174.Lander HM. Milbank AJ. Tauras JM. Hajjar DP. Hempstead BL. Schwartz GD. Kraemer RT. Mirza UA. Chait BT. Burk SC. Quilliam LA. Redox regulation of cell signalling. Nature. 1996;381:380–381. doi: 10.1038/381380a0. [DOI] [PubMed] [Google Scholar]
- 175.Lando D. Peet DJ. Gorman JJ. Whelan DA. Whitelaw ML. Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lau FC. Shukitt-Hale B. Joseph JA. Nutritional intervention in brain aging: reducing the effects of inflammation and oxidative stress. Subcell Biochem. 2007;42:299–318. [PubMed] [Google Scholar]
- 177.Le Bras M. Clement MV. Pervaiz S. Brenner C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol Histopathol. 2005;20:205–219. doi: 10.14670/HH-20.205. [DOI] [PubMed] [Google Scholar]
- 178.Lee J-M. Calkins MJ. Chan K. Kan YW. Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- 179.Lee SR. Kwon KS. Kim SR. Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J Biol Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 180.Lee SR. Yang KS. Kwon J. Lee C. Jeong W. Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 181.Leppa S. Eriksson M. Saffrich R. Ansorge W. Bohmann D. Complex functions of AP-1 transcription factors in differentiation and survival of PC12 cells. Mol Cell Biol. 2001;21:4369–4378. doi: 10.1128/MCB.21.13.4369-4378.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Leslie NR. The redox regulation of PI 3-kinase-dependent signaling. Antioxid Redox Signal. 2006;8:1765–1774. doi: 10.1089/ars.2006.8.1765. [DOI] [PubMed] [Google Scholar]
- 183.Li F. Sonveaux P. Rabbani ZN. Liu S. Yan B. Huang Q. Vujaskovic Z. Dewhirst MW. Li CY. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell. 2007;26:63–74. doi: 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lillig CH. Holmgren A. Thioredoxin and related molecules: from biology to health and disease. Antioxid Redox Signal. 2007;9:25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 185.Lin B. Kolluri SK. Lin F. Liu W. Han Y-H. Cao X. Dawson MI. Reed JC. Zhang X-K. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor nur77/TR3. Cell. 2004;116:527–540. doi: 10.1016/s0092-8674(04)00162-x. [DOI] [PubMed] [Google Scholar]
- 186.Liu H. Nishitoh H. Ichijo H. Kyriakis JM. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol Cell Biol. 2000;20:2198–2208. doi: 10.1128/mcb.20.6.2198-2208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Liu Q. Berchner-Pfannschmidt U. Moller U. Brecht M. Wotzlaw C. Acker H. Jungermann K. Kietzmann T. A Fenton reaction at the endoplasmic reticulum is involved in the redox control of hypoxia-inducible gene expression. Proc Natl Acad Sci U S A. 2004;101:4302–4307. doi: 10.1073/pnas.0400265101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liu R-M. Choi J. Age-associated decline in [gamma]-glutamylcysteine synthetase gene expression in rats. Free Radic Biol Med. 2000;28:566–574. doi: 10.1016/s0891-5849(99)00269-5. [DOI] [PubMed] [Google Scholar]
- 189.Liu X-M. Peyton KJ. Ensenat D. Wang H. Hannink M. Alam J. Durante W. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc Res. 2007;75:381–389. doi: 10.1016/j.cardiores.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Liu Y. Kern JT. Walker JR. Johnson JA. Schultz PG. Luesch H. A genomic screen for activators of the antioxidant response element. Proc Natl Acad Sci U S A. 2007;104:5205–5210. doi: 10.1073/pnas.0700898104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lizundia R. Chaussepied M. Huerre M. Werling D. Di Santo JP. Langsley G. c-Jun NH2-terminal kinase/c-Jun signaling promotes survival and metastasis of B lymphocytes transformed by Theileria. Cancer Res. 2006;66:6105–6110. doi: 10.1158/0008-5472.CAN-05-3861. [DOI] [PubMed] [Google Scholar]
- 192.Loeb LA. Wallace DC. Martin GM. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc Natl Acad Sci U S A. 2005;102:18769–18770. doi: 10.1073/pnas.0509776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lu B. Wang L. Stehlik C. Medan D. Huang C. Hu S. Chen F. Shi X. Rojanasakul Y. Phosphatidylinositol 3-kinase/Akt positively regulates Fas (CD95)-mediated apoptosis in epidermal Cl41 cells. J Immunol. 2006;176:6785–6793. doi: 10.4049/jimmunol.176.11.6785. [DOI] [PubMed] [Google Scholar]
- 194.Lu H. Forbes RA. Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. 2002;277:23111–23115. doi: 10.1074/jbc.M202487200. [DOI] [PubMed] [Google Scholar]
- 195.Lu YP. Lou YR. Yen P. Newmark HL. Mirochnitchenko OI. Inouye M. Huang MT. Enhanced skin carcinogenesis in transgenic mice with high expression of glutathione peroxidase or both glutathione peroxidase and superoxide dismutase. Cancer Res. 1997;57:1468–1474. [PubMed] [Google Scholar]
- 196.Madrid LV. Mayo MW. Reuther JY. Baldwin AS., Jr Akt stimulates the transactivation potential of the relA/p65 subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem. 2001;276:18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 197.Maiorino M. Aumann KD. Brigelius-Flohe R. Doria D. van den Heuvel J. McCarthy J. Roveri A. Ursini F. Flohe L. Probing the presumed catalytic triad of a selenium-containing peroxidase by mutational analysis. Z Ernahrungswiss. 1998;37(suppl 1):118–121. [PubMed] [Google Scholar]
- 198.Maki CG. Huibregtse JM. Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53(1) Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- 199.Mallis RJ. Buss JE. Thomas JA. Oxidative modification of H-ras: S-thiolation and S-nitrosylation of reactive cysteines. Biochem J. 2001;355:145–53. doi: 10.1042/0264-6021:3550145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Marshall HE. Stamler JS. Inhibition of NF-kappa B by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 201.Matheu A. Maraver A. Klatt P. Flores I. Garcia-Cao I. Borras C. Flores JM. Vina J. Blasco MA. Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 202.Matoba S. Kang JG. Patino WD. Wragg A. Boehm M. Gavrilova O. Hurley PJ. Bunz F. Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 203.Matsuzawa A. Ichijo H. Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid Redox Signal. 2005;7:472–481. doi: 10.1089/ars.2005.7.472. [DOI] [PubMed] [Google Scholar]
- 204.Matthews JR. Wakasugi N. Virelizier JL. Yodoi J. Hay RT. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992;20:3821–3830. doi: 10.1093/nar/20.15.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mattson MP. Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid Redox Signal. 2006;8:1997–2006. doi: 10.1089/ars.2006.8.1997. [DOI] [PubMed] [Google Scholar]
- 206.McCubrey JA. LaHair MM. Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8:1775–1789. doi: 10.1089/ars.2006.8.1775. [DOI] [PubMed] [Google Scholar]
- 207.Meng TC. Fukada T. Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 208.Metzen E. Zhou J. Jelkmann W. Fandrey J. Brune B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell. 2003;14:3470–3481. doi: 10.1091/mbc.E02-12-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Migliaccio E. Giorgio M. Mele S. Pelicci G. Reboldi P. Pandolfi PP. Lanfrancone L. Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 210.Mitomo K. Nakayama K. Fujimoto K. Sun X. Seki S. Yamamoto K. Two different cellular redox systems regulate the DNA-binding activity of the p50 subunit of NF-kappa B in vitro. Gene. 1994;145:197–203. doi: 10.1016/0378-1119(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 211.Monks TJ. Xie R. Tikoo K. Lau SS. Ros-induced histone modifications and their role in cell survival and cell death. Drug Metab Rev. 2006;38:755–767. doi: 10.1080/03602530600959649. [DOI] [PubMed] [Google Scholar]
- 212.Morita K. Saitoh M. Tobiume K. Matsuura H. Enomoto S. Nishitoh H. Ichijo H. Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 2001;20:6028–6036. doi: 10.1093/emboj/20.21.6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Morito N. Yoh K. Itoh K. Hirayama A. Koyama A. Yamamoto M. Takahashi S. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene. 2003;22:9275–9281. doi: 10.1038/sj.onc.1207024. [DOI] [PubMed] [Google Scholar]
- 214.Moss RW. Do antioxidants interfere with radiation therapy for cancer? Integr Cancer Ther. 2007;6:281–292. doi: 10.1177/1534735407305655. [DOI] [PubMed] [Google Scholar]
- 215.Motohashi H. Katsuoka F. Engel JD. Yamamoto M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc Natl Acad Sci U S A. 2004;101:6379–6384. doi: 10.1073/pnas.0305902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Mottet D. Dumont V. Deccache Y. Demazy C. Ninane N. Raes M. Michiels C. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J Biol Chem. 2003;278:31277–31285. doi: 10.1074/jbc.M300763200. [DOI] [PubMed] [Google Scholar]
- 217.Mu ZM. Yin XY. Prochownik EV. Pag, a putative tumor suppressor, interacts with the Myc Box II domain of c-Myc and selectively alters its biological function and target gene expression. J Biol Chem. 2002;277:43175–43184. doi: 10.1074/jbc.M206066200. [DOI] [PubMed] [Google Scholar]
- 218.Muller FL. Lustgarten MS. Jang Y. Richardson A. Van Remmen H. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 219.Murata H. Ihara Y. Nakamura H. Yodoi J. Sumikawa K. Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 220.Muto A. Tashiro S. Tsuchiya H. Kume A. Kanno M. Ito E. Yamamoto M. Igarashi K. Activation of Maf/AP-1 repressor BacH2 by oxidative stress promotes apoptosis and its interaction with promyelocytic leukemia nuclear bodies. J Biol Chem. 2002;277:20724–20733. doi: 10.1074/jbc.M112003200. [DOI] [PubMed] [Google Scholar]
- 221.Nadeau PJ. Charette SJ. Toledano MB. Landry J. Disulfide bond-mediated multimerization of ask1 and its reduction by thioredoxin-1 regulate H2O2-induced c-jun NH2-terminal kinase activation and apoptosis. Mol Biol Cell. 2007;18:3903–3913. doi: 10.1091/mbc.E07-05-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nakamura H. Nakamura K. Yodoi J. Redox regulation of cellular activation. Annu Rev Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- 223.Nakamura T. Gu Z. Lipton SA. Contribution of glutamatergic signaling to nitrosative stress-induced protein misfolding in normal brain aging and neurodegenerative diseases. Aging Cell. 2007;6:351–359. doi: 10.1111/j.1474-9726.2007.00284.x. [DOI] [PubMed] [Google Scholar]
- 224.Nakano H. Nakajima A. Sakon-Komazawa S. Piao JH. Xue X. Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006;13:730–737. doi: 10.1038/sj.cdd.4401830. [DOI] [PubMed] [Google Scholar]
- 225.Nawata R. Yujiri T. Nakamura Y. Ariyoshi K. Takahashi T. Sato Y. Oka Y. Tanizawa Y. MEK kinase 1 mediates the antiapoptotic effect of the Bcr-Abl oncogene through NF-kappaB activation. Oncogene. 2003;22:7774–7780. doi: 10.1038/sj.onc.1206901. [DOI] [PubMed] [Google Scholar]
- 226.Nemoto S. DiDonato JA. Lin A. Coordinate regulation of IkappaB kinases by mitogen-activated protein kinase kinase kinase 1 and NF-kappaB-inducing kinase. Mol Cell Biol. 1998;18:7336–7343. doi: 10.1128/mcb.18.12.7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Nowicki MO. Falinski R. Koptyra M. Slupianek A. Stoklosa T. Gloc E. Nieborowska-Skorska M. Blasiak J. Skorski T. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 228.Oberley TD. Oberley LW. Antioxidant enzyme levels in cancer. Histol Histopathol. 1997;12:525–535. [PubMed] [Google Scholar]
- 229.Ogryzko VV. Schiltz RL. Russanova V. Howard BH. Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 230.Oliner JD. Kinzler KW. Meltzer PS. George DL. Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 231.Omenn GS. Goodman GE. Thornquist MD. Balmes J. Cullen MR. Glass A. Keogh JP. Meyskens FL., Jr Valanis B. Williams JH., Jr. Barnhart S. Cherniack MG. Brodkin CA. Hammar S. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst. 1996;88:1550–1559. doi: 10.1093/jnci/88.21.1550. [DOI] [PubMed] [Google Scholar]
- 232.Omenn GS. Goodman GE. Thornquist MD. Balmes J. Cullen MR. Glass A. Keogh JP. Meyskens FL. Valanis B. Williams JH. Barnhart S. Hammar S. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 233.Ott M. Zhivotovsky B. Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007;14:1243–1247. doi: 10.1038/sj.cdd.4402135. [DOI] [PubMed] [Google Scholar]
- 234.Palmer LA. Doctor A. Chhabra P. Sheram ML. Laubach VE. Karlinsey MZ. Forbes MS. Macdonald T. Gaston B. S-nitrosothiols signal hypoxia-mimetic vascular pathology. J Clin Invest. 2007;117:2592–2601. doi: 10.1172/JCI29444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Pan S. Berk BC. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: key role for glutaredoxin in the death pathway. Circ Res. 2007;100:213–219. doi: 10.1161/01.RES.0000256089.30318.20. [DOI] [PubMed] [Google Scholar]
- 236.Pantano C. Reynaert NL. van der Vliet A. Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid Redox Signal. 2006;8:1791–1806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- 237.Park HS. Huh SH. Kim MS. Lee SH. Choi EJ. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proc Natl Acad Sci U S A. 2000;97:14382–14387. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Park HS. Lee JS. Huh SH. Seo JS. Choi EJ. Hsp72 functions as a natural inhibitory protein of c-Jun N-terminal kinase. EMBO J. 2001;20:446–456. doi: 10.1093/emboj/20.3.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Pelicano H. Carney D. Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Update. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 240.Pelicano H. Xu RH. Du M. Feng L. Sasaki R. Carew JS. Hu Y. Ramdas L. Hu L. Keating MJ. Zhang W. Plunkett W. Huang P. Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J Cell Biol. 2006;175:913–923. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Pennington JD. Wang TJ. Nguyen P. Sun L. Bisht K. Smart D. Gius D. Redox-sensitive signaling factors as a novel molecular targets for cancer therapy. Drug Resist Update. 2005;8:322–330. doi: 10.1016/j.drup.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 242.Pervaiz S. Pro-oxidant milieu blunts scissors: insight into tumor progression, drug resistance, and novel druggable targets. Curr Pharm Des. 2006;12:4469–4477. doi: 10.2174/138161206779010503. [DOI] [PubMed] [Google Scholar]
- 243.Pinchuk I. Schnitzer E. Lichtenberg D. Kinetic analysis of copper-induced peroxidation of LDL. Biochim Biophys Acta. 1998;1389:155–172. doi: 10.1016/s0005-2760(97)00139-2. [DOI] [PubMed] [Google Scholar]
- 244.Pineda-Molina E. Klatt P. Vazquez J. Marina A. Garcia de Lacoba M. Perez-Sala D. Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry. 2001;40:14134–14142. doi: 10.1021/bi011459o. [DOI] [PubMed] [Google Scholar]
- 245.Poeggeler B. Melatonin, aging, and age-related diseases: perspectives for prevention, intervention, and therapy. Endocrine. 2005;27:201–212. doi: 10.1385/ENDO:27:2:201. [DOI] [PubMed] [Google Scholar]
- 246.Poole LB. Karplus PA. Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 247.Poppek D. Grune T. Proteasomal defense of oxidative protein modifications. Antioxid Redox Signal. 2006;8:173–184. doi: 10.1089/ars.2006.8.173. [DOI] [PubMed] [Google Scholar]
- 248.Pouyssegur J. Mechta-Grigoriou F. Redox regulation of the hypoxia-inducible factor. Biol Chem. 2006;387:1337–1346. doi: 10.1515/BC.2006.167. [DOI] [PubMed] [Google Scholar]
- 249.Radi R. Peluffo G. Alvarez MN. Naviliat M. Cayota A. Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med. 2001;30:463–488. doi: 10.1016/s0891-5849(00)00373-7. [DOI] [PubMed] [Google Scholar]
- 250.Radisky DC. Levy DD. Littlepage LE. Liu H. Nelson CM. Fata JE. Leake D. Godden EL. Albertson DG. Nieto MA. Werb Z. Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Rahman I. Gilmour PS. Jimenez LA. MacNee W. Oxidative stress and TNF-alpha induce histone acetylation and NF-kap-paB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002;234–235:239–248. [PubMed] [Google Scholar]
- 252.Rahman I. Marwick J. Kirkham P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem Pharmacol. 2004;68:1255–1267. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 253.Ran Q. Liang H. Gu M. Qi W. Walter CA. Roberts LJ., 2nd Herman B. Richardson A. Van Remmen H. Transgenic mice over-expressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem. 2004;279:55137–55146. doi: 10.1074/jbc.M410387200. [DOI] [PubMed] [Google Scholar]
- 254.Rauhala P. Lin AM. Chiueh CC. Neuroprotection by S-nitrosoglutathione of brain dopamine neurons from oxidative stress. FASEB J. 1998;12:165–173. doi: 10.1096/fasebj.12.2.165. [DOI] [PubMed] [Google Scholar]
- 255.Rebrin I. Kamzalov S. Sohal RS. Effects of age and caloric restriction on glutathione redox state in mice. Free Radic Biol Med. 2003;35:626–635. doi: 10.1016/s0891-5849(03)00388-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Reinstein E. Ciechanover A. Narrative review: protein degradation and human diseases: the ubiquitin connection. Ann Intern Med. 2006;145:676–684. doi: 10.7326/0003-4819-145-9-200611070-00010. [DOI] [PubMed] [Google Scholar]
- 257.Requena JR. Fu MX. Ahmed MU. Jenkins AJ. Lyons TJ. Thorpe SR. Lipoxidation products as biomarkers of oxidative damage to proteins during lipid peroxidation reactions. Nephrol Dial Transplant. 1996;11(suppl 5):48–53. doi: 10.1093/ndt/11.supp5.48. [DOI] [PubMed] [Google Scholar]
- 258.Reynaert NL. Cless K. Korn SH. Vos N. Guala AS. Wouters EF. van der Vliet A. Janssen-Heininger YM. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci U S A. 2004;101:8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Reynaert NL. van der Vliet A. Guala AS. McGovern T. Hristova M. Pantano C. Heintz NH. Heim J. Ho YS. Matthews DE. Wouters EF. Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Rhee SG. Yang K-S. Kang SW. Woo HA. Chang T-S. Controlled elimination of intracellular H2O2: regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 261.Rivera A. Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J Biol Chem. 2005;280:29346–29354. doi: 10.1074/jbc.M504852200. [DOI] [PubMed] [Google Scholar]
- 262.Rosen DR. Siddique T. Patterson D. Figlewicz DA. Sapp P. Hentati A. Donaldson D. Goto J. O'Regan JP. Deng H-X. Rahmani Z. Krizus A. McKenna-Yasek D. Cayabyab A. Gaston SM. Berger R. Tanzi RE. Halperin JJ. Herzfeldt B. den Bergh RV. Hung W-Y. Bird T. Deng G. Mulder DW. Smyth C. Laing NG. Soriano E. Pericak-Vance MA. Haines J. Rouleau GA. Gusella JS. Horvitz HR. Brown RH. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 263.Sablina AA. Budanov AV. Ilyinskaya GV. Agapova LS. Kravchenko JE. Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Saitoh M. Nishitoh H. Fujii M. Takeda K. Tobiume K. Sawada Y. Kawabata M. Miyazono K. Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Salmeen A. Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid Redox Signal. 2005;7:560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- 266.Sarbassov DD. Guertin DA. Ali SM. Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 267.Sarkisian CJ. Keister BA. Stairs DB. Boxer RB. Moody SE. Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 268.Sattler M. Verma S. Shrikhande G. Byrne CH. Pride YB. Winkler T. Greenfield EA. Salgia R. Griffin JD. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275:24273–24278. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- 269.Schieke SM. Briviba K. Klotz LO. Sies H. Activation pattern of mitogen-activated protein kinases elicited by peroxynitrite: attenuation by selenite supplementation. FEBS Lett. 1999;448:301–303. doi: 10.1016/s0014-5793(99)00372-5. [DOI] [PubMed] [Google Scholar]
- 270.Schofield CJ. Ratcliffe PJ. Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun. 2005;338:617–626. doi: 10.1016/j.bbrc.2005.08.111. [DOI] [PubMed] [Google Scholar]
- 271.Schonhoff CM. Gaston B. Mannick JB. Nitrosylation of cytochrome c during apoptosis. J Biol Chem. 2003;278:18265–18270. doi: 10.1074/jbc.M212459200. [DOI] [PubMed] [Google Scholar]
- 272.Schopfer FJ. Baker PRS. Freeman BA. NO-dependent protein nitration: a cell signaling event or an oxidative inflammatory response? Trends Biochem Sci. 2003;28:646–654. doi: 10.1016/j.tibs.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 273.Schriner SE. Linford NJ. Martin GM. Treuting P. Ogburn CE. Emond M. Coskun PE. Ladiges W. Wolf N. Van Remmen H. Wallace DC. Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 274.Schwartz JL. The dual roles of nutrients as antioxidants and prooxidants: their effects on tumor cell growth. J Nutr. 1996;126:1221S–1227S. doi: 10.1093/jn/126.suppl_4.1221S. [DOI] [PubMed] [Google Scholar]
- 275.Serrano M. Lin AW. McCurrach ME. Beach D. Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 276.Shatrov VA. Sumbayev VV. Zhou J. Brune B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1alpha (HIF-1alpha) accumulation via redox-dependent mechanisms. Blood. 2003;101:4847–4849. doi: 10.1182/blood-2002-09-2711. [DOI] [PubMed] [Google Scholar]
- 277.Shaulian E. Schreiber M. Piu F. Beeche M. Wagner EF. Karin M. The mammalian UV response: c-jun induction is required for exit from p53-imposed growth arrest. Cell. 2000;103:897–907. doi: 10.1016/s0092-8674(00)00193-8. [DOI] [PubMed] [Google Scholar]
- 278.Shaulian E. Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 279.Sheehan D. Meade G. Foley VM. Dowd CA. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Shen HM. Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 281.Shi Y. Caspase activation: revisiting the induced proximity model. Cell. 2004;117:855–858. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 282.Shrivastava P. Pantano C. Watkin R. McElhinney B. Guala A. Poynter ML. Persinger RL. Budd R. Janssen-Heininger Y. Reactive nitrogen species-induced cell death requires Fas-dependent activation of c-Jun N-terminal kinase. Mol Cell Biol. 2004;24:6763–6772. doi: 10.1128/MCB.24.15.6763-6772.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Song JJ. Rhee JG. Suntharalingam M. Walsh SA. Spitz DR. Lee YJ. Role of glutaredoxin in metabolic oxidative stress: glutaredoxin as a sensor of oxidative stress mediated by H2O2. J Biol Chem. 2002;277:46566–46575. doi: 10.1074/jbc.M206826200. [DOI] [PubMed] [Google Scholar]
- 284.Soussi T. Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303–312. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 285.Squier TC. Redox modulation of cellular metabolism through targeted degradation of signaling proteins by the proteasome. Antioxid Redox Signal. 2006;8:217–228. doi: 10.1089/ars.2006.8.217. [DOI] [PubMed] [Google Scholar]
- 286.Srinivasan A. Lehmler HJ. Robertson LW. Ludewig G. Production of DNA strand breaks in vitro and reactive oxygen species in vitro and in HL-60 cells by PCB metabolites. Toxicol Sci. 2001;60:92–102. doi: 10.1093/toxsci/60.1.92. [DOI] [PubMed] [Google Scholar]
- 287.Stadtman ER. Role of oxidant species in aging. Curr Med Chem. 2004;11:1105–1112. doi: 10.2174/0929867043365341. [DOI] [PubMed] [Google Scholar]
- 288.Su H. Bidere N. Zheng L. Cubre A. Sakai K. Dale J. Salmena L. Hakem R. Straus S. Lenardo M. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- 289.Suh JH. Shenvi SV. Dixon BM. Liu H. Jaiswal AK. Liu R-M. Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci US A. 2004;101:3381–3386. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Sui H. Wang W. Wang PH. Liu LS. Protective effect of antioxidant ebselen (PZ51) on the cerebral cortex of stroke-prone spontaneously hypertensive rats. Hypertens Res. 2005;28:249–254. doi: 10.1291/hypres.28.249. [DOI] [PubMed] [Google Scholar]
- 291.Sumbayev VV. Yasinska IM. Regulation of MAP kinase-dependent apoptotic pathway: implication of reactive oxygen and nitrogen species. Arch Biochem Biophys. 2005;436:406–412. doi: 10.1016/j.abb.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 292.Sun J. Steenbergen C. Murphy E. S-Nitrosylation: no-related redox signaling to protect against oxidative stress. Antioxid Redox Signal. 2006;8:1693–1705. doi: 10.1089/ars.2006.8.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Szabo C. Ischiropoulos H. Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 294.Szatrowski TP. Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 295.Tan KP. Yang M. Ito S. Activation of Nrf2 by toxic bile acids provokes adaptive Defense responses to enhance cell survival at the emergence of oxidative stress. Mol Pharmacol Epub ahead of print. 2007 doi: 10.1124/mol.107.039370. [DOI] [PubMed] [Google Scholar]
- 296.Tanaka H. Matsumura I. Ezoe S. Satoh Y. Sakamaki T. Albanese C. Machii T. Pestell RG. Kanakura Y. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Mol Cell. 2002;9:1017–1029. doi: 10.1016/s1097-2765(02)00522-1. [DOI] [PubMed] [Google Scholar]
- 297.Tobiume K. Matsuzawa A. Takahashi T. Nishitoh H. Morita K. Takeda K. Minowa O. Miyazono K. Noda T. Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Toledano MB. Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A. 1991;88:4328–4332. doi: 10.1073/pnas.88.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Tomita Y. Marchenko N. Erster S. Nemajerova A. Dehner A. Klein C. Pan H. Kessler H. Pancoska P. Moll UM. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. 2006;281:8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 300.Tomko RJ., Jr Bansal P. Lazo JS. Airing out an antioxidant role for the tumor suppressor p53. Mol Intervent. 2006;6:23–25. doi: 10.1124/mi.6.1.5. [DOI] [PubMed] [Google Scholar]
- 301.Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–d391. doi: 10.2741/999. [DOI] [PubMed] [Google Scholar]
- 302.Trachootham D. Zhou Y. Zhang H. Demizu Y. Chen Z. Pelicano H. Chiao PJ. Achanta G. Arlinghaus RB. Liu J. Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 303.Trifunovic A. Wredenberg A. Falkenberg M. Spelbrink JN. Rovio AT. Bruder CE. Bohlooly YM. Gidlof S. Oldfors A. Wibom R. Tornell J. Jacobs HT. Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 304.Trifunovic A. Mitochondrial DNA and ageing. Biochim Biophys Acta. 2006;1757:611–617. doi: 10.1016/j.bbabio.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 305.Tsuzuki T. Nakatsu Y. Nakabeppu Y. Significance of error-avoiding mechanisms for oxidative DNA damage in carcinogenesis. Cancer Sci. 2007;98:465–470. doi: 10.1111/j.1349-7006.2007.00409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Turpaev KT. Reactive oxygen species and regulation of gene expression. Biochemistry (Moscow) 2002;67:281–292. doi: 10.1023/a:1014819832003. [DOI] [PubMed] [Google Scholar]
- 307.Tyner SD. Venkatachalam S. Choi J. Jones S. Ghebranious N. Igelmann H. Lu X. Soron G. Cooper B. Brayton C. Hee Park S. Thompson T. Karsenty G. Bradley A. Donehower LA. p53 Mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 308.Vafa O. Wade M. Kern S. Beeche M. Pandita TK. Hampton GM. Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 309.Valko M. Rhodes CJ. Moncol J. Izakovic M. Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 310.Valko M. Leibfritz D. Moncol J. Cronin MTD. Mazur M. Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 311.Van Laethem A. Nys K. Van Kelst S. Claerhout S. Ichijo H. Vandenheede JR. Garmyn M. Agostinis P. Apoptosis signal regulating kinase-1 connects reactive oxygen species to p38 MAPK-induced mitochondrial apoptosis in UVB-irradiated human keratinocytes. Free Radic Biol Med. 2006;41:1361–1371. doi: 10.1016/j.freeradbiomed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 312.Van Remmen H. Ikeno Y. Hamilton M. Pahlavani M. Wolf N. Thorpe SR. Alderson NL. Baynes JW. Epstein CJ. Huang TT. Nelson J. Strong R. Richardson A. Life-long reduction in Mn-SOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 313.Van Waes C. Nuclear factor-kappaB in development, prevention, and therapy of cancer. Clin Cancer Res. 2007;13:1076–1082. doi: 10.1158/1078-0432.CCR-06-2221. [DOI] [PubMed] [Google Scholar]
- 314.Vandermoere F. El Yazidi-Belkoura I. Adriaenssens E. Lemoine J. Hondermarck H. The antiapoptotic effect of fibroblast growth factor-2 is mediated through nuclear factor-kappaB activation induced via interaction between Akt and IkappaB kinase-beta in breast cancer cells. Oncogene. 2005;24:5482–5491. doi: 10.1038/sj.onc.1208713. [DOI] [PubMed] [Google Scholar]
- 315.Vanhaesebroeck B. Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346:561–576. [PMC free article] [PubMed] [Google Scholar]
- 316.Veis DJ. Sorenson CM. Shutter JR. Korsmeyer SJ. Bcl-2-deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell. 1993;75:229–240. doi: 10.1016/0092-8674(93)80065-m. [DOI] [PubMed] [Google Scholar]
- 317.Velu CS. Niture SK. Doneanu CE. Pattabiraman N. Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46:7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Venugopal R. Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998;17:3145–3156. doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 319.Vermeulen L. De Wilde G. Van Damme P. Vanden Berghe W. Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–1324. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Vermulst M. Bielas JH. Kujoth GC. Ladiges WC. Rabinovitch PS. Prolla TA. Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 321.Victor VM. Rocha M. Targeting antioxidants to mitochondria: a potential new therapeutic strategy for cardiovascular diseases. Curr Pharm Des. 2007;13:845–863. doi: 10.2174/138161207780363077. [DOI] [PubMed] [Google Scholar]
- 322.Vila M. Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:365–375. doi: 10.1038/nrn1100. [DOI] [PubMed] [Google Scholar]
- 323.Vlasova , II Tyurin VA. Kapralov AA. Kurnikov IV. Osipov AN. Potapovich MV. Stoyanovsky DA. Kagan VE. Nitric oxide inhibits peroxidase activity of cytochrome c cardiolipin complex and blocks cardiolipin oxidation. J Biol Chem. 2006;281:14554–14612. doi: 10.1074/jbc.M509507200. [DOI] [PubMed] [Google Scholar]
- 324.Voehringer DW. Meyn RE. Redox aspects of Bcl-2 function. Antioxid Redox Signal. 2000;2:537–550. doi: 10.1089/15230860050192314. [DOI] [PubMed] [Google Scholar]
- 325.Wang GL. Jiang BH. Semenza GL. Effect of altered redox states on expression and DNA-binding activity of hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1995;212:550–556. doi: 10.1006/bbrc.1995.2005. [DOI] [PubMed] [Google Scholar]
- 326.Wang T. Arifoglu P. Ronai Z. Tew KD. Glutathione S-transferase P1–1 (GSTP1–1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem. 2001;276:20999–21003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- 327.Wang X. McCullough KD. Franke TF. Holbrook NJ. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem. 2000;275:14624–14631. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- 328.Wang Y. Zeigler MM. Lam GK. Hunter MG. Eubank TD. Khramtsov VV. Tridandapani S. Sen CK. Marsh CB. The role of the NADPH oxidase complex, p38 MAPK, and Akt in regulating human monocyte/macrophage survival. Am J Respir Cell Mol Biol. 2007;36:68–77. doi: 10.1165/rcmb.2006-0165OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Wek RC. Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. Epub ahead of print. 2007 doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 330.Welch HC. Coadwell WJ. Stephens LR. Hawkins PT. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 2003;546:93–97. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- 331.Weston CR. Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 332.Wood ZA. Schroder E. Robin Harris J. Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 333.Xia Y. Wang J. Xu S. Johnson GL. Hunter T. Lu Z. MEKK1 mediates the ubiquitination and degradation of c-Jun in response to osmotic stress. Mol Cell Biol. 2007;27:510–517. doi: 10.1128/MCB.01355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Xia Y. Wang J. Liu T-J. Yung WKA. Hunter T. Lu Z. c-Jun downregulation by HDAC3-dependent transcriptional repression promotes osmotic stress-induced cell apoptosis. Mol Cell. 2007;25:219–232. doi: 10.1016/j.molcel.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Xiao L. Lang W. A dominant role for the c-Jun Nh2-terminal kinase in oncogenic ras-induced morphologic transformation of human lung carcinoma cells. Cancer Res. 2000;60:400–408. [PubMed] [Google Scholar]
- 336.Xue W. Zender L. Miething C. Dickins RA. Hernando E. Krizhanovsky V. Cordon-Cardo C. Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Yamaguchi T. Sano K. Takakura K. Saito I. Shinohara Y. Asano T. Yasuhara H. Ebselen in acute ischemic stroke: a placebo-controlled, double-blind clinical trial: Ebselen Study Group. Stroke. 1998;29:12–17. doi: 10.1161/01.str.29.1.12. [DOI] [PubMed] [Google Scholar]
- 338.Yang DD. Kuan C-Y. Whitmarsh AJ. Rinocn M. Zheng TS. Davis RJ. Rakic P. Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 339.Yarian CS. Toroser D. Sohal RS. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech Ageing Dev. 2006;127:79–84. doi: 10.1016/j.mad.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Yasukawa T. Tokunaga E. Ota H. Sugita H. Martyn JA. Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280:7511–7518. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 341.Young TW. Mei FC. Yang G. Thompson-Lanza JA. Liu J. Cheng X. Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res. 2004;64:4577–4584. doi: 10.1158/0008-5472.CAN-04-0222. [DOI] [PubMed] [Google Scholar]
- 342.Yu CX. Li S. Whorton AR. Redox regulation of PTEN by S-nitrosothiols. Mol Pharmacol. 2005;68:847–854. doi: 10.1124/mol.104.010504. [DOI] [PubMed] [Google Scholar]
- 343.Zhao Y. Wang ZB. Xu JX. Effect of cytochrome c on the generation and elimination of O2*- and H2O2 in mitochondria. J Biol Chem. 2003;278:2356–2360. doi: 10.1074/jbc.M209681200. [DOI] [PubMed] [Google Scholar]
- 344.Zhong H. SuYang H. Erdjument-Bromage H. Tempst P. Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]