

SUMMARY
We present the x-ray structure of PimD, both substrate-free and in complex with 4,5-desepoxypimaricin. PimD is a cytochrome P450 monooxygenase with native epoxidase activity that is critical in the biosynthesis of the polyene macrolide antibiotic pimaricin. Intervention in this secondary metabolic pathway could advance the development of drugs with improved pharmacologic properties. Epoxidation by P450 typically includes formation of a charge-transfer complex between an oxoferryl π-cation radical species (Compound I) and the olefin π-bond as the initial intermediate. Catalytic and structural evidence presented here suggest that epoxidation of 4,5-desepoxypimaricin proceeds via a hydroperoxoferric intermediate (Compound 0). The oxygen atom of Compound 0 distal to the heme iron may insert into the double bond of the substrate to make an epoxide ring. Stereoelectronic features of the putative transition state suggest substrate-assisted proton delivery.
Keywords: P450, PimD, polyene antibiotics, pimaricin, mechanism of epoxidation, Compound 0
Polyene macrolide antibiotics constitute a large group of antifungal agents produced mainly by Streptomyces spp. that are widely used in human and veterinary medicine and in food preservation. They are characterized structurally by a large hydroxylated macrolactone ring incorporating a chromophore formed by a system of three to eight conjugated double bonds and a six-membered hemi-ketal ring with an exocyclic carboxyl group (Omura and Tanaka, 1984). Most polyene macrolides contain a single aminosugar, mycosamine (3-amino-3,6-dideoxy-D-mannose), attached to the macrolactone ring via a glycosidic bond. Common polyene macrolides used to treat fungal infections in humans are pimaricin (natamycin), amphotericin B and nystatin A1 (Fig. 1). Amphotericin is the most effective drug for the treatment of life-threatening systemic fungal infections, nystatin for superficial mycoses, and pimaricin for fungal keratitis, and also as a food preservative to prevent mold contamination of dairy products and meats.
Figure 1.

Chemical structures of the polyene macrolide antibiotics.
While the polyol segment of the macrolactone is flexible and hydrophilic, the polyene chromophore has both a rigid planar structure and lipophilic nature, resulting in a fully extended amphipathic rod-shaped molecule, which determines the characteristic physicochemical properties and mode of action of these antibiotics. The conjugation system of chromophore is susceptible to auto-oxidation and the production of free radicals, giving rise in part to the strong antifungal and antiparasitic properties of polyene macrolides (Brajtburg et al., 1990). Another major mode of their action stems from interactions with ergosterol in cytoplasmic membranes, causing disorganization of membrane structure leading to formation of transmembrane channels, leakage of ions and small molecules, followed by cell death (Aparicio et al., 2004; Baginski et al., 2006; Baginski et al., 2005; Zotchev, 2003). Polyene macrolides interact with cholesterol in mammalian membranes, although to a lesser extent than with ergosterol in fungal membranes. Nevertheless, serious side effects such as nephrotoxicity, cardiotoxicity and neurotoxicity result from the use of these antibiotics in humans (Schaffner, 1984). These undesirable effects limit the use of amphotericin in human medicine to those cases in which all other therapies have failed.
The polyene biosynthetic gene clusters encode modular polyketide synthases that assemble the macrolactone cores, cytochrome P450 enzymes and enzymes for biosynthesis and attachment of mycosamine (Aparicio et al., 2003). The P450 monooxygenase genes encoded by the polyene clusters for the three most used antibiotics, pimaricin (Streptomyces natalensis), amphotericin (Streptomyces nodosus) and nystatin (Streptomyces noursei), form two distinctive phylogenetic groups (Aparicio et al., 2003; Volokhan et al., 2006). The P450 enzymes from the first group are involved in the oxidation of the exocyclic methyl branch to the carboxyl group, whereas enzymes from the second group, including PimD, AmphL and NysL, are involved in oxidative modifications of the polyol segment in the three antibiotics. Their enzymatic functions have been established by gene disruption and in vitro reconstitution of enzymatic activities of purified enzymes (Byrne et al., 2003; Mendes et al., 2005; Mendes et al., 2001; Volokhan et al., 2006). While sharing ≥55% overall sequence identity, the three enzymes possess distinct regio-specificities and catalyze different chemical reactions: hydroxylation of the C-8 atom in amphotericin (AmphL) and the C-10 atom in nystatine (NysL), and epoxidation of the C4-C5 double bond in pimaricin (PimD) (Fig. 1) (Aparicio et al., 2003).
The consensus mechanism of epoxidation by P450 includes formation of a charge-transfer complex (also known as π-complex) between an oxoferryl π-cation radical species (FeIV=O porphyrin π-cation radical, termed Compound I) and the olefin π-bond as the initial intermediate (Guengerich, 2003; Meunier et al., 2004; Ortiz de Montellano and De Voss, 2002; Shaik et al., 2007; Sono et al., 1996). The reaction proceeds either by synchronous insertion of ferryl oxygen to the olefin π-bond, or by electron transfer in the charge-transfer complex followed by formation of the olefin π-cation radical and then epoxidation via either a radical or a cationic path. In support of the charge-transfer complex formation is the orientation of substrate in the P450 enzyme EpoK, involved in epoxidation of the anticancer agents epothilones (Nagano et al., 2003). An alternative mechanism for olefin epoxidation involving the hydroperoxoferric intermediate (FeIII-OOH, Compound 0) has been proposed based on experimental evidence (Chandrasena et al., 2004; Jin et al., 2003; Newcomb et al., 2003; Vaz et al., 1998) but is a subject of debate on theoretical grounds (Hirao et al., 2006; Jin et al., 2004; Meunier et al., 2004; Ogliaro et al., 2002; Shaik et al., 2007). Consensus has been reached that the sluggish electrophilic oxidant, Compound 0, is unable to compete with Compound I for double-bond epoxidation, although in the absence of the latter it may become a default oxidant. In this work we provide structural and catalytic evidence that due to the stereoelectronic reasons Compound I does not serve as an oxidant in PimD and epoxidation of 4,5-desepoxypimaricin may proceed via Compound 0 with the insertion of its oxygen atom distal to the heme iron into a double bond to form the epoxide ring. Substrate-assisted input of a proton via the specific transition state may govern the capacity of PimD to catalyze the less favored reaction.
RESULTS
Overall structures of PimD and 4,5-desepoxypimaricin
The structures of both substrate-free (2.1 Å) and 4,5-desepoxypimaricin-bound PimD (1.95 Å) feature the overall protein fold common to the P450 family with some conformational differences associated with the 4,5-desepoxypimaricin binding (Fig. 2A, B). Both forms superimposed with the r.m.s.d. of 0.73 Å for Cα atoms. Unlike the substrate-bound form, the substrate-free form is stabilized in the crystal lattice via formation of the intermolecular Cys10-Cys10 bond between the symmetry-related molecules.
Figure 2. Overall structure of PimD.

A and B, Superimposed structures of substrate-free (wheat) and 4,5-desepoxypimaricin-bound (light blue) PimD are shown with the α-helices labeled. The protein backbone is depicted by ribbon and the heme (orange) and 4,5-desepoxypimaricin by spheres. Desepoxypimaricin is colored according the elements with the carbon atoms yellow, oxygen red and nitrogen blue. A, Distal protein surface with respect to heme. B, Image is rotated ~ 90° toward viewer. C, Stereoscopic view of PimD with 4,5-desepoxypimaricin bound in the active site. For clarity, only a few residues (green) within 6 Å from 4,5-desepoxypimaricin are shown. Fragments of the protein backbone are shown as gray ribbon. Color schemes for 4,5-desepoxypimaricine and heme are as in A and B. Loops are labeled with the numbers for a range of the amino acid residues constituting the loop. 2Fo-Fc electron density map (blue wire mesh) is calculated with the 4,5-desepoxypimaricin coordinates omitted from the input. Images are generated using PYMOL (DeLano, 2002).
In the PimD active site 4,5-desepoxypimaricin has a rod-shaped conformation (Fig. 2C), similar to that determined for amphotericin B in organic solvent by x-ray crystallography (Ganis et al., 1971). The extended tetraene chromophore and the C2-C3 and C4-C5 conjugated double bonds constrain the single bonds of the short polyol segment of the 26-membered macrolactone ring to adopt an almost linear staggered conformation. Single bonds adjacent to the conjugated double bonds have a skewed conformation to satisfy the ring closure. Both the mycosamine pyranosidic ring and the six-membered hemi-ketal ring have chair conformations. The long axis of the ~17 Å “rod” runs virtually parallel to the I-helix projecting over three helical turns. The end harboring the hemi-ketal points toward the C-terminus of the I-helix, while the lactone end points toward the opening to the bulk solvent created by the C-helix, the N-terminus of the I-helix, and the short B-helix (Fig. 2B). The hydrophilic surface of the macrolide ring faces the I-helix, while the hydrophobic surface leans against the stretch of the residues A278 through P286 (Fig. 2C). The variability of this region between PimD, NysL and AmphL may directly correlate with the size of the polyene macrolide substrate (Fig. 3). The turn between the second and third strands in the β-sheet 4 (L381-G389) runs co-planar to and interacts with the mycosamine pyranosidic ring. These residues are missing from the electron density in the substrate-free form.
Figure 3. Sequence alignments between polyene macrolide monooxygenases.

Multiple sequence alignments between PimD (Streptomyces natalensis), NysL (Streptomyces noursei) and AmphL (Streptomyces nodosus) are shown. Accession numbers of the proteins in the Swiss-Prot/TrEMBL (http://us.expasy.org/sprot) database are given next to the name of the protein. Alignments were performed using CLUSTALW program online (Thompson et al., 1994). The figure was generated using ESPript (Gouet et al., 1999). The secondary structure annotation and residue numbering at the top correspond to PimD. Amino acid residues within 6 Å from the substrate in the active site are labeled with blue (clustered) and green (isolated) triangles. Iron proximal cystein ligand is marked with a red star. Grey stars highlight residues in alternate conformations. If aligned pair-wise, PimD is 57% identical to NysL, and 55% identical to AmphL. NysL and AmphL share 71% sequence identity and are more closely related.
Conformational changes associated with substrate binding
In PimD, the residues within 6 Å from the substrate are largely clustered in the four protein regions (marked by blue triangles in Fig. 3) including the BC-loop (Tyr76, Val77, Phe81, Leu82, Leu85), the I-helix (His226, Leu227, Gly230, Phe233, Ala234, Ser238, Ser241, Ile242, Asn245), the unstructured region between the K-helix and the fourth strand of the β-sheet 1 (Ala278, Arg279, Gly281, Gly282, Ser283, Val284, Leu285, Pro286), and the turn between the second and third strands of the β-sheet 3 (Leu381, Gly384, Gln385, Leu386, Ser387, Gly388 and Gly389) (boldface indicates residues invariant between PimD, NysL and AmphL). Four residues (green triangles), Leu13, Leu178, Leu311, and Thr338, are from isolated parts of the primary sequence. Substrate binding either stabilizes, or induces, the more compact “closed” protein conformation (colored cyan in Fig. 2A, B). Four protein regions move in concert upon substrate binding. The FG-loop shifts about 7 Å toward the active site, which brings the invariant Leu178 in range to interact with the tetraene chromophore of 4,5-desepoxypimaricin. The BC-loop moves inward about 3 Å, while the C-helix moves away from the active site to accommodate the inward relocation of the BC-loop. The opening at the N-terminal end of the I-helix is retained upon substrate binding (Fig. 2B). The newly ordered glycine-rich turn in β-sheet 3 (Leu381-Gly389) forms a lid over the active site, interacting with the pyranosidic ring of mycosamine (Fig. 2C). These interactions are important for substrate binding, as epoxidation is impaired in the absence of glycosylation with mycosamine (Caffrey et al., 2008).
4,5-Desepoxypimaricin binding
The substrate binds in the active site via multiple hydrophobic and H-bonding interactions with KD of 4 μM. The macrolactone ring is approximately orthogonal to the plane of the porphyrin macrocycle and shifted off the porphyrin center toward the heme propionate groups (Fig. 4A). The hemi-ketal ring provides two direct H-bonds to the amino acid side chains, including those between hydroxyl groups at C-11 and Ser241, and the exocyclic carboxyl group at C-12 and hydroxyl group of Ser283. The latter is invariant in all three enzymes, while the H-bonding functionality of Ser241 is invariant in NysL and conserved in AmphL via replacement with a threonine (Fig. 3). All other H-bonds between the protein and the substrate are mediated by water molecules. Although the heme Fe is high-spin, the water molecule (W2168) is bound along the iron distal coordination axis only 3.3 Å away. The axial location and proximity of W2168 to the heme Fe explain the spin equilibrium observed in solution for the PimD-4,5-desepoxypimaricin complex. At 100 μM 4,5-desepoxypimaricin, a concentration 25 times exceeding the KD value, only a partial shift to the heme Fe high-spin state is achieved (green trace in Fig. 4B), indicating that while in solution the low-spin form dominates equilibrium. However when crystallized, the substrate-bound PimD is in a high-spin state, as evidenced by the displacement of the Fe atom out of the plane of the porphyrin ring toward the proximal thiolate and the axial water residing out of range to form a coordination bond. W2168 is held in place by H-bonds to the substrate hydroxyl group at C-7 (distance 2.7 Å) and carbonyl oxygen of Ala234 (2.6 Å) virtually equidistantly from the C-4 (3.2 Å) and C-5 (3.6 Å) carbons thus projecting in the middle of the C4-C5 double bond.
Figure 4. 4,5-desepoxypimaricin interactions in the catalytic site.

A, The H-bonding interactions of 4,5-desepoxypimaricin are indicated by the dashed lines with the distances in Angstroms. The I-helix is traced by a grey ribbon. A fragment of the 2Fo-Fc electron density map (blue mesh) indicates rotation of the Ser238 side chain in toward the I-helix grove and H-bonding to the carbonyl oxygen of A234. Color schemes for the heme (van der Waals spheres) and 4,5-desepoxypimaricin (sticks) are as in Fig. 2 with the H atoms at the C4-C5 double bond shown in grey. B, UV-vis absorbance spectra are shown for PimD (5 μM) in the ferric low-spin state (red), ferrous CO-bound form (blue), and ferric substrate-bound form at 100 μM 4,5-desepoxypimaricin (green). The latter trace represents a mix between the low-spin and high-spin forms. All spectra were recorded at room temperature in 100 mM potassium phosphate, pH 7.5, and 10% glycerol.
Impairment of the proton delivery network
Position of W2168 suggests the role of substrate in the delivery of protons required for activation of molecular oxygen. This function is normally supported by the threonine residue, highly conserved across the P450 protein family, which plays a key role in the conversion of Compound 0 to Compound I (Imai et al., 1989; Martinis et al., 1989). However, this residue is substituted by Ser238 in PimD and the related polyene macrolide monooxygenases shown in Figure 3. Surprisingly, in both substrate-free and substrate-bound PimD, the side chain of Ser238 is rotated in towards the I-helix groove and it H-bonds to the carbonyl oxygen of the highly conserved Ala234, resulting in distortion of the α-helical H-bonding pattern (Fig. 4A). Such a distortion is typically caused by a water molecule bound in the I-helix groove, which is absent in the PimD structure. Thus, the hydroxyl of Ser238 replaces the function of the normally present water and is thereby excluded from the proton delivery network. This arrangement resembles that of the macrolide monooxygenase EryF, with the qualification that in EryF the alanine which substitutes for the highly conserved threonine is incapable of H-bonding. That role is filled by the hydroxyl group of the EryF substrate, deoxyerythronolide B, which together with the carbonyl oxygen of the highly conserved alanine, H-bond to the water molecule in the vicinity of the heme Fe in the ferric substrate-bound state (Cupp-Vickery et al., 1996).
Structure-derived mechanism of epoxidation
The plane of the conjugated C2-C3 and C4-C5 double bonds in 4,5-desepoxypimaricin forms a ~125° angle to the porphyrin plane with the C4-H σ-bond pointing directly to the heme Fe, while the trans C5-H σ-bond points in the opposite direction (Fig. 5A–C). Accordingly, the π-orbitals of this conjugated system point away from the iron at a ~125° angle to the heme orthogonal, as shown by the blue arrow in Fig. 5A, B. This topology is unfavorable for the synchronous oxygen insertion or the formation of the charge-transfer complex. Instead, both the direction of the C4-H σ-bond and the 3.7 Å distance between the H and Fe atoms are favorable for hydroxylation by the hydrogen abstraction and oxygen rebound mechanism (Fig. 5C). However, the exceptionally high bond dissociation energy associated with proton abstraction from a vinyl carbon prevents it from happening (Bach and Dmitrenko, 2004).
Figure 5. Structure-based mechanism of epoxidation.
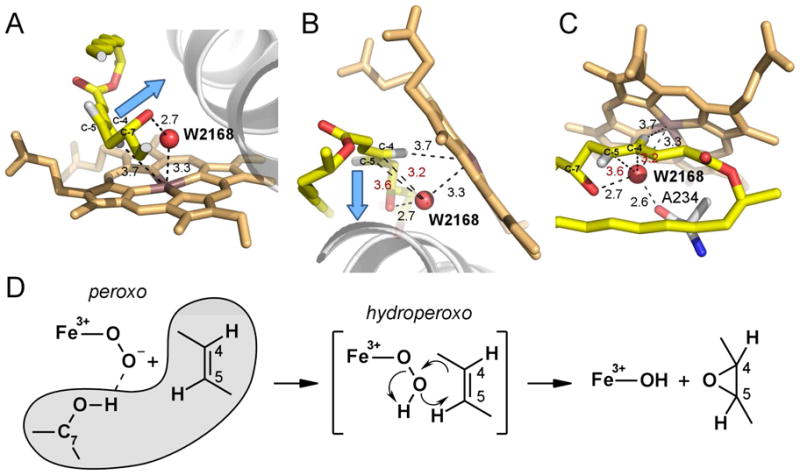
Three different views of the disposition of atoms in the O2-scission site are shown in A, B and C to emphasize orientation of the to-be-epoxidized double bond C4-C5 and position of W2168 (red sphere) with respect to each other and the heme iron. A clipped fragment of 4,5-desepoxypimaricin accommodating the reaction site is shown in yellow with oxygen atoms in red and hydrogen atoms in grey. Ala234 is shown with carbon atoms in grey. Blue arrow points are collinear with the C4-C5 π-orbitals. Fragment of the I-helix is shown as a grey ribbon. Distances are in Angstroms. In red are the distances between W2168 and the C4 or C5 carbons. D, Epoxidation reaction scheme. Substrate atoms are outlined in grey.
Based on the atom disposition in Fig. 5A–C, epoxidation of 4,5-desepoxypimaricin by PimD could be explained by a concerted substrate-assisted mechanism in which the hydroperoxoferric intermediate, Compound 0, acts as an oxidant inserting its distal oxygen atom into the C4-C5 double bond (Fig. 5D). In this scheme, upon reduction with the first electron of the PimD-substrate complex, molecular oxygen binds to the heme Fe, expelling W2168 from its place-holding position, in a series of events similar to what has been observed by x-ray crystallography for the ferrous O2-bound EryF-substrate complex (Nagano et al., 2005). Following the same scenario, the distal oxygen atom would occupy a position suited for insertion into the double bond, and at the H-bonding distance to the C7-OH group of 4,5-desepoxypimaricin. Thus, the next intermediate, a highly nucleophilic peroxoferric species formed upon reduction with the second electron, would be positioned favorably to abstract a proton from the C7-OH group, resulting in significant lengthening of the O-O peroxide bond, thus bringing the distal oxygen atom even closer to the π-orbitals. Taken together this would allow formation of the electrophilic hydroperoxoferric transient intermediate with a bonding geometry favoring insertion of the distal oxygen atom via concerted cyclic six-electron rearrangement of the transition state (Fig. 5D) to give the diastereomerically pure product with the epoxy protons in trans configuration (Ceder et al., 1977).
Catalytic evidence of Compound 0 reactivity
The reaction mechanism in Figure 5D suggests that the catalytic conversion of 4,5-desepoxypimaricin may be achieved via the peroxide shunt pathway in the P450 catalytic cycle, where hydrogen peroxide provides both the oxygen atom and the electrons to bypass the requirements for molecular oxygen and NADPH-derived reducing equivalents. Given that organic peroxides are not capable of forming Compound 0 and inserting the distal oxygen atom, we reasoned that only hydrogen peroxide would form an epoxide product. Indeed, formation of pimaricin via the shunt pathway was detected only for hydrogen peroxide, while peracetic acid and iodosobenzene failed to generate the product (Fig. 6A). However, the overall loss of substrate/product material and the heme bleaching were observed for all tested peroxides, suggesting non-specific degradation of the heme and polyene chromophores by the free-radical species formed in the course of the reaction, perhaps as a result of action of the high-valent iron-oxo species, including the powerful oxidant Compound I. To prove this assumption, the same series of reactions was carried out in the presence of the strong antioxidant ascorbic acid, known to reduce free-radical species, thus neutralizing their action. In the presence of ascorbic acid, heme bleaching was strongly inhibited, as expected, and overall recovery of material notably increased in all reactions (Fig. 6B). More importantly, the yield of the epoxidated product pimaricin increased in the hydrogen peroxide reaction, while ascorbic acid failed to rescue PimD epoxidation capability with organic peroxides (Fig. 6A). Unlike shunt pathway, enzymatically driven epoxidation was not affected by ascorbic acid suggesting that no uncoupled formation of free-radicals occurred in the system (data not shown). Collectively, these results strongly suggest that 4,5-desepoxypimaricin epoxidation in the hydrogen peroxide reaction may occur exclusively via a hydroperoxoferric intermediate, Compound 0.
Figure 6. PimD-catalyzed epoxidation via peroxide shunt pathway.
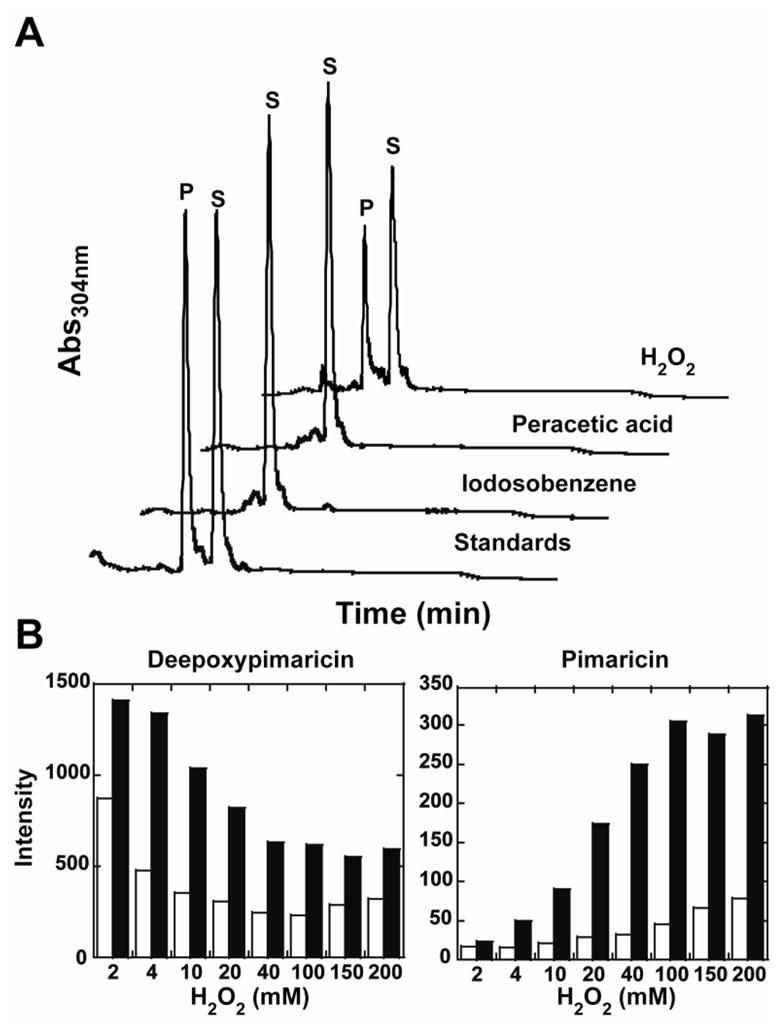
A, High-pressure liquid chromatography traces at 304 nm corresponding to reactions of PimD (10 μM) with 4,5-desepoxypimaricin (100 μM) in the presence of 10 mM ascorbic acid driven by either H2O2 (100 mM), peracetic acid (2 mM) or iodosobenzene (1 mM). Mix of the authentic 4,5-desepoxypimaricin (S) and pimaricin (P) was used as a standard. A new peak is only observed in the presence of H2O2 that corresponds to the epoxidation of the substrate to pimaricin. B, protection effect of 10 mM ascorbic acid (filled bars) on the overall recovery of the substrate 4,5-desepoxypimaricin (left panel) and the product pimaricin (right panel) from the oxidative damage by free-radicals generated upon PimD reaction with H2O2. Empty bars represent recovery of the substrate and product in absence of ascorbic acid.
Reactions of PimD with iodosobenzene and peroxynitrite
The formation of the high-valent iron-oxo species upon interaction with oxidants was addressed by stopped-flow spectroscopy combined with global analysis by singular value decomposition. As accumulation of the active species in the reaction with hydrogen peroxide was slower than bleaching of the heme, the faster reactions with iodosobenzene or peroxynitrite were monitored (Fig. 7). Data resulted from the rapid mix of PimD with iodosobenzene (Fig. 7A) fit satisfactorily to a three-step kinetic model, A→ B → C → D, with the rate constants k1 = 0.271 ± 0.001s−1, k2 = 1.141 ± 0.011 s−1, and k3 = 0.0581 ± 0.0001 s−1. Simpler models were also tested, but failed to give satisfactory fits. The species A corresponds to the initial low-spin ferric protein with a Soret band at 414 nm, species B and C were assigned to oxygen containing intermediates, and D results from the heme bleaching process. The first reaction (k1=0.271 s−1) leads to species B with a Soret band at 423 nm with a shoulder centered at 406 nm, perhaps due to contamination by species C. The spectrum of B bears no similarity to the spectrum of Compound I reported in the literature (Kellner et al., 2002; Raner et al., 2006; Sheng et al., 2008; Spolitak et al., 2005), but closely matches the spectrum of Compound II (Fe4+-OH) observed by Spolitak et al. (Spolitak et al., 2008), as indicated by the increase in absorbance at 435 nm for the first few seconds (Fig. 7A, inset) and the prominent α and β bands at 561 and 527 nm, respectively (Fig. 7B). The species B then decays at a rate of k2=1.141 s−1 to the intermediate C, which reaches maximum concentration (60% conversion) over 9 s (Fig. 7B, inset). The spectrum of C with a Soret band at 410 nm and a broad band centered at 625 nm closely resembles that of Compound ES (Fe4+=O with the protein-based free radical), observed with P450cam and P450BM3 (Raner et al., 2006; Spolitak et al., 2005). A similar result was obtained for peracetic acid. Overall, the reactivity of PimD toward alkylhydroperoxides is notably reduced compared to that of P450cam variants (Spolitak et al., 2005; Spolitak et al., 2006; Spolitak et al., 2008), P450BM3 (Raner et al., 2006) and CYP119 (Kellner et al., 2002; Sheng et al., 2008), and no spectral evidence for Compound I were detected. Instead, we observed notable amounts of Compound II, suggesting that the environment of the PimD active site favors the homolytic cleavage of the ferric-peroxo bond. Alternatively, Compound II could result from the fast reduction of Compound I with excess of peroxide.
Figure 7. Stopped-flow analysis of PimD interactions with iodosobenzene and peroxynitrite.
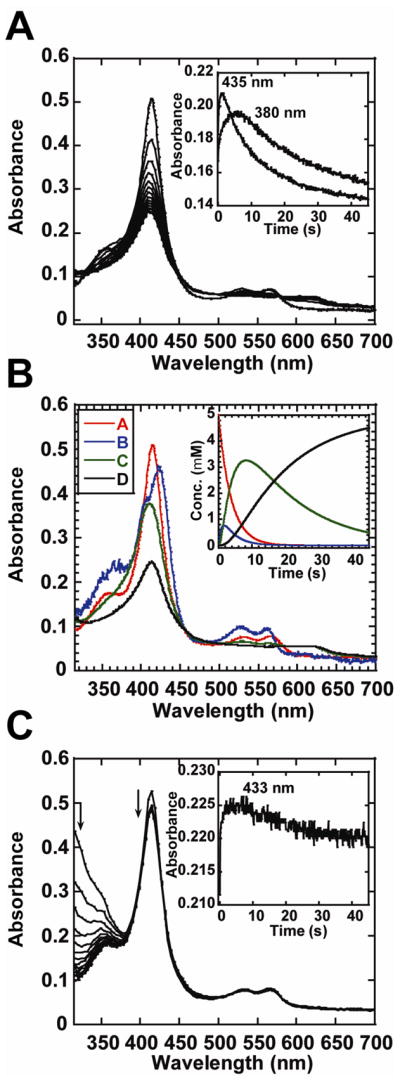
A, Rapid-scan absorbance spectra for the first 45 s of reaction between PimD (5 μM) and iodosobenzene (150 μM) selected in 3 s intervals are shown. Inset: kinetics recorded at 435 and 380 nm. B, singular value decomposition analysis of data in A using A→ B → C → D kinetic model, with k1 = 0.271 s−1, k2 = 1.141 s−1, and k3 = 0.0581 s−1. Inset: time dependence of the different forms of PimD in the reaction with iodosobenzene. C, rapid-scan absorbance spectra for the first 45 s of reaction between PimD (5 μM) and peroxyntrite (250 μM) selected in 3 s intervals are shown. Inset: kinetics recorded at 433 nm. All data were collected at 10°C in 100 mM potassium phosphate, pH 7.4.
Newcomb et al. reported the use of peroxynitrite for the production of a putative Compound II through homolytic cleavage of the O-O bond (Newcomb et al., 2008; Newcomb et al., 2006), which led to the development of a photooxidation method for the formation of Compound I. In this context, we looked at the ability of PimD to form Compound II by mixing the resting ferric protein with an excess of peroxynitrite. As shown in Fig. 7C, PimD reacted poorly with peroxynitrite with only a slight decrease of the Soret band, as opposed to the rapid formation of a spectrally distinct intermediate characterized by a Soret band at 433 nm in CYP119 (Newcomb et al., 2006). However, careful inspection of kinetics at 433 nm indicated a slight increase in absorbance during the first 5.5 s followed by a slow decay (Fig. 7C inset), suggesting that PimD could form Compound II, albeit very inefficiently. The failure to detect Compound I and the barely detectable accumulation of the non-catalytically-competent Compound II upon reaction with surrogate oxygen donors, are consistent with an impaired proton delivery system in PimD.
DISCUSSION
Oxidative modification of the polyol segment in pimaricin, amphotericin and nystatin is the final biosynthetic step performed by the highly homologous P450 monooxygenases PimD, AmphL and NysL, respectively. Common structural features conserved between pimaricin, amphotericin and nystatin suggest that all three polyenes bind in the same orientation, with the lactone end pointing toward the opening of the active site, providing space to compensate for the variable size of the macrolactone ring. On the basis of the PimD structure, we postulate that the substrate enters the binding channel from the N-terminal end of the I-helix, with the ionized exocyclic carboxyl group leading the way via interactions with the conserved positive charges of the Arg65 and Arg223 in the mouth of the channel, and then with the guanidinium group of Arg279, which deeply protrudes into the active site cavity void of the substrate. Upon substrate binding, the Arg279 side chain retracts and rotates >6 Å away from the carboxyl group (Fig. 2C), suggesting, at most, weak electrostatic interactions. Thus ionization of the exocyclic carboxyl group should not be essential for catalysis. This assumption accords with the observation that decarboxy-methyl polyene analogs of pimaricin, amphotericin and nystatin lacking negative charge on the exocyclic carboxyl group are capable of being biosynthetically generated (Brautaset et al., 2008; Caffrey et al., 2008). These analogs are of practical interest as suppression of this negative charge reduces the toxicity of polyene antibiotics (Cheron et al., 1988).
An impaired proton delivery system in PimD and the orientation of 4,5-desepoxypimaricin in the active site are inconsistent with the generally accepted mechanism of olefin epoxidation. The π-orbitals of the C4-C5 double bond are pointing away from and are too distant from the heme Fe to effect the oxygen insertion by the oxoferryl species, Compound I (Fig. 5A–C). This topology is in marked contrast with the x-ray structure reported for the biosynthetic P450 EpoK (Nagano et al., 2003). Strong intrinsic constraints on molecular topology of 4,5-desepoxypimaricin due to the relatively small size of the macrolactone ring accommodating (i) lactone function conjugated with the pair of double bonds, C2-C3 and C4-C5, (ii) the C9-C13 hemi-ketal heterocycle in chair conformation and (iii) conjugated tetraene C16-C23 virtually exclude local dynamic fluctuations that could possibly place the π-orbitals orthogonal to the heme plane. The structure of pimaricin, calculated with both a set of geometrical restraints derived from 1H NMR data in methanol and intrinsic constraints obtained from complete stereochemical information, points to a single conformer for the macrolactone ring (Volpon and Lancelin, 2002). Thus we must accept that either the substrate rotates in the active site as a rigid body or that epoxidation occurs via a mechanism other than that of EpoK. As the space in the active site is limited, extensive protein-substrate interactions would prevent global repositioning of such a large and complex molecule. Alternatively, a mechanism for olefin epoxidation by a hydroperoxoferric intermediate, Compound 0, does satisfy the stereoelectronic configuration of the PimD-4,5-desepoxypimaricin complex and is in accord with the catalytic activity of hydrogen peroxide and lack of the catalytic activity of organic peroxides in the P450 shunt pathway.
Although Compound I is the most reactive intermediate in the P450 catalytic cycle, the steric and stereoelectronic factors that limit the heme macrocycle’s ability to interact with bulky substrates invoke the reactivity of Compound 0. The small substrate molecules selected for model studies allow the steric factor to be disregarded in the interpretation of results. But for a bulky, rigid substrate the steric factor apparently represents an obstacle that may prevent Compound I from acting on 4,5-desepoxypimaricin. As Compound 0 precedes Compound I in the catalytic cycle and converts in a barrier-free reaction to Compound I upon protonation followed by O-O bond cleavage with loss of water (Harris and Loew, 1998; Ogliaro et al., 2002), Compound 0 would be formed and spent prior to formation of Compound I. Under physiological conditions, this would prevent uncoupled formation of free-radicals and protect protein, substrate and for that matter a living S. natalensis cell from massive oxidative damage observed in the shunt reaction in vitro where H2O2 provides the two extra protons facilitating formation of Compound I. Thus, formation of Compound I would be diminished if the delivery of protons were impaired or if Compound 0 were to be spent in the course of the reaction prior to the formation of Compound I. The synergism of both processes apparently drives PimD epoxidation down the less favorable pathway.
SIGNIFICANCE
Our key challenge was to explain the mechanism of the epoxidation reaction by PimD based on the crystal structure of the enzyme-substrate complex. Our structure shows that impaired proton-delivery pathway and the substrate position in the catalytic site are incompatible with the oxygen insertion by the oxoferryl π-cation radical intermediate, Compound I. We suggest an alternative mechanism for the 4,5-desepoxypimaricin epoxidation that involves the hydroperoxoferric intermediate, Compound 0. Because steric and stereoelectronic constraints permit only the oxygen atom of Compound 0 distal to the heme iron to be inserted into the double bond to make an epoxide ring, we used a variety of organic peroxides in the shunt pathway of the P450 catalytic cycle to proof that Compound I is not an oxidant in PimD epoxidation reaction. Our data suggest that the hydroperoxoferric intermediate may be the major, if not the only oxidant in this reaction. Compound I or other high-valent iron-oxo species may be formed in the shunt pathway but are spent in the non-specific reactions manifested in bleaching of heme and the polyene chromophore. The action of a hydroperoxoferric intermediate as a second oxidant in the electronically activated reactive site extends the repertoire of P450 catalysts in enzymatic oxidations in secondary metabolic pathways. These transformations are integral both in the creation of antibiotic molecules with complex oxygenation patterns and also in drug metabolism.
EXPERIMENTAL PROCEDURES
Material and reagents
PimD was overproduced in HMS174(DE3) Escherichia coli strain with an N-terminal His6 affinity tag and purified to electrophoretic homogeneity. Expression vector pMVM5 was constructed based on the pQE-30 (Qiagen) vector as described elsewhere (Mendes et al., 2005). E. coli transformants were grown at 37 °C and 240 rpm agitation until OD590 was 0.4–0.5 in Luria-Bertani medium supplemented with 1 mM thiamine, 50 μg/mL ampicillin and trace elements. PimD expression was induced by the addition of isopropyl-B-D-thiogalactopyranoside (IPTG, final concentration 0.2 μM) and δ-aminolevulinic acid, a precursor of heme biosysnthesis (final concentration 1 mM). Following induction, temperature was decreased to 25 °C and agitation to 140 rpm. After 20 hours the cells were harvested and lysed by sonication. Insoluble material was removed from the crude extract by centrifugation (40 min at 35,000 rpm). The supernatant was subjected to a series of chromatographic steps, including nickel-nitrilotriacetic acid (Ni-NTA) agarose (QIAGEN), followed by SP-Sepharose (Amersham Biosciences) in the flow-through regime, and then by Q-Sepharose (Amersham Biosciences). The protein was eluted from Q-Sepharose using a 0 to 0.5 M NaCl gradient. Fractions containing P450 were combined and concentrated using a Centriprep concentrating device (Millipore), and stored at −80 °C.
Catalytic competence of PimD was confirmed with the enzymatic spinach ferredoxine/ferredoxine reductase system as originally reported (Mendes et al., 2005). Briefly, conversion of 4,5-desepoxypimaricin into pimaricin was accomplished by combining 1 μM PimD, 100 μM 4,5-desepoxypimaricin, 100 μg/ml spinach ferredoxin, 0.2 unit/ml spinach ferredoxin–NADP+ reductase, 1.4 mM NADPH, 10 mM glucose 6-phosphate and 8 units/ml glucose-6-phosphate dehydrogenase in 50 mM Tris-Cl, pH 7.5. The reaction was stopped by the addition of 1 volume of methanol, centrifuged at 10,000 rpm for 3 min and the supernatant was analyzed by HPLC as described below. Under these conditions >80% of substrate was converted into epoxide product in 90 min.
4,5-desepoxypimaricin was obtained from ΔPimD mutant S.natalensis 6D4 culture broths following published procedures (Mendes et al., 2001). After 2 days of growth at 300 rpm and 28 °C, the supernatant was extracted with one volume of methanol. This was repeated once, and the solvent was evaporated to yield a dry powder. The residue thus obtained was dissolved in 100 ml methanol, treated with 30 ml acetone, and centrifuged to remove precipitated material. The supernatant was then lyophilized, resuspended in methanol, and diluted with one volume of water. Final purification of 4,5-desepoxypimaricin was carried out with a Waters 600 HPLC with a diode array ultraviolet detector set at 304 nm, fitted with a LiChroCART LiChrospher 100 RP-18 (10 μm; 10 × 250 mm) column. Elution was with a gradient (10 ml/min) of 100% methanol (methanol concentration: 50% 0–3 min, up to 90% 3–12 min, 90% 12–20min, down to 50% 20–25 min, 50% 25–30 min).
Spectroscopic binding assay
4,5-desepoxypimaricin binding assays were performed by spectrophotometric titration in 100 mM KPO4, pH 7.5 containing 10% glycerol using a Cary dual beam UV-visible scanning spectrometer (Varian). Stock solution of 4,5-desepoxypimaricin was prepared in DMSO. The concentration of PimD was determined at 450 nm from the difference spectra between the carbon monoxide-bound ferrous and water bound ferric forms, with an extinction coefficient of 91,000 M−1cm−1. To account for the absorbance of 4,5-desepoxypimaricin, 1 ml of protein (1 μM) in buffer was placed in the first chamber of two split cuvettes, and 1 ml of buffer was placed in the second chamber. After background scanning, equal volumes (1 μl) of 4,5-desepoxypimaricin solution were titrated into both the first chamber of the sample cuvette containing protein and the second chamber of the reference cuvette containing only buffer, resulting in concentration increases from 1 μM to 15 μM in 1 μM increments. The same volume of DMSO was added into the alternate chambers to correct for organic solvent effect. Difference spectra were recorded from 300 to 500 nm. The KD value was determined using GraphPad PRISM software (Graphpad Software Inc.) to fit titration data to rectangular hyperbola according to the functions ΔA = Amax(S/KD+S), where E is total enzyme and S total substrate concentration, Amax the maximal absorption shift at saturation, and KD the dissociation constant for the enzyme-ligand complex.
Crystallization, data collection, structure determination
Crystallization conditions were determined using commercial high throughput screening kits available in deep-well format (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP LabTech) operating with 96-well plates, and a hanging drop crystallization protocol. Optimization of conditions, if necessary, was carried out manually in 24-well plates. The protein was from 1.1 mM frozen stock in 20 mM Tris-HCl, pH 7.5, 200 mM NaCl, and 0.5 mM EDTA. Prior to crystallization, the protein was diluted to 0.2–0.4 mM by mixing with 10 mM Tris-HCl, pH 7.5, alone or supplemented with 2 mM 4,5-desepoxypimaricin. Crystals of the ligand-free PimD grew in 4-μl crystallization drop containing 1.5 M ammonium sulfate, 2% PEG 400, and 0.1 M HEPES, pH 8.0. Crystals of the PimD-4,5-desepoxypimaricin complex were harvested directly from the 0.2-μl drop of the 96-well screening plates containing 0.2 M sodium malonate, pH 7.0, and 20% PEG 3350. Prior to data collection, the crystals were cryo-protected by plunging them into a drop of reservoir solution supplemented with 20% ethylene glycol, and flash frozen in liquid nitrogen. Diffraction data were collected at 100–110 K at beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, USA. Data indexing, integration, and scaling were conducted using MOSFLM (Leslie, 1992) and the ELVES software suite (Holton and Alber, 2004). Crystal structure of the substrate-free PimD was determined to a resolution of 2.1 Å by molecular replacement using diffraction data processed in C2221 with Rmerge of 8.8% and the atomic coordinates of CYP105A1 of Streptomyces griseolus (PDB ID code: 2ZBZ) (Sugimoto et al., 2008) as a search model. The initial PimD model was built by using BUCCANEER program (1994; Cowtan, 2006). Refinement was performed by using REFMAC5 program (1994; Murshudov et al., 1997) until R and Rfree converged to 18.6% and 23.9%, respectively. Ramachandran statistics indicates 96.8% residues in preferred region, 2.7% in allowed region and 0.5% (2 residues) outliers, as calculated by COOT (Emsley and Cowtan, 2004). The refined substrate-free coordinates were used as the molecular replacement model to determine the co-crystal structure with 4,5-desepoxypimaricin to a resolution of 1.95 A with R and Rfree of 16.5% and 21.9%, respectively, using diffraction data processed in C2 with Rmerge of 7.1%. Ramachandran statistics indicates 98.6% residues in preferred region and 1.4% residues in allowed region (Table 1).
TABLE 1.
Data collection and refinement statistics.
| Protein | PimD | |
|---|---|---|
| Ligand | Substrate-free | 4,5-desepoxypimaricin |
| PDB ID | 2X9P | 2XBK |
| Data collection | ||
| Space group | C2221 | C2 |
| Cell dimensions | ||
| a, b, c (Å) | 118.6, 139.4, 70.0 | 99.9, 99.7, 58.5 |
| α, β, γ (°) | 90, 90, 90 | 90, 111.92, 90 |
| Molecules in AU | 1 | 1 |
| Wavelength | 1.1159 | 1.1159 |
| Resolution (Å) | 2.1 | 1.95 |
| Rsym or Rmerge (%) | 8.8 (56.6)1 | 7.1 (52.3) |
| I/σI | 12.3 (2.9) | 10.0 (2.3) |
| Completeness (%) | 99.7 (100.0) | 99.8 (100.0) |
| Redundancy | 5.9 (5.9) | 4.1 (4.1) |
| Refinement | ||
| No. reflections | 32420 | 36749 |
| Rwork/Rfree (%) | 18.6/23.9 | 15.0/21.4 |
| No. atoms | ||
| Protein | 2955 | 3147 |
| Heme | 43 | 43 |
| Ligand | none | 46 |
| Water | 177 | 272 |
| Mean B value | 31.7 | 30.8 |
| B-factors | ||
| Protein | 31.6 | 29.3 |
| Heme | 22.2 | 17.6 |
| Ligand | N/A | 27.7 |
| Water | 34.8 | 36.2 |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.025 | 0.027 |
| Bond angles (°) | 1.97 | 1.90 |
Values in parentheses are for highest-resolution shell.
N/A stays for not applicable
Conversion of desepoxypimaricin to pimaricin using the peroxide shunt pathway
Reaction mixtures (total volume, 100 μl) contained 10 μM PimD, 100 μM 4,5-desepoxypimaricin in 50 mM Tris-HCl buffer (pH 7.5). The reaction was initiated by the addition of H2O2 (0–200 mM), peracetic acid (PAA, 0–2 mM) or iodosobenzene (0–1 mM). Control incubations were performed without enzyme or peroxide. To reduce oxidative destruction of the heme, substrate, or product by Compound I, sodium ascorbate was added to the reaction mixtures at 10 mM, when indicated. The reactions were carried out for 30 min at 25 °C, terminated by the addition of 1 volume of methanol and centrifuged for 3 min at 10,000×g. Supernatants were collected and analyzed directly by HPLC as follows. Quantitative detection of pimaricin or 4,5-desepoxypimaricin was performed with an Agilent 1200 HPLC equipped with a diode array detector set at 304 nm and connected to a Symmetry reverse-phase-C18 column (3.5 μm; 4.6 mm×150 mm). Elution was performed at flow rate of 0.5 ml/min with a linear gradient (50–90%) of methanol. The entire column cycle was as following: 50% methanol, 0–3 min; increase methanol concentration up to 90%, 3–12 min, 90% methanol, 12–20 min; decrease methanol concentration down to 50%, 20–25 min; 50% methanol, 25–30 min. Retention times for pimaricin and 4,5-desepoxypimaricin were 14.3 and 15.5 min, respectively.
Stopped-Flow Spectrophotometry
Rapid mixing experiments were conducted with a Hi-Tech Scientific instrument (Bradford on Avon, U.K.) equipped with a photodiode array detector. All the reaction kinetics were measured at 10°C. Solutions of ferric PimD (10 μM) in 100 mM KPi (pH 7.4) were mixed with buffered solutions of iodosobenzene (from 50 to 150 μM), or with 500 μM peroxynitrite dissolved in 10 mM NaOH. Upon each mixing, total of 300 spectra were collected over various time scales. Rate constants were estimated by globally fitting the kinetic data at different ligand concentrations to the various models using singular value decomposition analysis implemented in ProK software (Applied Photophysics, Leatherhead, UK). The kinetic constants obtained from the fitting had uncertainties of ≤5%.
Acknowledgments
We thank Prof. Paul Ortiz de Montellano for valuable discussions, Mr. Potter Wickware for critical reading of the manuscript, Dr. Chiung-Kuang Chen and the staff members of beamline 8.3.1, Dr. James Holton, George Meigs and Jane Tanamachi, the Advanced Light Source at Lawrence Berkeley National Laboratory for assistance with data collection. This work was supported by NIH RO1 grant GM078553 (to L.M.P.) and Spanish Ministry of Science and Innovation grant BIO2007-67585 (to J.F.A.). H.O. was supported by NIH RO1 grant GM25515 (to Paul R. Ortiz de Montellano). J.S.A. was supported by F.P.U. fellowships AP2005-3644 from the Ministry of Science and Education, Spain. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Data deposition note: The atomic coordinates and structure factors (codes 2X9P and 2XBK) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
Author contributions: L.M.P. and H.O. designed research and analyzed data; P.M.K. performed research; J.F.A. and J.S.A. contributed new reagents, L.M.P. wrote the manuscript.
Conflict of interest statement: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Collaborative Computational Project Number 4. Acta Crysallogr D. 1994;50:760–763. [Google Scholar]
- Aparicio JF, Caffrey P, Gil JA, Zotchev SB. Polyene antibiotic biosynthesis gene clusters. Appl Microbiol Biotechnol. 2003;61:179–188. doi: 10.1007/s00253-002-1183-5. [DOI] [PubMed] [Google Scholar]
- Aparicio JF, Mendes MV, Anton N, Recio E, Martin JF. Polyene macrolide antibiotic biosynthesis. Curr Med Chem. 2004;11:1645–1656. doi: 10.2174/0929867043365044. [DOI] [PubMed] [Google Scholar]
- Bach RD, Dmitrenko O. Strain energy of small ring hydrocarbons. Influence of C-H bond dissociation energies. J Am Chem Soc. 2004;126:4444–4452. doi: 10.1021/ja036309a. [DOI] [PubMed] [Google Scholar]
- Baginski M, Czub J, Sternal K. Interaction of amphotericin B and its selected derivatives with membranes: molecular modeling studies. Chem Rec. 2006;6:320–332. doi: 10.1002/tcr.20096. [DOI] [PubMed] [Google Scholar]
- Baginski M, Sternal K, Czub J, Borowski E. Molecular modelling of membrane activity of amphotericin B, a polyene macrolide antifungal antibiotic. Acta Biochim Pol. 2005;52:655–658. [PubMed] [Google Scholar]
- Brajtburg J, Powderly WG, Kobayashi GS, Medoff G. Amphotericin B: current understanding of mechanisms of action. Antimicrob Agents Chemother. 1990;34:183–188. doi: 10.1128/aac.34.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautaset T, Sletta H, Nedal A, Borgos SE, Degnes KF, Bakke I, Volokhan O, Sekurova ON, Treshalin ID, Mirchink EP, et al. Improved antifungal polyene macrolides via engineering of the nystatin biosynthetic genes in Streptomyces noursei. Chem Biol. 2008;15:1198–1206. doi: 10.1016/j.chembiol.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Byrne B, Carmody M, Gibson E, Rawlings B, Caffrey P. Biosynthesis of deoxyamphotericins and deoxyamphoteronolides by engineered strains of Streptomyces nodosus. Chem Biol. 2003;10:1215–1224. doi: 10.1016/j.chembiol.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Caffrey P, Aparicio JF, Malpartida F, Zotchev SB. Biosynthetic engineering of polyene macrolides towards generation of improved antifungal and antiparasitic agents. Curr Top Med Chem. 2008;8:639–653. doi: 10.2174/156802608784221479. [DOI] [PubMed] [Google Scholar]
- Ceder O, Hansson B, Rapp U. Pimaricin. VIII. Structural and configurational studies by electron impact and field desorption mass spectrometry, 13C (25.2 MHz) and 1H (270 MHz)-NMR spectroscopy. Tetrahedron. 1977;33:2703–2714. [Google Scholar]
- Chandrasena RE, Vatsis KP, Coon MJ, Hollenberg PF, Newcomb M. Hydroxylation by the hydroperoxy-iron species in cytochrome P450 enzymes. J Am Chem Soc. 2004;126:115–126. doi: 10.1021/ja038237t. [DOI] [PubMed] [Google Scholar]
- Cheron M, Cybulska B, Mazerski J, Grzybowska J, Czerwinski A, Borowski E. Quantitative structure-activity relationships in amphotericin B derivatives. Biochem Pharmacol. 1988;37:827–836. doi: 10.1016/0006-2952(88)90168-2. [DOI] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Cupp-Vickery JR, Han O, Hutchinson CR, Poulos TL. Substrate-assisted catalysis in cytochrome P450 eryF. Nature Struct Biol. 1996;3:632–637. doi: 10.1038/nsb0796-632. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Ganis P, Avitabile G, Mechlinski W, Schaffner CP. Polyene macrolide antibiotic amphotericin B. Crystal structure of the N-iodoacetyl derivative. J Am Chem Soc. 1971;93:4560–4564. doi: 10.1021/ja00747a037. [DOI] [PubMed] [Google Scholar]
- Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Cytochrome P450 oxidations in the generation of reactive electrophiles: epoxidation and related reactions. Arch Biochem Biophys. 2003;409:59–71. doi: 10.1016/s0003-9861(02)00415-0. [DOI] [PubMed] [Google Scholar]
- Harris DL, Loew GH. Theoretical investigation of the proton assisted pathway to formation of cytochrome P450 Compound I. J Am Chem Soc. 1998;120:8941–8948. doi: 10.1021/ja954101m. [DOI] [PubMed] [Google Scholar]
- Hirao H, Kumar D, Shaik S. On the identity and reactivity patterns of the “second oxidant” of the T252A mutant of cytochrome P450cam in the oxidation of 5-methylenenylcamphor. J Inorg Biochem. 2006;100:2054–2068. doi: 10.1016/j.jinorgbio.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci U S A. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai M, Shimada H, Watanabe Y, Matsushima-Hibiya Y, Makino R, Koga H, Horiuchi T, Ishimura Y. Uncoupling of the cytochrome P-450cam monooxygenase reaction by a single mutation, threonine-252 to alanine or valine: possible role of the hydroxy amino acid in oxygen activation. Proc Natl Acad Sci U S A. 1989;86:7823–7827. doi: 10.1073/pnas.86.20.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Bryson TA, Dawson JH. Hydroperoxoferric heme intermediate as a second electrophilic oxidant in cytochrome P450-catalyzed reactions. J Biol Inorg Chem. 2004;9:644–653. doi: 10.1007/s00775-004-0575-7. [DOI] [PubMed] [Google Scholar]
- Jin S, Makris TM, Bryson TA, Sligar SG, Dawson JH. Epoxidation of olefins by hydroperoxo-ferric cytochrome P450. J Am Chem Soc. 2003;125:3406–3407. doi: 10.1021/ja029272n. [DOI] [PubMed] [Google Scholar]
- Kellner DG, Hung SC, Weiss KE, Sligar SG. Kinetic characterization of compound I formation in the thermostable cytochrome P450 CYP119. J Biol Chem. 2002;277:9641–9644. doi: 10.1074/jbc.C100745200. [DOI] [PubMed] [Google Scholar]
- Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 ESF-EAMCB Newslett Protein Crystallogr. 1992;26 [Google Scholar]
- Martinis SA, Atkins WM, Stayton PS, Sligar SG. A conserved residue of P450 involved in haem-oxygen stability and activation. J Am Chem Soc. 1989;111:9252–9253. [Google Scholar]
- Mendes MV, Anton N, Martin JF, Aparicio JF. Characterization of the polyene macrolide P450 epoxidase from Streptomyces natalensis that converts de-epoxypimaricin into pimaricin. Biochem J. 2005;386:57–62. doi: 10.1042/BJ20040490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes MV, Recio E, Fouces R, Luiten R, Martin JF, Aparicio JF. Engineered biosynthesis of novel polyenes: a pimaricin derivative produced by targeted gene disruption in Streptomyces natalensis. Chem Biol. 2001;8:635–644. doi: 10.1016/s1074-5521(01)00033-3. [DOI] [PubMed] [Google Scholar]
- Meunier B, deVisser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nagano S, Cupp-Vickery JR, Poulos TL. Crystal structures of the ferrous dioxygen complex of wild-type cytochrome P450eryF and its mutants, A245S and A245T: investigation of the proton transfer system in P450eryF. J Biol Chem. 2005;280:22102–22107. doi: 10.1074/jbc.M501732200. [DOI] [PubMed] [Google Scholar]
- Nagano S, Li H, Shimizu H, Nishida C, Ogura H, Ortiz de Montellano PR, Poulos TL. Crystal structures of epothilone D-bound, epothilone B-bound, and substrate-free forms of cytochrome P450epoK. J Biol Chem. 2003;278:44886–44893. doi: 10.1074/jbc.M308115200. [DOI] [PubMed] [Google Scholar]
- Newcomb M, Halgrimson JA, Horner JH, Wasinger EC, Chen LX, Sligar SG. X-ray absorption spectroscopic characterization of a cytochrome P450 compound II derivative. Proc Natl Acad Sci U S A. 2008;105:8179–8184. doi: 10.1073/pnas.0708299105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb M, Hollenberg PF, Coon MJ. Multiple mechanisms and multiple oxidants in P450-catalyzed hydroxylations. Arch Biochem Biophys. 2003;409:72–79. doi: 10.1016/s0003-9861(02)00445-9. [DOI] [PubMed] [Google Scholar]
- Newcomb M, Zhang R, Chandrasena RE, Halgrimson JA, Horner JH, Makris TM, Sligar SG. Cytochrome P450 compound I. J Am Chem Soc. 2006;128:4580–4581. doi: 10.1021/ja060048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogliaro F, de Visser SP, Cohen S, Sharma PK, Shaik S. Searching for the second oxidant in the catalytic cycle of cytochrome P450: a theoretical investigation of the iron(III)-hydroperoxo species and its epoxidation pathways. J Am Chem Soc. 2002;124:2806–2817. doi: 10.1021/ja0171963. [DOI] [PubMed] [Google Scholar]
- Omura S, Tanaka H. Production, structure and antifungal activity of polyene macrolides. In: Omura S, editor. Macrolide antibiotics: chemistry, biology and practice. New York: Academic Press; 1984. pp. 351–405. [Google Scholar]
- Ortiz de Montellano PR, De Voss JJ. Oxidizing species in the mechanism of cytochrome P450. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- Raner GM, Thompson JI, Haddy A, Tangham V, Bynum N, Ramachandra Reddy G, Ballou DP, Dawson JH. Spectroscopic investigations of intermediates in the reaction of cytochrome P450(BM3)-F87G with surrogate oxygen atom donors. J Inorg Biochem. 2006;100:2045–2053. doi: 10.1016/j.jinorgbio.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Schaffner CP. Polyene macrolides in clinical practice: pharmacology and adverse and other effects. In: Omura S, editor. Macrolide antibiotics, chemistry, biology and practice. New York: Academic Press; 1984. pp. 457–507. [Google Scholar]
- Shaik S, Hirao H, Kumar D. Reactivity patterns of cytochrome P450 enzymes: multifunctionality of the active species, and the two states-two oxidants conundrum. Nat Prod Rep. 2007;24:533–552. doi: 10.1039/b604192m. [DOI] [PubMed] [Google Scholar]
- Sheng X, Horner JH, Newcomb M. Spectra and kinetic studies of the compound I derivative of cytochrome P450 119. J Am Chem Soc. 2008;130:13310–13320. doi: 10.1021/ja802652b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- Spolitak T, Dawson JH, Ballou DP. Reaction of ferric cytochrome P450cam with peracids: kinetic characterization of intermediates on the reaction pathway. J Biol Chem. 2005;280:20300–20309. doi: 10.1074/jbc.M501761200. [DOI] [PubMed] [Google Scholar]
- Spolitak T, Dawson JH, Ballou DP. Rapid kinetics investigations of peracid oxidation of ferric cytochrome P450cam: nature and possible function of compound ES. J Inorg Biochem. 2006;100:2034–2044. doi: 10.1016/j.jinorgbio.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Spolitak T, Dawson JH, Ballou DP. Replacement of tyrosine residues by phenylalanine in cytochrome P450cam alters the formation of Cpd II-like species in reactions with artificial oxidants. J Biol Inorg Chem. 2008;13:599–611. doi: 10.1007/s00775-008-0348-9. [DOI] [PubMed] [Google Scholar]
- Sugimoto H, Shinkyo R, Hayashi K, Yoneda S, Yamada M, Kamakura M, Ikushiro S, Shiro Y, Sakaki T. Crystal structure of CYP105A1 (P450SU-1) in complex with 1alpha,25-dihydroxyvitamin D3. Biochemistry. 2008;47:4017–4027. doi: 10.1021/bi7023767. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz ADN, McGinnity DF, Coon MJ. Epoxidation of olefins by cytochrome P450: Evidence from site-specific mutagenesis for hydroperoxo-iron as an electrophilic oxidant. Proc Natl Acad Sci U S A. 1998;95:3555–3560. doi: 10.1073/pnas.95.7.3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volokhan O, Sletta H, Ellingsen TE, Zotchev SB. Characterization of the P450 monooxygenase NysL, responsible for C-10 hydroxylation during biosynthesis of the polyene macrolide antibiotic nystatin in Streptomyces noursei. Appl Environ Microbiol. 2006;72:2514–2519. doi: 10.1128/AEM.72.4.2514-2519.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpon L, Lancelin JM. Solution NMR structure of five representative glycosylated polyene macrolide antibiotics with a sterol-dependent antifungal activity. Eur J Biochem. 2002;269:4533–4541. doi: 10.1046/j.1432-1033.2002.03147.x. [DOI] [PubMed] [Google Scholar]
- Zotchev SB. Polyene macrolide antibiotics and their applications in human therapy. Curr Med Chem. 2003;10:211–223. doi: 10.2174/0929867033368448. [DOI] [PubMed] [Google Scholar]