

Abstract
The extracellular signal-regulated kinase (ERK) isoforms, ERK1 and ERK2, are believed to be key signaling molecules in nociception and nociceptive sensitization. Studies using inhibitors targeting the shared ERK1/2 upstream activator, mitogen-activated protein kinase kinase (MEK), and transgenic mice expressing a dominant-negative form of MEK have established the importance of ERK1/2 signaling. However, these techniques do not discriminate between ERK1 and ERK2. To dissect the function of each isoform in pain, we used mice with a targeted genetic deletion of ERK1 [ERK1 knock-out (KO)] to test the hypothesis that ERK1 is required for behavioral sensitization in rodent pain models. Despite activation (phosphorylation) of ERK1 after acute noxious stimulation and in models of chronic pain, we found that ERK1 was not required for formalin-induced spontaneous behaviors, complete Freund's adjuvant-induced heat and mechanical hypersensitivity, and spared nerve injury-induced mechanical hypersensitivity. However, ERK1 deletion did delay formalin-induced long-term heat hypersensitivity, without affecting formalin-induced mechanical hypersensitivity, suggesting that ERK1 partially shapes long-term responses to formalin. Interestingly, ERK1 deletion resulted in elevated basal ERK2 phosphorylation. However, this did not appear to influence nociceptive processing, since inflammation-induced ERK2 phosphorylation and pERK1/2 immunoreactivity in spinal cord were not elevated in ERK1 KO mice. Additionally, systemic MEK inhibition with SL327 (α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)benzeneacetonitrile) attenuated formalin-induced spontaneous behaviors similarly in wild-type and ERK1 KO mice, indicating that unrelated signaling pathways do not functionally compensate for the loss of ERK1. Together, these results suggest that ERK1 plays a limited role in nociceptive sensitization and support a predominant role for ERK2 in these processes.
Introduction
Extracellular signal-regulated kinases (ERKs), ERK1 and ERK2, are mitogen-activated protein kinases (MAPKs) (Pearson et al., 2001) that have been identified as critical players in sensitization to noxious stimuli after peripheral inflammation and nerve damage (Ji et al., 1999, 2002; Karim et al., 2001; Ciruela et al., 2003; Obata et al., 2003; Song et al., 2005). A variety of acute noxious stimuli and chronic pain models result in ERK1/2 activation (phosphorylation) at many levels of the nociceptive sensory system including dorsal root ganglion (DRG), spinal cord, and amygdala (Ji et al., 2009). The use of inhibitors that block activation of both ERK1 and ERK2 by inhibiting their shared upstream MAP kinase kinases (MEK1/2) and transgenic expression in neurons of a dominant-negative form of MEK1, which suppresses MEK1/2–ERK1/2 signaling, have demonstrated that ERK1/2 is necessary for nociceptive sensitization (Ji et al., 1999, 2002, 2009; Karim et al., 2001, 2006; Song et al., 2005; Hu et al., 2006; Seino et al., 2006).
Although much is known about MEK1/2–ERK1/2 signaling, little is known about the specific functions of each ERK isoform. Functional redundancy has been a working model because the isoforms are 90% homologous (Boulton et al., 1991) and no isoform-specific inhibitors currently exist. However, there is emerging evidence that ERK1 and ERK2 may be functionally distinct. ERK1 knock-outs (ERK1 KOs) are viable but exhibit behavioral abnormalities correlated with altered synaptic plasticity in striatum (Pagès et al., 1999; Selcher et al., 2001; Mazzucchelli et al., 2002), whereas ERK2 knock-outs are embryonic lethal at embryonic day 8.5 (Krens et al., 2006). Alternative methods for targeting ERK2, including reduced expression from a hypomorphic mutant allele and conditional deletion in telencephalon, have revealed a requirement for ERK2 in several learning and memory paradigms (Satoh et al., 2007; Samuels et al., 2008). In cell culture, genetic targeting or RNA interference (RNAi) experiments suggest specific roles for ERK1 and ERK2 (Mazzucchelli et al., 2002; Vantaggiato et al., 2006). Evidence to support this hypothesis includes the observations that ERK1 exclusively interacts with the MEK–ERK signaling scaffold MP1 (Schaeffer et al., 1998) and the fact that differences in amino acid sequence between ERK1 and ERK2 occur in domains that may affect MEK1/2 binding, ERK dimerization, and subcellular localization (Boulton et al., 1991; Zhang et al., 1994; Cobb and Goldsmith, 2000). Indeed, ERK1 and ERK2 have different rates of shuttling between the cytoplasm and nucleus because of sequence differences in the N terminus (Marchi et al., 2008).
Currently, the importance of ERK1 in nociceptive sensitization remains unknown. Therefore, we tested the necessity of ERK1 in acute noxious sensitization and in models of chronic inflammatory and neuropathic pain using ERK1 KO mice. Although ERK1 is activated in these models, genetic deletion of ERK1 had a minimal impact on these ERK-dependent behaviors. Interestingly, ERK1 deletion increased basal ERK2 phosphorylation without affecting inflammation-induced changes in ERK2 phosphorylation. Our observations indicate that ERK1 is not required for nociceptive sensitization and suggest that ERK2 plays a predominant role.
Materials and Methods
Animals.
All experiments were performed according to the guidelines of the National Institutes of Health and were approved by the Animal Care and Use Committee of Washington University School of Medicine. Mice were housed with a 12 h light/dark cycle and ad libitum access to food and water. Targeted deletion of ERK1 was achieved by homologous recombination in a process described in detail previously (Nekrasova et al., 2005). Briefly, the targeting vector deleted exons 1–6 of mapk3, which includes the kinase active site and phosphorylation loop, and replaced them with a Neo cassette flanked with loxP sites. Embryonic stem cells from 129 Sv mice were transfected and selected for homologous recombination. Successfully targeted clones were injected into CD1 blastocysts, which were implanted in pseudopregnant CD1 females. Chimeras with germ-line transmission were obtained on the CD1 background. The mapk3 mutation was subsequently transferred to the C57BL/6 background by backcrossing >10 times. C57BL/6 mapk3 mutant heterozygotes were crossed, producing mice heterozygous for the mapk3 mutant allele, homozygous for the mutant allele (ERK1 KO), and homozygous for the wild-type mapk3 allele (WT). Seven- to 9-week-old ERK1 KO and WT male littermates from these crosses were used in all experiments. The experimenter was blind to the genotype and treatment in all behavioral experiments.
Measurement of heat and mechanical thresholds.
All behavioral tests were performed in rooms isolated from other activity and with white noise present. Tests were conducted at room temperature (∼25°C) during the second half of the light cycle. Before testing, mice were allowed to acclimate to the behavioral room and apparatus for 2–3 h. After acclimation, measurements of paw withdrawal thresholds to heat and mechanical stimuli were performed. Measurements were consistently made when mice were calm. Modular clear Plexiglas behavior chambers (width, 10 cm; length, 10 cm; height, 15 cm), which could be placed on different floor surfaces, were used with one mouse per chamber. Responses to heat were measured using a modified Hargreaves test (Hargreaves et al., 1988). Mice were placed in behavior chambers on a 390G Plantar Test Apparatus (IITC Life Science) with a glass plate floor heated to ∼30°C. Radiant heat was applied to the plantar surface of the hindpaw using a focused beam of light with an active intensity of 14% maximal, a cutoff of 20 s, and idle intensity of 5% maximal. The 100% beam intensity heats to ∼250°C. To measure paw withdrawal latency, a timer was automatically initiated at the onset of heat application (active intensity), and on paw withdrawal, the heat source was returned to idle intensity automatically stopping the timer. Five independent measurements, separated by 15 min alternating between right and left paws, were used to determine baseline withdrawal latency. For time points hours after paw injections, two independent measurements were averaged. On subsequent days, three independent measurements were used. To measure responses to mechanical stimuli, mice were placed in behavioral chambers on an elevated wire mesh floor. Calibrated von Frey filaments (North Coast Medical) were then applied through the mesh to the plantar surface of the hindpaw between the anterior and posterior foot pads, except for the spared nerve injury (SNI) experiment in which filaments were applied to the lateral surface of the hindpaw in sural nerve territory. Filaments were pressed into the skin until the filament bent, held for ∼1 s, and then removed. Withdrawal of the hindpaw from the mesh indicated a positive withdrawal response. Each filament was applied five times at a frequency of ∼0.5 Hz. The lightest filament (0.008 g) was initially used followed by progressively heavier filaments until the filament that evoked three withdrawals out of five applications was reached. The calibrated force of the filament was recorded as the paw withdrawal threshold. Three to five independent measurements were averaged to yield the baseline threshold for each hindpaw. Hours after paw injections, a single measurement was used, whereas on subsequent days, three independent measurements were averaged for each time point.
Formalin-induced spontaneous behaviors.
To measure spontaneous behavioral responses to formalin injection, mice were acclimated in behavior chambers with a Plexiglas floor for at least 1 h. Mice were then injected subcutaneously with 10 μl of 2% formalin (Sigma-Aldrich) in sterile 0.9% NaCl into the plantar surface of the hindpaw and immediately returned to the behavior chamber. Total time spent licking and lifting the injected paw was recorded in 5 min bins for 1 h. In experiments with the MEK inhibitor, α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)benzeneacetonitrile (SL327) (Tocris Bioscience), mice were allowed to acclimate as above, injected with SL327 or vehicle (DMSO), and then returned to the behavior chamber. Thirty minutes later, mice were injected into the plantar surface of the hindpaw with 3.5% formalin, returned to the behavior chamber, and video recorded from below with a webcam (Logitech) at a resolution of 960 × 720 for 1 h without the experimenter present. Subsequently, videos were scored for time spent licking, lifting, and flinching the injected paw.
Formalin and complete Freund's adjuvant-induced hypersensitivity.
Formalin-induced hypersensitivity to heat and mechanical stimuli was used as a model of persistent pain. Heat hypersensitivity to formalin was assayed by measuring baseline paw withdrawal latencies using a Hargreaves-style apparatus as described above on day 1. On day 2, mice were acclimated (2–3 h) and injected with 10 μl of 2% formalin (in sterile 0.9% NaCl). Threshold measurements from the injected (ipsilateral) and uninjected (contralateral) paws were made at 1, 2, and 3 h after injection. On subsequent postinjection days, mice were first allowed to acclimate (2–3 h), and then paw withdrawal thresholds were obtained. To measure mechanical hypersensitivity to formalin, a similar time course was performed except von Frey filaments were used as detailed above. Intraplantar injections of 10 μl of 3.5% formalin (in sterile 0.9% NaCl) were made and subsequent threshold measurements were obtained. A higher concentration of formalin was used in mechanical experiments because 2% formalin did not produce robust mechanical hypersensitivity in either WT or ERK1 KO mice (data not shown). Intraplantar injection of complete Freund's adjuvant (CFA) (10 μl, 1.0 mg/ml; Sigma-Aldrich) was used as a chronic inflammatory pain model. The same experimental design used in formalin-induced hypersensitivity experiments was used to assay mechanical and heat hypersensitivity after CFA injection.
Spared nerve injury model.
To measure mechanical hypersensitivity after spared nerve injury (Decosterd and Woolf, 2000), paw withdrawal thresholds from the lateral surface of each hindpaw were obtained using von Frey filaments with the up–down method (Chaplan et al., 1994). Baseline thresholds were measured on day 1 after 2–3 h of acclimation on wire mesh. On day 2, mice were anesthetized with pentobarbital, a small incision in the thigh was made, and the three main branches of the sciatic nerve were exposed. The common peroneal and tibial nerves were ligated with 8-0 silk suture and cut distal to ligation. One millimeter of distal nerve stump was removed, and the surgical wound was closed with 7 mm surgical clips. After 3 d of recovery, clips were removed. To measure mechanical hypersensitivity after surgery, mice were acclimated to the mesh (2–3 h), and mechanical withdrawal thresholds were measured from the sural nerve territory (lateral surface of hindpaw).
Sample preparation and Western blot analysis.
Mice were placed individually in cages and allowed to acclimate for 2–3 h to approximate behavioral testing conditions. For formalin and CFA experiments, mice were then injected subcutaneously into the plantar surface of the hindpaw with 10 μl of 3.5% formalin or vehicle (0.9% NaCl) in formalin experiments or 10 μl of CFA or 0.9% NaCl in CFA experiments. Mice were then killed using a guillotine, and spinal cords were rapidly removed by hydraulic extrusion. In dose-finding experiments with SL327, mice were acclimated as above, injected with SL327 or vehicle (DMSO), returned to their cage for 30 min, injected with 3.5% formalin, and killed 3 min after paw injection. Spinal cord levels lumbar 3–6 (L3–L6) were isolated, and ipsilateral and contralateral sides were separated. For CFA experiments, the dorsal half was isolated and flash-frozen on dry ice. For formalin experiments, the entire dorsoventral extent was flash-frozen. In experiments with uninjected mice, the lumbar enlargement was isolated and flash-frozen without additional dissection. In naive subjects, immediately after spinal cord extraction thoracic and lumbar DRGs were dissected in ice-cold PBS (Mediatech), pooled into a single sample, and immediately homogenized. Frozen spinal cord and fresh DRG samples were homogenized using a Dounce homogenizer in ice-cold buffer containing 20 mm Tris, pH 7.5, 1 mm EDTA, 1 mm Na4P2O7, 25 μg/ml aprotinin (Sigma-Aldrich), 25 μg/ml leupeptin (Sigma-Aldrich), 100 μm PMSF (Roche Applied Science), 1 μg/ml microcystin LR (Enzo Life Science), and 1 mm Na3VO4 (Sigma-Aldrich) in Milli-Q distilled water. Protein concentrations were determined with the BCA protein assay (Pierce Biotechnology) and SmartSpec 3000 spectrophotometer (Bio-Rad).
Protein homogenates were size separated by SDS-PAGE. For pERK/ERK or ERK/actin Westerns, 10 μg of protein was used. For c-Jun N-terminal kinase (JNK) and p38 Westerns, 20 μg of DRG homogenate was used, and 20–40 μg of spinal cord homogenate was used. For formalin and CFA experiments, precast Tris-HCl 4% stacking and 10% separating polyacrylamide midi-gels were used (Bio-Rad). For experiments with naive mice, 5% stacking and 10% separating polyacrylamide mini-gels were prepared based on the Laemmli system (Coligan et al., 2001). Gels were transferred to nitrocellulose membranes using a wet transfer system (Bio-Rad). Membranes were blocked for 1 h at room temperature (RT) with Odyssey blocking buffer (LI-COR Biosciences) and then simultaneously probed with two primary antibodies from different host species for 1 h at RT diluted in blocking buffer unless otherwise noted. Primary antibodies used include anti-pERK1/2 (mouse monoclonal; 1:1000; Cell Signaling), anti-ERK1/2 (rabbit polyclonal; 1:1000; Cell Signaling), anti-actin (1:2000; mouse monoclonal; Sigma-Aldrich), anti-JNK (rabbit polyclonal; 1:1000; Cell Signaling), anti-pJNK (mouse monoclonal; 1:1000; Cell Signaling), anti-p38 (rabbit polyclonal; 1:1000; Cell Signaling), anti-p-p38 (mouse monoclonal; 1:500; primary incubation for 4 h at RT; Cell Signaling), and anti-β-tubulin (mouse monoclonal; 1:20,000; Sigma-Aldrich). After several washes, membranes were then incubated for 1 h at RT with fluorescently conjugated secondary antibodies goat anti-rabbit IRDye800 (1:20,000; LI-COR Biosciences) and goat anti-mouse Alexa Fluor 680 (1:20,000; Invitrogen), which were detected with the Odyssey Infrared Fluorescence Imaging System (LI-COR Biosciences) for simultaneous detection of 700 and 800 nm wavelength fluorescent emissions corresponding to mouse and rabbit primary antibodies, respectively. Densitometry of bands in 700 (pERK1/2, pJNK, p-p38, β-tubulin, or actin) and 800 (ERK1/2, JNK, p38) channels was performed using the Odyssey Infrared Fluorescence Imaging System software (LI-COR Biosciences).
Immunohistochemistry.
Mice were placed individually in cages and allowed to acclimate for 2–3 h to approximate behavioral testing conditions. Three minutes after formalin injection (10 μl; 3.5% in 0.9% NaCl), mice were overdosed with Avertin anesthesia (1.25% working solution in 0.9% NaCl diluted from a stock of 1.0 mg/ml 2,2,2-tribromoethanol in tert-amyl alcohol, both from Sigma-Aldrich) by intraperitoneal injection. Mice were then transcardially perfused at a rate of 15–20 ml per minute with warm (∼37°C) PBS and then 150 ml of ice-cold paraformaldehyde (4% in PBS). Spinal cord was isolated, rinsed in PBS overnight at 4°C, and then cryoprotected in 30% sucrose/PBS for 2 d at 4°C. Transverse 30 μm sections were obtained using a cryostat and collected in ice-cold PBS. Floating sections were then stained for pERK1/2 using 3,3′-diaminobenzidine (DAB) secondary detection. All incubations were performed with gentle agitation at RT, unless otherwise noted, with several washes between steps. Endogenous peroxidase activity was suppressed by incubating in 10% methanol, 0.3% H2O2 (in PBS) for 30 min. Sections were blocked with 1% normal goat serum/0.02% Triton X-100 in PBS (1% NGST) for 1 h, and then incubated in anti-pERK1/2 (1:3000 in 1% NGST; rabbit polyclonal; Cell Signaling) overnight at 4°C. Sections were removed from 4°C and allowed to warm to RT for 1 h, after which sections were incubated with biotinylated goat anti-rabbit IgG (1:200 in 1% NGST; Vector Laboratories) for 1 h and then extrAvidin peroxidase (1:1000 in 1% NGST; Sigma-Aldrich) for 1 h. Finally, sections were incubated for 6 min with a DAB solution supplemented with nickel prepared from a DAB substrate kit (Vector Laboratories). Sections were placed on microscope slides and dehydrated by exposing slices to ethanol/water solutions with progressively increasing concentrations of ethanol and finally CitriSolv (Thermo Fisher Scientific). After dehydration, slides were coverslipped with distrene plasticizer xylene mounting media (Electron Microscopy Sciences). Bright-field and dark-field images were captured with a 10× objective using an Olympus compound microscope with a Photometrics CoolSnap HQ monochrome camera. Bright- and dark-field images from the same field were then superimposed in Photoshop (version 7.0; Adobe) and the lamina II–III boundary was traced on the dark-field image. The boundary was then applied to the bright-field image, and the resultant image was used to count positively staining cell profiles in lamina I–II using NIH ImageJ, version 1.42. During imaging and quantification, the experimenter was blind to genotype. Color images were obtained using a NanoZoomer automated microscope (Hamamatsu).
Statistical analysis.
Statistical analysis was performed using Excel (Microsoft) and Prism (GraphPad Software). Details of specific statistical tests are included in Results. Graphs showing means and SEM were graphed using Prism.
Results
Baseline sensory function remains normal in ERK1 KO mice
Although a large body of evidence indicates that ERK1/2 is important in nociception, little is known about the relative contribution of each individual isoform. To assess the role of ERK1 in nociceptive sensory function, we used mice with a targeted deletion of mapk3 (ERK1 KO), in which part of exon 1 and all of exons 2–6 are replaced with a pPGKneolox cassette (Nekrasova et al., 2005). Deletion of ERK1 was confirmed by Western blots of spinal cord and DRG tissue obtained from ERK1 KO mice (Fig. 1A,B). As expected, ERK1 immunoreactivity was undetectable in ERK1 KO mice (Fig. 1A,B) (unpaired t test, ***p < 0.001). Deletion of ERK1 had no effect on ERK2 protein expression (unpaired t test). These data confirm previous findings that ERK1 protein is eliminated from the CNS (Selcher et al., 2001; Nekrasova et al., 2005) and extends these findings to the peripheral nervous system by demonstrating ERK1 deletion without altered ERK2 expression.
Figure 1.
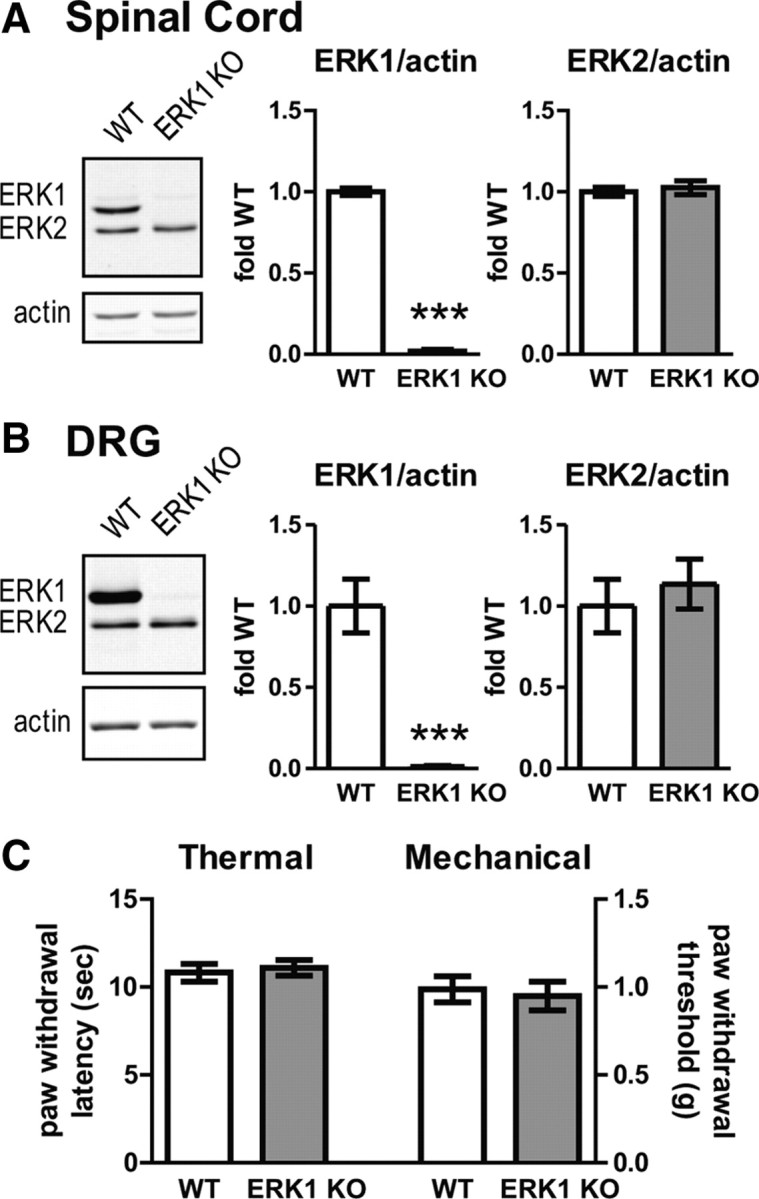
Disruption of mapk3 eliminates ERK1 expression in spinal cord and dorsal root ganglion without affecting heat or light touch thresholds. A, Spinal cords were isolated from WT (n = 13) and ERK1 KO (n = 14) littermates and analyzed by Western blot for ERK1/2 and the loading control, actin. To quantify the intensity of ERK bands, integrated intensities of each isoform were divided by actin integrated intensities and plotted as fold WT. B, DRGs from WT (n = 7) and ERK1 KO (n = 8) were dissected and analyzed by quantitative Western blot as in A. In both spinal cord and DRGs, there is a dramatic reduction in ERK1 expression without a significant change in ERK2 protein levels. C, No differences were observed in heat thresholds obtained by applying radiant heat to the hindpaws of WT (n = 10) and ERK1 KO (n = 10) littermates using a Hargreaves-style apparatus. No differences were observed in hindpaw mechanical withdrawal thresholds obtained with von Frey filaments from WT (n = 14) and ERK1 KO (n = 13) littermates. Error bars indicate SEM. ***p < 0.001, unpaired t test.
After confirming the absence of ERK1 in important nociceptive anatomic loci, we measured the baseline sensory function of ERK1 KO mice. The latency to paw withdrawal from a radiant heat source was comparable between ERK1 KO and WT mice (Fig. 1C). Additionally, paw withdrawal thresholds to mechanical stimulation with von Frey filaments in ERK1 KO mice were similar to WT littermates (Fig. 1C). In both cases, there was no statistically significant difference between WT and ERK1 KO mice (unpaired t test). Both sensory tests rely on reflexive withdrawal of the hindlimb to measure threshold responses. Since alterations in motor function secondary to ERK1 deletion could confound the results of these tests, we also screened the ERK1 KO mice for motor abnormalities. We found no difference in the performance of ERK1 KO and WT mice on the accelerating rotarod task or in exploratory behaviors in an open-field task (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Overall, these results indicate that genetic disruption of ERK1 does not affect baseline withdrawal responses from heat or mechanical stimuli.
ERK1 is not required for first or second phases of the formalin test
Previous reports indicate that spinal ERK1 is activated (phosphorylated) within minutes after intraplantar injection of the noxious chemical formalin and that inhibition of spinal ERK1/2 using intrathecally applied MEK inhibitors reduces the second phase of spontaneous formalin-induced nociceptive behaviors (Ji et al., 1999; Karim et al., 2001). Therefore, we hypothesized that ERK1 was necessary for full expression of spontaneous nociceptive behaviors after formalin injection. Surprisingly, ERK1 KO and WT littermates have indistinguishable formalin responses (Fig. 2). Both WT and ERK1 KO show a stereotypical biphasic response characterized by an immediate increase in licking and lifting (first phase), which subsides momentarily but gradually increases and persists over the after ∼30 min (second phase). There is no statistically significant difference between genotypes when considering the complete time course of the formalin response [two-way repeated-measures (RM) ANOVA: F = 0.64, DFn = 1, DFd = 308, p = 0.4304]. Moreover, the amount of time spent in nociceptive behaviors in the first phase (0–5 min) and the second phase (5–40 min) is similar between the genotypes (Fig. 2, unpaired t test). Overall, these data indicate that, even in the absence of ERK1, formalin can induce robust spontaneous behaviors that have previously been shown to require ERK1 and/or ERK2, suggesting a specific role for ERK2 in this process.
Figure 2.

Formalin-induced spontaneous nociceptive responses are preserved in ERK1 KO mice. The amount of time spent in spontaneous nociceptive behaviors after 2% formalin injection (licking and lifting of the injected paw) was recorded from WT (n = 16) and ERK1 KO (n = 14) littermates and plotted in 5 min bins. No statistically significant difference between WT and ERK1 KO was detected over the course of the 60 min trial or in total time spent in the first phase (0–5 min, bottom left graph) or second phase (5–40 min, bottom right graph) of nociceptive behaviors. Error bars represent SEM.
ERK1 KO mice develop robust hypersensitivity in long-term inflammatory and neuropathic pain models
ERK1 and/or ERK2 contribute to the development and maintenance of chronic inflammatory and neuropathic pain (Ji et al., 2009). To directly assess the contribution of ERK1 from all tissues to CFA-induced hypersensitivity, ERK1 KO mice were injected with CFA into the hindpaw and examined for hypersensitivity to radiant heat and mechanical stimuli over time. ERK1 KO mice became robustly hypersensitive to radiant heat applied to the paw ipsilateral to CFA injection (Fig. 3A). The degree of hypersensitivity in ERK1 KO mice was comparable with WT littermate controls (two-way RM ANOVA: F = 1.38, DFn = 1, DFd = 90, p = 0.2562). In the contralateral paw, there was no statistically significant hypersensitivity over time (two-way RM ANOVA: F = 1.96, DFn = 5, DFd = 90, p = 0.0923). Moreover, there was no statistically significant difference between the genotypes (two-way RM ANOVA: F = 2.26, DFn = 1, DFd = 90, p = 0.1504). CFA injection also produced dramatic hypersensitivity to mechanical stimulation that was indistinguishable between WT and ERK1 KO littermates (Fig. 3B). There was no statistically significant difference between genotypes in withdrawal thresholds in the ipsilateral paw (two-way RM ANOVA: F = 0.86, DFn = 1, DFd = 70, p = 0.3761) or the contralateral paw (two-way RM ANOVA: F = 4.41, DFn = 1, DFd = 70, p = 0.062). Overall, these data indicate that ERK1 KO mice show normal sensitization to mechanical and heat stimuli in the CFA model of long-term inflammatory pain.
Figure 3.

ERK1 is not necessary for CFA-induced hypersensitivity. A, To measure CFA-induced heat hypersensitivity, WT (n = 10) and ERK1 KO (n = 10) littermates were injected with CFA subcutaneously into the plantar surface of the hindpaw. Paw withdrawal latencies to a radiant heat stimulus were then obtained from both hindpaws, ipsilateral and contralateral to CFA injection, using a Hargreaves-style apparatus. Data were normalized to preinjection baseline paw withdrawal latencies within subject and plotted as percentage baseline. B, To measure CFA-induced mechanical allodynia, WT (n = 6) and ERK1 KO (n = 6) littermates were injected with CFA, and paw withdrawal thresholds to light touch were measured using von Frey filaments. In all cases, there was no statistically significant difference between genotypes. Error bars indicate SEM.
The mechanisms driving nociceptive sensitization in neuropathic pain are often distinct from inflammatory pain. Indeed, the time course and anatomic localization of ERK1/2 phosphorylation is different between neuropathic (Obata et al., 2004; Zhuang et al., 2005) and inflammatory (Ji et al., 2002; Obata et al., 2003) pain models. However, the relative importance of ERK1 or ERK2 in neuropathic pain remains unknown. To address this issue, we measured the responses of ERK1 KO mice in the SNI model, in which two branches of the sciatic nerve are transected (tibial and common peroneal branches), leaving the third branch, the sural nerve, intact. In skin innervated by the sural nerve ipsilateral to SNI, ERK1 KO and WT mice developed equivalent, robust mechanical hypersensitivity (Fig. 4). In the contralateral paw, withdrawal thresholds remained unchanged relative to baseline. There is no statistically significant difference between ERK1 KO and WT littermates in withdrawal thresholds ipsilateral (two-way RM ANOVA: F = 0.05, DFn = 1, DFd = 102, p = 0.8263) or contralateral (two-way RM ANOVA: F = 0.89, DFn = 1, DFd = 102, p = 0.3591) to the nerve injury. These results indicate that ERK1 KO mice are equally susceptible to SNI-induced mechanical hypersensitivity as WT littermates, suggesting that ERK1 is not required in this model of neuropathic pain.
Figure 4.

ERK1 KO and WT littermates develop similar mechanical hypersensitivity after peripheral nerve injury. One day after baseline measurements, WT (n = 9) and ERK1 KO (n = 10) littermates were anesthetized, and the tibial and common peroneal branches of the sciatic nerve were ligated then transected, leaving the sural branch intact. After the spared nerve injury surgery, paw mechanical withdrawal thresholds were measured ipsilateral (solid lines) and contralateral (dashed lines) to nerve injury. Baseline and postinjury withdrawal thresholds were determined by probing the lateral plantar surface of the paw, which is innervated by the sural nerve. Data are represented as percentage baseline within each subject. Error bars indicate SEM.
ERK1 KO mice show reduced heat hypersensitivity after intraplantar injection of formalin
Intraplantar injection of formalin produces well characterized immediate spontaneous behaviors, which give way to hypersensitivity to heat and mechanical stimuli hours after the injection (Fu et al., 2001; Zeitz et al., 2004; Hu et al., 2006). Inhibition of ERK1/2 activation in the amygdala has no effect on immediate spontaneous behaviors but, instead, dramatically reduces long-term formalin-induced hypersensitivity (Carrasquillo and Gereau, 2007). Therefore, we reasoned that, although there was no apparent role of ERK1 in formalin-induced spontaneous nociceptive behaviors, ERK1 may be involved in longer-term formalin-induced hypersensitivity. To test this hypothesis, we injected ERK1 KO and WT littermates with formalin and measured responses to heat and mechanical stimuli. Interestingly, ERK1 KO mice showed attenuated heat hypersensitivity 1 and 2 h after 2% formalin injection in the ipsilateral paw (Fig. 5A). A two-way repeated-measures ANOVA confirmed this difference, indicating a significant main effect of genotype (F = 9.69; DFn = 1; DFd = 56; p = 0.0076) with a Bonferroni post test revealing a significant difference at 1 and 2 h time points (p < 0.05). Neither WT nor ERK1 KO mice developed heat hypersensitivity in the contralateral paw. At the same time points after 3.5% formalin injection, ERK1 KO mice developed robust mechanical hypersensitivity ipsilateral to injection that was similar to WT littermates (Fig. 5B). Although 1 and 3 h time points appear elevated in ERK1 KO mice, there is no statistically significant difference between the genotypes (two-way RM ANOVA: F = 3.81, DFn = 1, DFn = 60, p = 0.0700). It is important to note that we used 3.5% formalin to measure mechanical hypersensitivity. In WT mice obtained from ERK1 heterozygous crosses, 2% formalin did not produce robust mechanical hypersensitivity (data not shown). Using 2% formalin, we observed no difference between ERK1 KO and WT littermates (data not shown). Overall, these data indicate that ERK1 KO mice do not fully express formalin-induced heat hypersensitivity, despite normal formalin-induced mechanical hypersensitivity.
Figure 5.

ERK1 contributes to the onset of heat hypersensitivity after intraplantar formalin injection. A, To assess the role of ERK1 in long-term formalin-induced heat hypersensitivity, WT (n = 8) and ERK1 KO (n = 8) littermates were injected with 2% formalin subcutaneously into the plantar side of the right hindpaw. Paw withdrawal latencies were measured before and after injection and are graphed as percentage of preinjection baseline. ERK1 KO mice have reduced hypersensitivity to heat at 1 and 2 h after formalin injection in the ipsilateral paw (two-way RM ANOVA with Bonferroni's post test, *p < 0.05). B, To measure formalin-induced mechanical hypersensitivity, WT (n = 8) and ERK1 KO (n = 9) littermates were injected with 3.5% formalin. Paw withdrawal thresholds were obtained using von Frey filaments. There were no statistically significant differences between WT and ERK1 KO. Error bars reflect SEM.
Deletion of ERK1 elevates ERK2 phosphorylation without affecting ERK2 expression
ERK1 and ERK2 are both phosphorylated by MEK1/2, giving rise to the possibility that eliminating one ERK isoform might alter the phosphorylation state of the remaining isoform. The effect of ERK1 deletion on ERK2 phosphorylation in spinal cord or DRG remains unknown. Therefore, we analyzed tissue from naive WT and ERK1 KO mice by Western blot to size separate the isoforms and quantify the basal phosphorylation state of each isoform independently (Fig. 6A). Interestingly, ERK1 KO mice have significantly elevated basal ERK2 phosphorylation compared with WT littermates in both spinal cord and DRG. It is important to note that, in these tissues, total ERK2 expression remains unaltered (Fig. 1). To determine whether the activity of structurally related MAP kinases may be affected by ERK1 deletion, we analyzed spinal cord and DRG homogenates from naive WT and ERK1 KO littermates for JNK and p38 expression and phosphorylation (Fig. 6B–D). No significant difference was detected between ERK1 KO and WT homogenates, suggesting that altered basal p38 or JNK activity cannot account for the minimal behavioral impact of ERK1 deletion. Overall, these data do indicate that, in spinal cord and DRG, ERK1 elimination results in enhanced basal phosphorylation of ERK2.
Figure 6.

In spinal cord and DRG, basal ERK2 phosphorylation increases in the absence of ERK1. A, Spinal cord (SC) homogenates from naive WT (n = 7) and ERK1 KO (n = 8) littermates were analyzed simultaneously for pERK1/2 and ERK1/2 by quantitative Western blot. Although there is no difference in total ERK2 expression in ERK1 KO spinal cord (Fig. 1), ERK2 phosphorylation is significantly elevated. Similar results were observed in whole-cell homogenates prepared from DRG (WT, n = 7; ERK1 KO, n = 8; unpaired t test, *p < 0.05). B–D, The same spinal cord and DRG homogenates were used to evaluate the expression and phosphorylation of related MAPKs, JNK and p38. The integrated intensities of JNK2/3, JNK1, and p38 bands were divided by corresponding loading control β-tubulin bands, normalized to the mean WT intensity, and expressed as fold WT. To evaluate phosphorylation, pJNK2/3, pJNK1, and p-p38 bands were divided by their corresponding total protein bands from the same samples, normalized, and then expressed as fold WT. No significant difference was detected between WT and ERK1 KO mice in either spinal cord or DRG (WT SC, n = 7; ERK1 KO SC, n = 8; WT DRG, n = 5; ERK1 KO DRG, n = 6; unpaired t test). WT data are represented with white bars, and ERK1 KO data are represented with gray bars. Error bars indicate SEM.
Inflammation-induced elevation of pERK2 is equivalent between ERK1 KO and WT littermates
Given that ERK2 phosphorylation is elevated in naive ERK1 KO mice, we hypothesized that noxious stimulation would lead to hyperphosphorylation of ERK2. To test this hypothesis, ERK1 KO and WT mice were injected with either formalin or saline and killed at the peak of formalin-induced ERK1/2 phosphorylation in spinal cord (3 min after injection) (Fig. 7A). After normalizing to the total ERK loading control, ipsilateral intensities were divided by contralateral intensities within each subject, to provide a within-animal control. As previously reported (Karim et al., 2001), injection of WT mice with formalin significantly increased pERK1 and pERK2 compared with WT saline-injected mice (Fig. 7A) (for pERK1: unpaired t test, p < 0.001; for pERK2: two-way ANOVA, F = 55.92, DFn = 1, DFd = 20, p < 0.0001). ERK1 KO mice also show significant elevation of pERK2 after formalin injection (two-way ANOVA: F = 55.92, DFn = 1, DFd = 20, p < 0.0001). Interestingly, there is no significant difference between WT and ERK1 KO littermates in formalin-induced pERK2 increases (two-way ANOVA: F = 2.11, DFn = 1, DFd = 20, p = 0.1617). These data suggest that ERK1 deletion does not affect the change in spinal ERK2 phosphorylation attributable to formalin-induced inflammation. Similar results were obtained when using the CFA model of inflammation (Fig. 7B). WT and ERK1 KO littermates were injected with CFA or saline and killed at the peak time point of CFA-induced ERK1/2 phosphorylation (30 min after injection) (Ji et al., 2002). Western blot analysis of the dorsal portion of L3–L6 spinal cords indicated a significant increase in ipsilateral pERK1 for WT and pERK2 for both WT and ERK1 KO mice injected with CFA compared with saline-injected mice (pERK1: unpaired t test, p < 0.001; pERK2: two-way ANOVA, F = 21.02, DFn = 1, DFd = 17, p = 0.0003). This CFA-induced increase in pERK2 was not different between WT and ERK1 KO mice (two-way ANOVA: F = 0.13, DFn = 1, DFd = 17, p = 0.7202). Similar results were obtained by normalizing pERK/ERK values to WT saline (supplemental Figs. S2, S3, available at www.jneurosci.org as supplemental material). Using this alternative analysis, ERK1 KO mice have elevated pERK2 in all conditions compared with WT littermates. However, the relative increase in pERK2 attributable to formalin or CFA appears similar between WT and ERK1 KO, as is reflected in Figure 7. Overall, these data indicate that ERK1 deletion does not affect inflammation-induced elevation of pERK2.
Figure 7.

Inflammation-induced ERK2 phosphorylation is not elevated in ERK1 KO mice. A, ERK1 KO and WT littermates were injected subcutaneously into the plantar surface of the hindpaw with saline or 3.5% formalin (n = 6 per group) and killed 3 min later. The dorsoventral extent of L3–L6 spinal cord was isolated and processed for pERK1/2 and ERK1/2 quantitative Western blot analysis. Ipsilateral pERK/ERK intensities were normalized to contralateral intensities from the same subject, and data were graphed as fold contralateral. Formalin injection led to significantly elevated pERK1 and pERK2 in WT mice. In ERK1 KO mice, the formalin-induced increase in pERK2 was similar to WT. B, ERK1 KO and WT littermates were injected subcutaneously into the plantar surface of the hindpaw with saline or CFA (n = 5–6 per group). After 30 min, the dorsal halves of L3–L6 spinal cords were obtained and analyzed for pERK1/2 and ERK1/2 by Western blot as above. As with formalin injection, CFA-injected WT mice had significantly elevated pERK1 and pERK2. Elimination of ERK1 had no significant impact on CFA-induced pERK2 elevation. Error bars represent SEM. Unpaired t test or two-way ANOVA with Bonferroni's post test: *p < 0.05, **p < 0.01, ***p < 0.001.
Formalin-induced pERK1/2 immunostaining is slightly reduced in ERK1 KO mice
Although increases in pERK2 after noxious stimulation appear unaffected by ERK1 deletion, it is possible that the anatomic localization of pERK2 is altered which may reflect compensatory changes in nociceptive circuitry arising from the loss of ERK1 during development. To address this, ERK phosphorylation was measured by immunohistochemistry with a phosphospecific ERK1/2 antibody, since current antibodies cannot discriminate between pERK1 and pERK2 because of high sequence homology in the phosphorylation loop. ERK1 KO mice were injected with formalin and L3–L4 spinal cord sections were stained for pERK1/2. Interestingly, formalin-injected ERK1 KO mice had a similar pattern of pERK1/2 immunoreactivity to WT littermates (Fig. 8A). For both genotypes, positively staining cell profiles and more diffuse fiber staining was evident in the superficial lamina of the dorsal horn ipsilateral to paw injection. The dorsoventral or mediolateral extent of pERK1/2 immunostaining was not appreciably different between genotypes. To quantify pERK1/2 staining, all pERK1/2-positive cell profiles found in the superficial dorsal horn (lamina I and lamina II), as defined by dark-field illumination, were counted from serial sections separated by at least 90 μm (Fig. 8B). In both WT and ERK1 KO littermates, significantly more pERK1/2-positive cell profiles appear ipsilateral to formalin injection than contralateral (two-way ANOVA: F = 56.68, DFn = 1, DFd = 4, p = 0.0017). Interestingly, the number of pERK1/2-positive cell profiles ipsilateral to formalin injection is significantly reduced in ERK1 KO mice compared with WT littermates (two-way ANOVA: F = 9.79, DFn = 1, DFd = 8, p = 0.0141). This likely reflects the loss of ERK1 phosphorylation after formalin, which is normally seen in WT mice (Fig. 7). Importantly, in ERK1 KO mice formalin-induced pERK1/2 immunostaining is not elevated and does not occur in unexpected laminae of the dorsal horn. Both observations would argue against the potential confounds of ERK2 hyperphosphorylation or developmental remodeling of nociceptive circuitry.
Figure 8.
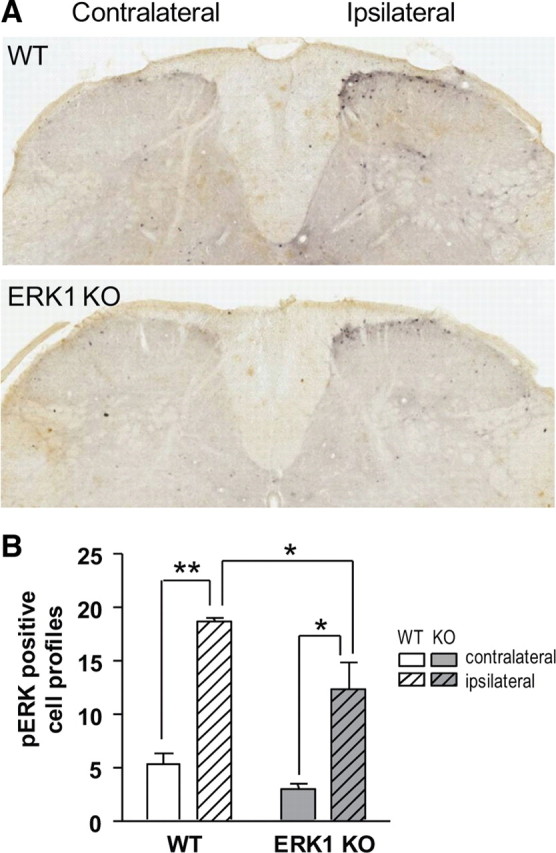
Noxious peripheral stimulation induces pERK1/2 immunoreactivity in superficial dorsal horn laminae despite the loss of ERK1. A, Three minutes after subcutaneous intraplantar injection of 3.5% formalin, WT and ERK1 KO littermates were killed and analyzed for pERK1/2 immunoreactivity in lumbar spinal cord. On the side of the spinal cord ipsilateral to paw injection, pERK1/2-positive cell profiles appear in the superficial lamina of formalin-injected WT and ERK1 KO mice. Diffuse staining is also apparent, possibly because of ERK1/2 phosphorylation in dendrites or afferent fibers. In both WT and ERK1 KO, there is no gross difference in the anatomic pattern of pERK1/2 staining. B, To quantify results from A, pERK1/2-positive cell profiles in laminae I–II were counted from six sections per animal with three animals per group. The mean number of cell profiles for each subject was then used to calculate the mean number for the entire group. Formalin stimulation significantly increases pERK1/2-positive cell profiles in both WT and ERK1 KO littermates. Two-way ANOVA with Bonferroni's post test: *p < 0.05, **p < 0.01. Error bars reflect SEM.
MEK inhibition attenuates formalin-induced spontaneous behaviors similarly in WT and ERK1 KO littermates
Although we observed no change in p38 or JNK expression or phosphorylation with ERK1 deletion, it remains possible that other signaling pathways compensate for the loss of ERK1 in the context of nociceptive sensitization. To address this possibility, we sought to test whether MEK inhibition could reduce formalin-induced spontaneous nociceptive behaviors in ERK1 KO mice, as has been reported in WT mice (Karim et al., 2001). Previous studies have used intrathecal delivery of MEK inhibitors. Since ERK1 is eliminated in all tissues of the ERK1 KO and since ERK1/2 activity is important for nociceptive sensitization in areas in addition to spinal cord, a more appropriate test would involve systemic MEK inhibition. SL327 is a structural analog of U0126 [1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene] that is blood-brain barrier permeable and has previously been used to inhibit MEK in the brain by systemic intraperitoneal administration (Atkins et al., 1998). However, it has not yet been used in the formalin test and has not been evaluated in spinal cord. Thus, we began our study by determining the dose of SL327 required to maximally inhibit formalin-induced ERK1/2 phosphorylation in spinal cord (Fig. 9A). Pretreatment with both 30 and 50 mg/kg SL327 intraperitoneally 30 min before 3.5% formalin injection to the hindpaw significantly reduced pERK1 and pERK2 in spinal cord tissue ipsilateral and contralateral to the injected paw (one-way ANOVA: pERK1 ipsi, F = 14.9, DFn = 3, DFd = 11, p = 0.0012; pERK2 ipsi, F = 29.85, DFn = 3, DFd = 11, p < 0.0001; pERK1 contra, F = 15.23, DFn = 3, DFd = 11, p = 0011; pERK2 contra, F = 36.42, DFn = 3, DFd = 11, p < 0.0001). Therefore, we used 50 mg/kg SL327 to evaluate the effect of systemic MEK inhibition on the formalin test in both WT and ERK1 KO littermates (Fig. 9B). SL327 at 50 mg/kg significantly attenuated formalin-induced spontaneous nociceptive behaviors in WT mice (two-way RM ANOVA: F = 8.09, DFn = 1, DFd = 198, p = 0.0108). Importantly, no difference was detected between WT and ERK1 KO mice injected with SL327 (two-way RM ANOVA: F = 0.91, DFn = 1, DFd = 187, p = 0.3528). Vehicle-injected ERK1 KO and WT littermates also behaved similarly (two-way RM ANOVA: F = 0.08, DFn = 1, DFd = 165, p = 0.7795), recapitulating results from Figure 2 in animals injected with DMSO. Overall, these data indicate that systemic MEK inhibition reduces spontaneous nociceptive behavior in the formalin test regardless of ERK1 deletion, suggesting that compensation from MEK-independent signaling pathways in ERK1 KO is negligible.
Figure 9.
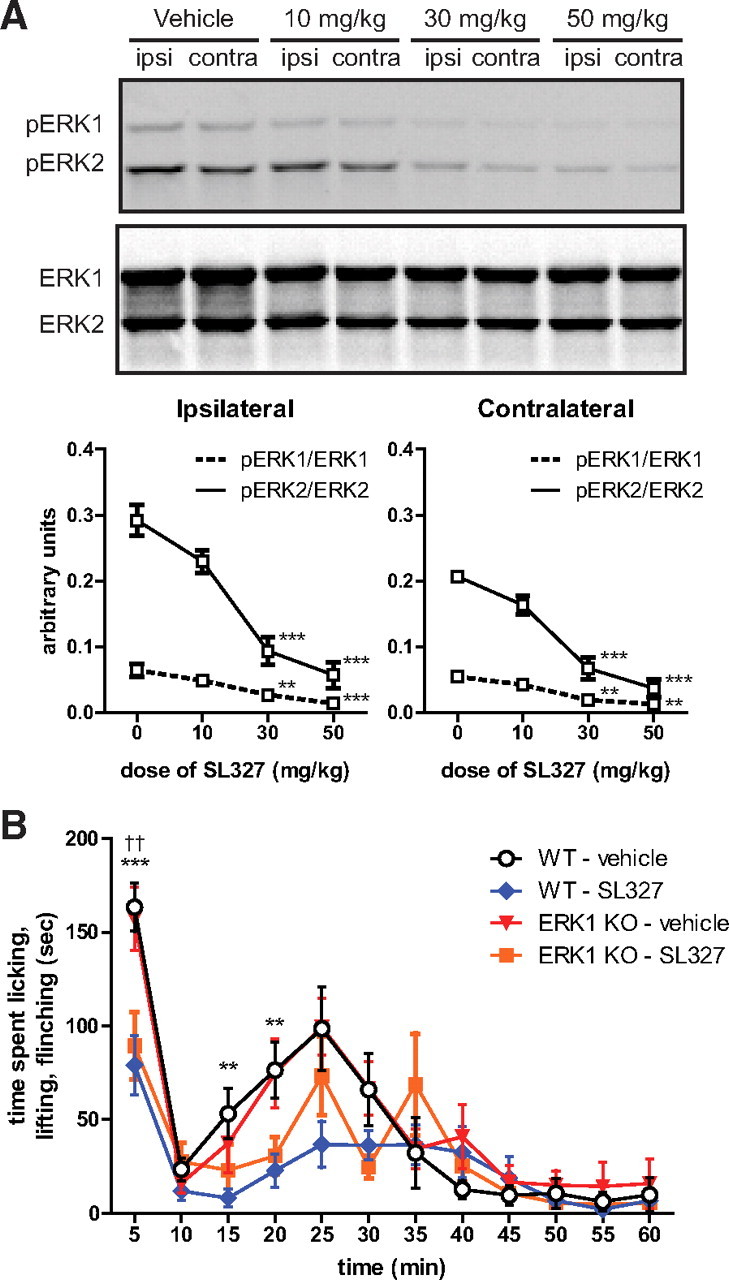
Systemic MEK inhibition attenuates formalin-induced spontaneous behaviors similarly in WT and ERK1 KO littermates. A, Several doses of the blood–brain barrier-permeant MEK inhibitor, SL327, or vehicle (DMSO) were administered intraperitoneally 30 min before hindpaw intraplantar formalin injection (3.5% in saline). Subjects (n = 3 per dose) were killed 3 min after formalin injection, and homogenates of spinal cord tissue ipsilateral and contralateral to the paw injection were analyzed for ERK phosphorylation. SL327 significantly reduced pERK1/ERK1 and pERK2/ERK2 at 30 and 50 mg/kg [one-way ANOVA with Dunnett's post test comparing to vehicle (0) within ERK isoform; **p < 0.01, ***p < 0.001]. B, ERK1 KO and WT mice were injected with either vehicle (1 ml/kg DMSO) or 50 mg/kg SL327 intraperitoneally (WT–vehicle, n = 9; WT–SL327, n = 11; KO–vehicle, n = 8; KO–SL327, n = 8). After 30 min, formalin (3.5% in saline) was injected subcutaneously into the plantar surface of the hindpaw, and spontaneous nociceptive behaviors were measured. SL327 significantly attenuated formalin-induced nociceptive behaviors in both WT and ERK1 KO littermates (two-way RM ANOVA with Bonferroni's post test; asterisks comparing WT–vehicle and WT–SL327, **p < 0.01, ***p < 0.001; daggers comparing KO–vehicle and KO–SL327, ††p < 0.01). No significant difference was detected between WT and ERK1 KO mice injected with SL327.
Discussion
Although it is clear that ERK1 and/or ERK2 drive behavioral sensitization in many pain models, little is known about the relative contribution of each isoform and whether they are redundant. This study is the first to address directly the role of ERK1 in rodent models of pain by characterizing the nociceptive function of ERK1 KO mice. We found that acute sensitivity to both noxious heat and the noxious chemical, formalin, remains intact despite the loss of ERK1 from all tissues. ERK1 deletion does not affect hypersensitivity attributable to chronic inflammation or nerve injury. Interestingly, long-term formalin-induced hypersensitivity to heat is slightly reduced by ERK1 deletion. Together, these data suggest ERK1 is not required for nociceptive sensitization.
Accumulating evidence from this study and elsewhere strongly suggests that spinal ERK2, and not ERK1, is critical for CFA-induced hypersensitivity. Targeting ERK1/2 nonspecifically with intrathecally applied MEK inhibitors attenuates CFA-induced hypersensitivity (Ji et al., 2002; Obata et al., 2003). Interestingly, RNAi-mediated ERK2 knockdown in spinal cord dramatically reduces CFA-induced behavioral sensitization (Xu et al., 2008). However, because of the partial reduction of ERK2 expression achieved with this method, there was a relatively small effect on ERK2 phosphorylation. ERK2 knockdown also caused an elevation of ERK1 phosphorylation in the spinal cord. In the physiologically normal context of isoform coexpression, spinal ERK1 is phosphorylated after CFA injection to the paw (Ji et al., 2002; Obata et al., 2003; Adwanikar et al., 2004; Xu et al., 2008), raising the possibility that ERK1 plays some role in behavioral sensitization. The current study directly addresses this question and demonstrates that ERK1 deletion has no effect on CFA-induced heat or mechanical hypersensitivity. Given the combined results of targeting MEK, ERK2, and now ERK1, it is likely that spinal ERK2 specifically drives CFA-induced hypersensitivity.
In the present study, we observed that ERK1 deletion is accompanied by increased ERK2 phosphorylation in the spinal cord and DRG of naive mice. This could arise if ERK isoforms normally compete for binding and phosphorylation with their shared upstream kinase MEK. Without ERK1 expression, MEK binds and exclusively phosphorylates the remaining isoform ERK2. Similar observations have been made in other systems, supporting such a model of MEK–ERK1/2 interaction. In NIH/3T3 cells, RNAi-mediated knockdown of one isoform elevates the phosphorylation state of the remaining isoform (Vantaggiato et al., 2006; Lefloch et al., 2008). However, the degree of isoform competition varies with context. In vivo, ERK1 deletion only affects ERK2 phosphorylation in certain non-neuronal tissues, such as the thymus (Nekrasova et al., 2005), but not in hippocampus, cortex, and striatum (Mazzucchelli et al., 2002). However, primary cultures of cortex or striatum from ERK1 KO embryos exhibit ERK2 hyperphosphorylation after stimulation with either glutamate or KCl without altered phosphorylation in untreated cultures. It appears that MEK–ERK1/2 interactions depend on cellular context, relative activity of MEK–ERK signaling, and perhaps developmental context given the differences between embryonic cultures and adult mice (Mazzucchelli et al., 2002).
In all studies using isoform-specific knock-out or knockdown, the functional importance of ERK isoform hyperphosphorylation is difficult to separate from the effect of deleting the targeted isoform. In ERK1 KO mice, ERK2 hyperphosphorylation in striatal cultures is correlated with augmented long-term synaptic potentiation in striatum and enhanced place preference conditioning for morphine (Mazzucchelli et al., 2002). Similar ERK2 hyperphosphorylation occurs in hippocampal cultures from ERK1 KO embryos (Mazzucchelli et al., 2002), but there is minimal impact of ERK1 deletion on hippocampal plasticity and, behaviorally, no effect on hippocampus-dependent tasks such as contextual fear conditioning and passive avoidance (Selcher et al., 2001). In the present study, elevated ERK2 phosphorylation is not associated with altered baseline withdrawal responses to noxious heat or mechanical stimulation, and there is no gain-of-function in nociceptive sensitization. Additionally, spinal pERK2 elevation above baseline after intraplantar formalin or CFA is similar between WT and ERK1 KO. This suggests that elevated basal ERK2 phosphorylation is unrelated to nociceptive sensitization. Basal phosphorylation may occur in non-nociceptive cells of the spinal cord, or it may be an artifact of tissue preparation or homogenization. In both cases, deletion of ERK1 would lead to ERK2 hyperphosphorylation only in this non-nociceptive-specific pool of ERK.
An alternative interpretation is that ERK2 hyperphosphorylation actually relates to nociception and functionally compensates for the loss of ERK1, masking a physiologic role for ERK1. We do not favor this interpretation for several reasons. First, knockdown of spinal ERK2 also increases basal ERK1 phosphorylation without affecting baseline sensory responses but with a profound effect on CFA-induced hypersensitivity (Xu et al., 2008). If ERK isoforms were interchangeable in the nociceptive spinal cord, then one would expect ERK1 to compensate for the loss of ERK2, which did not occur. Second, pERK1/2 immunohistochemistry is actually reduced in ERK1 KO mice, indicating that elevated pERK2 cannot make up for the loss of ERK1 in total pERK1/2 immunoreactivity. If isoforms were completely redundant, this net loss of pERK1/2 would be expected to affect formalin-induced spontaneous behaviors. Nevertheless, it remains difficult to exclude a role for ERK1 without directly testing ERK2 function. The converse is also true, and, as such, the current study solidifies our understanding of spinal ERK2 function in the CFA model, as outlined above.
To our knowledge, the present study is the first to examine the role of ERK isoforms in the formalin model of chemical nociception. Moreover, our experiments with the systemically active MEK inhibitor, SL327, provide additional evidence that ERK1/2 activity is required for the behavioral response to formalin. Since SL327 exerts similar effects in WT and ERK1 KO mice, it seems likely that ERK2 is specifically required in this behavioral model, although this must be directly tested. It is tempting to hypothesize that the underlying cellular mechanisms of the formalin test would also rely on ERK2 and not ERK1. These mechanisms may include sensitization processes in spinal cord that amplify incoming nociceptive information via enhanced synaptic strength and intrinsic excitability (Ikeda et al., 2006; Sandkühler, 2009), both of which involve ERK1/2 (Ji et al., 2009). Formalin also increases primary afferent activity, which likely contributes to immediate, phase 1 behavioral responses (Puig and Sorkin, 1996). Since SL327 decreases the first phase of the formalin test and since ERK1 appears to be dispensable for formalin-induced spontaneous behaviors, our results predict that ERK2 specifically contributes to elevated afferent activity, perhaps by increasing excitability via direct phosphorylation of voltage-gated sodium channels (Stamboulian et al., 2010).
Noxious stimulation or inflammation induces hypersensitivity to heat, which is primarily attributable to peripheral sensitization of primary afferent nociceptors (Levine and Alessandri-Haber, 2007). Previous studies have demonstrated a role for nociceptor ERK1/2 in inflammation-induced peripheral sensitization using MEK inhibitors (Dai et al., 2002; Obata et al., 2003; Zhuang et al., 2004; Seino et al., 2006). ERK1/2 may contribute to heat hypersensitivity by modulating TRPV1 (Zhuang et al., 2004; Firner et al., 2006), which is a molecular sensor of noxious heat that is required for CFA-induced heat hypersensitivity (Caterina et al., 2000; Levine and Alessandri-Haber, 2007). Our observation that ERK1 is not required for CFA-induced heat hypersensitivity suggests that ERK2 may specifically modulate TRPV1, a possibility that must be directly tested. Interestingly, our results also indicate that formalin-induced heat hypersensitivity is in part mediated by ERK1. The discrepancy between CFA- and formalin-induced heat hypersensitivity reported here may be the result of discrete parallel signaling pathways within nociceptive terminals that are sensitive to either CFA or formalin. Given that ERK1 KO mice develop heat hypersensitivity at later time points after formalin injection, it is also possible that ERK1 contributes to heat hypersensitivity but ultimately ERK2 is sufficient to induce sensitization.
In addition to spinal cord and DRG, ERK1/2 is activated in many regions of the nociceptive system after noxious stimulation including the amygdala (Carrasquillo and Gereau, 2007). In amygdala, microinjection of MEK inhibitors does not affect acute formalin-induced spontaneous behaviors but does attenuate mechanical hypersensitivity. In the current study, we found that ERK1 deletion does not affect formalin-induced mechanical hypersensitivity. This fits well with our previous observations that formalin did not significantly induce ERK1 phosphorylation in amygdala (Carrasquillo and Gereau, 2007). However, since amygdala MEK inhibition affected both ERK1 and ERK2 phosphorylation, it was difficult exclude a role for ERK1. The current study extends our knowledge of the role of the amygdala in nociception by providing evidence that ERK1 is not required for formalin-induced mechanical hypersensitivity.
Results from the present study indicate that ERK1 is not required for formalin-induced spontaneous behavior, chronic inflammatory, and neuropathic pain models, suggesting a predominant isoform-specific role for ERK2. Isoform-specific functions may also occur in humans, since copy number variation of each isoform yields distinct neurological deficits. ERK1 deletion or duplication is associated with autism (Kumar et al., 2008), whereas reduced ERK2 expression is associated with microcephaly, cognitive deficits, and developmental delay (Newbern et al., 2008). The relevance to the human nervous system suggests that dissecting the roles of ERK1 and ERK2 will be important for understanding and treating chronic pain conditions.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS48602 (R.W.G.) and F30NS061398 (B.J.A.). Additional support was provided by National Institutes of Health Neuroscience Blueprint Interdisciplinary Center Core Grant P30 NS057105 (Washington University). We thank C. Qui and S. Vogt for mouse colony maintenance. B.J.A. is immensely grateful for the help and encouragement of Y. Carrasquillo, H. Hu, and S. Chiechio, and also thanks M. Morales, B. Kolber, M. Montana, and S. Davidson for their critical reading of this manuscript.
References
- Adwanikar H, Karim F, Gereau RW., 4th Inflammation persistently enhances nocifensive behaviors mediated by spinal group I mGluRs through sustained ERK activation. Pain. 2004;111:125–135. doi: 10.1016/j.pain.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Ciruela A, Dixon AK, Bramwell S, Gonzalez MI, Pinnock RD, Lee K. Identification of MEK1 as a novel target for the treatment of neuropathic pain. Br J Pharmacol. 2003;138:751–756. doi: 10.1038/sj.bjp.0705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb MH, Goldsmith EJ. Dimerization in MAP-kinase signaling. Trends Biochem Sci. 2000;25:7–9. doi: 10.1016/s0968-0004(99)01508-x. [DOI] [PubMed] [Google Scholar]
- Coligan JE, Dunn BM, Pleogh HL, Speicher DW, Wingfield PT, editors. Current protocols in protein science. New York: Wiley; 2001. [Google Scholar]
- Dai Y, Iwata K, Fukuoka T, Kondo E, Tokunaga A, Yamanaka H, Tachibana T, Liu Y, Noguchi K. Phosphorylation of extracellular signal-regulated kinase in primary afferent neurons by noxious stimuli and its involvement in peripheral sensitization. J Neurosci. 2002;22:7737–7745. doi: 10.1523/JNEUROSCI.22-17-07737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Firner M, Greffrath W, Treede RD. Phosphorylation of extracellular signal-related protein kinase is required for rapid facilitation of heat-induced currents in rat dorsal root ganglion neurons. Neuroscience. 2006;143:253–263. doi: 10.1016/j.neuroscience.2006.07.047. [DOI] [PubMed] [Google Scholar]
- Fu KY, Light AR, Maixner W. Long-lasting inflammation and long-term hyperalgesia after subcutaneous formalin injection into the rat hindpaw. J Pain. 2001;2:2–11. doi: 10.1054/jpai.2001.9804. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW., 4th The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jäger T, Sandkühler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RW, 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Hu HJ, Adwanikar H, Kaplan D, Gereau RW., 4th Impaired inflammatory pain and thermal hyperalgesia in mice expressing neuron-specific dominant negative mitogen-activated protein kinase kinase (MEK) Mol Pain. 2006;2:2. doi: 10.1186/1744-8069-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krens SF, Spaink HP, Snaar-Jagalska BE. Functions of the MAPK family in vertebrate-development. FEBS Lett. 2006;580:4984–4990. doi: 10.1016/j.febslet.2006.08.025. [DOI] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, Christian SL. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Lefloch R, Pouysségur J, Lenormand P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol. 2008;28:511–527. doi: 10.1128/MCB.00800-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta. 2007;1772:989–1003. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Marchi M, D'Antoni A, Formentini I, Parra R, Brambilla R, Ratto GM, Costa M. The N-terminal domain of ERK1 accounts for the functional differences with ERK2. PLoS One. 2008;3:e3873. doi: 10.1371/journal.pone.0003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pagès G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouysségur J, Brambilla R. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/s0896-6273(02)00716-x. [DOI] [PubMed] [Google Scholar]
- Nekrasova T, Shive C, Gao Y, Kawamura K, Guardia R, Landreth G, Forsthuber TG. ERK1-deficient mice show normal T cell effector function and are highly susceptible to experimental autoimmune encephalomyelitis. J Immunol. 2005;175:2374–2380. doi: 10.4049/jimmunol.175.4.2374. [DOI] [PubMed] [Google Scholar]
- Newbern J, Zhong J, Wickramasinghe RS, Li X, Wu Y, Samuels I, Cherosky N, Karlo JC, O'Loughlin B, Wikenheiser J, Gargesha M, Doughman YQ, Charron J, Ginty DD, Watanabe M, Saitta SC, Snider WD, Landreth GE. Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proc Natl Acad Sci U S A. 2008;105:17115–17120. doi: 10.1073/pnas.0805239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Tachibana T, Fukuoka T, Tokunaga A, Yoshikawa H, Noguchi K. Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci. 2003;23:4117–4126. doi: 10.1523/JNEUROSCI.23-10-04117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Mizushima T, Fukuoka T, Tokunaga A, Noguchi K. Differential activation of MAPK in injured and uninjured DRG neurons following chronic constriction injury of the sciatic nerve in rats. Eur J Neurosci. 2004;20:2881–2895. doi: 10.1111/j.1460-9568.2004.03754.x. [DOI] [PubMed] [Google Scholar]
- Pagès G, Guérin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouysségur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Puig S, Sorkin LS. Formalin-evoked activity in identified primary afferent fibers: systemic lidocaine suppresses phase-2 activity. Pain. 1996;64:345–355. doi: 10.1016/0304-3959(95)00121-2. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- Seino D, Tokunaga A, Tachibana T, Yoshiya S, Dai Y, Obata K, Yamanaka H, Kobayashi K, Noguchi K. The role of ERK signaling and the P2X receptor on mechanical pain evoked by movement of inflamed knee joint. Pain. 2006;123:193–203. doi: 10.1016/j.pain.2006.02.032. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song XS, Cao JL, Xu YB, He JH, Zhang LC, Zeng YM. Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats. Acta Pharmacol Sin. 2005;26:789–798. doi: 10.1111/j.1745-7254.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Nav1.7 and alters its gating properties. J Neurosci. 2010;30:1637–1647. doi: 10.1523/JNEUROSCI.4872-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 2006;5:14. doi: 10.1186/jbiol38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Garraway SM, Weyerbacher AR, Shin SJ, Inturrisi CE. Activation of the neuronal extracellular signal-regulated kinase 2 in the spinal cord dorsal horn is required for complete Freund's adjuvant-induced pain hypersensitivity. J Neurosci. 2008;28:14087–14096. doi: 10.1523/JNEUROSCI.2406-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitz KP, Giese KP, Silva AJ, Basbaum AI. The contribution of autophosphorylated alpha-calcium-calmodulin kinase II to injury-induced persistent pain. Neuroscience. 2004;128:889–898. doi: 10.1016/j.neuroscience.2004.07.029. [DOI] [PubMed] [Google Scholar]
- Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 Å resolution. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]