

Abstract
CpG dinucleotides contribute to epigenetic mechanisms by being the only site for DNA methylation in mammalian somatic cells. They are also mutation hotspots and ∼5-fold depleted genome-wide. We report here a study focused on CpG sites in the coding regions of Hox and other transcription factor genes, comparing methylated genomes of Homo sapiens, Mus musculus, and Danio rerio with nonmethylated genomes of Drosophila melanogaster and Caenorhabditis elegans. We analyzed 4-fold degenerate, synonymous codons with the potential for CpG. That is, we studied “silent” changes that do not affect protein products but could damage epigenetic marking. We find that DNA-binding transcription factors and other developmentally relevant genes show, only in methylated genomes, a bimodal distribution of CpG usage. Several genetic code-based tests indicate, again for methylated genomes only, that the frequency of silent CpGs in Hox genes is much greater than expectation. Also informative are NCG-GNN and NCC-GNN codon doublets, for which an unusually high rate of G to C and C to G transversions was observed at the third (silent) position of the first codon. Together these results are interpreted as evidence for strong “pro-epigenetic” selection acting to preserve CpG sites in coding regions of many genes controlling development. We also report that DNA-binding transcription factors and developmentally important genes are dramatically overrepresented in or near clusters of three or more CpG islands, suggesting a possible relationship between evolutionary preservation of CpG dinucleotides in both coding regions and CpG islands.
Keywords: DNA methylation, epigenetics, evolution, gene duplication
CpG dinucleotides are of special interest for several reasons. In somatic cells of mammals and other vertebrates, cytosine DNA methylation is almost entirely in CpGs (1, 2) and is an epigenetic mechanism essential for normal development (3, 4). The C in CpGs is highly mutable, with C to T (and complementary G to A) transitions being the most common mutations. The ≈30-fold increased mutation rate for CpG is generally thought to be due to the enzymatic methylation of CpGs, with the formation of 5-methylcytosine (mC). Deamination of mC then leads to enhanced mutagenesis (5, 6). Most likely for this reason the frequency of CpGs in the mammalian genome is on average ∼5-fold below expectation based on genome-wide nucleotide composition. Importantly, some regions of the genome are not depleted of CpGs and, if >200 bp, are called CpG islands (CGIs) (7, 8). As a hallmark feature, CGIs are usually unmethylated. However, some CGIs show tissue-specific methylation, and much evidence indicates that methylated CpG sites (mCpG) in promoters, enhancers, and other regulatory regions do play an essential role in embryonic development, gene imprinting, and X chromosome inactivation (3, 9, 10). For the above reasons there have been numerous studies of CpGs in promoters (11, 12). Over 50% of promoters are in CGIs and there is a strong inverse correlation, especially in cancer (13), between promoter CpG methylation and transcription. Much evidence indicates that methylated CpG-rich promoters are locked in the off state (3, 14).
The focus of this paper is different. We have investigated CpG usage in protein-coding regions. In coding regions a different system seems to be at work. Although on average 5-fold depleted in frequency, those CpGs within genes tend to be highly methylated (15), and it is now clear that such methylation not only is compatible with transcription but also may be positively correlated with transcription level (10, 14, 15). The biological significance of intragenic CpG methylation is only beginning to be appreciated and its impact on gene expression and development is still poorly understood. Furthermore, it remains unclear in general whether there is (and, if so, how strong) a link between epigenetic marking via methylation of CpGs in genes coding regions and major factors of evolution, mutations and natural selection. We have addressed these questions by comparing CpG-associated nucleotide frequencies in coding regions of Hox genes and Hox-like genes in methylated vs. nonmethylated genomes. Previous reports of tissue-specific intragenic CpG methylation of Hox clusters with possible contribution to their epigenetic regulation (16, 17) influenced this choice, as did our suggestion that epigenetic silencing should enhance the rate of evolution by gene duplication (18–20). We focused our study on synonymous variability of CpG dinucleotides in coding regions. The advantage of studying CpGs in protein-coding regions, not regulatory regions, is the opportunity to use the genetic code (Fig. 1) in the special way described below.
Fig. 1.

Genetic code. Colored are the eight quartets of codons with a completely degenerate third position (4d codons). Colored in blue are four quartets (Ser, Pro, Thr, and Ala) that include the CpG-containing NCG codons with silent mutation prone G. Four other quartets (colored in green) do not contain such silent CpGs and were used as controls.
Methylation of cytosines makes mCpGs of both strands hypermutable (5, 6). The most frequent mutation is a mC→T that, if in the coding strand, appears as a CpG→TpG transition and, if in the transcribed strand, is converted (in one round of replication) into a complementary CpG→CpA transition on the coding strand. Also, CpGs represent a potential site for epigenetic regulation by methylation and therefore could be under surveillance of selection. It should be noted that preservation of CpGs over evolutionary time can be by direct selection for mCpG or/and by indirect selection for hypomethylation in the germ line, such as may be the case for CGIs. For coding regions, one would expect to reveal either type of pro-epigenetic selection by studying synonymous mutations in CpGs. They do not change protein products of the gene but could alter RNA structure or epigenetic marking.
By the genetic code (Fig. 1), there are two kinds of synonymous changes in CpG sites: One affects G in the third position of NCG codons, and the other affects the C in CpGs formed by two neighboring codons, NNC followed by GNN. For brevity, we call both of these silent G- or silent C-containing sites silent CpGs. If selection preserves them for some epigenetic purpose, we would predict that the codons NCG and NNC followed by GNN would be overrepresented when compared with their synonymous variants.
Over evolutionary time the genome is expected to reach equilibrium between depletion of old and formation of new silent CpGs. Thus departure from the expected equilibrium frequency is evidence for selection. This result is exactly what we found for the Hox and some other transcription factor genes: significant overrepresentation of silent CpGs in Homo sapiens and other methylated genomes, but, importantly, not in nonmethylated genomes. This line of investigation, applied genome-wide, led to the finding that CpG usage in synonymous, 4-fold degenerate (4d) codons (Fig. 1) shows a bimodal distribution. Most coding regions are 5-fold depleted, in keeping with the long-known 5-fold underrepresentation of CpG in the mammalian genome, but homeodomain gene family members and some, but not all, DNA-binding transcription factors are very different, showing relatively little CpG depletion. We also find that DNA-binding transcription factor genes and developmentally important genes are strikingly overrepresented in clusters of CGIs.
Results
Bimodal Distribution and Preservation of CpGs in Hox and Other Transcription Factor Coding Regions.
To enable genome-wide study of CpG depletion or preservation in protein-coding regions, we made use of 4d codons. As shown in Fig. 1, there are eight amino acids encoded by 4d codons: Leu, Val, Ser, Pro, Thr, Ala, Arg, and Gly. We calculated CpGnorm, as a measure of observed CpG usage relative to that expected in synonymous codons, with values closer to 1.0 indicating preservation (Materials and Methods). Note that CpGnorm is normalized for, and independent of, G+C content and applies only to coding regions.
As shown in Fig. 2, most H. sapiens genes are distributed around CpGnorm = 0.32, consistent with the known (21) depletion of CpG in the entire genome (see below). However, there is a tail to larger values, and H. sapiens Hox genes are quite different, centered around CpGnorm = 0.8. Moreover, the distribution for all homeodomain-containing genes is clearly bimodal, with about half being similar to the Hox distribution. A high CpGnorm distribution pattern is not unique to homeodomain-containing genes. The entire class of transcription factor genes shows the bimodal distribution (Fig. S1), with DNA-binding factors showing a more pronounced shift to high CpGnorm. Clearly many transcription factor genes are similar to the Hox family in the preservation of CpGs in coding regions. However, a closer analysis of DNA-binding factors reveals that, in contrast to Hox and other homeodomain-containing proteins, zinc finger proteins, which are extremely common mammalian transcription factors, are indistinguishable from the whole genome distribution (Fig. 2).
Fig. 2.

Frequency distributions of CpGnorm of gene coding regions (Materials and Methods): (A) H. sapiens; (B) D. melanogaster. The number of genes for each case is shown in the key. All curves are normalized to have area = 1.
The preservation of CpG dinucleotides in 4d codons is most pronounced in the region of Hox genes that overlap with CGIs, although there is some preservation (CpGnorm = 0.6) even outside of CGIs (Fig. S2).
Fig. 2B and Fig. S3 show CpGnorm analysis of other organisms. It is clear that Drosophila melanogaster and Caenorhabditis elegans are quite different from H. sapiens, Mus musculus, and Danio rerio, with a unimodal distribution of CpGnorm centered close to 0.86 and showing little difference between all genes and Hox genes. Thus this type of analysis shows a general, distinctive difference between methylated and nonmethylated genomes. In contrast to vertebrates, the nonmethylated genomes do not show compartmentalization into high and low CpG classes when 4d codon analysis is applied to protein-coding regions.
Estimation of CpG Depletion in Methylated Genomes.
Using the CpGdepl measure (Materials and Methods), we find that the depletion of silent CpGs in Human Hox genes is very small, CpGdepl = 1.2, in contrast to the entire coding part of the genome for which the silent CpGs are ∼3-fold underrepresented (CpGdepl = 3.1). The latter result is lower than the overall-genome (∼5-fold) underrepresentation. The reason is that in any silent CpG dinucleotide from gene coding regions, only one of two nucleotides, either G or C, is prone to a silent mutation in contrast to introns or intergenic regions. The correct, per site, estimate of CpG depletion is roughly two times larger, meaning that for most genes underrepresentation of silent CpGs in the protein-coding region is virtually the same as in the whole genome. The Hox and other transcription factors are notably different.
Excess of NCG Codons Indicates Preservation of Silent CpGs in Coding Regions of Vertebrate Hox Genes.
Four amino acids, Ser, Pro, Thr, and Ala (colored blue in Fig. 1), have CpG-containing NCG codons with a “silent” G at the third position. Four other quartets (Leu, Val, Arg, and Gly) (colored green in Fig. 1) serve as controls because none of their codons contain silent CpGs. Fig. 3A shows variations in usage of 4d codons in H. sapiens Hox genes measured (in percent) with respect to their average genome values; positive and negative values mean their over- and underrepresentation, respectively. For Hox genes, all 4d codons ending with C or G are somewhat overrepresented, perhaps for reasons discussed in the next section. But beyond this, NCG codons are in obvious excess, which is suggestive of selection. Fig. 3B shows data for 39 randomly chosen genes, the same number as in the Hox gene family (Table S1). No preference for synonymous codons is seen in this control.
Fig. 3.

Variation in usage of 4d codons in Hox genes compared with the average genome values (Materials and Methods). (A) H. sapiens. (B) Randomly chosen H. sapiens protein-coding genes. A total of 39 genes have been retrieved from the H. sapiens genome, the same number as in the Hox clusters. (C) Hox genes of D. melanogaster. Different colors mark these eight 4d-encoded amino acids with the first two nucleotides shown in parentheses. The horizontal line in the boxes represents the codon bias calculated from the original dataset (Materials and Methods). Upper and lower ends of boxes represent the SE of the bootstrap distribution. Whiskers represent the 0.95 adjusted bootstrap percentile (BCa) interval (bootstrap value = 100,000).
In sharp contrast to H. sapiens, D. melanogaster does not show a difference between Hox genes and the entire genome (Fig. 3C). The same striking differences were seen in other comparisons of methylated vs. nonmethylated genomes: rodent M. musculus and fish D. rerio vs. nematode C. elegans (Fig. S4).
Importantly, the preference of NCG codons seen for Hox genes of H. sapiens (Fig. 3A), M. musculus, and D. rerio (Fig. S4) is not due to a bias in nucleotide composition. First, if we assume that selection prefers not silent CpGs but simply G or C at the third codon position, then we should observe the same pattern of usage for control 4d codons (not CpG containing) of Leu, Val, Arg, and Gly. This is clearly not the case (Fig. 3). Second, in coding regions of H. sapiens Hox genes, the third position of 4d codons does show a strong bias to G or C (78 ± 1%) (Table S2). However, for complete genes (exons plus introns) and entire Hox clusters (with intergenic regions also included), the G or C bias is significantly smaller: 55 ± 4% and 52 ± 1%, respectively. This result suggests that the bias to C or G at the third position of 4d codons specifically characterizes Hox coding regions rather than the local genome regions where these Hox genes reside. Third, if codon usage in Hox genes were determined by the nucleotide frequencies, one would observe an excess of the NCC over NCG codons inasmuch as C is more frequent than G at the third position of all 4d codons in Hox genes: 45 ± 5% C vs. 33 ± 6% G (Table S2). Opposite to expectation, silent G clearly prevails over silent C in codons for Ser, Ala, Pro, and Thr, suggesting that the strong bias of codon usage in Hox genes is associated with CpG sites rather than with the G+C content. This result in turn suggests that the observed relatively high frequency of C in the third codon position of mammalian Hox genes (Fig. 3A and Fig. S4) may reflect formation of the CpG with the next codon, i.e., the NNC-GNN configuration.
CGA and CGG Codons.
These CpG-containing codons are of particular interest because C→A transversions convert them into the non-CpG codons AGA and AGG still coding for the same amino acid, arginine (Fig. 1). The hypothesis of selection maintaining mCpGs along the gene body predicts an excess of CG-containing codons over their AGA and AGG synonyms in CpG-methylated genomes but not in non-CpG–methylated genomes. As in the previous case with NCG codons (Fig. 3 and Fig. S4), we estimated variations in usage of these arginine codons in Hox genes relative to their usage in entire genomes. The result turned out to be consistent with the prediction. In methylated human Hox genes, AGA and AGG are underrepresented (−54.6 ± 7.5% and −24.1 ± 7.2%, respectively) whereas CGC is overrepresented (+104.7 ± 12.9%). By contrast, in nonmethylated Drosophila, usage of arginine codons in Hox genes is virtually not different from their usage at a whole-genome level. This result again suggests that only in CpG-methylated genomes, selection preserves CGG and CGA codons from synonymous C→A transversions.
Excess of NCC-GNN↔NCG-GNN Transversions in Hox Coding Regions.
Usually, C→G/G→C transversions at CpG sites are rare compared with C→A/G→T transversions and especially C→T/G→A transitions. For example, in the TP53 tumor suppressor gene from H. sapiens cancers, silent G→C and C→G at CpG sites comprise only 10.5% in contrast to 32.6% of C→A/G→T and 56.9% of C→T/G→A (International Agency for Research on Cancer database). We find that for certain sequences in Hox genes these numbers are different.
Fig. 4 illustrates the unique feature of NCC GNN sites: a C→G transversion at the third position of the first codon destroys an old CpG but at the same time creates a new CpG shifted only one position to the left. Mirror symmetrically, the same is true for a new CpG shifted to the right by a reverse G→C transversion in the NCG-GNN structure. In contrast, a C→G in a NCC codon (or, symmetrically, the reverse G→C in NCG) not followed by GNN creates (or eliminates) a CpG site without any compensatory change. Therefore, if selection preserves the CpG profile in coding regions of Hox genes, one would predict a significant increase of the C→G (G→C) frequency in the first case (NCC-GNN and NCG-GNN) and, on the contrary, a significant decrease of these transversions in the second case (NCC-HNN and NCG-HNN, H equals not G) compared with three other types of base substitutions. This difference is precisely what we observe for aligned coding regions of M. musculus and H. sapiens Hox genes (see diagrams in Fig. 4). For example, C↔T transitions decrease from 62 to 44% and C↔G transversions increase from 24 to 38%.
Fig. 4.

Conservation of the CpG profile in the coding region of Hox genes. Shown is an illustration of the effect of C↔G transversion in NCC-GNN and NCG-GNN pairs in comparison with NCC-HNN and NCG-HNN (H equals not G) pairs of neighboring codons. The pie charts show the observed frequencies (in percent) of C↔G (green), C↔T (red), or C↔A (cyan) substitutions between H. sapiens and M. musculus. See text for details.
Hox and Other Transcription Factors Are Located in Clusters of CpG Islands.
Genome-wide analyses have shown that exons often overlap with CGIs (12), and the synonymous substitution rate of CpG-containing codons is substantially reduced in regions of overlap (10, 12, 22). We noticed that CGIs are distributed throughout the Hox A locus and often overlap with exons. This observation prompted an analysis of CGIs. To determine how CGIs are distributed in the genome, we developed an algorithm that enables an analysis of clustering (SI Text and Fig. S5). A CGI cluster is defined as a set of CGIs with distance between consecutive CGIs less than a given threshold (T). Genes belong to a CGI cluster if they totally or partially overlap with a CGI cluster. Consistent with the known nonrandom distribution of genes in the genome and the existence in the mammalian genome of isochores (23), defined as large regions of similar G+C content, we find that CGIs are not randomly distributed; instead they often occur in clusters. For example, the 11 Hox A genes are located in a large cluster of CGIs (Fig. S6). In fact, all of the Hox loci are located in CGI clusters of three or greater, a feature that, to our knowledge, has not previously been noted. Given this result, we asked what genes tend to be in CGI clusters. Table 1 and Table S3 shows Gene Ontology (GO) results for clusters of three or greater, with T = 10,000 bp and CGI length 500 bp. It is clear that transcription factors, especially DNA-binding transcription factors, are dramatically overrepresented (P value = 9 × e−66) in CGI clusters of three or greater. Another high-scoring category is “regulation of gene expression” (P value = 8 × e−26). Similar results were obtained for T = 5,000 and 15,000.
Table 1.
Top five enriched Gene Ontology (GO) terms for genes overlapping with CpG island clusters
| GO accession | GO biological process term | Enrichment | P value |
| GO:0043565 | Sequence-specific DNA binding | 3.593 | 9.1e-66 |
| GO:0007389 | Pattern specification process | 3.191 | 6.11e-11 |
| GO:0007420 | Brain development; | 2.971 | 8.98e-10 |
| GO:0003700 | Transcription factor activity | 2.700 | 3.33e-52 |
| GO:0009790 | Embryonic development | 2.628 | 4.74e-11 |
For a complete list see Table S3.
Promoters are known to be associated with CGIs, so one possibility to be considered is that the association with CGI clusters just reflects this fact. However, CGI-associated promoters are enriched for general housekeeping genes (12, 14, 24) and only weakly enriched for transcription factor and developmental genes (Table S3). When we subtract genes in CGI clusters from the gene ontology analysis of total CGI-associated genes, transcription factors and developmental genes no longer register as significantly enriched (Table S3). Thus, housekeeping genes are associated with single CGIs, but many genes involved with embryonic development, especially DNA-binding transcription factors, have a special relationship with CGI clusters.
Discussion
In this paper we focused on CpG dinucleotides in coding regions, and we made four main observations. First, genome-wide analysis of CpG abundance in 4d codons, normalized for G+C, gives a distribution in which most coding regions show the expected depletion (CpGnorm = 0.32), but ∼10% of protein-coding genes show much less depletion (CpGnorm > 0.6). These CpG-rich cases include Hox and other homeobox-containing genes. In contrast, coding regions of zinc finger-containing transcription factors are CpG poor (CpGnorm ≈ 0.27) (Fig. 2). Second, a more detailed analysis of CpG usage in Hox genes indicates that CpGs are strongly preserved in coding regions and this preservation does not depend on G+C content (Figs. 3 and 4). Third, the mammalian genome is organized so that a high percentage of DNA-binding transcription factors and genes involved in development are part of large regions marked by clusters of CGIs (Table 1). Fourth, organisms such as D. melanogaster and C. elegans, which do not have DNA methylation, do not show any of these features, suggesting that epigenetic marking of CpGs by DNA methylation is at the root of these differences (Figs. 2 and 3, Figs. S3 and S4). We conclude that the special preservation over evolutionary time of CpGs in a small portion (∼10%) of coding regions is due to pro-epigenetic selection. This selection can be due to either one or both of (i) a function(s) for mCpG in some coding regions and (ii) protection from mutational depletion, for example, by hypomethylation in the germ line.
Pro-epigenetic Selection.
Undoubtedly, methylated mCpGs are major marks for epigenetic regulation, affecting chromatin structure and gene regulation. Until very recently, one would assign these functions mainly to the mCpGs of noncoding DNA (promoters, enhancers, insulators, etc.). However, several findings suggest that mCpG in gene bodies has a function(s). First, recent genome-wide methylation studies revealed a positive correlation between transcription levels and gene-body methylation levels (2, 10, 14, 25). Second, by comparing M. musculus and H. sapiens genomes, Medvedeva et al. (22) found that the synonymous substitution rate of CpG-containing codons is substantially reduced where protein-coding exons overlap CGIs. Third, the sea squirt Ciona intestinalis has a genome about equally divided between methylated and unmethylated domains, with most gene bodies in the methylated domain (10). Fourth, the DNA of the honey bee, Apis mellifora, contains methylated DNA and Elango et al. have found that its genome is equally divided into high-CpG and low-CpG classes (26). These authors suggested that exons are the primary target of DNA methylation and found that the high-CpG genes in A. mellifora are enriched for genes associated with developmental processes. Finally, our detailed analysis of codon usage in developmentally important Hox genes clearly establishes that CpGs in these protein-coding regions are in some way preserved from mutational depletion.
CpG usage in coding regions could affect RNA structure stability, so this reason for preservation cannot be ruled out. Kondrashov et al. (27) calculated that synonomous sites are under weak selection for G and C. But the strong selection we find for Hox genes suggests something more. Bird and his colleagues proposed that methylation of CpGs within CpG-rich coding regions, such as found in the sea squirt, may reduce inappropriate transcription (10). Also noncoding RNAs of intragenic origin could function as antisense or as precursors of miRNAs; that is, they could be an important part of complicated systems regulating gene expression during development. An antisense transcript of the M. musculus Hoxa11 gene is a particularly intriguing example (28). Transcripts produced from the antisense strand overlap the gene. Repression of the antisense transcript by the Hoxa11 protein or mutual strand-symmetric repression cannot be excluded as well (29, 30). Indeed, frequent C and/or G at the third position of codons on the coding (sense) strand could notably increase not only the 2D stability of mRNA but also the probability of long ORFs on the complementary (antisense) strand. For example, the antisense strand of HoxA11 genes does have quite long reading frames for putative antisense protein(s) (28), although not that long as in the cases of actual sense–antisense coding (see, for example, ref. 31). At any rate, it is clear that these two, antisense- and mCpG-mediated, mechanisms are not alternative—they might both, in a mutually tuned manner, be involved in regulation of gene expression. Indeed, multiple methylated CpGs along the coding sequence would change the interface between the gene body and its regulators. Feedback self-regulation was suggested long ago (32) and is quite characteristic for the Hox genes (28).
The key regulators of Hox gene transcription are Polycomb group (PcG) proteins that belong to the zinc finger family. Remarkably, coding regions of zinc finger genes show a CpGnorm distribution that is similar to most coding regions and in sharp contrast with Hox genes (Fig. 2). Indeed, it looks as if silent CpGs are under surveillance of a particularly strong pro-epigenetic selection in coding regions of the genes that not only regulate transcription of functionally subordinate genes but are themselves targets for such regulation. Further in silico studies of entire gene networks of transcription factors are required to find out how common is this difference in the CpGnorm distribution (a signature of pro-epigenetic selection) between gene regulators and gene targets of regulation. Genes in CGI clusters are of interest in this regard.
Some regions are protected from methylation. Promoters have been most studied in this regard. The majority of promoters are contained within CGIs and are relatively rich in CpG, comprising the HCG class noted by Saxonov et al. (12). Most HCG promoters are not methylated in somatic cells and, although experimental data are limited, are commonly thought also to be unmethylated in the germ line. Being hypomethylated, these promoters and CGIs would not be subject to hypermutagenesis at CpG, thus explaining the lack of depletion over evolutionary time (22). The rate of mutation in HCG promoters is, indeed, lower than in most noncoding regions (12). This mechanism still requires selection because the question becomes: What is preventing the methylation of these CGIs and promoters? One hypothesis is that these promoters are protected by the binding of certain protein factors such as Sp1 (22) or Cfp1 (33). However, most coding regions that are associated with HCG promoters are not protected from CpG depletion, so something must be different about the coding regions of high-CpGnorm genes.
A working hypothesis that reconciles several observations is that some genes, especially those in CGI clusters, such as the Hox family, are not methylated in the germ line. These genes are not intrinsically resistant to methylation, as they show tissue-specific methylation in somatic cells (2, 14). However, they may indeed be protected in the germ line. It is known, for example, that the Hox genes are hypomethlated in sperm, whereas most genes are highly methylated (2). Future work should involve analysis of the methylation status of the high-CpGnorm class of genes during gametogenesis.
CGI Clusters.
A striking finding is that DNA-binding transcription factors and other developmentally related genes are strongly associated with clusters of three or more CGIs, whereas housekeeping genes are associated with single CGIs (Table 1 and Table S3). This result raises the possibility that clustering of CGIs is somehow part of the mechanism protecting some genes important for development from CpG methylation in the germ line but not in somatic cells.
Gene Regulation, Gene Duplication, and Evolution.
The major transitions in evolution (34) seem to have been all crucially influenced by “soft” epigenetic inheritance (35, 36). In particular, the role of flexible epigenetic reactivation might be very critical in evolutionary survival of young gene duplicates (18–20). Apparently, the Hox genes are of interest in this regard (18–20).
There are several clusters of Hox genes in methylated genomes of vertebrates (e.g., clusters of Hox A, B, C, and D in mammals), but only one in nonmethylated genomes of invertebrates (e.g., Antennapedia–Bithorax cluster in D. melanogaster). Thus, each Hox gene is represented by several paralogs of closely related sequence and function in methylated genomes, in contrast to a single such gene in nonmethylated genomes. Presumably the clustering of structurally and functionally similar genes as well as presence of several such clusters is the result of gene and cluster duplications followed by divergence of function. Mathematical modeling has shown that tissue-specific epigenetic silencing and/or changes in expression greatly aid retention of functional duplicates (20), especially for organisms such as vertebrates, which have a relatively small effective population size. The duplication event may stimulate epigenetic silencing (ES) of excessive gene copies to reduce possible dosage-based and/or other expression imbalances caused by the duplication. It should be noted that if the duplicates are identical, ES does not need to distinguish them, but may just stochastically affect one or the other. Importantly, silencing is reversible; therefore, in a different tissue, in a stage of development, or even in the next generation, ES may affect the other twin gene. Either way, stochastic epigenetic silencing makes visible to selection first one duplicate and then the other, and this is all that is needed to preserve them both. The important point is that selection must be applied soon after duplication to avoid degradation of the duplicate to a nonfunctional pseudogene. This line of reasoning and the findings reported here suggest that CpG methylation, including exonic methylation, may favor the retention of duplicates by aiding the rapid emergence of tissue-specific expression soon after duplication. This idea again suggests that the intragenic CpGs studied in this paper could be involved in developmentally important regulatory circuits, consistent with the observed pro-epigenetic selection.
Materials and Methods
The sequence data for protein-coding genes and information on Gene Ontology were retrieved from the Ensembl database v. 56 by custom Perl API scripts. The list of genes containing specific protein domain was also retrieved from the Ensembl database v. 56, using the appropriate InterPro entries.
The gene alignment was in two steps: First we aligned the amino acid sequences using the MUSCLE release 3.6, and then from this amino acid sequence alignment we reconstructed the nucleotide one using a Perl script based on aa_to_dna_aln function included in BioPerl package release 1.6.0.
For statistical analysis of 4d codon variation within the corresponding quartets, we used the R software version 2.9.2. Primary data were retrieved from the Codon Usage Database at http://www.kazusa.or.jp/codon/. For each particular codon, we calculated its variation as 100 × (UH − UG)/ UG, where UH and UG are its frequencies (measured in percent, relative to the other three synonymous codons in the quartet) averaged for Hox genes (UH) and the whole genome (UG), respectively.
For simulation studies, the Hox gene replicas were generated using a custom Perl script that retains the same amino acid sequence but chooses the codons proportionally to their genomic frequencies. For each simulated Hox gene replica, the number of CpG sites at 4d codons was calculated and compared with the number of CpGs in the real Hox gene.
To study the general frequency pattern of CpG sites in 4d codons, we used, as a first approximation, the approach described in ref. 27. All 4d codons (blue and green in Fig. 1) were divided into four nonoverlapping groups in which the third (silent) nucleotide was (i) preceded by C (i.e., can be denoted as postC), (ii) followed by G (preG), (iii) preceded by C and followed by G (postCpreG), and (iv) neither preceded by C nor followed by G (nonCpG). The first three are CpG-prone groups. In cases with multiple transcripts, we always selected for analyses the longest one. As an integral measure of selection acting in favor of silent CpGs despite their high mutability in methylated genomes, we used the CpGnorm index defined as the ratio of the observed numbers of CpGs in 4d CpG-prone sites of the gene (CpGobs), divided by the number expected from the C and G content in 4d nonCpG sites; i.e.,
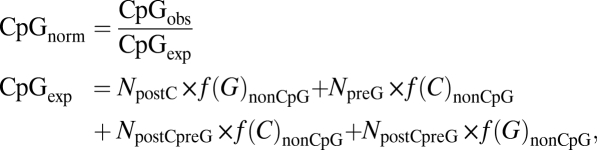 |
where NpostC, NpreG, and NpostCpreG are the total numbers of postC, preG, and postCpreG sites in the gene, and f(C)nonCpG [f(G)nonCpG] is the fraction of C (G) at non-CpG sites.
The reverse ratio, CpGexp/CpGobs, can be used as a measure of CpG mutational depletion, CpGdepl. In this case, we assume that the frequencies of C and G in non-CpG sites roughly reflect the frequencies of C and G at CpG sites in the ancestral state, before their methylation-induced hypermutability. The assumption seems reasonable because we use for estimation of both CpGnorm and CpGdepl only 4d codons in which all mutations at the third position are amino acid sequence neutral. Thus, if the silent CpG sites from blue codon quartets were not methylated, they would be mutagenically equipotent with the silent non-CpG sites from green codon quartets (Fig. 1).
A kernel density plot was used to represent the distribution of CpGnorm values for different sets of genes. The function “density” in the R package with default option was used to evaluate the kernel density.
For CGI cluster analysis, information on CGI location in each chromosome was retrieved from Ensembl database v. 57. CGI clusters are defined as described in SI Text. Genes belong to a CGI cluster if they totally or partially overlap with a CGI cluster. Overrepresented Gene Ontology categories were identified using Gene Ontology Statistics (GOstat) bioinformatics software, applying Benjamini correction for multiple testing (37).
The complete list of CpGnorm values can be obtained as a spreadsheet from S.B., S.N.R., or A.D.R.
Supplementary Material
Acknowledgments
S.N.R. holds the Susumu Ohno Chair in Theoretical Biology and S.B. is a Susumu Ohno Distinguished Fellow.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1010506107/-/DCSupplemental.
References
- 1.Bird AP. Gene number, noise reduction and biological complexity. Trends Genet. 1995;11:94–100. doi: 10.1016/S0168-9525(00)89009-5. [DOI] [PubMed] [Google Scholar]
- 2.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 4.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 5.Rideout WM, 3rd, Coetzee GA, Olumi AF, Jones PA. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science. 1990;249:1288–1290. doi: 10.1126/science.1697983. [DOI] [PubMed] [Google Scholar]
- 6.Yang AS, Jones PA, Shibata A. In: The Mutational Burden of 5-Methylcytosine. Russo VEA, Martienssen RA, Riggs AD, editors. Plainview, NY: Cold Spring Harbor Lab Press; 1996. pp. 77–94. [Google Scholar]
- 7.Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- 8.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reik W, Walter J. Genomic imprinting: Parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki MM, Bird A. DNA methylation landscapes: Provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 11.Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci USA. 1993;90:11995–11999. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA. 2006;103:1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rauch TA, Wu XW, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci USA. 2009;106:671–678. doi: 10.1073/pnas.0812399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones PA. The DNA methylation paradox. Trends Genet. 1999;15:34–37. doi: 10.1016/s0168-9525(98)01636-9. [DOI] [PubMed] [Google Scholar]
- 16.Illingworth R, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinn JL, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodin SN, Parkhomchuk DV, Riggs AD. Epigenetic changes and repositioning determine the evolutionary fate of duplicated genes. Biochemistry (Mosc) 2005;70:559–567. doi: 10.1007/s10541-005-0149-5. [DOI] [PubMed] [Google Scholar]
- 19.Rodin SN, Parkhomchuk DV, Rodin AS, Holmquist GP, Riggs AD. Repositioning-dependent fate of duplicate genes. DNA Cell Biol. 2005;24:529–542. doi: 10.1089/dna.2005.24.529. [DOI] [PubMed] [Google Scholar]
- 20.Rodin SN, Riggs AD. Epigenetic silencing may aid evolution by gene duplication. J Mol Evol. 2003;56:718–729. doi: 10.1007/s00239-002-2446-6. [DOI] [PubMed] [Google Scholar]
- 21.Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8:1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Medvedeva YA, et al. Intergenic, gene terminal, and intragenic CpG islands in the human genome. BMC Genomics. 2010;11:48. doi: 10.1186/1471-2164-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. doi: 10.1016/s0378-1119(99)00485-0. [DOI] [PubMed] [Google Scholar]
- 24.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 25.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 26.Elango N, Hunt BG, Goodisman MAD, Yi SV. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc Natl Acad Sci USA. 2009;106:11206–11211. doi: 10.1073/pnas.0900301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kondrashov FA, Ogurtsov AY, Kondrashov AS. Selection in favor of nucleotides G and C diversifies evolution rates and levels of polymorphism at mammalian synonymous sites. J Theor Biol. 2006;240:616–626. doi: 10.1016/j.jtbi.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 28.Lemons D, McGinnis W. Genomic evolution of Hox gene clusters. Science. 2006;313:1918–1922. doi: 10.1126/science.1132040. [DOI] [PubMed] [Google Scholar]
- 29.Grewal SIS, Rice JC. Regulation of heterochromatin by histone methylation and small RNAs. Curr Opin Cell Biol. 2004;16:230–238. doi: 10.1016/j.ceb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Hsieh JT, et al. Tumor suppressive role of an androgen-regulated epithelial cell adhesion molecule (C-CAM) in prostate carcinoma cell revealed by sense and antisense approaches. Cancer Res. 1995;55:190–197. [PubMed] [Google Scholar]
- 31.Rodin AS, Rodin SN, Carter CW., Jr. On primordial sense-antisense coding. J Mol Evol. 2009;69:555–567. doi: 10.1007/s00239-009-9288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.García-Bellido A. Genetic control of wing disc development in Drosophila. Ciba Found Symp. 1975;0:161–182. doi: 10.1002/9780470720110.ch8. [DOI] [PubMed] [Google Scholar]
- 33.Thomson JP, et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082–1086. doi: 10.1038/nature08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szathmáry E, Maynard Smith J. The Major Transitions in Evolution. Oxford: Oxford Univ Press; 1995. [Google Scholar]
- 35.Jablonka E, Lamb MJ. The evolution of information in the major transitions. J Theor Biol. 2006;239:236–246. doi: 10.1016/j.jtbi.2005.08.038. [DOI] [PubMed] [Google Scholar]
- 36.Jablonka E, Lamb MJ. Soft inheritance: Challenging the modern synthesis. Genet Mol Biol. 2008;31:389–395. [Google Scholar]
- 37.Beissbarth T, Speed TP. GOstat: Find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics. 2004;20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.