

Abstract
The biosynthesis of chloramphenicol requires a β-hydroxylation tailoring reaction of the precursor L-p-aminophenylalanine (L-PAPA). Here, it is shown that this reaction is catalyzed by the enzyme CmlA from an operon containing the genes for biosynthesis of L-PAPA and the nonribosomal peptide synthetase CmlP. EPR, Mössbauer, and optical spectroscopies reveal that CmlA contains an oxo-bridged dinuclear iron cluster, a metal center not previously associated with nonribosomal peptide synthetase chemistry. Single-turnover kinetic studies indicate that CmlA is functional in the diferrous state and that its substrate is L-PAPA covalently bound to CmlP. Analytical studies show that the product is hydroxylated L-PAPA and that O2 is the oxygen source, demonstrating a monooxygenase reaction. The gene sequence of CmlA shows that it utilizes a lactamase fold, suggesting that the diiron cluster is in a protein environment not previously known to effect monooxygenase reactions. Notably, CmlA homologs are widely distributed in natural product biosynthetic pathways, including a variety of pharmaceutically important beta-hydroxylated antibiotics and cytostatics.
Keywords: beta-hydroxylation, dinuclear iron cluster, nonheme oxygenase, spectroscopy, nonribosomal peptide synthetase
The incorporation of nonproteinogenic amino acids is a cornerstone of the structural and functional diversity of natural products, including a wide range of antibiotics and cytostatics (1). Often, the “bioactivity” of the final synthesized natural product depends upon the action of tailoring enzymes, which may be independent enzymes or modules of a nonribosomal peptide synthetase (NRPS). One commonly encountered modification is the formation of β-hydroxy amino acids. The resulting hydroxylated amino acid is more hydrophilic than its precursor and often participates in secondary reactions (2), including further oxidation (3), glycosylation (4, 5), methylation (6), phosphorylation (7), and macrolactonization during antibiotic heterocyclization (8). Many of the nonribosomal peptides that are modified in this fashion are mainstays of the pharmaceutical industry for use as chemotherapeutics and antimicrobial agents, such as the widely used antitumor drug bleomycin and the teicoplanin group of glycopeptide antibiotics (5). Hence, the elucidation of a unique mechanism for this oxygenation reaction represents a singularopportunity for novel pharmaceutical design and engineering.
Amino acid β-hydroxylation in natural product biosynthetic pathways has been shown to be catalyzed by either nonheme iron-containing alpha-ketoglutarate (αKG)-dependent dioxygenase enzymes (7, 9) or cytochrome P450 monooxygenases (10). The high C–H bond strength at the β-carbon of the amino acid (∼85 kcal/mol) necessitates the formation of a potent oxidant, which is accommodated by the formation of activated and highly reactive metal-oxygen adducts by these enzymes, FeIV-oxo (11) and FeIV-oxo heme π-cation radical (reviewed in refs. 12 and 13), respectively.
Another class of metallo-oxygenase that has been directly shown to form a high-valent Fe-oxygen species during catalysis utilizes an oxygen-bridged dinuclear iron cluster in the resting active site (14). The reaction cycle of these enzymes was first studied in methane monooxygenase (MMO) (15–18). Based on this known chemistry of the diiron-containing oxygenase class, it is likely that amino acid β-hydroxylation could readily be achieved, but no such activity has been reported. Accordingly, we reexamined the gene sequences of all NRPS-containing pathways known to produce β-hydroxylated amino acids. Many of these pathways were found to contain members of a highly homologous and uncharacterized enzyme family, which are devoid of the primary sequence signatures of either the αKG-dependent or P450 oxygenases. Rather, they contain sequences that are reminiscent of dinuclear metal-binding motifs.
To determine whether this class of enzymes utilizes a dinuclear metal cluster to carry out amino acid β-hydroxylation, the CmlA gene of the previously characterized chloramphenicol biosynthetic pathway was selected for study. CmlA in Streptomyces venezuelae is hypothesized to carry out the key β-hydroxylation of L-p-aminophenylalanine (L-PAPA) tethered on the NRPS CmlP as shown in Fig. 1 (19). Here we have characterized the CmlA active site metal center using UV/Vis, EPR, and Mössbauer spectroscopies. In addition, transient kinetic and analytical methods have been used to establish the substrate specificity and function of the enzyme. These studies reveal a previously unrecognized class of enzymes for amino acid β-hydroxylation as well as a role and active site architecture for dinuclear iron-containing oxygenases.
Fig. 1.

Role of CmlA in the chloramphenicol biosynthesis pathway. Proposed overall scheme for chloramphenicol biosynthesis in S. venezuelae from the L-PAPA precursor. CmlA is proposed to catalyze β-hydroxylation of L-PAPA after it is covalently bound to the T domain of CmlP by a thioester linkage.
Results
Heterologous Expression and Absorption Spectra of CmlA.
The chloramphenicol biosynthesis operon of S. venezuelae consists of 11 open reading frames (20). The putative β-hydroxylase for L-PAPA, CmlA, is located directly upstream of the NRPS CmlP. The cloning, heterologous expression, and purification of CmlA are described in detail in Materials and Methods and resulted in a high yield of enzyme. Following one-step metal affinity chromatography, SDS-PAGE of the purified enzyme showed the presence of a single polypeptide with a mass similar to that predicted by its primary sequence (∼60 kDa) (Fig. S1). Inductively coupled plasma emission mass spectrometry (ICP) showed that iron is the only metal present in the enzyme in more than trace amounts. The stoichiometry of protein to iron is 1∶2.06 ± 0.04 irrespective of the metal content of the growth medium (Table S1).
The absorption spectrum of CmlA reveals a broad feature between 300 and 340 nm (ε ∼ 3.5 mM-1 cm-1) (Fig. 2A, dashed trace and Inset). This spectrum rules out the possibility that CmlA is a heme-containing P450 monooxygenase. Absorption features of this type are typical of oxygen-bridged dinuclear nonheme iron systems and are attributable to an oxo-to-FeIII charge transfer transition (21). The addition of 2 M buffered sodium azide to the enzyme results in the formation of a chromophore with broad absorption bands at 345 and 450 nm (Fig 2A, solid trace), as observed for azide-complexes of numerous other proteins containing oxo-bridged diiron clusters, including ribonucleotide reductase (RNR), stearoyl-ACP desaturase (Δ9D) (21), and hemerythrin (22).
Fig. 2.

Absorption spectra show that CmlA is a diiron cluster-containing enzyme. (A) Absorption spectrum of CmlA as isolated (dashed and Inset) and upon reaction with 2 M sodium azide (solid). (B) Reductive titration of CmlA with sodium dithionite. The arrow shows the direction of change. The inset shows that 2 reducing equivalents are required to fully reduce the enzyme.
Anaerobic reductive titration of the resting enzyme using sodium dithionite (Fig. 2B) in the presence of methyl viologen as a mediator shows that two reducing equivalents are required to fully reduce the enzyme and bleach the 340 nm charge transfer band. This agrees with the quantification of metals bound to the enzyme. Moreover, it is characteristic of an oxygen-bridged diiron cluster, as opposed to a sulfur-bridged diiron cluster or a P450 system, which could accept only one electron, or the mononuclear FeII of an αKG-linked system, which could accept none.
Mössbauer and EPR Studies of the CmlA Enzyme.
Mössbauer and EPR studies provide an unambiguous demonstration of the presence of an oxo-bridged diiron cluster in CmlA. Fig. 3A shows 4.2 K Mössbauer spectra of oxidized (trace 1,2) and dithionite-reduced (trace 3) 57Fe-enriched CmlA. The spectrum of the oxidized sample taken in the absence of an external magnetic field (Fig. 3A, spectrum 1) consists of a doublet with quadrupole splitting, ΔEQ ≈ 1.4 mm/s, and isomer shift, δ = 0.51 mm/s characteristic of high-spin (S = 5/2) FeIII. The B = 8.0 T spectrum of Fig. 3A, spectrum 2 can be simulated well (solid line) by assuming that the magnetic splittings can be solely attributed to the applied field B (i.e. the spectrum lacks paramagnetic hyperfine structure), and thus it reflects a system with ground state spin S = 0. Upon reduction (Fig. 3A, spectrum 3), all iron in the sample is transformed into species with isomer shift, δ ≈ 1.26 mm/s, a shift characteristic of high-spin (S = 2) FeII. When considered together with the iron stoichiometry and titration data presented above, these data show that oxidized CmlA contains a metal center comprising two high-spin FeIII sites antiferromagnetically coupled to yield a cluster ground state with spin S = 0.
Fig. 3.

Mössbauer and EPR characterization of the diiron cluster of CmlA. (A) Mössbauer spectra of oxidized (1, 2) and reduced (3) CmlA recorded at 4.2 K, in applied magnetic fields B as indicated. Solid line in (1) is a spectral simulation for two (similar) doublets with ΔEQ and δ values given in Table 1. The solid line in (2) is a spectral simulation assuming that the ground state spin of the diiron center has S = 0; sign (ΔEQ) and η values obtained are listed in Table 1. The oxidized protein contains a minor mononuclear FeIII contaminant. The solid line in (3) is a simulation for two doublets (1∶1 intensity ratio) having the ΔEQ values of Table 1. (B) EPR spectra of CmlA in oxidized as isolated (1), partially reduced mixed-valent (2) and fully reduced (3) oxidation states. The EPR measurement conditions are given in SI Materials and Methods. The small signals near g = 4.3 and 2 are due to adventitious iron and mediator dyes, respectively.
We (23) and others (24) have demonstrated that one can determine the exchange coupling constant, J, of diiron(III) centers by studying spectra in applied magnetic fields at variable temperature (The spectra are quite sensitive to J provided J < 90 cm-1.) A fit to an 8.0 T spectrum taken at 80 K (shown and analyzed in Fig. S2) suggests that J > 90 cm-1 (convention  ). The value of J suggests that the diiron center of CmlA is oxo-bridged (J = 180–260 cm-1 (21) rather than a hydroxo-bridged [J < 20 cm-1 (25)], which is in accord with the relatively intense optical spectrum of the diferric enzyme. The best fits to the dataset of oxidized CmlA were obtained by allowing two cluster sites in a 1∶1 ratio with slightly different ΔEQ-values (Table 1).
). The value of J suggests that the diiron center of CmlA is oxo-bridged (J = 180–260 cm-1 (21) rather than a hydroxo-bridged [J < 20 cm-1 (25)], which is in accord with the relatively intense optical spectrum of the diferric enzyme. The best fits to the dataset of oxidized CmlA were obtained by allowing two cluster sites in a 1∶1 ratio with slightly different ΔEQ-values (Table 1).
The shape of the fully reduced CmlA spectrum, Fig. 3A (trace 3), is best described by a superposition of two similar doublets in a 1∶1 ratio with high-energy lines that perfectly overlap at 2.66 mm/s. The parameters listed in Table 1 are typical of high-spin FeII with octahedral N/O coordination. In this respect, the CmlA center is quite similar to those observed for other diiron proteins, especially those rich in oxygen ligands (δ > 1.2 mm/s) such as MMO and toluene 4-monooxygenase (T4MO). The site with ΔEQ = 2.90 mm/s has a distinctly smaller δ value, suggesting that it has one more nitrogen ligand (probably histidine) than the other site. We have studied and analyzed Mössbauer spectra of reduced CmlA between 2 and 120 K in applied magnetic fields up to 8.0 T. Analysis shows that the two ferrous sites are antiferromagnetically coupled, with J ≈ 12 cm-1. For this J-value, the exchange coupling is of the same magnitude as the zero-field splittings of the two iron sites, resulting in a ground state of mixed-spin of predominantly S = 0 character. Our simulations indicate that reduced CmlA should not exhibit a parallel mode EPR signal at X-band [as observed for diferrous MMO, for example (26)], and indeed we have not observed such a signal. The observation of high-spin FeII upon reduction is in accord with the interpretation that CmlA contains a diiron center, ruling out an interpretation that the diamagnetic species observed in the as-isolated protein is due to a low-spin (S = 0) FeII site(s).
Table 1.
Quadrupole splittings and isomer shifts of oxidized and reduced CmlA
| Fe site 1 | Fe site 2 | |||||
| ΔEQ(mm/s)* | η† | δ(mm/s) | ΔEQ(mm/s) | η | δ(mm/s) | |
| Oxidized | − 1.30(4) | 0.5 | 0.51(1) | − 1.50(4) | 0.5 | 0.51(1) |
| Reduced | + 2.75(4) | 0 | 1.30(3) | + 2.90(4) | 0 | 1.21(3) |
*Signs of ΔEQ and values for η were determined from spectra taken at B = 8.0 T.
†η = (Vxx-Vyy)/Vzz is the asymmetry parameter of the electric field gradient tensor.
The as-isolated FeIIIFeIII state of CmlA is EPR silent (Fig. 3B, top spectrum) as expected from the Mössbauer spectra (Fig. 3A, spectrum 1). The addition of a substoichiometric amount of reductant to CmlA, in the presence of the mediator phenazine methosulfate, yields an S = 1/2 EPR signal with g-values of 1.95, 1.80, and 1.75 (Fig. 3B, spectrum 2). This type of rhombic signal, with all g-values < 2 is a distinguishing feature of the FeIIIFeII mixed-valent state of diiron cluster-containing proteins with O/N ligand donors; e.g. MMO, (25, 27). It arises from the S = 1/2 ground state of an antiferromagnetically coupled pair of high-spin FeIII (S = 5/2) and FeII (S = 2) ions. The signal from CmlA maximized during titration at ∼0.35 spins/2Fe. The reduction of CmlA with excess sodium dithionite in the presence of methyl viologen resulted in a state in which CmlA was EPR-silent as monitored in either perpendicular or parallel mode EPR (Fig. 3B, spectrum 3). The behavior monitored with EPR suggests that the two redox couples in the series FeIIIFeIII → FeIIIFeII → FeIIFeII are approximately equal.
The exchange coupling in the EPR active, partially reduced state of CmlA was quantified by determining the power saturation dependence of the g < 2 signal as a function of temperature. The signal intensity obeyed typical Curie law dependence, showing that it arises from the electronic ground state (Fig. S3). When the data were analyzed as described in SI Materials and Methods, the antiferromagnetic exchange coupling constant was determined to be J = 22 cm-1, a value suggesting that the oxo bridge of the diferric cluster is protonated to form a hydroxo bridge between the two Fe sites in the mixed-valent cluster. This serves to maintain a constant net charge. The EPR parameters of mixed-valent CmlA are shown to be similar to those of other diiron enzymes with hydroxo-bridged clusters in Table S2.
Stopped-Flow Kinetic Studies and Product Determination.
The substrate for antibiotic tailoring enzymes such as CmlA in NRPS pathways may be either the free amino acid or the amino acid after adenylation and subsequent covalent attachment on the thiolation (T) domain of the NRPS as a thioester. Important clues as to the substrate for CmlA derived from studies of the kinetic parameters of ATP pyrophosphate exchange by the NRPS CmlP with various amino acids by Walsh and coworkers (19). It was shown that among the amino acids tested, the highest rate of adenylation (A) domain exchange activity occurred with L-PAPA. This finding agreeds with in vivo isotope labeling experiments by Vining and coworkers that demonstrated L-PAPA as the specific amino acid precursor in chloramphenicol maturation (28, 29).
To directly elucidate the substrate specificity of CmlA, transient kinetic experiments were pursued in concert with product identification by HPLC/MS. Oxygen activation in oxygenase reactions requires a source of electrons (e.g., NADH), and often, additional reductase and electron transfer components. Examination of the sequenced chloramphenicol biosynthesis operon (20) failed to reveal the presence of a redox partner for the CmlA enzyme. Consequently, single turnover experiments utilizing chemically reduced CmlA were pursued to establish the reaction catalyzed by the enzyme.
Mixing reduced diferrous CmlA with buffer containing saturating O2 resulted in exceedingly slow oxidation of the diferrous cluster (k = 0.004 s-1 at 4 °C), (Fig. 4A, bottom trace and Inset), which was well approximated by a single exponential kinetic phase. The slow rate of iron oxidation was unperturbed upon mixing with free-amino acids, including L-PAPA and p-nitro-phenylalanine. No metabolites deriving from these potential substrates were detected in HPLC studies of the reaction products (Fig. 4B, top trace). These findings for small molecule substrates suggest the possibility that CmlA specifically hydroxylates L-PAPA only after loading onto the NRPS T domain.
Fig. 4.

Identification of the substrate and product of the CmlA reaction. (A) Stopped-flow absorption spectroscopy of the reaction of fully reduced CmlA with O2 in the presence of equimolar L-PAPA (bottom trace and Inset) or L-PAPA tethered to CmlP (B) LC-MS EIC chromatograms of the reaction of reduced CmlA with free L-PAPA and  (top two traces) or CmlP bound L-PAPA and
(top two traces) or CmlP bound L-PAPA and  (middle trace) or
(middle trace) or 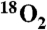 (bottom three traces) monitored at the indicated m/z (L-PAPA = 181, 16O-L-PAPS = 197, 18O-L-PAPS = 199).
(bottom three traces) monitored at the indicated m/z (L-PAPA = 181, 16O-L-PAPS = 197, 18O-L-PAPS = 199).
The CmlP NRPS was cloned, and heterologously coexpressed with a broad specificity phosphopanthetheine transferase (SfP) from Bacillus subtilis (30), and purified (Fig. S1). Loading of L-PAPA onto the T domain was accomplished as described earlier (19). When using a two-fold excess of the L-PAPA-loaded CmlP as the substrate, the rate constant for oxidation of diferrous CmlA increased more than 1,000-fold (k ∼ 12 s-1), indicating that this is likely to be the natural substrate (Fig. 4A, top trace). A similar degree of activation was not observed in stopped-flow studies with apoCmlP (not phosphopantetheinylated) or with holoCmlP in which L-PAPA was not covalently attached, indicating that the enhanced oxidation was not simply due to a structural rearrangement elicited upon binding of diferrous CmlA to the NRPS itself.
Chemical studies were pursued to determine the product of the CmlA-CmlP-L-PAPA reaction. Detection of the NRPS-linked amino acids and oxidation products was facilitated by the heterologous expression of a truncated construct of CmlP, consisting of only the A and T modules of the NRPS (CmlPAT). The ability of the CmlPAT construct to be phosphopantetheinylated and catalyze adenylation and successive aminoacyl transfer of L-PAPA to the T domain was verified by electrospray mass spectral analysis of the protein (Fig. S4), which showed the expected mass changes upon phosphopantetheinylation (+340 Da) and L-PAPA aminoacyl transfer (+164 Da), respectively (Fig. S4).
Reaction of reduced CmlA with L-PAPA-CmlPAT following by base-catalyzed release of the tethered amino acid and separation by HPLC showed a significant conversion (> 90%) of L-PAPA to a polar aromatic reaction product with a shorter retention time (compare Fig. 4B, top and bottom traces). This product was not observed upon addition of the diferric (as isolated) enzyme, establishing that the enzyme is active in the diferrous form. MS experiments revealed that the product has an m/z 16 amu larger than L-PAPA, consistent with a single hydroxylation at the β-position to form p-aminophenylserine (L-PAPS). When the reaction was conducted in an  atmosphere, all of the product was m/z 18 amu larger than L-PAPA (Fig. 4B, compare bottom two traces) confirming that the CmlA enzyme utilizes a monooxygenation mechanism, and that oxygen is derived from O2 rather than water.
atmosphere, all of the product was m/z 18 amu larger than L-PAPA (Fig. 4B, compare bottom two traces) confirming that the CmlA enzyme utilizes a monooxygenation mechanism, and that oxygen is derived from O2 rather than water.
Discussion
The metal quantification and spectroscopic studies described here show that CmlA as isolated contains a spin-coupled, oxo-bridged diiron cluster and specifically rule out other types of metal centers commonly found in oxygenases. It is evident from single-turnover kinetic studies that the enzyme is functional in the diferrous state and that its substrate is L-PAPA covalently bound to the peptidyl carrier T module of the NRPS CmlP. Characterization of the product from this reaction shows that one oxygen atom is incorporated exclusively from O2, demonstrating that CmlA is a monooxygenase. Together these studies show that a diiron monooxygenase is functional in an NRPS antibiotic assembly line, establishing a unique paradigm for tailoring reactions in these fascinating and often complex pathways. These and other aspects of this newly recognized enzyme class are discussed in the following sections.
The identification of CmlA as a catalytically distinct diiron monooxygenase prompted the search for homologous enzymes involved in a similar function. A BLAST query of bacterial genomic sequences revealed at least 40 highly conserved genes with > 40% identity to CmlA, including a number in known antibiotic biosynthesis pathways in which a β-hydroxyamino acid is formed. These include the biosynthetic gene clusters for the glycopeptide antibiotics A40926 (31), A47934 (5), the teicoplanin family of antibiotics (32, 33), and bleomycin (34).
Analysis of the primary sequence of CmlA and its homologs reveals the presence of two domains, although in some homologs a thioester reductase (which may take the place of the Cmlp reductase domain in the chloramphenicol operon) is additionally fused at the C-terminus (Fig. 5A). Otherwise, the 530 amino acid region homologous to the sequence of CmlA can be divided into two halves of roughly equal size. The N-terminal domain does not show significant similarity to any known enzyme structure. However, the C-terminal closely resembles the metallo β-lactamase family of enzymes. The overall αββα fold of metallo-β−lactamases represents a significant structural departure from the 4-α-helix bundle core structure that has been found for all oxygenases with diiron clusters in previous studies (18) with the possible exception of human deoxyhypusine hydroxylase, which may use a series of α-hairpin turns to support the cluster (35).
Fig. 5.

Overall structure of CmlA and its proposed mechanism. (A) Schematic organization of the family of enzymes homologous to CmlA. (B) Proposed scheme for diiron ligation in CmlA based on spectroscopy and sequence prediction (C) Proposed catalytic cycle for CmlA based on stopped-flow spectroscopy and  labeling experiments.
labeling experiments.
The metallo-lactamase motif provides a convenient structural framework for the binding of a dinuclear center, utilizing a His-X-His-X-Asp-His consensus sequence, which is also present in CmlA. Whereas the site typically comprises two zinc ions, minor variations in this site (and downstream residues) can affect metal binding specificity and support the binding of an Fe∶Zn heteronuclear cluster or a diiron cluster (reviewed in ref. 36). These subtle variations result in a wide variety of reactions, including hydrolysis, dioxygen reduction, and NO reduction. However, these enzymes are not known to catalyze substrate hydroxylation or to generate high-valent reactive oxygen species. The class of antibiotic biosynthesis diiron enzymes, typified by CmlA, therefore, represents functional and evolutionary expansion of the lactamase folding motif to include enzymes capable of dioxygen activation and subsequent monooxygenation chemistry.
A sequence alignment of the putative metal-binding site of CmlA and homologs is presented in Table 2 and compared to other members of the metallo-lactamase family. Whereas the high-degree of sequence conservation precludes the unambiguous assignment of metal-binding residues (a full sequence alignment of this region is presented in Fig. S5), it is likely that the metallo-lactamase signature sequence metal ligands also form the basic framework for the CmlA diiron site. The other three ligands of the metallo-lactamase cluster, His, Cys/Ser, His, are found downstream in the sequence at intervals of ∼60, ∼20, and ∼40 residues, respectively. In the case of the CmlA family, there are completely conserved Asp and Glu residues in the sequence at approximately the correct spacing. These additional carboxylate ligands would be consistent with the spectroscopic studies reported here and with the ligand environments of the known diiron cluster-containing oxygenases. Accordingly, the Mössbauer spectrum of reduced CmlA more closely resembles those of Δ9D, MMO, and RNR (1 N and 3–5 O ligands per iron) with a similar isomer shift δ≥1.24 (21, 25, 37). Diferrous enzymes with more nitrogen ligands such as hemerythrin (2 N and 3 O ligands per iron, δ = 1.14 mm/s) and AlkB (4 N ligands per iron, δ = 1.05–1.15 mm/s) produce substantially smaller isomer shift values (38, 39). Based on the conserved sequence motif of CmlA homologs and these spectroscopic parameters, a model for the dinuclear iron site is presented in Fig. 5B. Structural and mutagenesis studies will be required to confirm this tentative assignment. However, the more oxygen rich environment seems to play a role in promoting oxygen activation rather than hydrolysis or oxygen reduction chemistry.
Table 2.
Partial Clustal-W alignment of the proposed dinuclear binding motif in CmlA with other homologs in NRPS antibiotic biosynthetic pathways
| CmlA and homologs | ||||||||
| Species | Antibiotic | Consensus Sequence | ||||||
| Streptomyces venezuelae | chloramphenicol | H305-N-H-Q-D-H | x63 | E | x25 | D | x26 | E |
| Actinoplanes teichomycetius | teicoplanin | H301-G-H-S-D-H | x65 | E | x25 | D | x26 | E |
| Streptomyces verticillus | bleomycin | H316-A-H-H-D-H | x66 | E | x25 | D | x26 | E |
| Nonomuraea sp. ATCC 39727 | A40926 | H301-G-H-S-D-H | x65 | E | x25 | D | x26 | E |
| Streptomyces toyacaensis | A47934 | H301-G-H-S-D-H | x65 | E | x25 | D | x26 | E |
| Other dinuclear metal-binding enzymes with a lactamase fold | ||||||||
| Enzyme or family (function) | Metal specificity | Consensus sequence | ||||||
| Metallo-lactamase (hydrolysis) | Zn∶Zn | H30–120-x-H-x-D/C/R/S-H | x57–70 | H | x18–26 | C/S | x37–41 | H |
| Glyoxylase (hydrolysis) | Fe∶Zn | H54-x-H-x-D-H | x50 | H | x22 | D | x38 | H |
| ROO (oxygen reductase) | Fe∶Fe | H79-x-E-x-D-H | x61 | H | x22 | D | x60 | H |
| FprA (NO reductase) | Fe∶Fe | H81-x-E-x-D-H | x61 | H | x18 | D | x60 | H |
BLAST analysis was performed against GenBank bacterial sequences. Putative residues involved in metal ion binding are designated in red. For comparison, other dinuclear enzymes that utilize a lactamase folding motif with differing metal-binding specificities are also shown.
There is no clear chemical rationale for utilizing a diiron cluster for β-hydroxyamino acid formation, rather than mononuclear FeII of αKG-linked oxygenases or heme of P450s. Each of these metal centers is capable of producing the required metal-oxygen adducts with sufficient oxidizing potential to oxidize the ∼85 kcal/mol Cβ bond of L-amino acids. The reactive species for each of these families of enzymes has been shown to containing an Fe(IV)-oxo or Fe(IV)- μ-oxo moiety (11–13, 17, 18). Thus, it is likely that the diiron cluster of CmlA also forms such a species. Direct evidence for this species might be obtained from transient kinetic studies or the observation of partial solvent exchange once the O–O bond of O2 is broken. Neither approach has yet revealed evidence for a high-valent intermediate. This is not surprising because the only spectroscopically characterized dinuclear FeIV species [MMO compound Q (15, 17, 18)] has only been observed in the unique, completely sequestered, active site of that enzyme. Moreover, no exchange with solvent oxygen is observed, presumably due to solvent exclusion or short lifetime of the intermediate, or both (40, 41).
The stopped-flow and single-turnover studies show that CmlA requires the binding of the amino acid to the phosphopantetheine arm of the peptidyl carrier protein (PCP) domain and that this is both necessary and sufficient for the initiation of catalysis. A similar scenario is observed in diiron cluster- containing desaturases, in which the stearoyl substrate linked to the ACP carrier protein is an absolute requirement for catalysis (42). Another particularly relevant example is found in the αKG-dependent halogenases associated with NRPS pathways (43). Thus, these ternary protein–protein–substrate interactions appear to be very important in regulating oxygen reactivity in tailoring enzymes, presumably to ensure high specificity.
Catalysis is envisioned to involve a set of intermediates similar to those characterized in MMO (Fig. 5C) (15, 17, 18). Dioxygen is proposed to initially bind to the diferrous cluster to form a peroxodiferric intermediate, followed by O–O bond heterolysis to afford a high-valent intermediate (similar to the bis-μ-oxo-bridged dinuclear FeIV cluster of MMO compound Q). Previous studies of MMO show that this species would be capable of abstracting a hydrogen atom from Cβ of L-PAPA to form a substrate radical and a formal hydroxyl radical bound in the cluster. Subsequent hydroxyl radical rebound would form the T-domain-tethered L-PAPS.
In summary, we report a previously unrecognized and widely distributed family of monooxygenases that employ a unique mechanism for antibiotic tailoring. This expands the repertoire of diiron monooxygenases into natural product biosynthesis pathways. The family utilizes a protein fold not previously encountered in oxygen activating enzymes, offering the opportunity to investigate fundamental aspects of monooxygenase chemistry in a different protein environment. The present results may also lead to methods to recognize similar reactions in other antibiotic biosynthetic pathways and to design novel therapeutics.
Materials and Methods
Cloning and Enzyme Preparation.
The procedures for cloning, expression, and mutagenesis of the cmlA and cmlP genes are described in the SI Text along with the procedures for purification of CmlA and CmlP in native and posttranslationally modified forms.
Spectroscopy.
The procedures for optical, Mössbauer, and EPR spectroscopies of CmlA are described in the SI Text.
Stopped-Flow Kinetics.
Stopped-flow experiments were performed using an Applied Photophysics Ltd. SX.18.MV stopped-flow spectrophotometer. 100 μM CmlA was made anaerobic and reduced with one equivalent of Na2S2O4 and 0.5 equivalents methyl viologen before loading into a stopped-flow syringe. Reduced CmlA was rapidly mixed with an equimolar amount of either L-PAPA or L-PAPA-loaded CmlP equilibrated with O2 saturated buffer. The rate of oxidation of CmlA at 340 nm was fit to a single exponential kinetic process using Pro-K software (Applied Photophysics).
Single-Turnover Experiments and Product Determination.
L-PAPA-loaded CmlPAT and CmlA (400 μM each) were made anaerobic in a sealed 500 μL reaction volume. The CmlA was reduced with equimolar Na2S2O4 and 0.5 equivalents methyl viologen. Then, 1 mL of O2(g) ( or 97% isotopically enriched
or 97% isotopically enriched 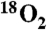 ) was added and the sample was allowed to react for 10 min. Samples were immediately quenched with 70% trichloroacetic acid to a final concentration of 10% and incubated on ice. The precipitated protein was collected at 16,000 rpm at 4 °C and the supernatant discarded. Protein pellets were washed twice with diethyl ether:ethanol (3∶1), and once with diethyl ether. The pellet was dried at 37 °C for 10 min and stored at -20 °C. Thioester cleavage was performed by dissolving the pellets with 500 μL 0.2 M potassium hydroxide for 15 min at 70 °C while stirring. The dissolved fraction was evacuated to dryness and the amino acid pellet dissolved in a minimal volume of H2O prior to LC-MS analysis. LC-MS analysis was performed on a Hewlett Packard 1100LC using a Waters HILIC silica column (2.1 × 150 mm × 3 μm) with an isocratic elution of acetonitrile:100 mM ammonium formate (87∶13) in 0.1% formic acid at a flow rate of 0.25 mL/ min. Mass monitoring was performed using a Bruker Microtof orthogonal quadrupole time of flight mass spectrometer scanning from 50 to 1000 m/z.
) was added and the sample was allowed to react for 10 min. Samples were immediately quenched with 70% trichloroacetic acid to a final concentration of 10% and incubated on ice. The precipitated protein was collected at 16,000 rpm at 4 °C and the supernatant discarded. Protein pellets were washed twice with diethyl ether:ethanol (3∶1), and once with diethyl ether. The pellet was dried at 37 °C for 10 min and stored at -20 °C. Thioester cleavage was performed by dissolving the pellets with 500 μL 0.2 M potassium hydroxide for 15 min at 70 °C while stirring. The dissolved fraction was evacuated to dryness and the amino acid pellet dissolved in a minimal volume of H2O prior to LC-MS analysis. LC-MS analysis was performed on a Hewlett Packard 1100LC using a Waters HILIC silica column (2.1 × 150 mm × 3 μm) with an isocratic elution of acetonitrile:100 mM ammonium formate (87∶13) in 0.1% formic acid at a flow rate of 0.25 mL/ min. Mass monitoring was performed using a Bruker Microtof orthogonal quadrupole time of flight mass spectrometer scanning from 50 to 1000 m/z.
Supplementary Material
Acknowledgments.
We thank Courtney Aldrich and Dan Wilson for providing the plasmids for Sfp coexpression and overexpression and information regarding protocols for NRPS loading. We also thank Joseph Dalluge for his assistance in developing mass spectrometry protocols for amino acid detection. This work was supported by National Institute of Health Grants GM40466 (J.D.L), GM40466-S1(J.D.L.), and EB001475 (to E.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1007953107/-/DCSupplemental.
References
- 1.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 2.Chen H, et al. Aminoacyl-S-enzyme intermediates in β-hydroxylations and α, β-desaturations of amino acids in heptide antibiotics. Biochemistry. 2001;40:11651–11659. doi: 10.1021/bi0115434. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Hubbard BK, O’Connor SE, Walsh CT. Formation of β-hydroxy histidine in the biosynthesis of nikkomycin antibiotics. Chem Biol. 2002;9:103–112. doi: 10.1016/s1074-5521(02)00090-x. [DOI] [PubMed] [Google Scholar]
- 4.Lu W, et al. Characterization of a regiospecific epivancosaminyl transferase GtfA and enzymatic reconstitution of the antibiotic chloroeremomycin. Proc Natl Acad Sci USA. 2004;101:4390–4395. doi: 10.1073/pnas.0400277101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pootoolal J, et al. Assembling the glycopeptide antibiotic scaffold: the biosynthesis of A47934 from Streptomyces toyocaensis NRRL15009. Proc Natl Acad Sci USA. 2002;99:8962–8967. doi: 10.1073/pnas.102285099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miao V, et al. The lipopeptide antibiotic A54145 biosynthetic gene cluster from Streptomyces fradiae. J Ind Microbiol Biot. 2006;33:129–140. doi: 10.1007/s10295-005-0028-5. [DOI] [PubMed] [Google Scholar]
- 7.Neary JM, et al. An asparagine oxygenase (AsnO) and a 3-hydroxyasparaginyl phosphotransferase (HasP) are involved in the biosynthesis of calcium-dependent lipopeptide antibiotics. Microbiology. 2007;153:768–776. doi: 10.1099/mic.0.2006/002725-0. [DOI] [PubMed] [Google Scholar]
- 8.Ciabatti R, et al. Ramoplanin (A-16686), a new glycolipodepsipeptide antibiotic. III. Structure elucidation. J Antibiot. 1989;42:254–267. doi: 10.7164/antibiotics.42.254. [DOI] [PubMed] [Google Scholar]
- 9.Strieker M, Nolan EM, Walsh CT, Marahiel MA. Stereospecific synthesis of threo- and erythro-beta -hydroxyglutamic acid during kutzneride biosynthesis. J Am Chem Soc. 2009;131:13523–13530. doi: 10.1021/ja9054417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Walsh CT. Coumarin formation in novobiocin biosynthesis: β-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI. Chem Biol. 2001;8:301–312. doi: 10.1016/s1074-5521(01)00009-6. [DOI] [PubMed] [Google Scholar]
- 11.Price JC, et al. The first direct characterization of a high-valent iron intermediate in the reaction of an alpha-ketoglutarate-dependent dioxygenase: A high-spin FeIV complex in taurine/alpha-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 12.Groves JT. High-valent iron in chemical and biological oxidations. J Inorg Biochem. 2006;100:434–447. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Makris TM, von Koenig K, Schlichting I, Sligar SG. The status of high-valent metal oxo complexes in the P450 cytochromes. J Inorg Biochem. 2006;100:507–518. doi: 10.1016/j.jinorgbio.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 14.Lee SK, et al. A transient intermediate of the methane monooxygenase catalytic cycle containing an FeIVFeIV cluster. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]
- 15.Baik M-H, Newcomb M, Friesner RA, Lippard SJ. Mechanistic studies on the hydroxylation of methane by methane monooxygenase. Chem Rev. 2003;103:2385–2419. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- 16.Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Finding intermediates in the O2 activation pathways of non-heme iron oxygenases. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S-K, Nesheim JC, Lipscomb JD. Transient intermediates of the methane monooxygenase catalytic cycle. J Biol Chem. 1993;268:21569–21577. [PubMed] [Google Scholar]
- 18.Wallar BJ, Lipscomb JD. Dioxygen activation by enzymes containing binuclear non-heme iron clusters. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 19.Pacholec M, Sello JK, Walsh CT, Thomas MG. Formation of an aminoacyl-S-enzyme intermediate is a key step in the biosynthesis of chloramphenicol. Org Biomol Chem. 2007;5:1692–1694. doi: 10.1039/b703356g. [DOI] [PubMed] [Google Scholar]
- 20.He J, Magarvey N, Piraee M, Vining LC. The gene cluster for chloramphenicol biosynthesis in Streptomyces venezuelae ISP5230 includes novel shikimate pathway homologues and a monomodular non-ribosomal peptide synthetase gene. Microbiology. 2001;147:2817–2829. doi: 10.1099/00221287-147-10-2817. [DOI] [PubMed] [Google Scholar]
- 21.Fox BG, Shanklin J, Somerville C, Münck E. Stearoyl-acyl carrier protein Δ9 desaturase from Ricinus communis is a diiron-oxo protein. Proc Natl Acad Sci USA. 1993;90:2486–2490. doi: 10.1073/pnas.90.6.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reem RC, Solomon EI. Spectroscopic studies of the binuclear ferrous active site of deoxyhemerythrin: Coordination number and probable bridging ligands for the native and ligand-bound forms. J Am Chem Soc. 1987;109:1216–1226. [Google Scholar]
- 23.Pikus JD, et al. Recombinant toluene-4-monooxygenase: Catalytic and Mössbauer studies of the purified diiron and Rieske components of a four-protein complex. Biochemistry. 1996;35:9106–9119. doi: 10.1021/bi960456m. [DOI] [PubMed] [Google Scholar]
- 24.Sage JT, et al. Mössbauer analysis of the binuclear iron site in purple acid phosphatase from pig allantoic fluid. J Am Chem Soc. 1989;111:7239–7247. [Google Scholar]
- 25.Fox BG, et al. Mössbauer, EPR, and ENDOR studies of the hydroxylase and reductase components of methane monooxygenase from Methylosinus trichosporium OB3b. J Am Chem Soc. 1993;115:3688–3701. [Google Scholar]
- 26.Hendrich MP, Münck E, Fox BG, Lipscomb JD. Integer-spin EPR studies of the fully reduced methane monooxygenase hydroxylase component. J Am Chem Soc. 1990;112:5861–5865. [Google Scholar]
- 27.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 28.McGrath RM, et al. p-Aminophenylalanine and threo-p-aminophenylserine. Specific precursors of chloramphenicol. Biochem Biophys Res Commun. 1967;29:576–581. doi: 10.1016/0006-291x(67)90524-4. [DOI] [PubMed] [Google Scholar]
- 29.Siddiqueullah M, McGrath R, Vining LC. Biosynthesis of chloramphenicol. II. p-Aminophenylalanine as a precursor of the p-nitrophenylserinol moiety. Can J Biochem. 1967;45:1881–1889. doi: 10.1139/o67-221. [DOI] [PubMed] [Google Scholar]
- 30.Lambalot RH, et al. A new enzyme superfamily—the phosphopantetheinyl transferases. Chem Biol. 1996;3:923–936. doi: 10.1016/s1074-5521(96)90181-7. [DOI] [PubMed] [Google Scholar]
- 31.Sosio M, et al. The gene cluster for the biosynthesis of the glycopeptide antibiotic A40926 by Nonomuraea species. Chem Biol. 2003;10:541–549. doi: 10.1016/s1074-5521(03)00120-0. [DOI] [PubMed] [Google Scholar]
- 32.Li T-L, et al. Biosynthetic gene cluster of the glycopeptide antibiotic teicoplanin. Characterization of two glycosyltransferases and the key acyltransferase. Chem Biol. 2004;11:107–119. doi: 10.1016/j.chembiol.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 33.McCafferty DG, et al. Chemistry and biology of the ramoplanin family of peptide antibiotics. Biopolymers. 2002;66:261–284. doi: 10.1002/bip.10296. [DOI] [PubMed] [Google Scholar]
- 34.Du L, et al. The biosynthetic gene cluster for the antitumor drug bleomycin from Streptomyces verticillus ATCC15003 supporting functional interactions between nonribosomal peptide synthetases and a polyketide synthase. Chem Biol. 2000;7:623–642. doi: 10.1016/s1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- 35.Vu VV, et al. Human deoxyhypusine hydroxylase, an enzyme involved in regulating cell growth, activates O2 with a nonheme diiron center. Proc Natl Acad Sci USA. 2009;106:14814–14819. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomes CM, et al. Functional control of the binuclear metal site in the metallo-beta -lactamase-like fold by subtle amino acid replacements. Protein Sci. 2002;11:707–712. doi: 10.1110/ps.31202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynch JB, Juarez-Garcia C, Münck E, Que L., Jr Mössbauer and EPR studies of the binuclear iron center in ribonucleotide reductase from Escherichia coli. A new iron-to-protein stoichiometry . J Biol Chem. 1989;264:8091–8096. [PubMed] [Google Scholar]
- 38.Kurtz DM., Jr Oxo- and hydroxo-bridged diiron complexes: A chemical perspective on a biological unit. Chem Rev. 1990;90:585–606. [Google Scholar]
- 39.Shanklin J, et al. Mössbauer studies of alkane ω-hydroxylase: Evidence for a diiron cluster in an integral-membrane enzyme. Proc Natl Acad Sci USA. 1997;94:2981–2986. doi: 10.1073/pnas.94.7.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nesheim JC, Lipscomb JD. Large isotope effects in methane oxidation catalyzed by methane monooxygenase: Evidence for C-H bond cleavage in a reaction cycle intermediate. Biochemistry. 1996;35:10240–10247. doi: 10.1021/bi960596w. [DOI] [PubMed] [Google Scholar]
- 41.Ruzicka F, Huang DS, Donnelly MI, Frey PA. Methane monooxygenase catalyzed oxygenation of 1,1-dimethylcyclopropane. Evidence for radical and carbocationic intermediates. Biochemistry. 1990;29:1696–1700. doi: 10.1021/bi00459a005. [DOI] [PubMed] [Google Scholar]
- 42.Reipa V, Shanklin J, Vilker V. Substrate binding and the presence of ferredoxin affect the redox properties of the soluble plant Δ9-18∶0-acyl carrier protein desaturase. Chem Commun. 2004:2406–2407. doi: 10.1039/b409595b. [DOI] [PubMed] [Google Scholar]
- 43.Matthews ML, et al. Substrate-triggered formation and remarkable stability of the C–H bond-cleaving chloroferryl intermediate in the aliphatic halogenase, SyrB2. Biochemistry. 2009;48:4331–4343. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.