

Abstract
Recent evidence suggests that oxidative stress contributes significantly to the regulation of hematopoietic cell homeostasis. In particular, red blood cells and hematopoietic stem cells are highly sensitive to deregulated accumulation of reactive oxygen species (ROS). Unchecked ROS accumulation often leads to hemolysis, that is, to destruction and shortened life span of red blood cells. In addition, the process of erythroid cell formation is sensitive to ROS accumulation. Similarly, ROS buildup in hematopoietic stem cells compromises their function as a result of potential damage to their DNA leading to loss of quiescence and alterations of hematopoietic stem cell cycling. These abnormalities may lead to accelerated aging of hematopoietic stem cells or to hematopoietic malignancies. Antioxid. Redox Signal. 10, 1923–1940.
Introduction
Oxidative stress is due to the effects of reactive oxygen species (ROS). It has been estimated that several thousands DNA-damaging events occur in every cell of the human body every day, and a significant portion of this is caused by ROS (24). In the mitochondrial respiratory chain for production of energy, O2 molecules accept four electrons to form two molecules of water. In this process, partially reduced O2 species form the ROS (<5%). Some ROS contain unpaired electrons and are therefore referred to as free radicals. The acceptance of a single electron by O2 generates superoxide −O2. The mitochondrial respiratory chain is a major source of −O2. Superoxide is not an effective oxidant, but it impairs mitochondrial function by oxidizing the Fe-S cluster of many enzymes [reviewed in (6, 27, 33)].
Once formed, superoxide undergoes rapid dismutation both spontaneously and by a family of enzymes (superoxide dismutase, SOD) to form hydrogen peroxide, H2O2, and O2 (Fig. 1). H2O2 is removed by three general mechanisms: (a) it is catalyzed by two enzymes, catalase and glutathione (GSH) peroxidase, to H2O and O2; (b) it is converted by myeloperoxidase in neutrophils to hypochlorous acid (HOCl), a physiologically toxic product and a strong oxidant that acts as a bactericidal agent in phagocytic cells; and (c) H2O2 is converted in a spontaneous reaction, catalyzed by Fe2+ and called the Fenton reaction, to the highly reactive hydroxyl radical ·OH (·OH + RH → H2O + R). Lipid per-oxidation is readily achieved by hydroxyl radicals but not by hydrogen peroxide.
FIG. 1.

Reactive oxygen species. Cells generate aerobic energy by reducing molecular oxygen (O2) to water. During the metabolism of oxygen, superoxide (−O2) is occasionally formed. Superoxide is rapidly dismutated to hydrogen peroxide (H2O2), which is converted by glutathione peroxidase or catalase to water. MPD (myeloperoxidase) converts H2O2 in neutrophils to hypochlorous acid (HOCl), a strong oxidant that acts as a bactericidal agent in phagocytic cells. During a Fenton reaction, H2O2 is converted in a spontaneous reaction catalyzed by Fe2+ to the highly reactive hydroxyl radical ·OH.
Another important class of scavengers are glutathiones, which appear both in oxidized (GSSG) and reduced (GSH) forms. A third class of antioxidant enzymes is glutathione-S-transferase and the auxiliary enzyme, glutathione reductase, which use NADPH to regenerate GSH from GSSG. ROS, in particular the hydroxyl radical, can react with all biologic macromolecules (lipids, proteins, nucleic acids, and carbohydrates). Similar to its response to DNA damage, the cellular response to ROS-induced damage is either to arrest the cellular life cycle to allow the damage to be repaired or to initiate programmed cell death (6, 27, 33).
Hematopoietic cells appear to be particularly vulnerable in the presence of unchecked accumulation of ROS, because deficiencies in several ROS scavengers result in either anemia that is severe or even lethal in some cases and/or malignancies of hematopoietic tissues (36, 41, 61, 66, 87, 121) (see Table 1). Although ROS affect many hematopoietic cell lineages, this review is limited to the effect of ROS in the regulation of hematopoietic stem and erythroid cells. The review focuses on the impact of oxidative stress on the regulation of erythropoiesis (Part I). In the next section (Part II), the regulation of oxidative stress by FoxO transcription factors in hematopoietic cells, specifically in erythroid cells, is reviewed. Finally in Part III, what is known of the control of oxidative stress in hematopoietic stem cells and the impact of ROS on hematopoietic stem cell activity, aging, niche interactions, and potential neoplastic transformation are discussed.
Table 1.
Genetically Modified Mice with an Oxidative Stress Phenotype
| Targeted gene | Phenotype | Reference |
|---|---|---|
| P45NF-E2−/− | High rate of mortality, bleeding, defective megakaryocyte maturation, severe platelet deficiency, low hypochromic anemia, reticulocytosis, methemoglobin, increased ROS, reduced lifespan, reduced catalase activity in red blood cells. | 14 |
| Nrf1−/− | Embryonic lethality, impaired erythropoiesis due to defective fetal liver microenvironment, oxidative stress in hapatocytes | 15, 23 |
| Nrf2−/− | Anemia, with signs of damaged erythroid cells, decreased platelets, ROS increase | 64 |
| SOD2−/− | Mitochondrial injury, damage to cardiac muscle, neuronal tissues and metabolic abnormalities including acidosis and lipid accumulation, post natal lethality, reconstituted mice with SOD2-deficient fetal liver: ROS-mediated hemolytic anemia, signs of protein oxidation | 33, 34, 63, 68 |
| SOD3−/− | Exacerbation of oxidative stress | 11 |
| Peroxiredoxin I−/− | Several malignancies including lymphomas, fibrosarcomas, histiocytic sarcomas and osteosarcomas increased ROS in erythrocytes, protein oxidation, hemoglobin instability, Heinz body formation, decreased erythrocyte lifespan | 85 |
| Peroxiredoxin I−/+ | Increased frequency of malignancies | 85 |
| Peroxiredoxin II−/− | Compensated regenerative hemolytic anemia, splenomegaly, high reticulocytosis, increased Epo levels | 66 |
| Peroxiredoxin III−/− | Mitochondrial ROS | 19 |
| Glutathione peroxidase 1−/− | Normal | 40 |
| Glutathione peroxidase 4−/− | Embryonically lethal | 130 |
| Catalase−/− | Normal | 41 |
| AHSP−/− | Hemolytic anemia, increased ROS, signs of oxidative damage | 57 |
| ATM−/− | Ataxia, telangiectasia, neuronal degeneration, immunodeficiencies, genomic instability, predisposition to malignancies including lymphomas, ROS accumulation in hematopoietic stem cell pool in aging mice, bone marrow failure with age | 4, 47 |
| FoxO1, 3, 4−/− | Decreased hematopoietic stem cell pool, loss of quiescence and increased cell cycle of hematopoietic stem cells, myeloproliferation, ROS accumulation in hematopoietic stem cells, decrease of SOD3 and catalase | 111 |
| FoxO3−/− | Decreased hematopoietic stem cell pool, ROS in hematopoietic stem cells, loss of quiescence, G2/M arrest of hematopoietic stem cells, ROS-mediated myeloproliferation, decrease of SOD2, catalase, GPX-1, ROS-mediated chronic hemolysis, decrease rate of erythroid cell maturation, increased loss of hematopoietic stem cell activity with aging | 79, 125 |
Part I
Regulation of Oxidative Stress in Red Blood Cells and Precursors
Regulation of oxidative stress is particularly important to erythropoiesis, the process that results in the production of mature erythrocytes, from proliferation, survival, and differentiation of erythroid cell progenitors in response to erythropoietin (Epo) binding to erythropoietin receptor (EpoR) [reviewed in (107)] (Fig. 2). Erythroid precursors synthesize and accumulate hemoglobin as they mature. In addition, insertion of iron to heme in mitochondria of erythroid precursors provides potential for the Fenton reaction. Circulating erythrocytes carry oxygen bound to hemoglobin and as such are highly prone to oxidative damage (122). Consequently, erythrocytes are exposed to one of the highest levels of oxidative-stress conditions in the body. In agreement with this, compromised protection from ROS, as seen in genetic enzymatic deficiencies involving pathways that maintain intracellular reductive molecules or in molecules that protect globin, results in diseases of red blood cells, with shortened life span and hemolysis, leading to anemias (36, 41, 61, 66, 121). Not surprisingly, erythroid cells in healthy individuals contain a strong arsenal of antioxidant enzymes that protect the cells against oxygen radicals (54). These include superoxide dismutases, which convert superoxide to oxygen peroxide, catalase and glutathione peroxidase, which convert hydrogen peroxide to water, and nonenzymatic scavengers such as glutathione, peroxiredoxin, ascorbic acid, and carotenoids. Accumulation of ROS also has a significant impact on the senescence of circulating erythrocytes and their limited life span (7). Because mature erythrocytes are enucleated, important transcriptional regulation must take place before enucleation and release of erythrocytes to the peripheral circulation.
FIG. 2.
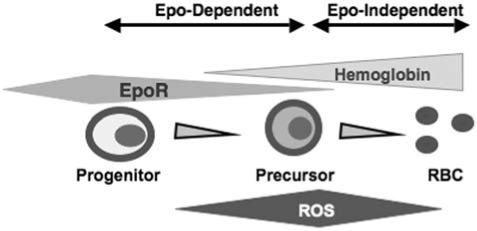
Schematic of erythroid cell differentiation. The most mature erythroid progenitor (colony-forming unit–erythroid cell, CFU-E) expresses the highest levels of EpoR during erythropoiesis. CFU-Es divide to produce erythroid precursor cells, and EpoR expression is downregulated during this process on erythroid precursors. Erythroid cells accumulate hemoglobin as they mature. It is noteworthy that whereas erythroid cells generated from the first divisions of CFU-E require Epo, Epo becomes dispensible for the maturation of subsequent erythroid cells.
P45NF-E2, Nrf1, and Nrf2 Transcription Factors
The 5′-flanking regions of some antioxidant genes, such as heme oxygenase, contain a common cis-acting regulatory antioxidant response element (ARE), the activation of which partly mediates the antioxidative stress response [(99), and reviewed in (51)].
The cap ’n’ collars (CNCs) of the basic zipper superfamily of transcription factors are identified by their ability to bind and activate the hypersensitive sites of the globin locus control region (1, 12, 13, 88). This family includes p45NF-E2, NF-E2–related factor (Nrf) 1, Nrf2, and Nrf3, and more distantly, Bach1 and Bach2. P45NF-E2 expression is restricted to hematopoietic cells. Some members of this family are identified as important in the activation of ARE and regulation of oxidative stress in erythropoiesis.
P45NF-E2−/− mice display a high rate of mortality because of bleeding as a result of defective megakaryocyte maturation, severe platelet deficiency, and a low hypochromic anemia associated with reticulocytosis (105, 106). At the steady state, P45NF-E2−/− red blood cells accumulate increasing amounts of ROS, as compared with their wild-type counterparts and, in particular, of hydrogen peroxide (14). This leads to abnormal oxidation of hemoglobin in the form of methemoglobin and increased osmotic fragility in P45NF-E2−/− red blood cells. Specifically, deformability of P45NF-E2−/− red blood cells is abolished in the presence of hydrogen peroxide.
The rate of red cell production increases dramatically in response to clinical or physiologic stimuli that threaten the tissue oxygenation, owing to enhanced proliferation of erythroid progenitors, in a process known as stress erythropoiesis (107). A classic approach to induce stress erythropoiesis is the use of phenylhydrazine. Phenylhydrazine treatment induces oxidative denaturation of hemoglobin, resulting in rapid destruction and significant loss of erythrocytes (hemolysis), which leads to anemia, characterized by a decrease of hemoglobin concentration and hematocrit levels (48). In response to phenylhydrazine-induced hemolytic anemia, bone marrow and spleen erythropoiesis are stimulated, releasing prematurely erythrocyte precursors, reticulocytes, into the bloodstream. Therefore, the reticulocyte content of blood reflects the severity of the hemolytic anemia and the degree of compensatory response of bone marrow and spleen to phenylhydrazine. Phenylhydrazine treatment dramatically increased the degree of anemia in P45NF-F2−/− mice, suggesting an impairment in response to stress erythropoiesis. The P45NF-E2−/− red blood cells display reduced life span and reduced expression of catalase activity (14). These studies suggest an important role for P45NF-E2 in the regulation of oxidative stress in red blood cells.
Two p45NF-E2–related Nrf1 and Nrf2 transcription factors are expressed ubiquitously and are also implicated in the regulation of antioxidant genes. Loss of Nrf1 results in abnormal fetal liver erythropoiesis, anemia, and embryonic lethality (15). Fetal liver is the major site of erythropoiesis between days 11 and 15 of embryogenesis in mice. A majority (>85%) of liver cells at that stage are erythroid. The erythroid abnormalities in Nrf1−/− embryos are sustained by an impaired microenvironment in Nrf1−/− E13 fetal liver (23). This view was supported by experiments showing that fetal liver Nrf1−/− erythroid progenitors were unable to mature to produce enucleated red blood cells in their own environment in vivo. Nonetheless, their ability to mature and produce hemoglobinized colonies of erythroid cells in frequencies comparable to those of their wild-type counterparts was intact in the petri dish in vitro. In addition, embryonic stem cells containing two disrupted Nrf-1 alleles contributed efficiently to blood cells in chimeric animals. These findings are consistent with the notion that the erythropoietic defect in Nrf1−/− embryos is due to a defective fetal liver environment and is not intrinsic to erythroid cells (23). Further studies showed increased oxidative stress, reduced expression of antioxidant enzymes, and apoptosis of fetal liver cells (23). In agreement with these findings, Nrf1−/− fibroblasts are highly sensitive to toxic oxidant compounds (64).
Nrf2 is widely expressed, and as does Nrf1, has antioxidant functions. Nrf2 promotes survival by modulating antioxidant and apoptosis pathways. Nrf2−/− mice are viable and develop normally but are sensitive to redox-inducing agents (16, 17). Nrf2−/− mice develop splenomegaly and oxidative stress–mediated hemolytic anemia as they age (66). Nrf2−/− splenocytes show a decrease of ARE-driven gene expression. The Nrf2−/− anemia is associated with signs of damaged erythroid cells and relative loss of platelets. Nrf2−/− cells are highly sensitive to hydrogen peroxide in culture. Increased IgG- and IgM-bound erythrocytes in Nrf2−/− mice suggest that immune-mediated phagocytosis in the spleen of Nrf2−/− mice is increased (66). Together these findings suggest that loss of Nrf2 sensitizes red blood cells to oxidative stress. ROS in Nrf2−/− erythrocytes enhances the immune reaction in the spleen, leading to their phagocytosis, resulting in the release of proteins and inflammatory compounds that triggers antigenic response. It is, therefore, proposed that the Nrf2-ARE pathway may serve as a therapeutic target for prevention of some degenerative and malignant diseases (66).
Superoxide Dismutase (SOD)
Superoxide dismutases (SODs) are enzymes that convert superoxide to hydrogen peroxide [reviewed in (134)]. These enzymes are found in the cytoplasm (CuZnSOD, SOD1), mitochondria (MnSOD, SOD2), and extracellular fluids (EC-SOD3, SOD3) (134). Although loss of SOD1 does not interfere with normal development and does not appear to affect hematopoiesis overtly (94), loss of SOD2 leads to defective hematopoiesis, and loss of SOD3 affects Epo production.
Loss of mitochondrial SOD2 leads to lethality on two different genetic Backgrounds, albeit at distinct time points. SOD2-deficient mice exhibit pathologic evidence of mitochondrial injury, damage to cardiac muscle and neuronal tissues, and metabolic abnormalities including acidosis and lipid accumulation (65, 70). SOD2-deficient mitochondrial dysfunctions are associated with decrease of iron–sulfur clusters containing enzymes of the respiratory chain and tri-carboxylic acid cycle, a partial defect in HMG-CoA lyase and extensive ROS damage to the DNA (78).
To assess the impact of SOD2 mutation on hematopoiesis, the hematopoietic compartment of lethally irradiated wild-type mice was reconstituted with SOD2 mutant fetal liver cells (36). This approach evaluates the potential of mutant hematopoietic stem cells in the donor pool to generate hematopoietic cells of all lineages in the recipient mice, in which the endogenous hematopoiesis is ablated by irradiation. These experiments showed that lethally irradiated mice reconstituted with SOD2-deficient fetal liver hematopoietic cells display hemolytic anemia with compensatory enhanced erythropoiesis, extensive damage to redox regulation, adenosine triphosphate synthesis and red cell metabolism, and presence of iron deposits in mature erythroid cells.
Oxidized proteins accumulate in SOD2-deficient erythrocytes, leading to their decreased life span (36). Interestingly, several subunits of mitochondrial ATP synthase were increased in SOD2−/− erythroid cells either because of too much demand or because of poor mitochondrial function secondary to oxidative damage (35). The anemia in recipient mice is corrected by in vivo use of SOD/catalase mimetic, strongly suggesting that oxidative stress mediated the hemolytic anemia in these chimeric mice (35). It is noteworthy that the hemolytic anemia in SOD2-deficient reconstituted mice resembles, in many respects, human sideroblastic anemia (76). In response to oxidative damage, proteins involved in pathways that reverse oxidative damage are increased, and their upregulation may be further enhanced by antioxidant treatment. In agreement with this, antioxidant treatment of SOD2-deficient erythrocytes increases levels of peroxiredoxin 2 (see later). SOD2-deficient erythroid cells may display a late defect in heme biosynthesis involving iron incorporation, as suggested by elevated zinc protoporphyrin in these cells (35).
Importantly, loss of SOD2 [reviewed in (134)] did not interfere with lymphoid or myeloid hematopoietic reconstitution in lethally irradiated mice (35, 36). SOD2 deficiency did not affect long-term reconstitution or radioprotection of fetal liver hematopoietic stem cells and was not associated with bone marrow failure in recipient mice. These studies suggest that loss of SOD2 does not affect the regulation of hematopoietic stem cells or the lineage commitment. These findings imply that SOD2 is not required for the regulation of oxidative stress in hematopoietic stem cells. However, these results (35) were derived from SOD2-deficient mouse fetal liver and thus exclude a role for SOD2 during embryonic hematopoietic stem cell development. Conditional genetic deletion of SOD2 may reveal further antioxidant functions for SOD2 in adult hematopoietic stem or progenitor cells or both.
Loss of SOD3 leads to exacerbation of oxidative stress (11). Experiments using SOD3-deficient mice under hypoxic conditions suggest that SOD3 is required for full induction of Epo in response to hypoxia (109). However, the impact of SOD3 on hematopoiesis is not clear (109).
Glutathione Peroxidase and Catalase
Glutathione peroxidases (GPXs) and catalase enzymes are thought to be a major antioxidant defense in erythroid cells. These enzymes use nicotinamide adenine dinucleotide phosphate (NADPH) generated by hexose monophosphate shunt through glucose 6-phosphate dehydrogenase as the electron donor and glutathione as the direct ROS scavenger. However, deficiencies in catalase or glutathione peroxidases have not been found to affect erythroid cells. Catalase-deficient mice develop normally (43).
The four known glutathione peroxidases all contain selenocysteine at their active sites (3). GPX-1 encodes for mitochondrial GPX, is ubiquitously expressed, and is the most abundant glutathione peroxidase. GPX-4 is also ubiquitously expressed and is a key enzyme in the detoxification of lipid hydroperoxides. GPX-1–deficient mice are normal, whereas GPX-4–deficient mice die during early development (42, 131). Whether the loss of GPX-4 has any effect on erythroid cells is not clear. Reduced levels of GPX-4 result in an increased life span in mice (93), by possibly delaying or reducing the occurrence, latency, and/or severity of diseases such as fatal lymphomas. These observations are attributed to the increased sensitivity to oxidative stress–mediated apoptosis in GPX-4 heterozygote mice (93).
Peroxiredoxins
Peroxiredoxins form a family of small antioxidant proteins that contain essential catalytic cysteine residues that scavenge ROS and are involved in cellular responses to ROS. Six members of the peroxiredoxin family of antioxidant proteins are known.
The redox-sensitive cysteine residues are oxidized in reaction with hydrogen peroxide to form an intermolecular disulfide with neighboring Cys-SH–containing proteins. This disulfide is reduced specifically by thioredoxin. Interestingly, peroxiredoxins are able to eliminate the intracellular H2O2 generated in response to growth factors and block NF-κB activation (57).
Mammalian peroxiredoxin I (MSP23 for macrophage 23-KDa stress protein) (47) is induced by serum stimulation and oxidative stress. Loss of peroxiredoxin I leads to development of severe hemolytic anemias and several malignancies, including lymphomas, fibrosarcomas, histiocytic sarcomas, and osteosarcomas in homozygous mice and increased frequency of malignancies in heterozygotes (87). Hemolytic anemias in peroxiredoxin I–deficient mice are characterized by increased ROS accumulation in erythrocytes, protein oxidation, hemoglobin instability, Heinz body formation, and decreased erythrocyte life span.
Loss of peroxiredoxin II results in a hemolytic anemia that is compensated by enhanced erythroid progenitor activity in the bone marrow and spleen, characterized by splenomegaly, high reticulocytosis, and compensatory increased Epo levels (68). These compensatory mechanisms limit the extent of anemia. Red blood cell proteins were oxidized with signs of increased iron deposit in the liver of splenectomized peroxiredoxin II−/− mice. When peroxiredoxin I−/− red blood cells were treated with H2O2, levels of methemoglobin, in which the iron in hemoglobin is in Fe3+ unstable form, were increased (68). Decreased levels of antioxidant activity were associated with oxidation of specific cysteine residues in some peroxiredoxin II−/− red blood cell proteins. Like SOD2, peroxiredoxin III is mitochondria specific and provides much of the mitochondrial antioxidant protection in converting hydrogen peroxide into water (19).
By using MitoTracker Red, a mitochondria-specific probe, and DHR123, a nonfluorescent mitochondrial ROS tracker that is oxidized to the fluorescent rhodamine 123, it was shown that ROS accumulate in peroxiredoxin III–deficient mitochondria. Peroxiredoxin III appears to be associated with the regulation of mitochondria-mediated apoptosis. Whether peroxiredoxin III has any role in the normal regulation of hematopoiesis is unclear at this time.
Peroxiredoxins may be directly involved in the regulation of cell cycle. Peroxiredoxin I and II can be directly phosphorylated by several cyclin-dependent kinases including cdc2 kinase, and their activity is hence modulated (20, 96).
Peroxiredoxins I to IV (not V and VI) possess consensus sequences (Ser/Thr)-Pro-Xaa-(Lys/Arg) for phosphorylation by cyclin-dependent kinases (CDKs) CDK2, 4, and 6, and cdc2. Cdc2/Cyclin B control the mitosis; Cdc2 is high in the G2/M phase of the cell cycle, but not present in G1/S. Cdc2/Cyclin B phosphorylation of peroxiredoxin I dramatically reduces its peroxidase activity (to 20% of the nonphosphorylated form) (20). These results may suggest a transient increase in ROS during mitosis. A potential target of signaling is Cdc25C, a dual-specificity phosphatase that is a key activator of Cdc2/Cyclin B during mitosis. The active-site cysteines of protein tyrosine phosphatases (His-Cys-Xaa-Xaa-Xaa-Xaa-Xaa-Arg) are sensitive to oxidation by hydrogen peroxide because their ionization is promoted by histidine and arginine residues. It is thus proposed that as the mitosis progresses and the nuclear envelope breaks down, cdc2, activated at the G2/M phase, phosphorylates peroxiredoxin I and II, resulting in inactivation of peroxiredoxins. Repression of peroxiredoxin activity during mitosis leads to ROS build-up, which induces inactivation of Cdc25, which in turn halts the positive-feedback loop formed by Cdc25 and cdc2 (20). Stepwise phosphorylation and inactivation of peroxiredoxins and subsequent ROS accumulation may provide a system to balance ROS impact on the cell cycle.
Hemoglobin-stabilizing Protein (AHSP)
Hemoglobin A is a tetramer that consists of two pairs of α and β globin protein subunits, with each monomer bound to a heme moiety. The synthesis of hemoglobin A is highly coordinated to minimize the accumulation of free α or β hemoglobin that are toxic to the cell. In β-thalassemia, a common inherited anemia in which mutations in the β globin result in impaired production of β hemoglobin, excessive accumulation of unpaired α globin is particularly damaging. AHSP is a specific molecular chaperone for α hemoglobin that maintains α hemoglobin in a stable state before its incorporation into hemoglobin A. AHSP-ablated mice exhibit erythrocytes with abnormal morphology that contain denatured hemoglobins (59). These, combined with studies of AHSP crystal structure, strongly suggest that AHSP is required for normal erythropoiesis by stabilizing free α hemoglobin, therefore blocking the deleterious effects of free α hemoglobin precipitation (32, 59). Loss of AHSP leads to hemolytic anemia with globin-chain precipitates [reviewed in (120)], increased ROS accumulation, and signs of oxidative damage in AHSP−/− erythrocytes (61). Although the anemia was compensated for with increased production of erythroid precursors, the transition from immature to mature erythrocytes was partially blocked with increased apoptosis in erythroid-precursor cells.
Whether increased ROS affects the erythroid precursor in AHSP−/− mice is not known, but it is likely. Importantly, loss of AHSP exacerbates β-thalassemia, suggesting a genetic interaction between the two (61). These studies further illustrate the damaging effects of ROS on erythroid cells. They may also provide a model for analysis of the impact of ROS on erythroid cell maturation (Fig. 2).
GATA-1 Transcription Factor
GATA-1 zinc-finger transcription factor is the master regulator of erythroid gene expression. Many, if not all, erythroid genes possess a consensus GATA-1 binding motif in their regulatory regions, and these genes require GATA-1 for their expression. Although GATA-1 has not been directly implicated in the regulation of erythroid cell oxidative stress, transgenic mice overexpressing GATA-1 mutant lacking its N-finger domain exhibit a hemolytic syndrome that is highly sensitive to oxidative stress in erythroid cells (85). Despite GATA-1 involvement in the regulation of P45NF-E2, expression of P45NFE2 was normal in the spleen or bone marrow of GATA-1− mice (85). Whether the GATA-1 N-finger domain is involved in the regulation of erythroid oxidative stress is not known. It is noteworthy that DNA binding of many transcription factors, including zinc-finger proteins, is modulated when critical cysteine residues are oxidized or alkalized (125). Whether the DNA binding of erythroid transcription factors such as GATA-1 is subject to redox regulation is not clear.
Summary, Part I
In brief, altered regulation of oxidative stress has deleterious affects on mature erythroid cells. Circulating red blood cells respond rapidly to environmental stress, such as hypoxia, dehydration, or oxidative stress. The unique structure of red blood cell membrane composed of ubiquitously expressed and erythroid-specific proteins allows these cells to withstand the shear stress and pass through capillary vessels. Alteration of protein structures and/or lipid or glucose metabolism of red blood cells leads to hemolysis and anemia.
Studies summarized here show that accumulation of ROS in erythroid cells often results in shortened red blood cell life span and hemolysis that may or may not be associated with anemias. Increased proliferation and differentiation of erythroid progenitors ensure compensation for premature loss of erythroid cells. Findings discussed here further support the notion that deficiency in oxidative-stress regulation leads to hemolysis in genetic disorders such as deficiency in glucose 6-phosphatede hydrogenase (G6PD). G6PD is an enzyme that converts NADP to NADPH. NADPH maintenance of reduced glutathione (GSH) is required for ROS detoxification; thus, deficiency in G6PD leads to destruction of red blood cells and anemia.
In addition, ROS may coordinate erythroid cell precursor cycle and differentiation, suggesting that mechanisms of ROS production and detoxification may be of major importance in the regulation of the erythroid cell rate of maturation. This is particularly important in hemoglobinopathies and other diseases of red blood cells in which compensatory erythropoiesis is the key to the prognosis of the patient.
Part II
FoxO Transcription Factors and the Regulation of Oxidative Stress in Hematopoiesis
The FoxO family of transcription factors was recently shown to have essential functions in the regulation of oxidative stress in hematopoietic stem and erythroid cells (75, 81, 113). FoxO1, FoxO3, FoxO4, and FoxO6 belong to the forkhead family of winged helix transcription factors and are the mammalian homologues of DAF-16 [abnormal DAuer Formation-16, reviewed in (38)], which regulates the life span downstream of insulin-receptor DAF-2 in Caenorhabditis elegans. Activation of DAF-16 (Fig. 3) results in a significant increase in C. elegans lifespan, partly through its mediation of defense against oxidative stress (69, 72). Likewise, dFOXO regulates oxidative stress in Drosophila (55).
FIG. 3.
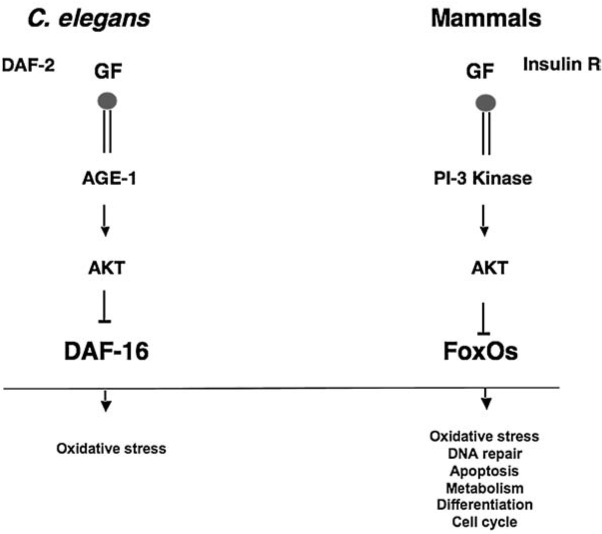
FoxO role in the regulation of oxidative stress is evolutionarily conserved. FoxO are the mammalian homologues of DAF-16 in Caenorhabditis elegans. These transcription factors are regulated by the highly conserved IGF-1 (Insulin)/PI3-kinase/AKT signaling pathway that suppress the activity of FoxO. Activation of DAF-16 enhances the worm's life span in part through resistance to oxidative stress. In mammals, active FoxO plays key roles in the regulation of transcription of several antioxidant enzymes, as well as the cell cycle, DNA repair, apoptosis, and cell-differentiation proteins.
Whereas FoxO6 appears to be specific to neuronal tissues, FoxO1, FoxO3, and FoxO4 are ubiquitously expressed. Similar to DAF-16, activation of FoxO3 in cultured fibroblasts and neuronal cells (62, 86), as well as in primary mouse hematopoietic stem and erythroid cells (75, 127) results in resistance to oxidative stress. This FoxO3 effect is mediated at least partly via upregulated transcription of the ROS-scavenging enzymes SOD2 and catalase (62, 86). In particular, FoxO3 binds directly in vivo to SOD2 promoter in quiescent cultured human colon carcinoma cells, as demonstrated by the chromatin immunoprecipitation (ChIP) assay (62). Regulation of SOD2 in cycling cultured cells may, however, be mediated by NF-κB and not by FoxO3 (108), suggesting that in proliferating cells, FoxO3 may not mediate the transcriptional regulation of SOD2.
FoxO3 responds to cellular stress (including oxidative stress) by inducing cell-cycle arrest, repair of damaged DNA, and apoptosis via upregulation of genes that are important regulators of these processes (Fig. 3) (9, 26, 37, 77, 114). In this regard, FoxO3 has a function to that of p53 tumor-suppressor protein, with which it shares several downstream targets and mechanisms of regulation (129, 130, 133).
FoxO proteins are downstream targets of the PI3-kinase/AKT signaling pathway (38). The PI3-kinase-AKT signaling pathway is highly conserved from worms to mammals (Fig. 3). FoxO transcription factors are direct substrates of AKT serine threonine kinase in response to cellular stimuli such as growth factors (like insulin) and oncoproteins (Fig. 4). The binding of growth factors to the receptor tyrosine kinases stimulates recruitment and activation of PI3-kinase, which in turn activates the AKT/SGK family of serine threonine kinases among several other kinases.
FIG. 4.
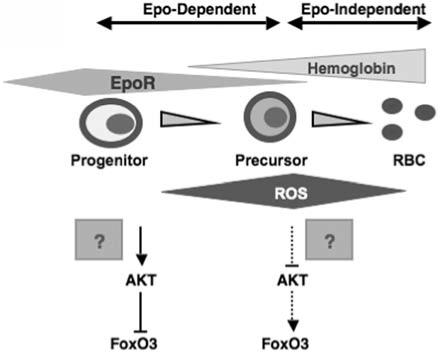
Model for the regulation of FoxO3 in erythroid cells. As postprogenitor erythroid precursors differentiate, hemoglobin is synthesized, and the expression of EpoR is decreased. AKT is active in erythroid progenitors, where it inhibits FoxO3. In erythroid precursors, downregulation of AKT signaling as a result of reduced expression of EpoR, coupled with accumulation of hemoglobin, produces ROS and activates nuclear localization of FoxO3. FoxO3 nuclear activity may coordinate the cell cycle and maturation by modulating ROS accumulation in erythroid precursor cells.
Phosphorylated FoxO binds to 14-3-3 adaptor proteins and translocates to the cytosol, where, away from their transcriptional targets, it is unable to induce gene expression (9). Conversely, stress stimuli or inhibition of the PI3-kinase/AKT by cytokine withdrawal results in nuclear localization and activation of FoxO (38). At least in fibroblasts, ROS activate Ral small GTPase that mediates JNK-dependent phosphorylation of FoxO on residues distinct from the ones phosphorylated by AKT.
The JNK-dependent phosphorylation of FoxO results in its nuclear localization and activation (31). This function of JNK is highly conserved among species. Importantly, by regulating FoxO, JNK antagonizes stress, including ROS-induced stress, to extend the life span in Drosophila and C. elegans (90, 118, 119). It is not known whether JNK regulates FoxO in hematopoietic cells. Oxidative stress–regulated mammalian sterile 20-like kinase 1 (MST-1) binds and phosphorylates directly FoxO and enhances FoxO's activity (69). This phosphorylation may be mediated by JNK. Similarly oxidative stress enhances FoxO monoubiquitination (117), association with β-catenin (30), and Sir2/SIRT1 deacetylase (10), all of which enhance FoxO's activity in some respects. It remains to be shown whether these FoxO regulations occur in hematopoietic cells and whether they similarly affect hematopoiesis.
Expression of FoxO3 increases significantly during primary erythroid cell maturation (75). As primary erythroid progenitor cells mature, FoxO3 translocates to the nucleus and becomes transcriptionally active, suggesting that FoxO3 may play a role in the regulation of erythroid cell maturation. Indeed, loss of FoxO3 resulted in significant ROS accumulation in erythrocytes, signs of oxidative damage like erythrocyte protein oxidation, and ROS-mediated reduced life span of circulating erythrocytes (75).
Specifically, in vivo treatment of FoxO3−/− mice with N-acetylcysteine, a precursor of glutathione and a generic ROS scavenger, improved significantly the FoxO3−/− red blood cell life span (75). These studies showed that FoxO3-null mice exhibit an oxidative stress–mediated compensated chronic hemolysis (75). FoxO3 is the first identified EpoR-regulated transcription factor that is critical for regulating oxidative stress in in vivo erythropoiesis.
In addition to oxidative damage in mature enucleated red blood cells, loss of FoxO3 leads to a decreased rate of erythroid cell maturation that is likely to be mediated by oxidative stress (75). The FoxO3-null erythroid precursor cells showed a mitotic arrest in G1 that was induced by the antioxidant activation of p53 and upregulation of CDK inhibitor p21CIP1/WAF1/Sdi1 (75). Similar types of increase in the size of the erythroid precursor cell compartment, as in FoxO3-null mice, are observed in murine models of β-thalassemia in which oxidative stress may play a role (25). These data predict (a) a model in which production of ROS as a result of hemoglobin accumulation coupled with downregulation of EpoR signaling, including the repressive effect of AKT on FoxO3 activity, results in nuclear translocation and activation of FoxO3 (75) (Figs. 2 and 4); (b) FoxO3 nuclear activity coordinates cell cycle and maturation by modulating ROS accumulation in erythroid precursor cells (40, 75) (Fig. 5). Based on these Findings, we anticipate a potential function for FoxO3 in the deregulated erythropoiesis of β-thalassemia and/or in other hemoglobinopathies and diseases of red blood cells.
FIG. 5.
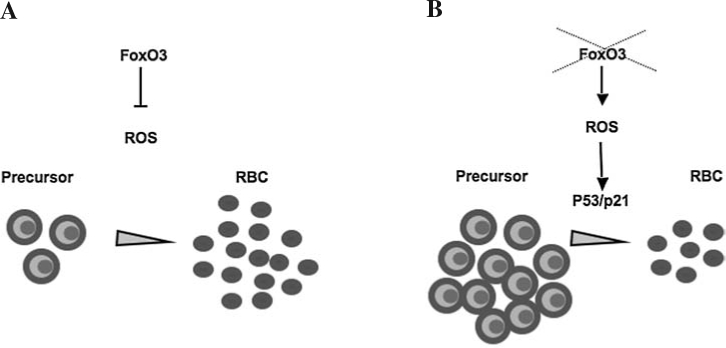
FoxO3 regulation of ROS coordinates the erythroid cell cycle and differentiation and determines the rate of erythroid cell maturation. (A) Nuclear FoxO3 activity in erythroid precursors represses ROS under normal conditions to coordinate the erythroid cell cycle and maturation. (B) In the absence of FoxO3, ROS accumulate in erythroid precursors, leading to the activation of p53/p21CIP1/WAF1/Sdi1, resulting in arrest of precursors in G1, reduced mature cells, and overproduction of precursors as a result of progenitor activity.
The function of FoxO3 in repressing oxidative stress is possibly mediated by its transcriptional regulation of antioxidant enzymes SOD2, catalase, glutathione, peroxidase-1 (GPX-1), and SOD1 in erythroid precursor cells (75). Evidence suggests that FoxO3 may directly regulate the expression of SOD2 and catalase during primary erythropoiesis: (a) endogenous FoxO activates SOD2 and catalase reporters containing FoxO binding sites during maturation of primary fetal liver erythroid cells; and (b) expression of SOD2 and catalase decreases significantly in FoxO3-null bone marrow erythroid cells (127). In addition to SOD2 and catalase enzymes, expression of glutathione peroxidase-1 and SOD1 also is suppressed in FoxO3-null primary bone marrow erythroid cells, suggesting that they may also be regulated by FoxO3 in these cells (127). Whether SOD1 and GPX-1 are direct targets of FoxO3 remains to be investigated.
Furthermore, expression of ataxia telangiectasia mutated (ATM) is highly suppressed in FoxO3-null erythroid cells (75). ATM is implicated in the regulation of oxidative stress but has not been known to play a function in erythropoiesis. Whether ATM regulates oxidative stress downstream of FoxO3 in erythropoiesis requires investigation.
The importance of FoxO3 regulation of oxidative stress in erythropoiesis is specifically highlighted in experiments in which treatment of FoxO3−/− mice with phenylhydrazine, which oxidizes hemoglobin, results in the death of FoxO3−/− mice at doses at which the wild-type counterparts survive (75). These results suggest that the death induced by phenylhydrazine treatment in FoxO3-deficient mice is likely the result of several combined mechanisms: (a) an excessive ROS-mediated destruction of circulating erythrocytes coupled with (b) a significant ROS-induced mitotic arrest, leading to (c) a significantly decreased number of mature FoxO3-null erythroid cells that together impede a timely hematopoietic response.
Summary, Part II
A common feature of loss of many of the erythroid antioxidant molecules or enzymes is the development of hemolytic anemia as a result of lack of antioxidant protection. These studies illustrate that unbalanced accumulation of ROS limits the life span of mature red blood cells. Although the direct causal effect of FoxO regulation of oxidative stress and mammalian organismal life span remains to be established, it is clear that the erythroid cell life span is controlled by FoxO3 regulation of oxidative stress. In addition, findings from the FoxO3-null mice suggest that oxidative stress leads to the deregulated coordination of cell cycle and maturation in bone marrow erythroid-precursor cells, which may in turn play a significant role in the regulation of stress erythropoiesis in a disease state. Oxidative stress–mediated regulation of the cell cycle and maturation in FoxO3-null erythroid cell maturation programs raise the question as to whether similar alterations in coordination of cell cycle/maturation occur in peroxiredoxin I, II, or SOD2 mutant models of erythroid cell differentiation or in hemoglobinopathies and other diseases of red blood cells in which ROS play a role.
Part III
Regulation of Oxidative Stress in Hematopoietic Stem Cells
Hematopoietic stem cells are mostly dormant, cytokine resistant, and self-renew seldom to produce differentiated cells. Hematopoietic stem cells have a low metabolic activity that spares them the damage of metabolic products such as ROS and DNA replication. However, increased ROS may result in accumulation of DNA damage and unscheduled activation of senescence mechanisms in the stem cell compartment in the long term. It is becoming increasingly clear that deregulated accumulation of ROS in hematopoietic stem cells leads to abnormal hematopoiesis (81, 113, 127). Thus, tight regulation of oxidative stress in hematopoietic stem cells is essential for normal control of homeostasis in hematopoietic tissues.
ATM Regulation of ROS in Hematopoietic Stem Cells
Loss-of-function mutations in the ataxia/telangiectasia mutated gene (ATM) results in a clinical phenotype characterized by ataxia, telangiectasia, neuronal degeneration, immunodeficiencies, genomic instability, predisposition to lymphomas and other malignancies, and extreme sensitivity to ionizing radiation (4, 56). Whereas ROS are found accumulated in several cell types and tissues and antioxidant enzymes are deregulated in ATM-mutant cells, the mechanism of ATM regulation of ROS is not clear (5, 56, 92). It is noteworthy that overexpression of ATM restores the normal levels of antioxidant transcripts in FoxO3-null primitive hematopoietic cells in which these transcripts are significantly reduced (127).
ATM is essential for the regulation of genomic stability. ATM serine threonine protein kinase is a critical enzyme in the regulation of stress response to DNA damage, specifically in double-strand DNA-break repair (5). Studies by Suda and colleagues (49) showed recently that regulation of ROS by ATM is essential for hematopoietic stem cell self-renewal, because loss of ATM results in ROS-mediated depletion of the hematopoietic stem cell pool, leading to bone marrow failure in old mice. Accumulation of ROS in ATM-null hematopoietic stem cells from old mice leads to upregulation of p16INK4a tumor suppressor and preferential activation of retinoblastoma protein, resulting in suppression of hematopoietic stem cell activity (49). Treatment with antioxidant reagent N-acetylcysteine (NAC) or with an MAPK inhibitor restores the quiescence and reconstitution capacity of ATM-null hematopoietic stem cells (50). Importantly, these findings provided the first evidence for a role for ROS in the regulation of hematopoietic stem cell activity.
FoxO Regulation of ROS in Hematopoietic Stem Cells
Under certain harsh environmental conditions, such as when food is scarce or the surrounding is too crowded, DAF-16 is activated to promote dauer formation (diapause) in C. elegans (72, 89). The dauer, which is in juvenile form and reproductively immature, is a reversible condition in which the metabolic rate is highly reduced and the life span is markedly enhanced. Disruptions of the C. elegans DAF-2/Age-1/AKT1/2 (homologous to insulin–insulin-like growth factor (IGF) receptor/PI3-kinase/AKT) signaling pathway leads to constitutive entry into dauer stage and extends markedly the worm's life span (67, 71–73, 83, 89, 91). These phenotypes are abolished by DAF-16 mutation, indicating that DAF-16 is the downstream effector of this pathway. DAF-16 regulation of longevity is mediated at least partly by its control of genes involved in stress resistance, including oxidative stress (67, 69, 82, 83, 91, 123). In many respects, the characteristics of the dauer are reminiscent of the “quiescent” state of hematopoietic stem cells. Quiescence is a major stem-cell attribute that strikingly resembles the dauer phase, in that it ensures the lifelong preservation of the stem cell pool. Thus, we postulated that FoxO transcription factors play an important role in the regulation of stem cells and asked whether, as with DAF-16 in the dauer formation, FoxO3 protects quiescence of hematopoietic stem cells by regulating oxidative stress. Indeed, recent findings demonstrate that FoxO (113), and, in particular, FoxO3 regulation of oxidative stress (127), is essential for maintenance of hematopoietic stem cell homeostasis (81, 127).
Like ATM, FoxO transcription factors are mediators of the antioxidant response in hematopoietic stem cells (113). Loss of three FoxO (FoxO1, FoxO3, and FoxO4) led to defective hematopoietic stem cell numbers and activity that was associated with increased accumulation of ROS, as measured by DCF-DA assay (113).
Although FoxO-null mice also displayed myeloproliferation, the relative increased accumulation of ROS was limited to the hematopoietic stem cell compartment and was not observed in myeloid progenitors (113). FoxO-null hematopoietic stem cells exit quiescence and enter the cell cycle, suggesting that ROS enhance cell-cycle entry and loss of quiescence in hematopoietic stem cells (113). Taken together, these studies suggest a critical role for FoxO transcription factors in the repression of ROS and regulation of hematopoietic stem cell quiescence.
Among the FoxOs, FoxO3 appears to play the principal role in the regulation of the hematopoietic stem cell pool (81, 127). In the hematopoietic system, FoxO3 is the most highly expressed FoxO in the bone marrow (75) and is the principal active (nuclear) FoxO in hematopoietic stem cells (81, 127). Although both FoxO1 and FoxO3 are expressed in hematopoietic stem cells (FoxO4 is barely detectable in HSC), FoxO3 is almost entirely nuclear, suggesting that FoxO3 is the principal active FoxO in these cells (127, 128). Several antioxidant enzymes, in particular SOD2, catalase (and glutathione peroxidase-1), and cell-cycle regulators that are direct targets of FoxO, are significantly and profoundly down-regulated in FoxO3-deficient hematopoietic stem cells, and their expression is restored by reintroduction of FoxO3 in FoxO3-null primitive hematopoietic cells (81, 127), suggesting that other FoxOs do not fully complement loss of FoxO3 in the regulation of cell-cycle and antioxidant genes in hematopoietic stem cells. In addition, loss of FoxO3 leads to significant defects in hematopoietic stem cell frequency and activity in young and old mice (possibly depending on the mouse strain) in a ROS-sensitive manner (81, 127). These combined findings suggest that FoxO3 is critical for the regulation of oxidative stress in hematopoietic stem cells, and FoxO3 modulation of oxidative stress mediates its regulation of hematopoietic stem cell activity (127).
In contrast to the triple deletion of FoxO (113), single FoxO3 deletion results in loss of quiescence (81, 127) associated with arrest of hematopoietic stem cells in the G2/M phase of the cell cycle (127). The activity of primitive FoxO3-null hematopoietic stem cells was restored with in vitro antioxidant NAC treatment (127). Similarly, NAC treatment corrected the number of multipotential colony-forming unit spleen (CFU-S) cells in FoxO3−/− mice in vivo (Yalcin and Ghaffari, unpublished data). The G2/M arrest in FoxO3-null hematopoietic stem cells is associated with marked ROS-mediated upregulation of p53 and p21CIP1/WAF1/Sdi1 and ROS-independent modulation of several genes involved in the regulation of the cell cycle, such as cyclin B (127). The exact mechanism of G2/M arrest in FoxO3−/− hematopoietic stem cells remains to be established (127). The p53 activation in FoxO3−/− hematopoietic stem cells resulted in up-regulation of sestrin 2 (SESN2) and GADD45. These findings are especially interesting because GADD45 is also positively regulated by FoxO3 (114). Nonetheless, enhanced expression of SESN2 an antioxidant molecule involved in regeneration of peroxiredoxins, and the DNA-repair transcript for GADD45, both transcriptional targets of p53 (58, 100), suggest that mechanisms of protection from ROS-mediated DNA damage are stimulated via p53 in FoxO3−/− hematopoietic stem cells. These findings provide an insight into the cooperation of p53 and FoxO (133) in primary cells in vivo.
These results highlight the essential function of regulation of oxidative stress in the maintenance of the hematopoietic stem cell pool and quiescence. In addition, they underline the critical role that both ATM and FoxO, specifically FoxO3, play in these processes (Fig. 6). FoxO3-deficient hematopoietic stem cells exhibit a phenotype less pronounced but similar to that of hematopoietic stem cells with deficiency in phosphatase and tensin homology (PTEN) that is a negative regulator of PI3-kinase/AKT (132, 135). However, whether oxidative stress has any impact on PTEN-null hematopoiesis is not clear.
FIG. 6.
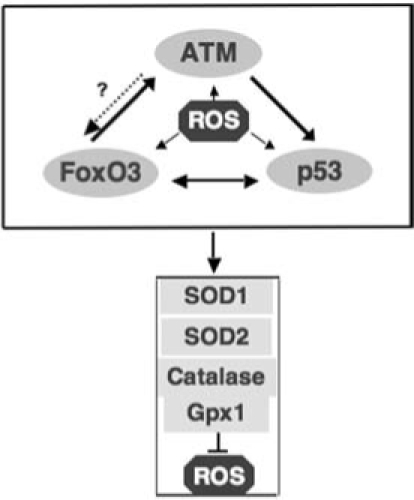
ATM/FoxO3/p53 axis of ROS control in hematopoietic stem cells. ROS activate ATM, FoxO3, p53 tumor suppressors, and each independently or in a cascade induces the expression of antioxidant enzymes to repress ROS.
FoxO3 and ATM in the Regulation of ROS in Hematopoietic Stem Cells
As discussed earlier, both ATM serine threonine kinase and FoxO3 are essential for the regulation of oxidative stress in hematopoietic stem cells. ATM is a critical regulator of the stress response, ROS-induced DNA damage (5), and hematopoietic stem cell self-renewal (49). ATM activates p53 via multiple pathways including direct phosphorylation. Thus, we were surprised to find that in FoxO3-null hematopoietic stem cells, in which p53 seems to be activated, both ATM transcript and protein (phosphorylated active Ser 1981 ATM and nonphosphorylated) expression were significantly reduced (127). The downregulation of ATM expression in FoxO3-null hematopoietic stem cells was ROS independent (127). Retroviral introduction of FoxO3 in FoxO3-deficient primitive hematopoietic cells rescued the expression of ATM; similarly, shRNA knockdown of FoxO3 in NIH 3T3 cells resulted in significant downregulation of ATM transcript and protein expression, supporting the view that FoxO3 is required for the regulation of optimal ATM expression. Importantly, ATM is sufficient to regulate expression of antioxidant enzymes in FoxO3-null hematopoietic stem cells (127). In agreement with these results, both ATM and FoxO3 rescued the expression of antioxidant enzymes in FoxO3-null hematopoietic stem cells and in ATM-null MEFs, suggesting that FoxO3 and ATM may regulate these enzymes independently. Further experiments are required to determine the genetic interaction of FoxO3 and ATM in the regulation of oxidative stress–mediated hematopoietic stem cell activity. In addition to the regulation of ATM expression, recent studies demonstrate that FoxO3 directly interacts with ATM and is critical for enhanced ATM phosphorylation and activation in response to DNA damage (115). Thus, given the key functions of both ATM and FoxO3 in the regulation of oxidative stress and DNA damage, it is conceivable that FoxO3 and ATM play a critical role in the regulation of oxidative stress–mediated DNA damage response.
These combined findings suggest a model in which FoxO3, together with ATM and p53, provides a strong tumor-suppressor network in protecting stem cells from damage that may undermine their genomic stability and result in their clonal expansion (model, Fig. 7). Sustained activation of these pathways may alternatively result in senescence, leading to the defects seen in FoxO3-null hematopoietic stem cells derived from aged mice (81).
FIG. 7.
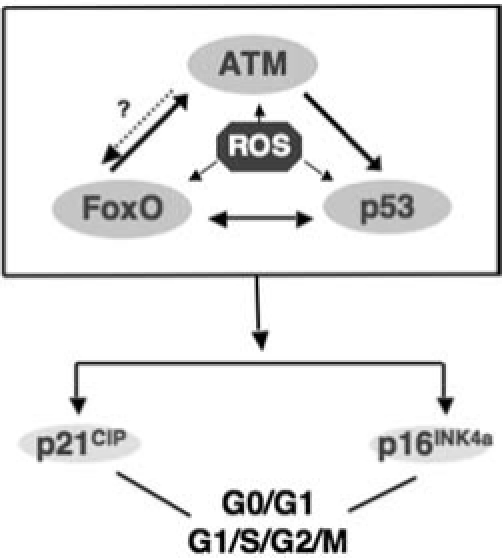
ROS activation of tumor suppressors in hematopoietic stem cells leads to senescence or genomic instability. Accumulation of ROS in hematopoietic stem cells activates tumor-suppressor pathways, presumably in response to damage, and in particular to DNA damage. This activation leads to senescence of hematopoietic stem cells. Cells that escape senescence may accumulate, increasing damage and perhaps mutations that undermine their genomic stability and provide them with a proliferative advantage leading to their clonal expansion.
Oxidative stress enhances the FoxO association with β-catenin, the principal effector of the Wnt signaling pathway (30). The FoxO/β-catenin association is critical for FoxO regulation of resistance to oxidative damage (30). Given the critical role of Wnt signaling in hematopoietic stem cell self-renewal (95), these findings raise the question as to whether FoxO play a role downstream of Wnt signaling in hematopoietic stem cells.
Oxidative stress also enhances the direct interaction of FoxO with Sir2/SIRT1, an NAD-dependent histone deacetylase that modulates the life span in various species (10, 74). It is noteworthy that in response to oxidative stress, Sir2/SIRT1 interacts directly with FoxO in cultured cells and enhances its resistance to oxidative stress while limiting its ability to induce apoptosis (10). Whether Sir2/SIRT1 regulation of FoxO has any function in modulating hematopoietic stem cell activity or aging or both is not known. Future elucidation of the mechanisms of regulation of FoxO3 in hematopoietic stem cells, in particular, potential interactions of FoxO3 with coactivators and corepressors, as well as identification of FoxO3 targets in stem cells, will be of fundamental importance for better understanding of aging of hematopoietic stem cells and malignant transformation in cancer stem cells.
ROS and the Hematopoietic Stem Cell Niche
Recent studies suggest that long-term repopulating cells reside in close proximity to the endosteal surface of the bone. Bone is thought to be a site of low oxygen tension, and the bone marrow niche where the hematopoietic stem cells are thought to reside is believed to be hypoxic (2). Because hypoxia maintains low levels of ROS accumulation, and generation of ROS is essential for activation of hypoxic response (8, 39) through stimulation of p38 MAPK (29), it is thought that ROS may be directly involved in the regulation of hematopoietic stem-cell niche interactions. It is conceivable that gradients of ROS may affect elements of the niche structure such as N-cadherin (44) and thus regulate the hematopoietic stem cell microenvironment in addition to ROS intrinsic effect on hematopoietic stem cells.
ROS, DNA Damage, and Hematopoietic Stem Cell Aging
ROS are generated throughout life and cause damage to macromolecules including DNA, proteins, and lipids (6). The rate of ROS production increases with age, partly because of age-associated alterations of the electron-transport chain. The replicative capacity and function of hematopoietic stem cells is reduced with age (21, 22, 98). In addition, hematopoietic stem cells reveal changes in homing, mobilization, and lineage choice (less lymphoid and more myeloid transcripts) with age (97, 98). At least some of these features are likely to be promoted by ROS accumulation in hematopoietic stem cells over time. In particular, loss of DNA-repair mechanisms in hematopoietic stem cells (97) may increase the sensitivity to harmful effects of ROS with age. Alternatively, ROS may contribute to alteration of expression or malfunction of molecules involved in DNA repair. Clues as to the deleterious impact of ROS in the alteration of hematopoietic stem cell function initially came from elegant studies by Suda and colleagues (49) carried in ATM-null mice. Accumulation of ROS in ATM−/− hematopoietic stem cells from old mice resulted in a major compromise of their self-renewal potential, leading to bone marrow failure in 24-week-old mice (49).
Like ATM, loss of FoxO3 leads to a marked decrease of hematopoietic stem cell reconstitution ability with age (81, 127). It is thus likely that the relative reduced function of ATM that is observed in FoxO3-null hematopoietic stem cells (127) mediates ROS accumulation, leading to the functional decline that is observed in these cells with age (81, 127). Both ATM and FoxO3 can induce the expression of antioxidant enzymes in FoxO3-null hematopoietic stem cells (127). However, it is likely that other signaling pathways will also contribute to the alterations of FoxO3-null hematopoietic stem cell activity. For instance, in ATM-null hematopoietic stem cells derived from aged mice, p16INK4a tumor suppressor is upregulated through a ROS-dependent mechanism, whereas upregulation of p16INK4a observed in FoxO3-null hematopoietic stem cells derived from young mice is ROS independent (49, 127). These results suggest that downregulation of ATM independent of ROS may mediate upregulation of p16INK4a in FoxO3-null hematopoietic stem cells. Upregulation of p16INK4a leads to the activation of retinoblastoma protein and cell-cycle arrest and is associated with hematopoietic stem cell aging (53). In addition to up-regulation of p16INK4a, the antioxidant activation of p53 tumor suppressor may also participate in mediating the functional decline with age in FoxO3-null hematopoietic stem cells. However, it is not known whether potential p53 regulation of hematopoietic stem cells is mediated by ROS (28). In addition, it is not clear whether deficiencies in DNA damage response also contribute to the phenotypes of ATM- or FoxO3-null hematopoietic stem cells (49, 115, 127).
Together these findings suggest that in hematopoietic stem cells, as in many other systems, ROS accumulation leads to the activation of tumor-suppressor pathways, possibly to offset the likelihood of damaged or mutated DNA being passed on to their progenies. Given FoxO and ATM involvement in response to and repair of DNA damage, it is highly likely that signaling pathways involving FoxO/ATM will be critical in the regulation of aging of hematopoietic stem cells. In response to ROS, the activated tumor-suppressor network may promote either cell-cycle arrest, as seen in FoxO3-null hematopoietic stem cells, (127) resulting in senescence that leads to defects of the hematopoietic stem cell compartment in aging mice (81), or apoptotic programs in hematopoietic stem cells. Alternatively, cells harboring an unrepaired genetic mutation that offers a growth or survival advantage may be positively selected and produce a mutant stem cell clone that leads to malignancies of blood cells. Thus, activation of tumor-suppressor networks in response to ROS accumulation may contribute to aging of hematopoietic stem cells.
ROS in Cellular Signaling of Normal and Neoplastic Hematopoiesis
Several lines of evidence support the notion that ROS directly or indirectly enhance cellular signaling [reviewed in (112)]. Stimulation of several growth-factor receptors, with ligands such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF), is associated with a transient increase in ROS production (3a, 84, 110).
Increase in growth factor–induced cell growth is also sustained by ROS inhibition of phosphatases that are involved in the repression of propagation of growth-factor receptor signaling, such as phosphatase and tensin homology (PTEN), protein tyrosine phosphatase 1B (PTP1B), and CDC25 (79, 80, 103, 104). Similarly, activation of oncogenes, such as Ras or c-Myc, is associated with increased hydrogen peroxide production (46, 111, 116). Unlike superoxide, hydrogen peroxide is membrane permeable, diffusible, and long lived. However, hydrogen peroxide is a weaker oxidant than superoxide. Hydrogen peroxide affects cellular signaling through protein modification, such as intramolecular disulfide bridge, sulphenyl-amide bond formation, direct activation of tyrosine kinases by Cys oxidation, or by inhibition of phosphatases [reviewed in (112)]. In turn, the catalytic activity of antioxidant enzymes such as peroxiredoxins, catalase, and glutathione peroxidase is modified by signaling molecules [reviewed in (96)], suggesting a dynamic balance between cellular signaling and regulation of oxidative stress.
Hematopoietic cytokines, such as IL-3, GM-CSF, G-CSF, Steel factor, and Thrombopoietin, induce cellular signaling through generation of hydrogen peroxide in hematopoietic cells (45, 102, 136). Expression of BCR-ABL oncoprotein in cultured hematopoietic cells is also associated with production of ROS. BCR-ABL–induced ROS accumulation results in inhibition of tyrosine phosphatases (60, 101), presumably leading to increased cell proliferation. These findings suggest that putative ROS-induced inhibition of phosphatases may contribute to leukemogenesis. In addition, ROS accumulation has been implicated in the mutagenesis of BCR-ABL oncoprotein, a mechanism through which BCR-ABL induces repression of the activity of imatinib tyrosine kinase inhibitor and therefore provides resistance to leukemia treatment (63).
ROS may also be involved in the cytotoxic effect of adaphostin, another molecule targeting BCR-ABL (18). These findings raise the possibility that in myelopoliferative diseases such as in chronic myeloid leukemia or polycythemia vera, in which an increase in the hematopoietic progenitor compartment and a high sensitivity to cytokines are observed, ROS may be involved. In agreement with this contention, we find enhanced myeloproliferation (a preleukemic condition) observed in FoxO3−/− mice (126) to be dependent on ROS that amplifies cytokine signaling and mediates increased proliferation (Marinkovic and Ghaffari; Yalcin and Ghaffari; Mungamuri and Ghaffari, unpublished data). Based on our unpublished findings (Yalcin and Ghaffari, unpublished observations), we propose a model in which the function of FoxO3 regulation of ROS is distinct in hematopoietic stem and progenitor cells (Fig. 8). These combined observations suggest that mechanisms regulating ROS may be exploited in the treatment of hematologic malignancies.
FIG. 8.
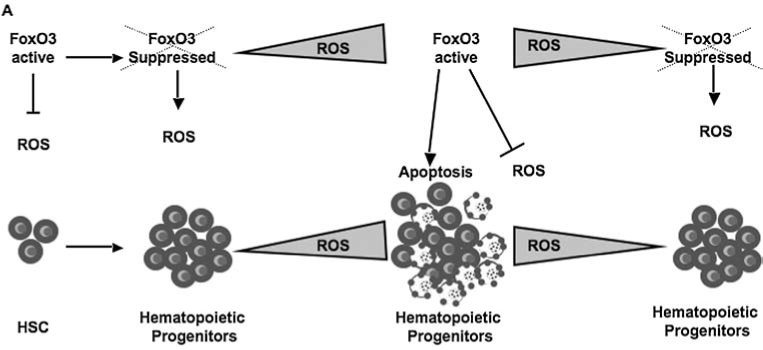
Modeling the function of FoxO3 in hematopoietic stem versus progenitor cells. The function of FoxO3 may be distinct in hematopoietic stem cells versus progenitor cells. In hematopoietic stem cells, FoxO3 is constitutively active to promote antioxidant resistance and quiescence. In hematopoietic progenitor cells that are subject to cytokines, ROS may play an active role in the regulation of cytokine-receptor signaling. In hematopoietic progenitors, FoxO3 is suppressed while ROS are under a certain threshold, above which, FoxO3 is activated to induce apoptosis. This model provides an intrinsic ROS-mediated dynamic balance between the degree of proliferation and survival of hematopoietic progenitors. Tight regulation of ROS via FoxO3 thwarts unlimited generation of hematopoietic progenitors.
Redox modulation of cysteine residues regulates DNA binding of several transcription factors, including p53, NF-κB, and AP-1 [reviewed in (112)]. Although a majority of these reports are not derived from experiments conducted in the hematopoietic system, they provide a potential extension of the number of mechanisms by which oxygen radicals regulate gene expression in normal and malignant hematopoiesis.
Although much remains to be learned about ROS regulation of hematopoietic cellular signaling, it is highly likely that ROS participate in the regulation of intensity and duration of cellular signaling. Analysis of gradients of ROS production suggests that stem cells are enriched within a population of hematopoietic cells that produce low concentrations of ROS (52). This population expresses high levels of calcium-sensing receptor (CaR) in ROS low population, calcium ions are required for homophilic interactions of N-cadherin on hematopoietic stem cells and osteoblasts. Thus, ROS modulations of N-cadherin may regulate the maintenance or mobilization of hematopoietic stem cells within the niche (44). ROS regulation of metalloproteases (ADMAS) (34, 124) may also contribute to leukemogenesis.
It is likely that ROS gradients contribute to the regulation of hematopoietic stem cell behavior: at low levels, ROS may modulate hypoxic response and microenvironment through, in part, alteration of expression or activity of adhesion molecules to maintain quiescence. At intermediate levels, ROS may affect the cell cycle and proliferation in part through direct modifications of molecules involved in these processes, and induce apoptosis by activating programmed cell death when accumulated at a high level. This provides an attractive model by postulating that modifications of ROS in response to intrinsic and extrinsic signals may adjust cellular behavior and regulate cell interactions with the surroundings.
Summary and Unresolved Issues
Recent studies demonstrate that control of ROS is essential for normal regulation of quiescence in hematopoietic stem cells and their cycling. FoxO3 and ATM play essential functions in the regulation of ROS in hematopoietic stem cells. In addition, ROS signaling may participate in the regulation of hematopoietic progenitor cell proliferation. Although exploration of the regulation and function of oxidative stress in hematopoietic stem cells has begun, many questions remain to be addressed. The source of ROS and metabolism of hematopoietic stem cells are not clear. ROS regulation of DNA damage in hematopoietic stem cells and progenitors and their relation to hematopoietic stem cell aging must be further explored. The relation between ROS in hematopoietic stem cells and within the niche and their cross-regulation of hypoxia remain to be examined. Molecular mechanisms of ROS control of the G0-G1 transition of hematopoietic stem cells are not known. In addition to p38 MAPK that has been implicated in the ROS response in hematopoietic stem cells, the signaling pathways that respond to ROS and regulate ROS remain to be mapped (is JNK involved?). ROS molecular targets in hematopoietic stem cells remain to be identified.
Acknowledgments
This work was supported by an American Cancer Society Research Scholarship (RSG LIB-110480), a Career Enhancement Award (K18 HL76510-01), Black Family Exploratory Research Award, Roche Foundation for Anemia Research (ROFAR) Award, and Mount Sinai School of Medicine Research Funds to S.G.
Abbreviations
AHSP, α-hemoglobin–stabilizing protein; ARE, antioxidant response element; ATM, ataxia telangiectasia mutated; CDK, cyclin-dependent kinase; EGF, epidermal growth factor; Epo, erythropoietin; FOXO, Forkhead Box O; GPX, glutathione peroxidase; G6PD, glucose 6-phosphate dehydrogenase; GSH, reduced glutathione; GSSG, oxidized glutathione; NAC, N-acetylcysteine; PDGF, platelet-derived growth factor; PTEN, phosphatase and tensin homology; PTP1B, protein tyrosine phosphatase 1B; ROS, reactive oxygen species; SESN2, sestrin 2; SOD, superoxide dismutase.
References
- 1.Andrews NC. Erdjument-Bromage H. Davidson MB. Tempst P. Orkin SH. Erythroid transcription factor NF-E2 is a haematopoietic-specific basic-leucine zipper protein. Nature. 1993;362:722–728. doi: 10.1038/362722a0. [DOI] [PubMed] [Google Scholar]
- 2.Arai F. Suda T. Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann N Y Acad Sci. 2007;1106:41–53. doi: 10.1196/annals.1392.005. [DOI] [PubMed] [Google Scholar]
- 3.Arthur JR. The glutathione peroxidases. Cell Mol Life Sci. 2000;57:1825–1835. doi: 10.1007/PL00000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3a.Bae YS. Kang SW. Seo MS. Baines IC. Tekle E. Chock PB. Rhee SG. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J Biol Chem. 1997;272:217–221. [PubMed] [Google Scholar]
- 4.Barlow C. Hirotsune S. Paylor R. Liyanage M. Eckhaus M. Collins F. Shiloh Y. Crawley JN. Ried T. Tagle D. Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 5.Barzilai A. Rotman G. Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair (Amst) 2002;1:3–25. doi: 10.1016/s1568-7864(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 6.Beckman KB. Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 7.Benz MH. Sa EJ. Pathobiology of the human erythrocyte and its hemoglobins. In: Livingstone C, editor. Hematology basic principles and practice. New York: Harcourt Brace; 2000. pp. 356–367. [Google Scholar]
- 8.Brunelle JK. Bell EL. Quesada NM. Vercauteren K. Tiranti V. Zeviani M. Scarpulla RC. Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Brunet A. Bonni A. Zigmond MJ. Lin MZ. Juo P. Hu LS. Anderson MJ. Arden KC. Blenis J. Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 10.Brunet A. Sweeney LB. Sturgill JF. Chua KF. Greer PL. Lin Y. Tran H. Ross SE. Mostoslavsky R. Cohen HY. Hu LS. Cheng HL. Jedrychowski MP. Gygi SP. Sinclair DA. Alt FW. Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 11.Carlsson LM. Jonsson J. Edlund T. Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci U S A. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan JY. Han XL. Kan YW. Cloning of Nrf1, an NF-E2-related transcription factor, by genetic selection in yeast. Proc Natl Acad Sci U S A. 1993;90:11371–11375. doi: 10.1073/pnas.90.23.11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan JY. Han XL. Kan YW. Isolation of cDNA encoding the human NF-E2 protein. Proc Natl Acad Sci U S A. 1993;90:11366–11370. doi: 10.1073/pnas.90.23.11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan JY. Kwong M. Lo M. Emerson R. Kuypers FA. Reduced oxidative stress response in red blood cells from p45NFE2-deficient mice. Blood. 2001;97:2151–2158. doi: 10.1182/blood.v97.7.2151. [DOI] [PubMed] [Google Scholar]
- 15.Chan JY. Kwong M. Lu R. Chang J. Wang B. Yen TS. Kan YW. Targeted disruption of the ubiquitous CNC-bZIP transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO J. 1998;17:1779–1787. doi: 10.1093/emboj/17.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan K. Han XD. Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan K. Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci U S A. 1999;96:12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandra J. Tracy J. Loegering D. Flatten K. Verstovsek S. Beran M. Gorre M. Estrov Z. Donato N. Talpaz M. Sawyers C. Bhalla K. Karp J. Sausville E. Kaufmann SH. Adaphostin-induced oxidative stress overcomes BCR/ABL mutation-dependent and independent imatinib resistance. Blood. 2006;107:2501–2506. doi: 10.1182/blood-2005-07-2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang TS. Cho CS. Park S. Yu S. Kang SW. Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 20.Chang TS. Jeong W. Choi SY. Yu S. Kang SW. Rhee SG. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J Biol Chem. 2002;277:25370–25376. doi: 10.1074/jbc.M110432200. [DOI] [PubMed] [Google Scholar]
- 21.Chen J. Astle CM. Harrison DE. Development and aging of primitive hematopoietic stem cells in BALB/cBy mice. Exp Hematol. 1999;27:928–935. doi: 10.1016/s0301-472x(99)00018-1. [DOI] [PubMed] [Google Scholar]
- 22.Chen J. Astle CM. Harrison DE. Genetic regulation of primitive hematopoietic stem cell senescence. Exp Hematol. 2000;28:442–450. doi: 10.1016/s0301-472x(99)00157-5. [DOI] [PubMed] [Google Scholar]
- 23.Chen L. Kwong M. Lu R. Ginzinger D. Lee C. Leung L. Chan JY. Nrf1 is critical for redox balance and survival of liver cells during development. Mol Cell Biol. 2003;23:4673–4686. doi: 10.1128/MCB.23.13.4673-4686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conger AD. Fairchild LM. Breakage of chromosomes by oxygen. Proc Natl Acad Sci U S A. 1952;38:289–299. doi: 10.1073/pnas.38.4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Franceschi L. Turrini F. Honczarenko M. Ayi K. Rivera A. Fleming MD. Law T. Mannu F. Kuypers FA. Bast A. van der Vijgh WJ. Brugnara C. In vivo reduction of erythrocyte oxidant stress in a murine model of beta-thalassemia. Haematologica. 2004;89:1287–1298. [PubMed] [Google Scholar]
- 26.Dijkers PF. Medema RH. Pals C. Banerji L. Thomas NS. Lam EW. Burgering BM. Raaijmakers JA. Lammers JW. Koenderman L. Coffer PJ. Forkhead transcription factor FKHRL1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 28.Dumble M. Moore L. Chambers SM. Geiger H. Van Zant G. Goodell MA. Donehower LA. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood. 2007;109:1736–1742. doi: 10.1182/blood-2006-03-010413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emerling BM. Platanias LC. Black E. Nebreda AR. Davis RJ. Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005;25:4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Essers MA. de Vries-Smits LM. Barker N. Polderman PE. Burgering BM. Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 31.Essers MA. Weijzen S. de Vries-Smits AM. Saarloos I. de Ruiter ND. Bos JL. Burgering BM. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng L. Gell DA. Zhou S. Gu L. Kong Y. Li J. Hu M. Yan N. Lee C. Rich AM. Armstrong RS. Lay PA. Gow AJ. Weiss MJ. Mackay JP. Shi Y. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell. 2004;119:629–640. doi: 10.1016/j.cell.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 33.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 34.Fischer OM. Hart S. Gschwind A. Prenzel N. Ullrich A. Oxidative and osmotic stress signaling in tumor cells is mediated by ADAM proteases and heparin-binding epidermal growth factor. Mol Cell Biol. 2004;24:5172–5183. doi: 10.1128/MCB.24.12.5172-5183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedman JS. Lopez MF. Fleming MD. Rivera A. Martin FM. Welsh ML. Boyd A. Doctrow SR. Burakoff SJ. SOD2-deficiency anemia: protein oxidation and altered protein expression reveal targets of damage, stress response, and antioxidant responsiveness. Blood. 2004;104:2565–2573. doi: 10.1182/blood-2003-11-3858. [DOI] [PubMed] [Google Scholar]
- 36.Friedman JS. Rebel VI. Derby R. Bell K. Huang TT. Kuypers FA. Epstein CJ. Burakoff SJ. Absence of mitochondrial superoxide dismutase results in a murine hemolytic anemia responsive to therapy with a catalytic antioxidant. J Exp Med. 2001;193:925–934. doi: 10.1084/jem.193.8.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghaffari S. Jagani Z. Kitidis C. Lodish HF. Khosravi-Far R. Cytokines and BCR-ABL mediate suppression of TRAIL-induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc Natl Acad Sci U S A. 2003;100:6523–6528. doi: 10.1073/pnas.0731871100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greer EL. Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 39.Guzy RD. Hoyos B. Robin E. Chen H. Liu L. Mansfield KD. Simon MC. Hammerling U. Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Hattangadi SM. Lodish HF. Regulation of erythrocyte lifespan: do reactive oxygen species set the clock? J Clin Invest. 2007;117:2075–2077. doi: 10.1172/JCI32559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hebbel RP. The sickle erythrocyte in double jeopardy: autoxidation and iron decompartmentalization. Semin Hematol. 1990;27:51–69. [PubMed] [Google Scholar]
- 42.Ho YS. Magnenat JL. Bronson RT. Cao J. Gargano M. Sugawara M. Funk CD. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem. 1997;272:16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- 43.Ho YS. Xiong Y. Ma W. Spector A. Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279:32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- 44.Hosokawa K. Arai F. Yoshihara H. Nakamura Y. Gomei Y. Iwasaki H. Miyamoto K. Shima H. Ito K. Suda T. Function of oxidative stress in the regulation of hematopoietic stem cell-niche interaction. Biochem Biophys Res Commun. 2007;363:578–583. doi: 10.1016/j.bbrc.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 45.Iiyama M. Kakihana K. Kurosu T. Miura O. Reactive oxygen species generated by hematopoietic cytokines play roles in activation of receptor-mediated signaling and in cell cycle progression. Cell Signal. 2006;18:174–182. doi: 10.1016/j.cellsig.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 46.Irani K. Xia Y. Zweier JL. Sollott SJ. Der CJ. Fearon ER. Sundaresan M. Finkel T. Goldschmidt-Clermont PJ. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 47.Ishii T. Yamada M. Sato H. Matsue M. Taketani S. Nakayama K. Sugita Y. Bannai S. Cloning and characterization of a 23-kDa stress-induced mouse peritoneal macrophage protein. J Biol Chem. 1993;268:18633–18636. [PubMed] [Google Scholar]
- 48.Itano HA. Hirota K. Hosokawa K. Mechanism of induction of haemolytic anaemia by phenylhydrazine. Nature. 1975;256:665–667. doi: 10.1038/256665a0. [DOI] [PubMed] [Google Scholar]
- 49.Ito K. Hirao A. Arai F. Matsuoka S. Takubo K. Hamaguchi I. Nomiyama K. Hosokawa K. Sakurada K. Nakagata N. Ikeda Y. Mak TW. Suda T. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 50.Ito K. Hirao A. Arai F. Takubo K. Matsuoka S. Miyamoto K. Ohmura M. Naka K. Hosokawa K. Ikeda Y. Suda T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 51.Jaiswal AK. Antioxidant response element. Biochem Pharmacol. 1994;48:439–444. doi: 10.1016/0006-2952(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 52.Jang YY. Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janzen V. Forkert R. Fleming HE. Saito Y. Waring MT. Dombkowski DM. Cheng T. DePinho RA. Sharpless NE. Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 54.Johnson RM. Goyette G., Jr Ravindranath Y. Ho YS. Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radic Biol Med. 2005;39:1407–1417. doi: 10.1016/j.freeradbiomed.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 55.Junger MA. Rintelen F. Stocker H. Wasserman JD. Vegh M. Radimerski T. Greenberg ME. Hafen E. The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol. 2003;2:20. doi: 10.1186/1475-4924-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamsler A. Daily D. Hochman A. Stern N. Shiloh Y. Rotman G. Barzilai A. Increased oxidative stress in ataxia telangiectasia evidenced by alterations in redox state of brains from Atm-deficient mice. Cancer Res. 2001;61:1849–1854. [PubMed] [Google Scholar]
- 57.Kang SW. Chae HZ. Seo MS. Kim K. Baines IC. Rhee SG. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J Biol Chem. 1998;273:6297–6302. doi: 10.1074/jbc.273.11.6297. [DOI] [PubMed] [Google Scholar]
- 58.Kastan MB. Zhan Q. el-Deiry WS. Carrier F. Jacks T. Walsh WV. Plunkett BS. Vogelstein B. Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 59.Kihm AJ. Kong Y. Hong W. Russell JE. Rouda S. Adachi K. Simon MC. Blobel GA. Weiss MJ. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature. 2002;417:758–763. doi: 10.1038/nature00803. [DOI] [PubMed] [Google Scholar]
- 60.Kim JH. Chu SC. Gramlich JL. Pride YB. Babendreier E. Chauhan D. Salgia R. Podar K. Griffin JD. Sattler M. Activation of the PI3K/mTOR pathway by BCR-ABL contributes to increased production of reactive oxygen species. Blood. 2005;105:1717–1723. doi: 10.1182/blood-2004-03-0849. [DOI] [PubMed] [Google Scholar]
- 61.Kong Y. Zhou S. Kihm AJ. Katein AM. Yu X. Gell DA. Mackay JP. Adachi K. Foster-Brown L. Louden CS. Gow AJ. Weiss MJ. Loss of alpha-hemoglobin stabilizing protein impairs erythropoiesis and exacerbates beta-thalassemia. J Clin Invest. 2004;114:1457–1466. doi: 10.1172/JCI21982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kops GJ. Dansen TB. Polderman PE. Saarloos I. Wirtz KW. Coffer PJ. Huang TT. Bos JL. Medema RH. Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 63.Koptyra M. Falinski R. Nowicki MO. Stoklosa T. Majsterek I. Nieborowska-Skorska M. Blasiak J. Skorski T. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–327. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kwong M. Kan YW. Chan JY. The CNC basic leucine zipper factor, Nrf1, is essential for cell survival in response to oxidative stress-inducing agents: role for Nrf1 in gamma-gcs(l) and gss expression in mouse fibroblasts. J Biol Chem. 1999;274:37491–37498. doi: 10.1074/jbc.274.52.37491. [DOI] [PubMed] [Google Scholar]
- 65.Lebovitz RM. Zhang H. Vogel H. Cartwright J., Jr Dionne L. Lu N. Huang S. Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JM. Chan K. Kan YW. Johnson JA. Targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia. Proc Natl Acad Sci U S A. 2004;101:9751–9756. doi: 10.1073/pnas.0403620101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee SS. Kennedy S. Tolonen AC. Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- 68.Lee TH. Kim SU. Yu SL. Kim SH. Park Do S. Moon HB. Dho SH. Kwon KS. Kwon HJ. Han YH. Jeong S. Kang SW. Shin HS. Lee KK. Rhee SG. Yu DY. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood. 2003;101:5033–5038. doi: 10.1182/blood-2002-08-2548. [DOI] [PubMed] [Google Scholar]
- 69.Lehtinen MK. Yuan Z. Boag PR. Yang Y. Villen J. Becker EB. DiBacco S. de la Iglesia N. Gygi S. Blackwell TK. Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 70.Li Y. Huang TT. Carlson EJ. Melov S. Ursell PC. Olson JL. Noble LJ. Yoshimura MP. Berger C. Chan PH. Wallace DC. Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 71.Libina N. Berman JR. Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- 72.Lin K. Dorman JB. Rodan A. Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 73.Lin K. Hsin H. Libina N. Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- 74.Lin SJ. Defossez PA. Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 75.Marinkovic D. Zhang X. Yalcin S. Luciano JP. Brugnara C. Huber T. Ghaffari S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest. 2007;117:2133–2144. doi: 10.1172/JCI31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martin FM. Bydlon G. Friedman JS. SOD2-deficiency sideroblastic anemia and red blood cell oxidative stress. Antioxid Redox Signal. 2006;8:1217–1225. doi: 10.1089/ars.2006.8.1217. [DOI] [PubMed] [Google Scholar]
- 77.Martinez-Gac L. Marques M. Garcia Z. Campanero MR. Carrera AC. Control of cyclin G2 mRNA expression by forkhead transcription factors: novel mechanism for cell cycle control by phosphoinositide 3-kinase and forkhead. Mol Cell Biol. 2004;24:2181–2189. doi: 10.1128/MCB.24.5.2181-2189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Melov S. Coskun P. Patel M. Tuinstra R. Cottrell B. Jun AS. Zastawny TH. Dizdaroglu M. Goodman SI. Huang TT. Miziorko H. Epstein CJ. Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci U S A. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meng TC. Buckley DA. Galic S. Tiganis T. Tonks NK. Regulation of insulin signaling through reversible oxidation of the protein-tyrosine phosphatases TC45 and PTP1B. J Biol Chem. 2004;279:37716–37725. doi: 10.1074/jbc.M404606200. [DOI] [PubMed] [Google Scholar]
- 80.Meng TC. Fukada T. Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 81.Miyamoto K. Araki KY. Naka K. Arai F. Tabuko K. Yamazaki S. Matsuoka S. Miyamoto T. Ito K. Ohmura M. Chen C. Hosokawa K. Nakauchi H. Nakayama K. Nakayama KI. Harada M. Motayama N. Suda T. Hirao A. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 82.Murakami S. Johnson TE. The OLD-1 positive regulator of longevity and stress resistance is under DAF-16 regulation in Caenorhabditis elegans. Curr Biol. 2001;11:1517–1523. doi: 10.1016/s0960-9822(01)00453-5. [DOI] [PubMed] [Google Scholar]
- 83.Murphy CT. McCarroll SA. Bargmann CI. Fraser A. Kamath RS. Ahringer J. Li H. Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 84.Nagayasu H. Hamada J. Kawano T. Konaka S. Nakata D. Shibata T. Arisue M. Hosokawa M. Takeichi N. Moriuchi T. Inhibitory effects of malotilate on invasion and metastasis of rat mammary carcinoma cells by modifying the functions of vascular endothelial cells. Br J Cancer. 1998;77:1371–1377. doi: 10.1038/bjc.1998.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nakano M. Ohneda K. Yamamoto-Mukai H. Shimizu R. Ohneda O. Ohmura S. Suzuki M. Tsukamoto S. Yanagawa T. Yoshida H. Takakuwa Y. Yamamoto M. Transgenic over-expression of GATA-1 mutant lacking N-finger domain causes hemolytic syndrome in mouse erythroid cells. Genes Cells. 2005;10:47–62. doi: 10.1111/j.1365-2443.2005.00814.x. [DOI] [PubMed] [Google Scholar]
- 86.Nemoto S. Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 87.Neumann CA. Krause DS. Carman CV. Das S. Dubey DP. Abraham JL. Bronson RT. Fujiwara Y. Orkin SH. Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 88.Ney PA. Andrews NC. Jane SM. Safer B. Purucker ME. Weremowicz S. Morton CC. Goff SC. Orkin SH. Nienhuis AW. Purification of the human NF-E2 complex: cDNA cloning of the hematopoietic cell-specific subunit and evidence for an associated partner. Mol Cell Biol. 1993;13:5604–5612. doi: 10.1128/mcb.13.9.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ogg S. Paradis S. Gottlieb S. Patterson GI. Lee L. Tissenbaum HA. Ruvkun G. The Forkhead transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- 90.Oh SW. Mukhopadhyay A. Svrzikapa N. Jiang F. Davis RJ. Tissenbaum HA. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of fork-head transcription factor/DAF-16. Proc Natl Acad Sci U S A. 2005;102:4494–4499. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ookuma S. Fukuda M. Nishida E. Identification of a DAF-16 transcriptional target gene, scl-1, that regulates longevity and stress resistance in Caenorhabditis elegans. Curr Biol. 2003;13:427–431. doi: 10.1016/s0960-9822(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 92.Peter Y. Rotman G. Lotem J. Elson A. Shiloh Y. Groner Y. Elevated Cu/Zn-SOD exacerbates radiation sensitivity and hematopoietic abnormalities of Atm-deficient mice. EMBO J. 2001;20:1538–1546. doi: 10.1093/emboj/20.7.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ran Q. Liang H. Ikeno Y. Qi W. Prolla TA. Roberts LJ., 2nd Wolf N. VanRemmen H. Richardson A. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J Gerontol A Biol Sci Med Sci. 2007;62:932–942. doi: 10.1093/gerona/62.9.932. [DOI] [PubMed] [Google Scholar]
- 94.Reaume AG. Elliott JL. Hoffman EK. Kowall NW. Ferrante RJ. Siwek DF. Wilcox HM. Flood DG. Beal MF. Brown RH., Jr Scott RW. Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 95.Reya T. Duncan AW. Ailles L. Domen J. Scherer DC. Willert K. Hintz L. Nusse R. Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 96.Rhee SG. Yang KS. Kang SW. Woo HA. Chang TS. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005;7:619–626. doi: 10.1089/ars.2005.7.619. [DOI] [PubMed] [Google Scholar]
- 97.Rossi DJ. Bryder D. Seita J. Nussenzweig A. Hoeijmakers J. Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 98.Rossi DJ. Bryder D. Zahn JM. Ahlenius H. Sonu R. Wagers AJ. Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci US A. 2005;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rushmore TH. Morton MR. Pickett CB. The antioxidant responsive element: activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266:11632–11639. [PubMed] [Google Scholar]
- 100.Sablina AA. Budanov AV. Ilyinskaya GV. Agapova LS. Kravchenko JE. Chumakov PM. The antioxidant function of the p53 tumorI suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sattler M. Verma S. Shrikhande G. Byrne CH. Pride YB. Winkler T. Greenfield EA. Salgia R. Griffin JD. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275:24273–24278. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- 102.Sattler M. Winkler T. Verma S. Byrne CH. Shrikhande G. Salgia R. Griffin JD. Hematopoietic growth factors signal through the formation of reactive oxygen species. Blood. 1999;93:2928–2935. [PubMed] [Google Scholar]
- 103.Savitsky PA. Finkel T. Redox regulation of Cdc25C. J Biol Chem. 2002;277:20535–20540. doi: 10.1074/jbc.M201589200. [DOI] [PubMed] [Google Scholar]
- 104.Seo JH. Ahn Y. Lee SR. Yeol Yeo C. Chung Hur K. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol Biol Cell. 2005;16:348–357. doi: 10.1091/mbc.E04-05-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shivdasani RA. Orkin SH. Erythropoiesis and globin gene expression in mice lacking the transcription factor NF-E2. Proc Natl Acad Sci U S A. 1995;92:8690–8694. doi: 10.1073/pnas.92.19.8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shivdasani RA. Rosenblatt MF. Zucker-Franklin D. Jackson CW. Hunt P. Saris CJ. Orkin SH. Transcription factor NF-E2 is required for platelet formation independent of the actions of thrombopoietin/MGDF in megakaryocyte development. Cell. 1995;81:695–704. doi: 10.1016/0092-8674(95)90531-6. [DOI] [PubMed] [Google Scholar]
- 107.Socolovsky M. Molecular insights into stress erythropoiesis. Curr Opin Hematol. 2007;14:215–224. doi: 10.1097/MOH.0b013e3280de2bf1. [DOI] [PubMed] [Google Scholar]
- 108.Storz P. Doppler H. Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol Cell Biol. 2005;25:8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Suliman HB. Ali M. Piantadosi CA. Superoxide dismutase-3 promotes full expression of the EPO response to hypoxia. Blood. 2004;104:43–50. doi: 10.1182/blood-2003-07-2240. [DOI] [PubMed] [Google Scholar]
- 110.Sundaresan M. Yu ZX. Ferrans VJ. Irani K. Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 111.Tanaka H. Matsumura I. Ezoe S. Satoh Y. Sakamaki T. Albanese C. Machii T. Pestell RG. Kanakura Y. E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Mol Cell. 2002;9:1017–1029. doi: 10.1016/s1097-2765(02)00522-1. [DOI] [PubMed] [Google Scholar]
- 112.Thannickal VJ. Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 113.Tothova Z. Kollipara R. Huntly BJ. Lee BH. Castrillon DH. Cullen DE. McDowell EP. Lazo-Kallanian S. Williams IR. Sears C. Armstrong SA. Passegue E. Depinho RA. Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 114.Tran H. Brunet A. Grenier JM. Datta SR. Fornace AJ., Jr DiStefano PS. Chiang LW. Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 115.Tsai WB. Chung YM. Takahashi Y. Xu Z. Hu MC. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat Cell Biol. 2008;10:460–467. doi: 10.1038/ncb1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Vafa O. Wade M. Kern S. Beeche M. Pandita TK. Hampton GM. Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 117.van der Horst A. de Vries-Smits AM. Brenkman AB. van Triest MH. van den Broek N. Colland F. Maurice MM. Burgering BM. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006;8:1064–1073. doi: 10.1038/ncb1469. [DOI] [PubMed] [Google Scholar]
- 118.Wang MC. Bohmann D. Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 119.Wang MC. Bohmann D. Jasper H. JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell. 2003;5:811–816. doi: 10.1016/s1534-5807(03)00323-x. [DOI] [PubMed] [Google Scholar]
- 120.Weiss MJ. Zhou S. Feng L. Gell DA. Mackay JP. Shi Y. Gow AJ. Role of alpha-hemoglobin-stabilizing protein in normal erythropoiesis and beta-thalassemia. Ann N Y Acad Sci. 2005;1054:103–117. doi: 10.1196/annals.1345.013. [DOI] [PubMed] [Google Scholar]
- 121.Winterbourn CC. Oxidative denaturation in congenital hemolytic anemias: the unstable hemoglobins. Semin Hematol. 1990;27:41–50. [PubMed] [Google Scholar]
- 122.Winterbourn CC. Oxidative reactions of hemoglobin. Methods Enzymol. 1990;186:265–272. doi: 10.1016/0076-6879(90)86118-f. [DOI] [PubMed] [Google Scholar]
- 123.Wolff S. Ma H. Burch D. Maciel GA. Hunter T. Dillin A. SMK-1, an essential regulator of DAF-16-mediated longevity. Cell. 2006;124:1039–1053. doi: 10.1016/j.cell.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 124.Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006;25:695–705. doi: 10.1007/s10555-006-9037-8. [DOI] [PubMed] [Google Scholar]
- 125.Wu X. Bishopric NH. Discher DJ. Murphy BJ. Webster KA. Physical and functional sensitivity of zinc finger transcription factors to redox change. Mol Cell Biol. 1996;16:1035–1046. doi: 10.1128/mcb.16.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yalcin S. Foxo3 modulation of ATM and oxidative stress mediates distinct functions in the regulation of hematopoietic stem and progenitor cell fate. Blood. 2007;110:1272a. [Google Scholar]
- 127.Yalcin S. Zhang X. Luciano JP. Marinkovic D. Vercherat C. Sarkar A. Grisotto M. Taneja R. Ghaffari S. Foxo3 is essential for the regulation of ATM and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem. 2008 doi: 10.1074/jbc.M800517200. (Epub ahead of print, in press) [DOI] [PubMed] [Google Scholar]
- 128.Yamazaki S. Iwama A. Takayanagi S. Morita Y. Eto K. Ema H. Nakauchi H. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J. 2006;25:3515–3523. doi: 10.1038/sj.emboj.7601236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang JY. Zong CS. Xia W. Yamaguchi H. Ding Q. Xie X. Lang JY. Lai CC. Chang CJ. Huang WC. Huang H. Kuo HP. Lee DF. Li LY. Lien HC. Cheng X. Chang KJ. Hsiao CD. Tsai FJ. Tsai CH. Sahin AA. Muller WJ. Mills GB. Yu D. Hortobagyi GN. Hung MC. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138–148. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang W. Dolloff NG. El-Deiry WS. ERK and MDM2 prey on FOXO3a. Nat Cell Biol. 2008;10:125–126. doi: 10.1038/ncb0208-125. [DOI] [PubMed] [Google Scholar]
- 131.Yant LJ. Ran Q. Rao L. Van Remmen H. Shibatani T. Belter JG. Motta L. Richardson A. Prolla TA. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 132.Yilmaz OH. Valdez R. Theisen BK. Guo W. Ferguson DO. Wu H. Morrison SJ. PTEN dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 133.You H. Mak TW. Crosstalk between p53 and FOXO transcription factors. Cell Cycle. 2005;4:37–38. doi: 10.4161/cc.4.1.1401. [DOI] [PubMed] [Google Scholar]
- 134.Zelko IN. Mariani TJ. Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 135.Zhang J. Grindley JC. Yin T. Jayasinghe S. He XC. Ross JT. Haug JS. Rupp D. Porter-Westpfahl KS. Wiedemann LM. Wu H. Li L. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 136.Zhu QS. Xia L. Mills GB. Lowell CA. Touw IP. Corey SJ. G-CSF induced reactive oxygen species involves Lyn-PI3-kinase-Akt and contributes to myeloid cell growth. Blood. 2006;107:1847–1856. doi: 10.1182/blood-2005-04-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]