

Abstract
The obesity epidemic is a global issue and shows no signs of abating, while the cause of this epidemic remains unclear. Marketing practices of energy-dense foods and institutionally-driven declines in physical activity are the alleged perpetrators for the epidemic, despite a lack of solid evidence to demonstrate their causal role. While both may contribute to obesity, we call attention to their unquestioned dominance in program funding and public efforts to reduce obesity, and propose several alternative putative contributors that would benefit from equal consideration and attention. Evidence for microorganisms, epigenetics, increasing maternal age, greater fecundity among people with higher adiposity, assortative mating, sleep debt, endocrine disruptors, pharmaceutical iatrogenesis, reduction in variability of ambient temperatures, and intrauterine and intergenerational effects, as contributing factors to the obesity epidemic are reviewed herein. While the evidence is strong for some contributors such as pharmaceutical-induced weight gain, it is still emerging for other reviewed factors. Considering the role of such putative etiological factors of obesity may lead to comprehensive, cause specific, and effective strategies for prevention and treatment of this global epidemic.
Introduction
The Prevalence of Obesity
The prevalence of obesity has increased substantially since the mid-20th century. Although there seems to have been an accelerated rate of increase somewhere around 1980, at least in the United States (Baskin et al., 2005; Ogden et al., 2007), evidence suggests that obesity has been increasing in prevalence for over one hundred years (Helmchen & Henderson, 2004). Within the United States, this increase has occurred in every age, race, sex and socioeconomic group. Although recent evidence suggests that the prevalence of obesity may have begun to asymptote within some segments of the U.S. (Ogden et al., 2008) and some other populations, there is no sign of any decreases in U.S. prevalence to date. Obesity has not only increased in the United States but also seems to have increased in virtually every country where detailed data are available (Caballero, 2007). Reasons for this increase are incompletely understood (Keith et al., 2006; Astrup et al., 2006; Bray & Champagne, 2005; Eisenmann, 2006).
The Hegemony of The Big Two
The two most commonly advanced reasons for the increase in the prevalence of obesity are certain food marketing practices and institutionally-driven reductions in physical activity, which we have taken to calling “the big two.” Elements of the big two include, but are not limited to, the “built environment”, increased portion sizes in commercially marketed food items, inexpensive food sources such as fast food, increased availability of vending machines with energy-dense items, increased use of high fructose corn syrup, and less physical education in schools. It is important to distinguish the big two from energy intake and physical activity energy expenditure or more loosely “diet and exercise” with which they are often inappropriately conflated. That is, when we question the strength of the evidence of the big two as contributors, or certainly the chief and near sole contributors to the obesity epidemic, we are not questioning the importance of energy intake and energy expenditure, including physical activity energy expenditure in influencing obesity levels. Rather, we are suggesting that a myopic emphasis on the “big two” has caused the popular media, and perhaps some researchers as well, to neglect the potential contributions of other factors to the balance between energy intake and expenditure. Our questioning of the big two stems from two points. First, the evidence supporting various elements of the big two as contributors to individual or population levels of obesity is often quite weak. Second, even though some elements of the big two do very likely play some role in influencing obesity levels, we believe that an unquestioned assumption of their preeminence has led to the possibly ill-advised expenditure of public effort and funds on programs aimed at reducing population levels of obesity that and has also reduced the exploration of other potential causes and the alternative obesity reduction programs that might stem from their identification.
The big two seem to be accorded special status in many dialogues and writings on obesity such that our usual healthy scientific skepticism is held back when considering them. Perhaps this is because the Big Two as explanations for obesity appeal to a prevalent anti-corporate sentiment (Crossley, 2002) or perhaps because they have an intuitive appeal based, in part, on their simplicity and the fact that they require little specialized knowledge to comprehend and deal with easily observable aspects of life with which all of us are familiar. Regardless of the cause, as scientists, we should retain our skepticism toward all hypotheses and our open-mindedness to new hypotheses.
What are some of the specific facts that enhance our skepticism of the big two as near-omnipotent causes of the obesity epidemic? We make no pretext of offering an exhaustive consideration of evidence for or against the big two, but rather highlight a few bullet points that reinforce our skepticism.
Restaurant Dining. Restaurant dining and fast-food restaurant dining in particular have been considered as major contributors to the obesity epidemic. Yet, Anderson and Matsa (2008) conducted an analysis of a nationally representative sample of 3-day food records and found that while diners at fast food restaurants ate roughly 200–300 kcal more during restaurant meals, they largely compensated by eating less at other occasions such that the net increase in energy intake associated with restaurant dining was extremely small (i.e., 24 kcal on days in which someone ate in a restaurant).
Physical Education. Some argue that a reduction in the frequency of physical education (PE) is a major contributor to obesity. Yet the evidence that PE frequency has decreased is itself questionable (Sturm, 2005) and some studies in children report that the frequency of participation in sport has increased (Salmon et al 2003). Regardless of changes in frequency of PE offerings or participation, much evidence suggests that standard PE classes have no appreciable impact on obesity levels (Cawley et al., 2007).
Sidewalks and the Built Environment. Some have suggested that aspects of the ‘built environment”, especially lack of sidewalks decreases walking which in turn increases obesity. Yet, when Miles et al (2008) compared “walking and obesity rates in two African-American neighborhoods that are similar in urban form but different in level of neighborhood disadvantage”, they found that “levels of leisure walking and physical activity were not higher, and rates of obesity were not lower in the non-poor neighborhood with better maintenance, more sidewalks and recreational facilities.”
High-Fructose Corn Syrup Consumption (HFCS). HFCS consumption (but not necessarily fructose per se) has increased substantially in the last several decades and has been speculated to be a contributor to the obesity epidemic (Bray et al 2004; Welsh et al 2005). Yet, a critical review (Forshee et al., 2007) and a recent position paper from the American Medical Association concluded “Because the composition of HFCS and sucrose are so similar, particularly on absorption by the body, it appears unlikely that HFCS contributes more to obesity or other conditions than sucrose” (American Medical Association, 2008).
Vending Machines. Vending machines have been discussed as a threat to childhood overweight and obesity and changes in school policy have been made to reflect this view (Sothern et al 2004). Yet little to no extant evidence indicates that vending machines have contributed to the problem (Faith et al., 2007).
These are just a few examples and we do not wish to imply that any of the hypothesized influences described in the bullet points above merit being summarily dismissed. Further research on many aspects of the big two is clearly warranted. Our point is merely that we do not have conclusive evidence that the big two or their individual elements are the preeminent contributors to the obesity. Despite the lack of solid evidence that clearly demonstrates the “culprits” are chiefly responsible for the obesity epidemic, researchers are quick to blame them even in the public eye. Cooper (2004), when advising politicians on policy making, stated “Yes, fast food and convenience foods are more prevalent today than ever before. And yes, portion sizes and caloric intake have increased. But that doesn’t mean that these are the only culprits in our growing battle with the bulge. The wholesale lack of physical activity is the primary reason [emphasis added] for our expanding waistlines”. Such confidence in one “culprit” is then often contradicted by public health proponent advancing their favored target for intervention.
Based on these confident statements by researchers, the mass media publishes articles with statements regarding obesity such as “There are several reasons for the rising rates, experts say. Contemporary life has become more sedentary, with more time spent in front of the computer and TV screens and less time engaging in rigorous activity.....Americans are also eating out more often and becoming accustomed to larger portions” (Taneeru 2006). This same article quoted Arkansas Governor Mike Huckabee, as saying “‘Kids today don’t come home and play in their neighborhoods and run and romp ‘til it’s dark… They come home, they get in behind locked doors so that their parents can know where they are and they turn on the computer or television or a video game and they sit with a bowl of chips in their lap and they eat and they sit.’”. As scientists we believe that we can and should do our part to offer a more sophisticated and data-based view of obesity.
The Complex Reality of Obesity and Other Putative Influences
Outside the circle of public health advocacy discussions, scientists widely and readily acknowledge that multiple factors contribute to obesity including but not necessarily limited to genetic, dietary, economic, psychosocial, reproductive, and pharmacologic factors. Our purpose here is to expand upon our brief discussions elsewhere (Keith et al., 2006) and offer a more thorough discussion of factors that may be contributing to the obesity epidemic beyond those conventionally included within the big two. We introduce one or two additional factors not covered by Keith et al. (2006). We do not discuss two factors covered in that previous article, namely reductions in smoking prevalence and demographic changes, because we believe that they are so well documented and supported that little further review of the evidence is needed at this time. For a useful recent consideration of the influence of smoking reduction on obesity, see (Baum 2008). While the role of decreased smoking in the obesity epidemic is an important observation, it does not change the fact that smoking is an unhealthful habit that we would not wish to promote. Similarly, demographic changes may play a role in obesity, but they are not subject to manipulation by reasonable public policy. For these reasons, we do not consider smoking and demographic changes further herein.
The Evidence We Will Cover
Although there is some variation from one putative cause to another, we generally seek evidence of the following types.
Ecological Correlation. This is generally the weakest form of evidence but worth reviewing because it provides a basis for hypothesis generation and can offer additional support to an overall body of evidence. Ecological correlative evidence would typically involve showing that the amount of the putative contributing factor has increased over time during the same period that obesity has increased or is positively correlated with obesity levels across populations (e.g., countries) within a time-period.
Individual Level Epidemiologic Correlation. This is a somewhat stronger form of evidence because we can, in principle, make better attempts to control for potential confounding factors. Such evidence, though stronger than ecological correlation, can still only demonstrate association and not causation.
Non-Human Experimental Evidence. Such data including that derived from cell line and model organism studies, are usually very convincing because they can provide us with evidence about causation and the mechanisms of action. However, such evidence always leaves extrapolation to humans open to question.
Human Experimental Evidence. Human experimental evidence (by experimental we mean studies in which subjects are randomly assigned to different levels of the independent variable under study) can offer evidence about causation in humans and are likely the best evidence we can obtain. Unfortunately, such evidence is often limited in the present context for several reasons. First it is often impractical or unethical to randomly assign people to be exposed to the factors under study (e.g., presumed toxins). Second, even when randomized experiments can be done it is often difficult to do them with sufficient fidelity on a large scale and for a sufficiently long period of time to permit confident conclusions about long-term population effects. Thus, the evidence available in this realm is often limited to small, short-term, or laboratory analogue studies.
For each of the putative causes we will review, we seek evidence in each of the categories described above to provide a picture of the strength or lack thereof of the entire body of evidence.
Infections & Obesity
General Statement of Putative Cause and Hypothesized Mechanism of Action
Although ten different microbes have been reported to cause obesity in various experimental models (Pasarica and Dhurandhar 2007), the possible contribution of infections in the etiology of human obesity is often overlooked. Considering the etiological role of infections in several other chronic diseases, (Dhurandhar 2001), a relationship between infections and obesity is plausible. The close interaction between the function of the immune system and adipose tissue adds to this plausibility. Adipocytes and macrophages share many similar functional characteristics, and they are in fact so similar that preadipocytes have the ability to differentiate into macrophages (Charriere et al 2003). Therefore, it is plausible for adipose tissue to expand in response to certain infections, as a direct consequence or by shifting of the organism to surplus energy state. In case of some infections, resulting adiposity could be due to a “bystander” effect, similar to the effect caused by helicobacter pylori which burrows under the mucus lining of stomach and causes gastritis and ulcers in the process (Dunn et al 1997).
Basic Science Evidence
Animal Evidence
Experimental infection of animal models with obesity-promoting microbes results in increased adiposity, demonstrating a direct cause-and-effect relationship. These experimental models range from non-human primates to insects and include rodents and chickens. Canine distemper virus (CDV) was the first reported obesity promoting virus (Lyons et al 1982). In mice infected with CDV, obesity develops after acute infection has abated and when no virus is detectable. This modus operendi supports the “hit and run” hypothesis that infection can have effects even when the active infection is no longer detectable (Bernard et al 1999; Nevels et al 2001). CDV decreases levels of melanin concentrating hormone (Veraeten et al 2001), and down-regulates leptin receptors in the hypothalamus, (Bernard et al 1999) which may promote positive energy balance in this model.
Other infectious agents have also been examined in this context. For example, Rous-associated virus-7 (RAV-7), an avian retrovirus, causes stunted growth, obesity and hyperlipidemia in chickens (Carter et al 1983a & b). The main effect of RAV-7 that may contribute to adiposity is a decrease in thyroid hormone levels (Duff et al 1969). In addition, Borna Disease virus has been found to cause obesity in rats (Gosztonyi et al 1995), by damaging the hypothalamus and by neuroendocrine dysregulations (Herden et al 2000), and scrapie agents have been reported to induce obesity in mice (Kim et al 1987; Carp et al 1998). Although the exact adipogenic mechanism is unknown, Scrapie agents disrupt normal glucose metabolism, with hyperglycemia resulting in disrupted transvascular glucose transport in some regions of the brain (Vorbrodt et al 2001), which may lead to functional dysregulation of hypothalamus and consequential adiposity.
Next, four adenoviruses were reported to promote obesity. Animals experimentally infected with SMAM-1, an avian adenovirus, or three human adenoviruses, adeneovirus-36 (Ad-36), Ad-5, and Ad-37 developed increased adiposity (Dhurandhar et al 1990; 1992; 2000; 2001; 2002; Whigham et al 2006; So et al 2005) compared to controls despite similar food intake.
SMAM-1 caused a 53% increase in visceral fat in chickens compared to the control group in just three weeks after inoculation (Dhurandhar et al 1990; 1992). Originally un-infected cage-mates of the experimentally infected chickens acquired the virus via horizontal transmission and developed visceral adiposity. Paradoxically, the increased adiposity due to SMAM-1 infection was accompanied by a reduction in serum cholesterol and triglycerides.
Like SMAM-1, Ad-36 increases adiposity in experimentally infected chickens, as well as rats, mice and marmosets (non-human primates) (Dhurandhar et al 2000; 2001; 2002; Pasarica et al 2006), without inducing detectable hyperphagia, and reduces serum cholesterol and triglycerides (Dhurandhar et al; 2000,Dhurandhar et al; 2001; 2002,). Compared to uninfected controls, Ad-36 infected marmosets showed a fourfold gain in body weight and a significant increase in total body fat (36±6 g vs 23±3 g). In the rat model, Ad-36 appears to exert both, central and peripheral effects (Pasarica et al 2006). Ad-36 decreased norepinephrine levels in the paraventricular nucleus, arcuate nucleus, dorso-medial hypothalamus and ventro-medial hypothalamus. Other neurotransmitters such as dopamine and 5-hydroxyindolacetic acid concentrations were also reduced in some of these brain regions. On the other hand, Ad-36 increased whole body insulin sensitivity and enhanced expression of genes involved in the adipogenic and de-novo lipogenesis pathway such as fatty acid synthase (FAS) and acetyl-CoA carboxylase (ACC-1) (Vangipuram et al 2007). Just four days following infection, Ad36 spreads to adipose tissue, liver, kidney, and brain, lowers inflammatory cytokine levels, and infected rats have increased epididymal fat pad weight (Pasarica et al 2008b). The adipogenic role of Ad36 was recently confirmed independently by another research group (Thomas et al 2008), who showed that Ad36 could be used as a tissue engineering tool for building adipose tissue.
Recently, more human adenoviruses were tested for their adipogenic potential in animal models. Six subgroups classify adenoviruses by genetic similarity. Adenovirus type 5 (Ad-5) induced adiposity in mice (So et al 2005), and Adenovirus 37 (Ad-37) increased adiposity in chickens, whereas other two human adenoviruses Ad-2 and Ad-31 are non-adipogenic in chickens (Whigham et al 2006). Thus, the adipogenic effect is not shared by all adenoviruses, but appears specific to certain serotypes. Adipogenic potential of remaining the 45 known human adenoviruses has not been tested.
Dragonflies (Libellula pulchella) infected with a common, non-invasive gregarine gut parasite (Apicomplexa: Eugregarinorida) showed symptoms similar to human metabolic syndrome (Schilder et al 2007). Infected dragonflies developed significantly higher thoracic lipid accumulation, an inability to oxidize fatty acids in muscle tissue, twofold higher hemolyph carbohydrate concentrations than uninfected insects, increased insulin resistance, and elevated markers of a chronic inflammatory state – symptoms similar to those of metabolic syndrome in humans. Although the factors secreted by the parasites were considered responsible for the phenomenon, the exact mechanism is unknown.
Even gut microflora have been reported to enhance adiposity. Introduction of normal gut microbiota into germ-free mice increases their body fat by 60% and induces insulin resistance (Backhed et al 2004) despite the lower food intake in this group. The introduction of gut microbiota increased harvest of monosaccharides from the gut and hepatic de novo lipogenesis. Fasting-induced adipocyte factor (Fiaf), a lipoprotein lipase (LPL) inhibitor released by the intestinal epithelium, is suppressed by introduction of microbiota. Disinhibition of LPL is thought to increase triglyceride storage in adipocytes and contribute to resulting adiposity. These researchers later discovered that microbiota of genetically obese mice (ob/ob mice) had a higher relative abundance of the firmicutes vs the bacteriodetes division of bacteria, and the reverse was true in the microbiota population of lean mice. When the gut of germ-free mice were colonized with microbiota of the genetically obese mice, the originally germ-free mice had a significantly increased level of total body fat than their germ-free counterparts colonized with microbiota from genetically lean mice (Turnbaugh 2006). Although increase in adiposity clearly followed gut colonization with microbiota, considering their role as “normal” intestinal flora, this model may not be considered an “infection”.
Role of gut bacteria in body weight and adiposity regulation was further studied by Cani et al (2007). Lipopolysaccharide (LPS) is a proinflammatory bacterial cell wall component that is normally produced by the death of gram negative bacteria and absorbed into the body. During sepeticemia, the usual levels of plasma LPS rise 10 to 50 fold and induce anorexia, among other metabolic effects. With less dramatic increase, LPS is able to induce weight gain and metabolic changes. Mice on a high fat diet had increased plasma LPS by about 2 fold, reduced gram-negative bacteriodetes in their gut, and increased body weight and insulin resistance (Cani et al 2007). Infusion of exogeneous LPS to mimic its plasma levels similar to a high fat diet also resulted in weight gain and impaired glycemic response. This study shows the role of bacterial products in regulation of body weight and metabolism.
In Vitro Evidence
In-vitro effects of few adipogenic pathogens have been studied; Ad-36 is perhaps the most studied among the group. This virus enhances differentiation and lipid accumulation of 3T3-L1 and human primary preadipocytes, an ability not possessed by all adenoviruses, as demonstrated by the use of Ad-2 as a negative control (Vangiparum et al 2004; Rathod et al 2009). Viral mRNA expression, but not necessarily DNA replication is required for this adipogenic effect (Rathod et al 2007). Even in absence of adipogenic cocktail MDI, Ad-36 activates phosphotidyl inositol kinase (PI3K) and cAMP pathways, increases cell replication, expression of several genes of adipogenic cascade such as PPARγ, CEBP/β and consequentially, induces differentiation and lipid accumulation in 3T3-L1 cells and human adipose derived stem cells (hASC) (Rogers et al 2007; Rathod et al 2009). In human primary adipose tissue derived stem cells, Ad36 robustly induces adipogenic commitment, differentiation and lipid accumulation (Pasarica et al 2008a).
E4 orf-1, a viral gene, is required and sufficient for this adipogenic effect of Ad-36 (Rogers et al, 2007). Moreover, Ad-36 reduces leptin expression and secretion in rodent fat cells, which may reduce the autocrine/paracrine inhibitory effect of leptin on preadipocyte differentiation. Interestingly, adenoviruses Ad-37 and Ad-9 also enhance 3T3-L1 differentiation and reduce leptin secretion (Vangipuram et al 2007), and Ad-31 also is able to increase 3T3-L1 differentiation in vitro (Whigham et al 2006)
In-vitro infection with Ad-36 increases glucose uptake by primary rodent adipocytes (Vangipuram 2007) and human adipose tissue explants or hASC (Rogers et al 2007, Vangipuram et al 2007) human primary skeletal muscle cells (Wang et al 2008) through a virally induced increase in PI3K activation. This may explain the increased insulin sensitivity observed in Ad-36 infected rats (Pasarica 2006). Overall, increased glucose uptake and de-novo lipogenesis, greater replication, differentiation and lipid accumulation in Ad-36 infected adipocyte progenitors and greater glucose uptake by skeletal muscle may contribute to adiposity with a healthy metabolic profile induced by the virus in-vivo.
In-vitro data for Ad-36 provides an example for determining molecular mechanisms of other adipogenic pathogens. Considering that ethical concerns preclude experimental infection of humans, such mechanistic in-vitro evidence is especially important for eventually determining the role of pathogens in human obesity.
Epidemiologic evidence
Obesity is associated with inflammation (Pickup et al 1997; 1998; Bistrian et al 2000), but specific causal relationships are as yet undetermined. Modest evidence shows that inflammation precedes obesity. Duncan et al 2003 showed that increase in pro-inflammatory markers could predict obesity in human participants (Duncan et al 2003). Macrophage colony stimulating factor (MCSF) is a proinflammatory marker increased in obesity, and overexpression of MCSF in adipose tissue increased adiposity in transgenic animals (Levine et al 1998). It is unknown if obesity promoting pathogens stimulate MCSF production leading to the growth of adipose tissue. Human and animal models show a role of adipose tissue in secreting pro-inflammatory cytokines (Maachi et al 2004; Trayhurn et al 2001), however, recent evidence suggests that increased pro-inflammatory cytokines is not a consequence of adiposity in at least some animal models. Whether inflammation is a cause or effect of obesity remains unresolved.
The issue appears to have inspired epidemiological reports of the relationship between infection, an obvious cause of inflammation, and obesity. For example, Fernandez-Real et al (2006) tested middle-aged men for antibodies to four common viral infections and determined a composite pathogen burden score based on these results. The composite score was significantly correlated with fat mass and percent fat mass (Figure 1). Overall, the composite pathogen burden score accounted for 9% of the variance in fat mass. Another study by the same group found that seropositivity to the same four common viruses was negatively associated with insulin sensitivity after controlling for BMI (Fernandez-Real et al 2006). The authors hypothesized that low grade inflammation may be caused by viral load, contributing to both obesity and insulin resistance.
Figure 1.

Association of infections with fat mass: Linear association between Quantitative Serological Index and percentage fat mass (top) and absolute fat mass (bottom). Reprinted with permission from Fernandez-Real et al 2007.
Specific infections may be linked to human obesity. Some studies report association of Chlamydia Pneumoniae with higher BMI in humans (Ekesbo et al 2000; Dart et al 2002), whereas others found no association (Blanc et al 2004; Falk et al 2002; Muller et al 2003). Interestingly the antibody positive subjects were older and of lower socioeconomic status, and had higher fasting insulin levels. The authors suggest that obesity might not only be a marker for lower socioeconomic status, but also for greater susceptibility to infection. This accords with evidence suggesting that obesity is associated with impaired immune function (Marti et al 2001).
Natural infection of Ad-36 is also associated with human obesity. Seventeen percent of the 500+ subjects screened, showed seropositivity to Ad-36, indicative of a past natural infection with the virus (Atkinson et al 2005). Thirty percent of the obese, but only 11% of the non-obese subjects in this sample were Ad-36 seropositive, demonstrating a preponderance of natural infection among the obese subjects. Moreover, the obese and non-obese seropositive groups were significantly heavier than their respective seronegative counterparts. Unlike Ad-36, seropostivity to Ad-2 or Ad-31. two non-adipogenic adenoviruses (Whigham et al 2006) showed equal distribution among the obese and non-obese groups and no association with BMI. This suggests that the differential Ad-36 seropositivity observed in obese subjects was not due to their increased propensity to catch infection. Instead, it is likely that Ad36 causatively contributed to their obesity. Furthermore, Ad-36 seropositivity was associated with lower levels of serum cholesterol and triglycerides, a response typical of animals experimentally infected by the virus (Dhurandhar et al 2000; 2001; 2002). Another study (Atkinson et al 2005) in the same report investigated the association of Ad-36 seropositvity with BMI in twins discordant for antibody status. The seropostive twins were heavier and fatter compared to their seronegative counterparts. This study is particularly compelling in favor of a causative role of Ad36 in human obesity because of the strong control offered by co-twin controls
Seropositivity to SMAM-1, an avian adenovirus, is also associated with higher BMI’s and as noted above, paradoxically lower serum lipids and cholesterol levels in obese subjects (Dhurandhar et al 1997). Twenty percent of obese subjects screened had antibodies to SMAM-1. Seropositive subjects had a significantly greater body weight (95.1±2.1 kg vs. 80.1±0.6kg; p<0.02), but 15% lower serum cholesterol and 60% lower serum triglycerides. Avian adenoviruses, being serologically different from their human counterparts, are considered to be incapable of infecting other species. The presence of antibodies to an avian adenovirus in humans challenges that view. Perhaps, the subjects of this study had antibodies to a human adenovirus which cross-reacts with SMAM-1. Prevalence of SMAM-1 seropositivity in general population has not been reported.
Experimental Evidence from Humans
For several reasons, establishing an unequivocal causative role for these pathogens in human obesity via experimentation is difficult. For ethical reasons, it does not seem that humans can be experimentally infected with these pathogens. Moreover, due to an insidious onset of obesity in most cases, linking a past infection to weight gain is not easy. The multifactorial etiology of obesity adds to the challenge. It is possible that certain pathogens exacerbate obesity by working in conjunction with other adipogenic factors but may easily be eclipsed by those factors. Therefore, determining the contribution of various pathogens in human obesity will have to depend on the collection and analysis of overwhelming indirect evidence. Elucidating molecular mechanisms of adipogenic actions of these pathogens may help in investigating the role of pathogens in human obesity. Cross-sectional, prospective and longitudinal studies of subjects with and without infections of candidate pathogens are required. Demonstration of coexistence of obesity and a candidate pathogen infection in family clusters or other cohabitants, or prospective follow-up of subjects with and without suspected infection may strengthen the evidence. In fact, prevalence of obesity in friends and family clusters was recently reported (Christakis et al 2007). This longitudinal study showed increased chances to “contract” obesity, if a spouse, sibling or friend became obese. This spread of obesity was not dependent on geographic location, but instead appeared to spread through social ties. This study does not prove a role of infection in the incidence of obesity, but the transfer through social ties is certainly consistent with the pattern one would expect to see in the spread of an infectious disorder.
Summary and Conclusions
Elucidation of the relationship between obesity and infections is only beginning, with Ad-36 being the most well studied example of obesity of infectious origin to date. Considering the role that adipose tissue has in mediating inflammatory function, as well as the ability of microbes and viruses to alter metabolism,, further research in this area may uncover many implications of infection in obesity.
Epigenetics and Obesity
General Statement of Putative Cause and Hypothesized Mechanism of Action
Just as genetic variation affects individual susceptibility to obesity (Rankinen et al., 2006), so too might interindividual epigenetic variation. Because epigenetic mechanisms are susceptible to environmental influences, especially during development, a potential causal pathway emerges in which some environmental factors that have been increasing in recent decades are commensurately deranging the establishment of epigenetic mechanisms that contribute to body weight regulation.
Epigenetics describes the study of mitotically heritable alterations in gene expression potential that are not caused by changes in DNA sequence (Jaenisch and Bird, 2003). Epigenetic mechanisms are established during prenatal and early postnatal development and function throughout life to maintain the diverse gene expression patterns of different cell types within complex organisms. Several molecular mechanisms including methylation of cytosines within CpG dinucleotides, various modifications of the histone proteins that package DNA in the nucleus, and cell-autonomous expression of myriad autoregulatory DNA binding proteins interact to perpetuate the regional chromatin conformation that dictates which genes will be transcriptionally competent in specific cell types (Feinberg, 2007). Literally meaning “above genetics,” epigenetics confers an extra level of information which is layered above the DNA sequence information and which, like the sequence, is replicated during cell division. In addition to this mitotic heritability, epigenetic mechanisms may also be meiotically heritable, conferring the potential for transgenerational epigenetic inheritance.
Our understanding of environmental influences on epigenetic processes remains rudimentary. Hence, we currently have a limited ability to propose specific environmental exposures whose increasing magnitude might influence epigenetic mechanisms at the population level and thereby contribute to the secular increase in obesity. Nonetheless, one exposure that is likely to be highly relevant is maternal obesity itself. The obesity epidemic is affecting all age groups, including women of childbearing age (Hedley et al., 2004). If the intrauterine environment of an obese woman induces developmental adaptations in her developing fetus that then predispose her offspring to obesity, feed-forward transgenerational amplification of obesity could result (Levin, 2000). Perhaps the strongest human data supporting this hypothesis are from children born before and after maternal weight loss by bariatric surgery: children born after maternal weight loss have a lower risk for obesity than do their siblings born before maternal weight loss (Kral et al., 2006). Such “metabolic imprinting” of body weight regulation could occur via epigenetic mechanisms (Waterland and Garza, 1999; Waterland, 2005). Indeed, a recent study in mice (Waterland et al, 2008) showed not only that maternal obesity promotes obesity in the next generation, but also that this effect is prevented by methyl donor supplementation, suggesting a role for DNA methylation in transgenerational amplification of obesity.
Transgenerational epigenetic inheritance has been documented in mice (Morgan et al., 1999) and has been suggested in recent human studies (Hitchins et al., 2007). It is important to note, however, that epigenetic models for amplification of obesity prevalence across generations need not involve transgenerational epigenetic inheritance. Maternal (F0) obesity may induce somatic epigenetic alterations in her (F1) offspring that confer susceptibility to obesity. Consequently, F1 females would have an increased likelihood of being obese during their pregnancies, resulting in a perpetuation or amplification of obesigenic epigenetic alterations in the F2 generation. Recent rat studies provide evidence for this model. Among inbred (genetically identical) rats, the level of maternal caregiving behavior in the early postnatal period modulates epigenetic mechanisms in the hippocampus of her offspring, causing persistent alterations in stress-responsiveness and other behaviors (Weaver et al., 2004). Female offspring of “low caregiving dams” themselves grow up to be relatively inattentive dams (Weaver et al., 2004), demonstrating inductive transfer of epigenetically based phenotypic variation across generations. In addition to such inductive effects passed through the mother, it is possible that bona fide transgenerational inheritance of induced epigenetic modifications could contribute to obesity risk. Importantly, not just maternal but also paternal environmental exposures theoretically could induce epigenetic modifications that pass through the male germ line to influence obesity risk in the offspring (Pembrey et al., 2006).
The hypothesized mechanism of action, in which environmental factors induce epigenetic alterations that increase susceptibility to obesity, is based on two postulates: 1) environment can induce persistent epigenetic alterations, and 2) epigenetic dysregulation can cause obesity. We will consider the data supporting these two postulates.
Environmental Influences on Epigenetic Regulation
Epigenetic mechanisms are intrinsically malleable and can be influenced by factors including diet, pharmacological agents, and environmental toxicants (Jirtle and Skinner, 2007). Most importantly, transient environmental influences during critical periods of development can induce permanent alterations in epigenetic gene regulation and associated phenotypes (Waterland and Michels, 2007). For example, methyl supplementation of female mice before and during pregnancy induces DNA hypermethylation at “metastable epialleles” such as agouti viable yellow (Avy) (Waterland and Jirtle, 2003) and axin fused (Waterland et al., 2006) in the offspring, with permanent phenotypic consequences. Maternal exposure to bisphenol A (a synthetic estrogen and ubiquitous industrial contaminant) before and during pregnancy has the opposite effect, inducing DNA hypomethylation at Avy and another metastable epiallele, CabpIAP (Dolinoy et al., 2007). In a rat model examining interactions among prenatal and early postnatal environmental exposures, both fetal undernutrition and postnatal leptin administration were shown to induce persistent changes in DNA methylation and expression of hepatic genes (Gluckman et al., 2007c). The previously mentioned study showing changes in offspring DNA methylation based on differences in maternal caregiving behavior (Weaver et al., 2004) provides yet another demonstration that, during early life, subtle environmental exposures can induce persistent and biologically important epigenetic alterations.
Epigenetics and Obesity
Data relating epigenetics to obesity are drawn from various sources. Animal models and human data illustrate that dysregulation of epigenetic processes can cause obesity. Recent molecular studies provide an additional link by showing that genes critical to energy balance are regulated by epigenetic mechanisms.
Cloned mice are born with normal birth weights but often develop adult-onset obesity accompanied by hyperinsulinemia and hyperleptinemia (Tamashiro et al., 2002). Cloning results in epigenetic abnormalities (Jaenisch and Bird, 2003); it is therefore possible that the obesity of cloned mice is attributable to epigenetic dysregulation. Avy mice provide a clear example of obesity resulting from epigenetic dysregulation. The Avy mutation causes ectopic expression of agouti protein, a paracrine signaling molecule that normally regulates the production of yellow pigment in fur. Agouti protein binds antagonistically to the melanocortin 4 receptor in the hypothalamus, inducing hyperphagic obesity (Wolff et al., 1999). Because of the epigenetic metastability of the Avy locus, genetically identical Avy/a mice vary dramatically in the degree of DNA methylation at Avy and, consequently, coat color and adult adiposity (Wolff et al., 1999).
Epigenetic dysregulation is also implicated in human obesity. A striking example is provided by Prader Willi syndrome (PWS), a developmental syndrome causing characteristic facial features, developmental delay, and hyperphagic obesity (Goldstone, 2004). PWS results from a lack of paternal expression of genomically imprinted genes on chromosome 15q11-q13. Whereas most PWS is caused by a paternal deletion for this region, about 25% of cases are caused by maternal uniparental disomy (i.e., inheriting both copies of chromosome 15 from the mother) (Goldstone, 2004). There is no genetic lesion in these cases; the inappropriate epigenetic silencing of both copies of the 15q11-q13 region leads to obesity.
If epigenetic dysregulation can cause obesity, it follows logically that epigenetic modifications will also sometimes prevent obesity. An epidemiologic study of parental inheritance of the human gene encoding insulin (INS) provides one such example. Several classes of INS alleles have been described on the basis of a variable number of tandem repeats (VNTR) at the locus. The very common class I VNTR allele appears to predispose to childhood obesity, but only when inherited from the father (Le Stunff et al., 2001). These data suggest that when inherited from the mother, the allele is epigenetically silenced, affording protection from the obesigenic effects of the class I VNTR polymorphism.
The latest version of the human obesity gene map (Rankinen et al., 2006) reported 11 human genes that cause monogenic obesity and 52 genomic regions harboring quantitative trait loci associated with human obesity. In addition to genetic variation, epigenetic variation at these same loci could affect body weight regulation. Mutations in the genes encoding leptin (LEP) and proopiomelanocortin (POMC) cause monogenic obesity in humans (Rankinen et al., 2006). The promoters of both genes contain regions with a high density of CpG sites. Although cytosine methylation at such CpG islands functions to silence genomically imprinted alleles and genes on the inactive X chromosome, most promoter region CpG islands are unmethylated in normal tissues (Shen et al., 2007). It is therefore surprising that both the LEP and POMC CpG islands exhibit tissue-specific methylation that correlates with expression. The LEP CpG island is slightly hypomethylated in human adipose tissue DNA relative to peripheral blood DNA (Stoger, 2006), consistent with the adipocyte-specific expression of leptin. It is possible that more striking tissue-specific hypomethylation would be observed in isolated primary adipocytes, because most DNA in adipose tissue is derived from non-leptin-expressing stromal-vascular cells. Indeed, a study of human cells in culture showed that the LEP CpG island in pre-adipocytes (which do not express leptin) is hypermethylated, and the leptin expression of differentiated adipocytes is correlated with LEP CpG island hypomethylation (Melzner et al., 2002). Similarly, the human POMC CpG island is hypomethylated in POMC-secreting human cells relative to cells that do not secrete POMC (Newell-Price et al., 2001). Studies such as these demonstrate that “obesity genes” are regulated by epigenetic mechanisms and pave the way for studies of their potential dysregulation in obesity.
Summary and Conclusions
Environmental factors during development can induce permanent alterations in epigenetic gene regulation, and epigenetic dysregulation can contribute to obesity. It is therefore plausible (if not likely) that environmental influences on epigenetic gene regulation contribute to the secular increase in obesity.
Maternal Age and Obesity
General Statement of the Putative Cause and Hypothesized Mechanisms of Action
Over the last 40 years, there has been a dramatic change in childbearing patterns, starting in Europe and the United States, but now being seen in many areas of the world, especially in countries playing an active role in the expansion of the global economy. As more women enter the workforce, marriage and childbearing are increasingly being postponed to facilitate completion of education and establishment in a professional career track before embarking on the demands of parenthood (DeVore, 1983; Heck et al., 1997; Nabukera et al., 2006; Ventura, 1989; CDC, 1989). This shift in the family paradigm has been enabled by advances in the field of assisted reproductive technologies (e.g., ovulation induction with gonadotrophins and in vitro fertilization) that have allowed women to achieve pregnancy, often their first, in their fifth decade of life. The mean pregnancy age has steadily increased throughout the world, and moreover, the mean age of first time mothers has risen, a factor further augmenting the impact of maternal age on obesity (Nabukera et al., 2006; Mathews and Hamilton, 2002). The impact of this change in the social structure and its effect on the traditional family in terms of attention given to children, the type of diet provided, and the amount of physical activity encouraged, among other things, has been proffered as a potential contributor to the increasing rates of childhood and adolescent obesity. All the while, the idea that maternal age itself might be a contributor to escalating rates of obesity has been neglected.
Animal models and epidemiologic investigations have demonstrated a direct and independent association between maternal age and obesity in offspring. Although all contributory pathways await elucidation, many mechanisms of influence are already well defined and contribute, both directly and indirectly, to the long-term risk of obesity in offspring. This section discusses the shifts witnessed in maternal age distribution over the last several decades, considers how these alterations may influence a developing fetus, and reviews the data that demonstrate an association between the age of the mother at birth and the likelihood of subsequent obesity in the offspring.
Basic Science Evidence: Animal Data
Ovine models have provided insight into the ontogeny of adipose tissue. The development of fetal adipose tissue begins during early fetal development and proceeds along distinct pathways with differentiation into white and brown adipocytes. During fetal development, most adipose tissue is composed of brown adipocytes. These cells are responsible for the production of uncoupling protein 1 (UCP1), and concentrations of this protein increase progressively and dramatically until birth, after which production declines to undetectable levels as a result of a biological switch allowing white adipocytes to then predominate (Symonds et al., 2004). UCP1 plays a key role in energy expenditure and thermogenesis and is considered the hallmark of brown adipose tissue metabolism (Hansen and Kristiansen, 2006; Ricquier, 2005).
Given the critical role that brown adipose tissue is thought to play in the modulation of obesity, any factors that influence the biological switch or the production of UCP1 may contribute to an increase in the risk of obesity (Hansen and Kristiansen, 2006; Ricquier, 2005). The regulation of this switching is influenced considerably by the maternal milieu, which is in part determined by maternal age (Symonds et al., 2004). Symonds and colleagues showed that at 1 year of age, sheep born to adult mothers have increased fat deposition compared with those born to juvenile ewes. Furthermore, the offspring of first time mothers had greater overall adipose tissue accretion accompanied by an accelerated loss of brown adipose tissue metabolism and a decline in tissue-specific UCP1. Because this accelerated decline in brown adipose metabolism may never return to a baseline level of function, it may be associated with a propensity for white adipose deposition in later life, consistent with the observation of Lowell and colleagues, who showed that mice lacking UCP1 develop obesity (Lowell et al., 1993).
Ecological Correlation Evidence
Increased Maternal Age
The fertility rate in the United States has declined from a peak in the 1960s of 3.8 births per woman to an average of 1.8 since the 1970s, a change that has largely been attributed to the postponement of marriage and the availability of effective contraception (Westoff, 1986). One manifestation of this change over the last 40 years has been a steady increase in the mean pregnancy age for all births as well as a rise in the mean age at the time of first births. Indeed, there has been a progressive and radical escalation in both the rates of birth and numbers of births to women 30 years and older (Mathews and Hamilton, 2002). Numerous other demographic studies have pointed to the same trend (Baird et al., 1991; Wadhera, 1989; Wadhera and Millar, 1991; Ventura, 1989; CDC, 1989; Martin et al., 2006), and the results, visible in Figures 2 and 3, are strikingly apparent.
Figure 2.
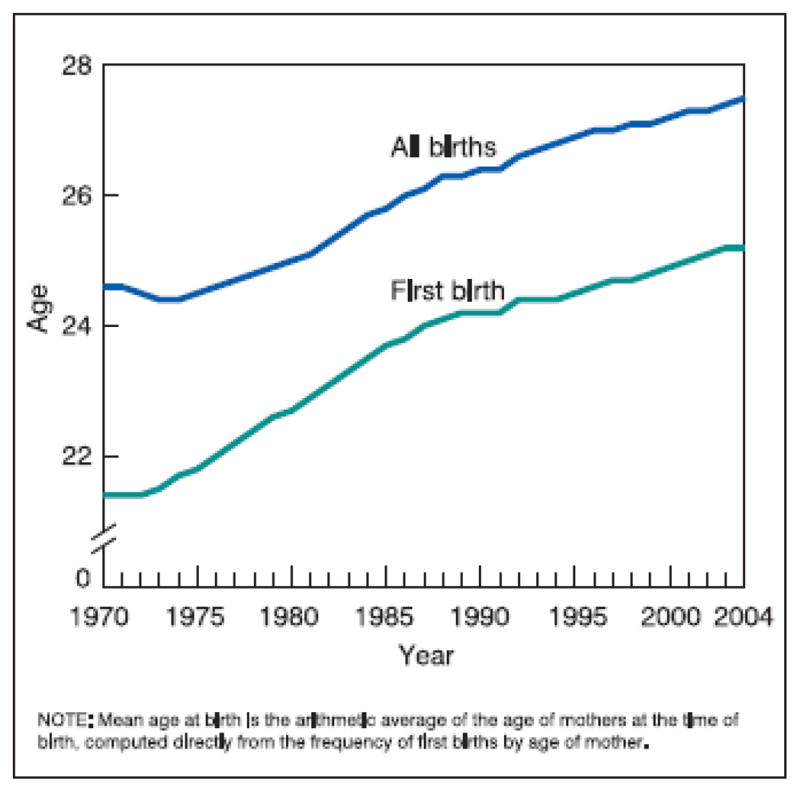
Mean maternal age for all births and first births in United States from 1970 – 2004.
Figure 3.

Percentage change in birth rates by age of mother from 1990 to 2004 in the United States.
These trends were first appreciated approximately 20 years ago. In the United States, the birth rate for women aged 30 to 34 increased from 52 per 1000 in 1975 to 71 per 1000 in 1987, and the proportion of women having their first child at 30 years of age or older also increased during this time period from 4% to 16% (Ventura, 1989; CDC, 1989; Mathews and Hamilton, 2002). During this time period, the number of first births to women 30 to 34 years of age and 35 to 39 years of age has quadrupled, and the first birth rate for each age group has more than doubled, from 7.3 to 17.5 per 1000 and 2.1 to 4.7 per 1000, respectively (Ventura, 1989).
Since the first recognition of this societal trend in the late 1980s and early 1990s, the changes have become more dramatic as the birth rates and maternal age means have continued on a similar steeply escalating trajectory. (Figure 2). Similar changes have been noted in other developed countries, including Japan, Sweden, Switzerland, and the Netherlands (Mathews and Hamilton, 2002).
In the United States in 2004, the most recent year for which final data are available, the number of births to women over 30 years of age rose to record levels, as did birth rates to these women. Between 1990 and 2004, the birth rate to US women 30 to 34 years of age increased 20% and the number of births increased 9%. More dramatically, the birth rate for women 35 to 39 increased 43% from 1990 to 2004, and the number of births to these women increased 50%, although the number of women this age increased only 5% during this time period. Similarly, the number of births to women 40 and older has more than doubled since 1990 (Martin et al., 2006) (Figure 3). The end result of these demographic shifts is that the mean maternal age at the time of birth in the United States rose 2.5 years between 1980 and 2004, from 25.0 to 27.5 years. A similar incremental increase occurred in mean age at first birth, which rose from 22.7 to 25.2 years (Martin et al., 2006).
Maternal Age and Obesity Associations
One of the first reports to establish an association between maternal age and childhood obesity was that of Wilkinson and colleagues. In a nested case-control study conducted as part of a large British prospective cohort study, these researchers found that the mothers of obese children were on average 3.5 years older at the time of birth than the mothers of normal-weight children (Wilkinson et al., 1977). Since then, several studies have demonstrated similar direct, independent, positive associations between maternal age at time of birth and multiple different parameters by which obesity is defined.
In a study of more than 8000 five- to eleven-year-old children, Duran-Tauleria and colleagues found a positive correlation of maternal age at the time of childbirth with both triceps and subscapular skinfold thickness in the offspring. In a multivariable model accounting for multiple covariates, maternal age independently explained 8% of the variability in these measurements and the rate of obesity (Duran-Tauleria et al., 1995). Blair and colleagues also assessed the relationship between maternal age and childhood fat accretion. In a longitudinal study of 871 New Zealand children, using bioelectrical impedance to calculate percentage body fat, Blair et al. found an association by maternal age at birth with an increase in body fat percentage at age seven in the offspring. Children born to women 30 years of age or older had a body fat composition that was 2.6 to 2.8 absolute percentage points higher than that of children born to women younger than 25 (Blair et al., 2007).
In addition to these studies that assessed individual measurements by which obesity could be defined, a similar association between maternal age and obesity can be seen when overall body composition is considered. Patterson and colleagues assessed data from more than 2400 girls aged 9 and 10 years followed as part of the National Heart, Lung, and Blood Institute’s Growth and Health prospective cohort study. Using a definition of obesity as a BMI at or greater than the 85th percentile for the age group, Patterson et al. found that the prevalence of obesity in girls at 9 and 10 years of age was directly related to maternal age. As maternal age at birth increased from the youngest group assessed (21 years or younger) to the eldest group (greater than 35 years), the prevalence of obesity increased (Figure 4). Moreover, in a multivariable model, Patterson and colleagues calculated that each 5-year increment in maternal age increased the likelihood for obesity by more than 14%, such that for girls born to women over 25 years of age, the risk for childhood obesity is increased by nearly a third and for those born to mothers 30 and over, the risk is nearly double (Patterson et al., 1997).
Figure 4.

Univariate relationship between maternal age and prevalence of obesity in 9 and 10 year old girls. Derived from data reported by Patterson et al.20
Part of the mechanism by which maternal age shapes the risk of obesity may be the effect of maternal age on birthweight. Older women are at risk for giving birth to infants at both ends of the birthweight distribution, i.e., both large and small for gestational age. Both of these groups are more likely to develop obesity. Compared with women 25 to 29 years of age, women 30 to 34 years of age were 24% more likely to give birth to large gestational age infants and those 35 or older were 42% more likely (Surkan et al., 2004). In a follow-up study of nearly 20,000 children, Stettler and colleagues found that each 100-g increase in birth weight above the median for gestational age was associated with an increase in the risk of overweight at 7 years by 7% (Stettler et al., 2002). Despite the above association between maternal age and large for gestational age neonates, low birth weight infants also occur more commonly among older mothers, and the rate of low birth weight infants has increased 2 to 3 fold over the last 15 years (Prysak et al., 1995; Nabukera et al., 2006; Yang et al., 2006). Whereas much of this risk has been thought to be associated with maternal comorbidities, even after adjustment for these confounders, the association persists (Cnattingius et al., 1992). Infants born at this lower end of the spectrum may manifest the thrifty phenotype and be predisposed to catch-up growth and later onset of obesity (Stettler et al., 2002). The impact of this factor on the etiology of obesity is addressed in the section, “Intrauterine and Intergenerational Effects.”
Although there is convincing evidence of an independent effect for maternal age on the risk of obesity, it would be naïve to believe that the effects of maternal age operate in isolation. As women delay child-bearing, the opportunities to accrue co-morbidities increase. As such, the prevalence of many of these conditions increases with maternal age (Bobrowski and Bottoms, 1995) and these co-morbidities may further increase the risk of obesity by the effect on the intrauterine environment, as discussed elsewhere in this article. Several studies have shown that maternal age modulated any protective effect of increasing birthweight against the adult onset of hypertension. Hardy and colleagues postulated that this may be due to the increased risk imparted by maternal pre-eclampsia, hypertension, and fetal growth restriction, all of which occur with increasing frequency in older mothers in general, especially older first time mothers, gravidas (Hardy et al., 2003). Similarly, Lawlor et al. reported an increase in systolic blood pressure at 5 years of age for each 5-year increase in maternal age above the mean, and, furthermore, found maternal age was one of the strongest independent predictors of childhood systolic blood pressure (Lawlor et al., 2004). Although not directly assessed in either of the above studies, the strong association between obesity and hypertension adds further to the evidence implicating maternal age as a contributor to childhood and adult obesity. Although the data implicating maternal age as an independent contributor to obesity in offspring are compelling, the potential synergistic role of maternal comorbidities, such as obesity, insulin resistance, and hypertension, should not be neglected.
Reproductive Fitness and Obesity
General Statement of the putative cause and hypothesized mechanisms of action
Herein, we define reproductive fitness as an individual’s or a population’s tendency (but not necessarily capacity) to reproduce and pass on their DNA. Measuring reproductive fitness directly is difficult, but fecundity, viewed here as the apparent capacity to produce offspring (i.e. birthrate), constitutes perhaps the most important and easily observed aspect of a population’s reproductive fitness. The propositions that would lead to the conclusion that reproductive fitness (essentially natural selection, Darwin (1859)) is contributing to recent increases in obesity prevalence are: (A) Adiposity levels in humans have a genetic component (i.e., are heritable); and (B) Individuals with a genetic predisposition toward higher levels of adiposity tend to reproduce at a higher rate than individuals with a predisposition toward lower levels of adiposity (Allison et al 2003).
The validity of proposition A is so well-established and well-documented that we will not review the evidence here. In brief, animal selection studies, and human twin, family, and adoption studies all consistently confirm the validity of this proposition. It has been estimated that within population genetic variations among people induce approximately 65% of the phenotypic variation among people in phenotypes like BMI (Segal et al 2002). For a detailed review, see Allison et al (2003). As such, our review focuses primarily on the validity of Proposition B which does not require that obesity causes greater fecundity. It only requires that the portion of the population having a greater genetic predisposition to obesity reproduces at a higher rate than others. This could occur because obesity causes greater fecundity, the genes causing obesity cause greater fecundity, people with stronger genetic predispositions to obesity are more fecund for any other reason (e.g., social, cultural, or economic factors), or any combination of these three.
Proposition (B) is actually something of an oversimplfication and special case of the true condition that must be met. The true and more general condition which is an easier standard to meet can be stated in terms of BMI and offspring distributions:
where τ is the BMI cutoff used to demarcate obesity (i.e., 30), g denotes a particular generation, V denotes the number of offspring, and BMIV and BMIU denote the BMI of an offspring and a parent, respectively.
Ecological Evidence
We found no studies in which the unit of analysis was a population that could suggest an ecological correlation relating fecundity and obesity predisposition.
Epidemiologic evidence
BMI and Fertility Impairment
Although these studies may seem counter to Proposition B, note that they refer in some sense to individual’s physiologic reproductive capacity but not necessarily their reproductive tendency (i.e., fecundity). Fecundity is a function of both physiologic reproductive capacity and other factors including willingness and ability to find a mate, copulation frequency, use of birth control, use of abortive techniques, and, in this context, producing conceptions that survive long-enough to be counted as obese or not and subsequently reproduce themselves.
BMI and Offspring Production
Studies in the United States have shown that an individual’s BMI is significantly and positively related to the number of offspring that the individual has produced (Weng et al 2004; Bastian et al 2005; Rosenberg et al 2003). Among nearly 12,000 African-American women enrolled in the Black Women’s Health Study, those having had at least one child between baseline measurement and the four years to follow-up gained more weight (~4.4 kg) than women who did not have any children (Rosenberg et al 2003). Similarly, Bastian et al (2005) noted a significant dose-response relationship (p <0.05) between having more children and increasing obesity rates among about 2,000 older American women of European ancestry. As for couples, in a nationally representative sample of 4,523 couples, Weng et al (2004) found the odds of obesity in the parents increased with each additional child they had for both women (OR = 1.07 with 95% CI: 1.04–1.10) and men (OR = 1.04 with 95% CI: 1.01–1.08). Bastian et al (2005) also reported an increase in odds of obesity in women for each additional child after excluding childless women (OR = 1.07 with 95% CI: 1.01–1.15). A recent study by Kim and colleagues (2006) showed positive correlations of overweight with parities both within and across countries/regions. Specifically, they analyzed recent data from the Demographic Health Surveys of 50 countries from 5 regions and found that, in 38 of the countries, the odds ratios of overweight were greater than 1 for women with 4 or more children compared to women with only a single child. The magnitude of the associations of parity to overweight was greatest in North Africa/West Asia and Latin America and Caribbean regions. The odd ratios of overweight were positively correlated with levels of country development (a development score was calculated from an analysis of 12 measurements of wealth, human development, and quality of life) even though there were negative correlations between total fertility and country development. However, these observations were retrospective and do not rule out the possibility that producing offspring leads to obesity and not the reverse.
Madrigal et al (2003) analyzed a sample of 290 Irish women married or widowed prior to World War II who had very little or no exposure to Western methods of contraception. They found a significant positive correlation between BMI and the number of offspring subsequently produced and also noted a significant positive Spearman correlation between the size of a woman’s family of orientation (number of her siblings + 1) and the number of offspring she produced (ρ = 0.15 with p = 0.009). This suggests that women with higher BMI tended to come from larger families and have larger families themselves (i.e., larger numbers of siblings and then offspring) and suggests that the association is not merely due to an effect of producing offspring increasing the risk of parental obesity. Likewise, in a study of US and Canadian college students and their parents, Ellis and Haman (2004) found that overweight and obese women tended to come from larger families (4.8 siblings) compared to women of normal or below normal weight (4.3 siblings).
Thus, there is a relationship between the number of offspring produced and BMI, particularly in women, but is there evidence to suggest that women are more likely to have a greater number of offspring because they are heavier? In the aforementioned study, Ellis and Haman collected self-reported data on BMI one year prior to pregnancy from the 5,986 mothers in their cohort and found a significant and positive correlation between BMI prior to pregnancy and the number of offspring (ρ = .11 with p ≤ 0.01). Although the effect appears modest, this study suggests that women with higher levels of adiposity are having more children than are individuals with a predisposition toward lower levels of adiposity.
Jokela et al conducted a prospective study of Finnish adolescents using BMI as a predictor of the number of children they would subsequently have after 21 years of follow-up (Jokela et al 2007). They found significant quadratic associations between adolescent BMI and number of offspring suggesting that, compared to those having normal adolescent BMI levels, underweight and overweight levels of adolescent BMI were associated with producing fewer offspring. This result appears to contradict the findings of Ellis and Haman, although they used adult BMI at one year prior to pregnancy as opposed to adolescent BMI to address the relationship between BMI and number of offspring. (Ellis et al 2004). Though Jokela et al (2007) adjusted for adult BMI, it was assessed after any pregnancies considered during the 21 years of follow-up. They dismiss reproductive differentials as an unlikely force behind the obesity epidemic, but go on to make an interesting observation which may support Proposition B:
“The present results suggest that stabilizing selection may be acting on BMI, since the low and high ends of BMI are selected against in terms of reproductive success. On the other hand, individuals with a BMI about one-half standard deviations above the mean were estimated to have 2–3% more children than those with mean BMI. If this pattern were to hold over generations, genetic factors might be shifting the mean BMI of the populations about 2 units upwards in future generations, albeit only at a modest rate.” (pp. 605)
This mean BMI shifting may relate to BMI increases recently reported by the CDC National Center for Health Statistics (NCHS). They estimated that average BMI among adult men and women in the United States have increased from about 25 in 1960 to 28 in 2002 (Ogden et al, 2004).
Experimental Evidence
Ethical constraints prohibit the conduct of either human laboratory studies or clinical trials to directly investigate the validity of Proposition B in humans. By laboratory experiments and husbandry, selection practices for greater phenotypic weight among a wide variety of animals has been well developed and documented (Crawford 1990; Barker 1967). Although the animals in these experiments may have phenotypic and genetic similarities to humans, the selection forces were controlled under experimental conditions and obviously cannot be directly translated to infer how selection would act on humans. These results do, however, strongly suggest that selection can affect biologically similar species and result in a population composed of individuals with increased weight.
Summary and Conclusions
Despite the implication by some studies that obesity might be detrimental to the potential for reproductive success, other studies have indicated that the overall effect of mild-to-moderate obesity on fecundity may be positive. This is plausible because: (1) obesity in women is associated with lower socioeconomic status (Lipowicz, 2003), which has been linked to producing more offspring (Salihu et al., 2004); (2) women that are too lean suffer impaired fertility (Frisch, 1987) and (3) other, currently unknown, biological, social or economic factors may drive a positive association between fecundity and a genetic predisposition to obesity. Indeed, the larger family sizes associated with obesity along with evidence that genetics influence obesity outcomes, suggests that individuals carrying genes conducive to obesity are reproducing at a greater rate than non-carriers, thereby contributing to increasing rates of obesity in the US and Canada (Ellis et al 2004).
The observational human studies mentioned above were not experiments conducted under controlled conditions and did not involve subjects randomized to either have children or to not have children. Thus, although the results of some studies examined are consistent with Proposition B (i.e., people with higher levels of adiposity are reproducing at a higher rate than are individuals with a predisposition toward lower levels of adiposity) they do not definitively establish the validity of this proposition.
Assortative Mating and Floor Effects
General Statement of the putative cause and hypothesized mechanisms of action
Assortative mating refers to any departure from random mating. Positive assortative mating occurs when mates are phenotypically more similar than one would expect by chance alone, whereas negative assortative mating occurs when mates are phenotypically more dissimilar than one would expect by chance alone. The term “assortative mating” is often used synonymously with “positive assortative mating” as it is much more common, and herein this convention will be used.
The consequences of assortative mating are complex and dependent on the “genetic architecture” of a particular trait. Given that obesity is a complex polygenic trait, models of assortative mating are quite complex. A consequence of assortative mating is an increase in the frequency of homozygotes in a population at the expense of heterozygotes, with an overall effect of increasing the total phenotypic variance. In the case of assortative mating among obese individuals, \the potential exists to increase the genetic predisposition to obesity among their offspring (genetic loading).
The propositions that would lead to the conclusion that assortative mating is contributing to increased obesity prevalence are:
human adiposity levels have a genetic component;
the Population Median Adiposity Index Has Been Below the Threshold for Defining Obesity During the Period of Population Growth of Obesity;
humans engage in positive assortment for adiposity.
If, furthermore, there are ‘floor effects’ such that there are countervailing forces that prevent a large portion of the population from becoming unusually thin, then the degree of positive skewness of the population distribution will increase and the mean level of adiposity will increase more dramatically. Note that it is not necessary for the degree of assortative mating to have increased over time for assortative mating to cause an increase in the prevalence of obesity over time. However, if an increase in assortative mating occurred, it might accelerate the increase in obesity levels.
Evidence that Human Adiposity Levels have a Genetic Component
There is good evidence that human obesity aggregates within families (Katzmarzyk et al 1999). Although there is still some disagreement as to the strength of the genetic effects on obesity, it is clear that human adiposity and a propensity towards weight gain is influenced by genes (Allison et al 2003; Katzmarzyk et al 2005). Heritability estimates are generally higher from twin studies, intermediate with nuclear family data, and lowest from adoption studies. In general, the most probable estimates of the heritability of body fat in humans range from about 25 – 75% (Maes et al 1997; Segal et al 2002). Obesity is a complex, multifactorial phenotype. As such, single gene mutations that are singly sufficient to cause human obesity are extremely rare. To date, only 176 cases of human obesity that are ostensibly due to mutations in 11 different genes have been reported (Rankinen et al 2006). Most genes involved in more common forms of obesity are viewed as “polygenes” - those which make a relatively small contribution and explain less than 5% of the phenotypic variance. More than 400 such sample associations have now been reported in the literature, with 22 genes supported by at least 5 studies (Rankinen et al 2006).
Evidence that the Population Median Adiposity Index Has Been Below the Threshold for Defining Obesity During the Period of Population Growth of Obesity
The importantce of this is that it (sub-proposition B above) is necessary for assortative mating to effectively increase the proportion above the threshold by increasing genetic variance. The current BMI threshold used internationally to define obesity is 30 kg/m2 (WHO 1998). It is clear that the population median is and has always been below this level (Fegal et al 2000).
Evidence that Humans Engage in Positive Assortment for Adiposity
Assortative mating is most often assessed in human studies by bivariate correlations among spousal pairs (Spuhler 1968). However, spousal similarity may also result from factors other than assortative mating. For example, spousal similarities may result from shared environmental effects, the differential survival of marriages in which spouses are more alike than not, or the effects of inbreeding (mating of biologically related individuals). Within this context, inbreeding may be considered an extreme form of assortative mating, in which the probability of increasing the genetic predisposition to obesity in the offspring of two obese parents could be very high.
Spousal resemblance has been documented for many human traits and physical characteristics, including body weight (Spuhler 1968). One study that attempted to control for the effects of cohabitation to determine an assortative mating effect per se on body weight was that of Allison et al (Allison 1996). In that study, there was a significant (r = 0.13; p = 0.023) spousal correlation for relative weight, based on data collected before marriage and cohabitation. Results from an Australian study provided evidence for significant spousal similarities in BMI and triceps skinfold, and the magnitude of the correlations did not increase with length of marriage duration. (Knuiman et al 1996). These results were supported in a study of the Canadian population in which the spousal correlation for BMI was 0.17 (p<0.05) in 1981 and 0.17 (p<0.05) after 7 years of follow-up provides further support for an assortative mating effect as the spousal similarity did not increase after several additional years of cohabitation (Katzmarzyk et al 1999).
More recently, Jacobson and colleagues, in their examination of the possible effects of assortative mating on obesity prevalence, demonstrated that a) spousal correlation for BMI was greatest for spousal pairs within the first 5 years of cohabitation and b) spousal pairs with greater than 5 years of cohabitation exhibited a slightly lower correlation (Figure 5) (Jacobson et al 2007). Overall, the age-adjusted spousal BMI correlation was 0.18 (95 percent confidence interval: 0.16, 0.20) in the study. Supporting the hypothesis that assortative mating is partially contributing to increasing obesity rates, the effect of parental obesity upon offspring BMI largely depended upon biologic relatedness with offspring of two obese biologic parents exhibiting greater odds of obesity as compared to offspring raised by two obese foster parents. Taken together, these results provide evidence of an assortative mating effect that cannot be explained by cohabitation or the differential survival of marriages among similar spouses.
Figure 5.

Spousal Spearman rank order correlations for body mass index among 8,663 spouse pairs, categorized by length of cohabitation. Figure reprinted from Jacobson et al. (Jacobson et al. 2007), with permission.
Hebebrand and colleagues (Hebebrand et al 2000) demonstrated that approximately 35% of parental pairs of extremely obese German youths were themselves above the 9th decile of the distribution for BMI. Similarly, data from the Canadian population demonstrated that parental pairs of obese (BMI ≥ 95th percentile) youth tended to cluster in the top of the distribution of BMI, whereas parental pairs of lean children (BMI ≤ 5th percentile) tended to cluster in the bottom portion of the distribution of BMI (Katzmarzyk et al 2002). These results provide circumstantial evidence that assortative mating may be playing a role in the obesity epidemic, but further research is required to test this hypothesis. The impact of assortative mating on obesity rates could occur quite quickly. There is overwhelming evidence from animal work that mating strategies can change the distribution of body weight, even in one generation (Helmink et al 2003).
Given that assortative mating for body weight phenotypes theoretically should increase total phenotypic variability, increases in the prevalence of both underweight and obesity should be observed; however, this would not be absolutely essential. We are aware of only one study that has indicated any data describing changes in the prevalence of underweight (Cotoyannis et al 2007). In their examination and comparisons of changes in the distribution of BMI in Canada and England from 1994–1995 to 2000–2001, Contoyanniss and Wildman (Cotoyannis et al 2007) showed that both populations showed polarization over time towards both tails (thinness and obesity) of the weight distribution. There is some evidence to suggest that the entire distribution of BMI has shifted to the right over time in the United States (Flegal et al 2000), and given that the prevalence of those with a BMI < 18 is only about 1.5% (Kuczmarski et al 1997), it would appear that the increases in the prevalence of obesity have not been paralleled by increases in the prevalence of underweight in the United States. There are clear floor effects on BMI in humans. In other words, there is a lower limit for BMI, under which life cannot be sustained. The lower limit of BMI compatible with human survival has been reported to be approximately 12 kg/m2 (Henry et al 1990; Henry et al 1994); however, data collected during the 1992–93 famine in Somalia indicated that adult humans (aged 25–80 y) can survive with a BMI as low as 10 kg/m2, as long as specialized medical care is provided (Collins et al., 1995). Nevertheless, the fact that there is a floor effect in BMI has likely accentuated the contribution of assortative mating to the obesity epidemic.
Sleep Debt and Obesity
General Statement of the Putative Cause and Hypothesized Mechanisms of Action
An impressive and still growing number of studies has noted the concomitant increased incidence of obesity with decreased amount of sleep in the population over the last 40 years (Figure 5). The association is observed across groups spanning diverse ages and ethnicities and has spurred animal and human mechanistic studies. The following lines of evidence suggest that biological mediators of appetite and energy homeostasis may be affected by sleep duration.
Basic Science Evidence
Experimental Evidence in Animals
The most robust effect of sleep deprivation in rodents is an increase in food intake without a corresponding increase in body weight (reviewed in Rechtschaffen et al., 2002). Weight loss is likely an effect of chronic and extensive sleep deprivation and/or the methods used to produce it. One exception to this pattern of weight loss was reported in a study of short-term sleep restriction in which rats had access to a carbohydrate-rich diet; following 72 hours of rapid eye movement (REM) sleep deprivation, rats gained weight (Bhanot et al., 1989). Increases in food intake following sleep deprivation in rodents are so well-established and consistent that sleep researchers frequently use the hyperphagia to gauge sleep deprivation intensity.
There is emerging evidence that the effects of sleep deprivation on feeding may be related, in part, to changes in appetite and energy-regulating peptides, though the evidence is still slender and sometimes contradictory Ghrelin is a gut-derived hormone released prior to feeding that stimulates hunger, feeding, and fat storage (van der Lely et al., 2004). In rats deprived of sleep for 5 hours, plasma ghrelin levels and food intake were increased in comparison to non-sleep-deprived controls (Bodosi et al., 2004). Leptin is another metabolic hormone that appears to be affected by sleep deprivation. It is secreted from adipose cells, attenuates food intake and increases energy expenditure (Halaas et al., 1995). Although leptin levels did not change significantly in the aforementioned 5-hour deprivation study in rats (Bodosi et al., 2004), another study reported that leptin concentrations were significantly suppressed and feeding was increased after chronic sleep deprivation for 16 days (Everson and Crowley, 2004). The varying results might have been due due to the length of the sleep deprivation (5 hrs versus 16 days); although reductions in plasma leptin were evident after 1–4 days of sleep deprivation (Everson and Crowley, 2004). Other peptides potentially involved in modulating the relationship between sleep deprivation and feeding include galanin and orexin. Galanin is a pituitary-secreted peptide known to stimulate food intake (Kyrkouli et al., 1986) and galanin mRNA is increased in brains of sleep-deprived rats (Lu et al., 2005), including in the hypothalamus (Fujihara et al., 2003; Toppila et al., 1995). The peptide orexin, synthesized in the lateral hypothalamus, stimulates food intake as well. In rodents, orexin levels are increased by sleep deprivation and decreased with recovery sleep (Pedrazzoli et al., 2004). Taken together, these basic studies reveal a potential link between sleep deprivation and systems involved in feeding regulation; sleep deprivation is consistently associated with changes in levels of hormones and peptides that lead to increased feeding that are reversed by sleep.
Experimental Evidence in Humans
Sleep debt exerts profound effects on metabolic hormones. These changes favor not only increased food intake and energy storage, but also potentially the development of diabetes and heart disease (Gottlieb et al., 2005; Spiegel et al., 2005; Bjorvatn et al., 2007; Flint et al., 2007; Knutson et al., 2007; Trenell et al., 2007; Van Cauter et al., 2007). Young men restricted to 4 hours of sleep a night for 6 days had a 40% decrease in the rate of glucose clearance and a 30% reduction in insulin response and glucose effectiveness, a measurement of the ability of glucose to mediate its own clearance independent of insulin (Van Cauter and Spiegel, 1999). The same group reported preliminary data that less severe but more chronic sleep deprivation (i.e. 8 days with 5 hrs in bed or 2 weeks with sleep restricted by 1.5 hrs) produced similar effects on glucose and insulin parameters (Knutson et al., 2007). Hence, a diabetic-like glucose/insulin profile, which is known to increase adiposity and weight gain (Knutson et al., 2007), is one mechanism by which sleep debt may be linked to obesity. This potential relationship is supported by the growing number of correlational studies showing significant associations between diabetes and reduced sleep (Gottlieb et al., 2005; Spiegel et al., 2005; Flint et al., 2007; Trenell et al., 2007).
Similar to rodent studies, experiments utilizing human subjects suggest that leptin may be a signal linking sleep debt to obesity. In young men sleep restricted to 4 hrs in bed for two consecutive nights, plasma leptin levels were decreased 18% in comparison to those observed when these same men were allowed 10 hours in bed on two consecutive nights (Spiegel et al., 2004b). Moreover, ratings of hunger and overall appetite for various food categories were increased 24% and 23%, respectively. Appetite for energy-dense, high-carbohydrate foods showed the greatest increase (Spiegel et al., 2004b). Another study found 5 hours of habitual sleep to be associated with 15.5% lower leptin levels in comparison to those habitually getting 8 hours of sleep (Taheri et al., 2004). Moreover, a dampened diurnal rhythm of leptin effect was also described in a study involving 10 male subjects who were sleep deprived for 88 hours (Mullington et al., 2003). As an adipose-secreted hormone, leptin not only decreases appetite and increases energy expenditure (Jeanrenaud and Rohner-Jeanrenaud, 2001), it is also affected by sleep time. Plasma leptin levels reach a maximum level midway through a normal nocturnal sleep period (Simon et al., 1998). Lower levels of plasma leptin related to sleep loss thus, might contribute to increased appetite. During partial sleep deprivation, thyroid stimulating hormone levels and the duration of its secretion have also been found to be blunted even when energy intake and activity levels were held constant (Spiegel et al., 2004a; Knutson et al., 2007). These changes in insulin, leptin, and thyroid stimulating hormone secretion thus could promote obesity by increasing appetite and decreasing energy utilization. Not all of the studies assessed hunger but of those that have, increased hunger accompanies the changes (Abraham et al., 1982).
Changes in ghrelin secretion appear to be another mechanism by which sleep debt may cause weight gain. In the aforementioned study in which young men were sleep restricted for two consecutive nights, sleep restriction was also associated with a 28% increase in plasma ghrelin levels (in 9 of the subjects tested) in comparison to the two consecutive nights with 10 hrs in bed (Spiegel et al., 2004b). As previously mentioned, these young men also showed increased appetite ratings thus, the increases in plasma ghrelin and concomitant decreases in plasma leptin, may have contributed to the reported increased appetite ratings (Spiegel et al., 2004b). Finally, Taheri et al. (2004) found 5 hours of habitual sleep to be associated with 14.9% higher ghrelin levels than those in persons habitually getting 8 hours of sleep (Taheri et al., 2004). Hence, abnormal increases in this hormone due to sleep debt could reasonably translate into greater appetite, food intake, and an increased risk of obesity.
Another explanation proposed to explain the association between sleep debt and obesity relates to general motor activity. It has been suggested that sleep debt causes tiredness, tiredness decreases activity, and decreased activity contributes to weight gain (Taheri, 2007). It appears, however, that the link between sleep debt and obesity may be independent of physical activity. In a sample of 7641 participants of the Finnish 2000 Health Examination Survey, controlling for the level of physical activity did not affect the significant association between sleep duration and obesity (Fogelholm et al., 2007). Moreover, a recent study reported that women that reported being short sleepers were more likely to have gained weight than those that self-reported to be longer sleepers, independent of levels of physical activity (Patel et al., 2006).
Sleep apnea and other sleep disorders of breathing produce sleep time restriction and have also been linked to increased occurrence of hypertension, insulin resistance, and recently to increased plasma neuropeptide Y levels, an appetite-stimulating peptide, all independently of body weight (Tasali and Van Cauter, 2002; Barcelo et al., 2004). A significant percent of the obese population suffers from sleep apnea (Young et al., 1993). This is disconcerting in that risks to endocrine health caused by sleep breathing disorders threaten to exacerbate already compromised metabolic regulation and control of normal body weight in this population.
It should be noted that despite studies linking mortality risk with decreased sleep time; less than 6 or 7 hrs a night has been associated with higher mortalilty rates (Heslop et al., 2002; Kripke et al., 2002). Research also indicates a relationship between mildly decreased sleep (generally to 6–7 hours/day) and lower mortality rates (see Alvarez and Ayas, 2004; Youngstedt and Kripke, 2004 for reviews). Several studies suggest a relationship between increased mortality risk and either too little or too much sleep (NSF, 2008; Hasler et al., 2004; Youngstedt and Kripke, 2004). It may be significant that the U-shaped function of BMI vs. hours of sleep per night (Taheri et al., 2004) mirrors that between mortality risk vs. hours of sleep per night (Kripke et al., 2002). How sleep duration might affect morbidity among the nonobese population remains unclear; however, the extent to which time taken away from sleeping is being spent in illness-provoking or health-risking activities should also be taken into account. Furthermore, the contribution of sleep quality vs. quantity also needs to be considered by controlling for sleep disturbances to more accurately assess an association between sleep and BMI. Lastly, genetic markers may be associated with increased propensity to gain weight with decreased sleep. Although insomnia is a very different phenomenon from sleep deprivation, it is interesting to note the one study that has linked genetic contributions of insomnia, sleepiness, and obesity. Watson et al. (2006) explored genetic contributions of sleep disturbances in over 1800 twin pairs and found that 10% of common additive gene effects accounted for 10% of the phenotypic association between obesity and insomnia (p<0.01). Similar gene model fitting studies are needed to assess the genetic contribution of reduced sleep to BMI.
Ecological Evidence
A Trend of Decreased Sleep Time in the Current Population
Epidemiological research studies as well as wide-scale polling by the National Sleep Foundation (NSF) suggest that American adolescents and adults are chronically sleep deprived (Bonnet and Arand, 1995; Broman et al., 1996; NSF, 2008; Wolfson and Carskadon, 1998; Spiegel et al., 1999). Before World War I, Americans were averaging 8.7 to 9 hours of sleep a night (Terman and Hocking, 1913; Tune, 1968). About 30 years ago, a decrease to 7.68 hours was reported (Bliwise, 1996); in over 1000 adults surveyed, 74.4% reported sleeping less than 8 hours a night (Taheri et al., 2004). Most recently, in the NSF 2008 sleep poll, adults reported sleeping on average 6 hours and 40 minutes per weeknight (NSF, 2008); 44% of adults reported sleeping less than 7 hours a night. The NSF survey also reported that individuals that self-reported to be short sleepers (less than 6 hours a night on workdays) were significantly more likely to be obese than those that self-reported to be normal or long sleepers (8 hours or more on workdays); 41% vs. 28% respectively (NSF, 2008). Causes attributed to the increasing sleep debt include insomnia, stress, social pressures, desire to get more work accomplished, night-shift work, medications used to treat colds, allergies, pain and cardiovascular problems, and modern-living habits such as late-night TV viewing and use of the internet (Bonnet and Arand, 1995; Broman et al., 1996; Spiegel et al., 1999; NSF, 2008; FDA, 2002). Children have later bedtimes but their morning waking time has stayed constant, a trend that some authors attribute to more liberal parental attitudes (Iglowstein et al., 2003). Increased TV viewing, especially at bedtime, is also thought to contribute to decreased quality of sleep in children (Owens et al., 1999).
Epidemiologic Evidence
Studies supporting an association between decreased sleep duration and increasing BMI are numerous enough to have produced several published reviews on the topic (Spiegel et al., 2005; Taheri, 2006; Knutson et al., 2007; Trenell et al., 2007; Van Cauter et al., 2007). While assessing the relative risk of diabetes among 70,076 female nurses over a decade (1986–1996), Ayas et al. found that whereas diabetes was associated with both short (5 or less hours/day) and long sleep duration (9 or more hours/day), BMI was associated with the increased incidence among the short-sleepers only (Ayas et al., 2003). In a study of over 900 mostly overweight patients (mean BMI of 30), increasing BMI was found to be associated with decreased hours of self-reported sleep per day (Vorona et al., 2005). Singh and colleagues reported that in 3158 individuals, those who slept 6 or fewer hours were at highest odds of being obese. In that study, more than twice the number of African Americans than Caucasians (18.7% vs. 7.4%) reported habitually sleeping 5 hours or less a night (Singh et al., 2005). It is noteworthy that in this study African Americans had substantially higher obesity rates than did European Americans. The possibility that differences in sleep duration can contribute to the higher incidence of obesity amoung minority populations is supported by a recent finding by Hale and Do (2007). In this study of over 32,000 adults in the United States, the investigators found a higher risk of short (<6) and of long (>9) sleep hours, both extremes attributed to increased rate of health problems, in black vs. white respondents.
Longitudinal studies also support the association between sleep debt and obesity. In a recent study, 68,000 nurses in the United States were followed over a 16 year period and those sleeping 5 and 6 hours a night gained significantly more weight than did those getting 7 to 9 hours of sleep per night (Patel et al., 2006). Results from 18,000 adults aged 32–59 years participating in NHANES I revealed that those sleeping less than 4 hours, 5 hours, and 6 hours a night were at a 73%, 50%, and 23% greater risk of being obese, respectively (Gangwisch and Heymsfield, 2004). A third longitudinal study followed 496 American adults over 13 years; the participants were tested four times between the ages of 27 and 40 years, and the results revealed a negative relation between sleep duration and both prevalence of obesity and BMI (Hasler et al., 2004). A fourth longitudinal sleep diary study of 1024 30–60-year-olds in the United States found a U-shaped relationship between average nightly sleep (6–9 hours) and BMI, with the lowest BMI corresponding to about 7.6 hours of sleep per night. In those sleeping less than 8 hours (74.4%), increases in BMI were significantly associated with decreased sleep time (Taheri et al., 2004).
Evidence from abroad includes a study of 1469 women in France where short sleep duration was associated with greater BMI (Cournot et al., 2004). Among 1772 adults and adolescents in Spain, those sleeping 9 or more hours had a lower incidence of obesity than did those sleeping 6 or less hours (Vioque et al., 2000). In over 3000 Scottish men and women, decreased sleep (<7 hours) was associated with increased BMI over a 4–7-year period (Heslop et al., 2002). A study interviewing 8091 older (55–101 years) adults from seven European countries found underweight to be common among the 5% sleeping the most (9 or more hours/day) and overweight or obesity to be common among the 5% sleeping the least (5 or more hours/day) (Ohayon, 2004). Among 4878 Brazilian truck drivers, those with the greatest BMI also reported the least sleep (Moreno et al., 2006).
Sleep debt may have the most potent and adverse effects on body weight in children and adolescents (Taheri, 2006). Childhood and adolescent overweight and obesity also increased substantially in prevalence between the 1960s and the 21st Century (NCHS, 2004; Ogden et al., 2008) and in a prospective study of 150 children in the United States from birth to 9.5 years of age, decreased hours of sleep at ages 3 and 4 years was a risk factor for overweight when surveyed at 9.5 years (Agras et al., 2004). Shortened sleep duration was related to obesity in 1031 French 5-year-olds (Locard et al., 1992). Among 6862 Bavarian 5–6-year-olds, less sleep was also related to increased BMI and body fat mass (von Kries et al., 2002). In 8170 Japanese children tested at 3 and then 6 years of age, short sleep duration was associated with overweight (Sugimori et al., 2004). An earlier study of 8274 Japanese 6–7-year-olds also found a strong “dose-response” relationship between short sleeping (<10 hours) and obesity (Sekine et al., 2002). Similarly, a 24-hour test among 383 multi-ethnic male and female adolescents in Texas aged 11–16 years revealed that obese adolescents had less sleep than the non-obese adolescents; for each less hour of sleep, the odds of obesity increased by 80% (Gupta, 2002). The significant relationship between increased TV viewing time and weight-gain in children (Andersen et al., 1998) may be due to decreased activity as well as possible sleep disruption (Owens et al., 1999). A sobering finding is that obesity in 7-year-olds could be predicted by decreased sleep as early as the age of 30 months (Reilly et al., 2005). Given that 50% of children in North America and 38% in Europe are projected to be overweight or obese by the year 2010 (Wang and Lobstein, 2006), the possible influence of sleep debt in children, including toddlers, cannot be ignored.
Summary and Conclusions
Public education of the benefits of sleep is currently targeted at preventing the dangers of automobile and on-the-job accidents as well as increasing performance in schools and the work place. The data reviewed suggest that sleep education might also impact the rising incidence of obesity. Increased sleep may also be promising as an antiobesity treatment; to address this, clinical trials are currently recruiting participants (NIDDK, 2007). Although not as imminent a threat as drowsy driving, obesity will exact a greater toll on human life as the second highest preventable cause of death.
Endocrine Disrupters and Obesity
General Statement of the putative cause and hypothesized mechanisms of action
Endocrine disrupting chemicals (EDCs), such as the flame retardant polybrominated diphenyl ether (PBDE), and the plasticizer bisphenol A (BPA), are very stable in the environment and many have been steadily increasing in levels in humans. PBDE, BPA, and other EDCs exposure have been implicated as contributing to obesity in both humans and model animals, possibly by interfering with estrogen and androgen signaling.
While adult exposure to EDCs has various dangerous consequences, mammalian embryos are also a primary concern because of their extreme sensitivity to perturbation by endocrine-like activities. Mammalian embryonic development involves fetal epigenetic programming in that hormonal signals during fetal development control not only the timing of gene expression but set the activity of genes and thus the functioning of organs and homeostatic systems for the remainder of life (Newbold et al 2004). For example, disruption of the activity of estrogen and testosterone signaling by EDCs during fetal development can lead to permanent changes in reproductive organ development, which can later in life impair homeostatic systems, such as the regulation of body fat and weight.
BPA is used to make plastic harder and is nearly ubiquitous in industrialized societies. BPA is also used to line cans, milk cartons, and other metal and paper-board containers commonly used to package foods and beverages. BPA may leach out of plastic storage containers to contaminate foods, beverages and drinking water. BPA content is greatest in clear, polycarbonate plastics, which were previously thought to be safe. It has been detectable in serum of pregnant women and cord serum taken at birth; is 5-fold higher in amniotic fluid at 15–18 weeks gestation, compared with maternal serum; and has been found in concentrations of up to 100 ng/g in placenta. Thus, the human population is widely exposed to BPA and it appears to accumulate in the fetus.
Other chemicals known as human endocrine disruptors include heavy metals, solvents, organophosphates, phthalates, dioxins (example 1, 2, 3, 4, 6, 7, 8-heptachloro dibenzo-p-dioxin), pesticides, and butyltins (example tri and/or tetra butyltins). Numerous pesticides have been reported to affect hormone synthesis and/or metabolism. There is particular concern about endocrine disrupting pesticides that are lipophilic, resistant to metabolism, and able to bioconcentrate up the food chain. This is because these substances become stored in body fats and can be transferred to the developing offspring via the placenta or via the egg. These include tributyltin (TBT) containing pesticides, DDT, atrazine, and several organochlorine pesticides. (see Table 1)
Table 1.
Sources, types, and examples of chemicals identified as potential endocrine disruptors (Adapted from Damstra, 2002)
| Sources | Types | Examples of Chemicals |
|---|---|---|
| Incineration | Industrial by-products | PCBs, dioxins |
| Atmospheric transport | Organochlorine pesticides | DDT, lindane, dieldrin |
| Agricultural runoff | Pesticides currently in use | Atrazine |
| Harbors | Antifoulants from paint applied to hulls of ships | TBT |
| Industrial/municipal effluents | Alkylphenols, natural estrogens | Nonylphenol, estradiol |
| Pulp mill effluents | Plant estrogens | Genistein |
| Consumer products | Flame Retardants | PBDEs |
| Consumer products | Plasticizers | Dibutyl phthalate |
Basic Science Evidence
In Vitro Evidence
The hormonal activity of endocrine disruptors is thought to occur through a variety of mechanisms (Fig 6). The most commonly proposed mechanism is by direct binding to nuclear receptors (NRs), such as the estrogen receptor (ER)(Fig 6 mechanism 1). The human genome contains at least 48 nuclear receptor family members, but the endogenous ligands are known only for a few of them (Chawla et al 2001). Nishikawa’s laboratory developed a high-throughput assay to identify ligands for NRs (Kanayama et al 2005). They found that the organontin endocrine disruptors, which are used as agricultural fungicides, rodent repellents, molluscicides, and in antifouling paints for ships and fishing nets, are high affinity agonist for two NR family members, PPARγ and the retinoic acid X receptor (RXR) (Kanayama et al 2005). Of possible relevance to their proposed contribution to the obesity epidemic is the fact that, like other PPARγ agonists, they efficiently promote adipocyte differentiation from pre-adipocytes (Kanayama et al 2005). A second proposed mechanism for endocrine disruptor function is as NR antagonists (Fig. 6, mechanism 2). For example, vinclozolin, a dicarboximide fungicide that is used on fruits, vegetables, ornamental plants, and turf grass, are known to be act as androgen receptor antagonists (Sohoni et al 1998). Also, Sohoni and Sumpter have shown that several endocrine disruptors that function as environmental estrogens, such as DDT, bisphenol A and butyl benzyl phthalate, are also anti-androgens (Sohoni et al 1998).
Figure 6.

The relationship between fewer hours of sleep per day (sleep debt) and the incidence of obesity from 1960 to the present. As average daily sleep times have decreased, the incidence of obese adults has increased. Data points adapted from (Terman et al. 1913; McGhie et al. 1962; Hammond 1964; Tune 1968; Tune 1969; Palmer et al. 1980; Hicks et al. 2001; Punjabi et al. 2003); National Sleep Foundation Polls 1995, 1998 – 2002, 2005; and CDC Polls 1962, 1974, 1980, 1994, 2000, 2002, and 2004.
Endocrine disruptors have also been proposed to function indirectly by inhibiting aromatases, such as the P450 family members CYP19 and CYP3A1, which, among their many functions, convert testosterone to estradiol (Fig. 6, mechanism 3). For example, Woodhouse and Cooke have recently shown that PCBs 28 and 105 and aroclor 1221 suppress the activity of CYP19 in an in vitro biochemical assay (Woodhouse et al 2004). Finally, endocrine disruptors can disrupt hormone levels by activating expression of the P450 enzymes (Fig. 6, mechanism 4). For example, Mikamo et al have shown that 7 of 54 tested endocrine disruptors, including methoxychlor and benzophenone, interacted with PXR, and induced CYP3A mRNA in the male rat liver (Mikamo et al 2003). In addition to the four proposed mechanisms of endocrine disruptor function in Figure 6, endocrine disruptors have also been shown to disrupt proper neuronal synapse formation (Shinomiya et al 2003), which could potentially affect release of brain-produced hormones that bind to nuclear receptors.
Animal Evidence
Exposure of the prepubertal mouse to a range of BPA doses induces a U-shaped curve for the endpoints of age of vaginal opening and uterine weight, two parameters that are the hallmarks of estrogen action, such that a more apparent effect is observed in the lower and higher doses relative to the medium range doses (EPA 1998). In animal studies, Newbold and others have shown that high-dose diethylstilbestrol (DES) exposures during pregnancy produce small to normal-size offspring that tend to stay small as adults, whereas low-dose exposures produce normal-size offspring that tend to fatten as they age (Mead 2004).
While disruption of normal hormonal regulation can contribute to obesity through myriad mechanisms, perhaps the most dramatic evidence for the role of the endocrine system in regulating adipose development is the effect of estrogen on white adipose tissue (WAT) regulation. In rodent models, ovariectomy increases WAT, and estrogen replacement therapy (ERT) decreases WAT (Wade et al 1985). Similarly, postmenopausal women have increased WAT and ERT restores WAT to lower levels (Tchernof et al 1998). The estrogen receptor-α knock out (αERKO) mouse model further argues for the role of estrogen in regulating WAT (Heine et al 2000). Both αERKO male and female mice develop insulin resistance and impaired glucose tolerance (Heine et al 2000). While there was no difference in brown adipose tissue (BAT) weight in αERKO mice of all ages, WAT increased with advancing age in male αERKO mice (Heine et al 2000). Surprisingly, the increase in WAT involves reduced energy expenditure rather than increased energy intake (Heine et al 2000).
Ecological Correlation Evidence
Evidence of increase in endocrine disruptors over the last few decades
Wittassek and collegeues (2007) showed that 98% of the urine samples metabolites of all phthalates were detectable in a German sample indicating a ubiquitous exposure throughout the last 20 years. It has been estimated the annually, on average, as much as 60 kilograms of PCDDs are produced in Canadian forest fires alone. By some estimates, this is approximately 10 times more than the amount formed in the 1976 Seveso Italy incident (Woodford et al 1994). In a report by Charles Benbrook, an estimate of 50 million pounds increase in pesticides (herbicides and insecticides) use had been recorded in accordance to the 550 million acres of genetically manufactured corn, soybeans and cotton planted in the US since 1996 (Benbrook 2003). More than 6 billion pounds were put into products in 2004 alone. BPA is also a high production volume chemical with considerable amounts produced annually in the United States alone. Not surprisingly, the U.S. Centers for Disease Control and Prevention found BPA in the urine of over 95% of nearly 400 U.S. adults and children tested..
Evidence of a correlation endocrine disruptors with obesity rates across countries or cultures
The escalation in obesity over the past few decades parallels the increase in concentration of several endocrine disruptors in the food chain (Nilsson 2000). For example, the concentration of PBDE in the milk of Swedish women from 1972 to 1998 has almost doubled every five years (Noren et al 2000). However, the rise in obesity in Sweden came quite late as compared to the rest of the world and really jumped from about 1990 to 2000 (Neovius et al., 2006). In the United States, the obesity rate among adults (30.6% in 2002) is the highest among all OECD countries, followed by Mexico (24.2% in 2000) and the United Kingdom (23% in 2003). Obesity rates in Continental European countries are lower, but are also rising.
There is also epidemiological and experimental evidence that industrial chemicals might cause an increase in obesity rates. Recently, a great deal of attention and interest has been directed toward the hypothesis that exposure to certain environmental chemicals might be capable of causing a spectrum of adverse effects as a result of endocrine modulation. Phthalates are known antiandrogens in experimental animal models. More than 75% of the U.S. population has measurable levels of phthalate metabolite in the urine (Silva et al 2004). Stahlhut and collegues (2007) found that the concentrations of several phthalate metabolites among NHANES populations to be positively and significantly correlated to abdominal obesity in adult U.S. males. Herman-Giddens and collegues (1997) analyzed data rating stages of sexual maturation in 17,077 girls ages 3 through 12 in the U.S. and found that girls appear to be maturing more rapidly, a trend attributed to increasing obesity, changes in the relationship between father and growing daughter, and environmental toxins. Furthermore, olestra which is a dietary fat substitute, decreased Arochlor 1254 contamination as well as facilitated weight loss in an obese diabetic male (Redgrave et al 2005). Additional studies predict that some chemicals “obesogens” may improperly regulate lipid metabolism and adipogenesis hence promoting obesity (Tabb et al 2007).
Pharmaceutical Iatrogenesis and Obesity
General Statement of the Putative Cause and Hypothesized Mechanisms of Action
Weight gain is associated with several commonly used medications, including psychotropic medications, antidiabetics, antihypertensives, steroid hormones and contraceptives, antihistamines, and protease inhibitors. The deleterious effects of drug-induced weight gain include increased risks for developing type II diabetes, hypertension, hyperlipidemia, and poor medication compliance. Although it is difficult to estimate the full impact of drug-induced weight gain, the recognition that some of the most widely prescribed classes of drugs can cause significant weight gain supports the hypothesis that drug-induced weight gain is contributing to the obesity epidemic.
Basic Science Evidence
In Vitro Evidence
There are data showing that pharmaceutical agents such as olanzapine can induce adipogenesis (Vestri et al., 2007; Yang et al., 2007; Minet-Ringuet et al., 2007).
Animal Evidence
Animal studies have proven to be unreliable predictors of weight gain in humans. Although increased weight gain has been described, paradoxical weight loss has also been observed with some drugs known to be associated with the greatest weight gain in humans. This may reflect the well-known differences in metabolic and neuroendocrine pathways regulating appetite and energy metabolism between humans and rodents. For example, chlomipramine, an antidepressant known to be associated with weight gain and increased food intake in humans, decreases food intake in animals (Calegari et al., 2007). Paroxetine, however, a serotonin reuptake inhibitor associated with weight gain in humans, causes weight loss and reduced food intake in animals (Konkle et al., 2003). The antipsychotics clozapine and olanzipine are well-known to cause hyperphagia and weight gain in humans. In animals, some studies (Cope et al., 2005) have shown similarity to humans (Cope et al., 2005, Minet-Ringuet et al., 2006), but in other cases there are significant differences (Cooper et al., 2007). These differences may be due to animal species and sex, dietary composition, and route of administration of the drugs.
Ecological Correlation Evidence
Increase in the Use of Weight Gain Inducing Pharmaceuticals
Data from NHANES demonstrate that the use of psychotropic medications among US adults has increased since 1988–1994, specifically, the use of antidepressants. The use of any psychotropic increased from 6.1% to 11.1% of the population between the third NHANES from 1988–1994 and NHANES 1999–2002. The age-adjusted prevalence of antidepressant use increased from 2.5% to 8.1%, more than a three-fold increase. The use of antipsychotics increased slightly from 0.8% to 1.0% of the population, but the use of newer atypical antipsychotics associated with weight gain increased as they became available during that time period (see Figure 9). Moreover, among children, antipsychotic use seems to have increased substantially (Olfson et al., 2006). Thus, it can be conservatively estimated from the population of the United States that more than 20 million Americans were taking antidepressants, with 2.5 million taking antipsychotics during NHANES 1999–2002 (Paulose-Ram et al., 2007). Office-based visits documenting the use of antidepressant pharmacotherapy for any purpose escalated from 16,534,268 in 1990 to 40,925,824 in 1998, a 147.5% increase, according to data from the US National Ambulatory Medical Care Survey for the time period of 1990 through 1998. Significant increases between time periods for antidepressant use were seen for all ages, gender, and race-ethnic groups (Skaer et al., 2000). More recent marketing data for the period 2002–2007 shows an increase in total antidepressant prescriptions from 200 million to 234 million, whereas new prescriptions increased from 83.5 to 97.5 million. Total prescriptions for antipsychotics increased over the same period from 33.6 to 47.2 million.
Figure 9.

Hermann RC, Yang D, Ettner SL, Marcus SC, Yoon C, Abraham M. Prescription of antipsychotic drugs by office-based physicians in the United States, 1989–1997. Reprinted with permission from Psychiatric Services. 2002;53(4):425–430.
The use of hypertension medications has, like the psychotropics, been on the rise. For example, more than 65 million Americans have hypertension and the prevalence is increasing (Fields et al., 2004). Between 1988–1994 and 1999–2002, antihypertensive medication use among hypertensive adults increased significantly from 57.3% to 62.9%. Antihypertensive medication use increased for men (47.5% versus 57.9%), non-Hispanic whites (58.8% versus 64.5%), non-Hispanic blacks (56.6% versus 65.2%), and persons with hypertension aged ≥70 years (65.3% versus 70.7%). The use of beta-blockers, known to be associated with weight gain, increased from 27.4% to 29.8% in treated hypertensives over the same period of time according to NHANES data (Gu et al., 2006). Thus, approximately 12 million Americans were being treated with beta-blockers in the early part of this decade.
Finally, according to the Centers for Disease Control and Prevention, 14.6 million Americans have diagnosed type 2 diabetes, and many treatments for type 2 diabetes cause weight gain that goes beyond restoration of normal body composition in a person with diabetes. US marketing data show that prescriptions for antidiabetic agents increased from 23.3 million to 26.8 million between 2002 and 2006. Furthermore, prescriptions for thiazolidinediones, a class with a propensity to weight gain, increased from 7.8 million in 2002 to 12.6 million in 2006. Similar trends were described throughout worldwide markets over the same period of time. In Asia, prescriptions for antidiabetics increased from 85.4 million to 101.9 million between 2002 and 2007. Prescriptions for thiazolidinediones increased from 5.7 million to 19.3 million over the same period.
In each case—psychotropics, antihypertensives, and antidiabetics—the absolute weight gain of individuals may be small, but the increasing prevalence of the disorders magnifies the impact of the drug-induced weight gain on population-wide obesity rates.
Epidemiologic Evidence
Allison et al. (1999) reviewed and compared the effects of a broad range of antipsychotic agents on body weight (Table 3). This meta-analysis of 81 reports estimated the weight change after 10 weeks of treatment at the standard dose for each agent. Compared with placebo treatment, which was associated with a mean weight reduction of 0.74 kg (1.64 lb), the mean weight changes with conventional antipsychotic agents ranged from a loss of 0.39 kg (0.89 lb) (molindone) to a gain of 3.19 kg (7.08 llb) (thioridazine/mesoridazine). Among the newer agents, clozapine produced the greatest weight increase and ziprasidone produced the least increase. Quetiapine was not included in the analysis because of limited data; however, a later report noted that 25% of patients gained at least 7% of their initial body weight (Kurzthaler and Fleischhacker, 2001).
Table 3.
Summary of the types of evidence supporting each putative contributor. Only studies with weight gain or obesity as a direct endpoint are counted in each category of evidence.
| Basic In Vitro Evidence | Animal Evidence | Ecological Correlation Evidence | Epidemiological Evidence | Experimental Evidence in Humans | |
|---|---|---|---|---|---|
| Infections | ✓ | ✓ | - | ✓ | - |
| Epigenetics | ✓ | ✓ | - | ✓ | - |
| Maternal Age | - | ✓ | ✓ | ✓ | - |
| Assortative Mating | - | ✓ | - | ✓ | - |
| Reproductive Fitness | - | ✓ | - | ✓ | - |
| Sleep Debt | - | ✓ | ✓ | ✓ | - |
| Endocrine Disruptors | ✓ | ✓ | ✓ | ✓ | - |
| Pharmaceutical Iatrogenesis | ✓ | ✓ | ✓ | ✓ | ✓ |
| Ambient Temperature | - | ✓ | ✓ | ✓ | - |
| Intrauterine and Intergenerational Effects | - | ✓ | ✓ | ✓ | - |
Pairwise comparisons among all of the antipsychotic agents indicated statistically significant differences among the drugs (Allison et al., 1999). The key point is that with the exception of ziprasidone, all of the newer agents were associated with significantly greater weight gain than was placebo after just 10 weeks of therapy. There is marked variation in the reported weight gain associated with long-term use of these atypical antipsychotic agents, ranging from virtually zero gain with ziprasidone to an average gain of 12 kg (27 lb) after 1 year of treatment with olanzapine (Allison et al., 1999). Risperidone produced less weight gain than did sertindole, olanzapine, or clozapine.
An epidemiologic survey of patients with bipolar disorder demonstrated that BMI and weight were each correlated with the number of weight gain-associated psychotropics to which patients had been exposed (McElroy et al., 2002). Fifty-eight percent of the patients with bipolar disorder were overweight, 21% were obese, and 5% were extremely obese, but it was difficult to distinguish an increase in prevalence because of the international nature of the survey.
Experimental Evidence From Humans
Weight gain induced by tricylic antidepressants has been recognized for many years, and amitryptyline in particular may be associated with the greatest weight gain. Massand (2000) and Leslie et al. (2007) evaluated the relative risk of weight gain associated with drugs within the major classes of antidepressant medications. The antidepressants vary considerably with respect to their long-term weight-gain potential. Several reports suggested that weight gain with tricylic antidepressants ranged from 0.57 kg (1.27 lb) to nearly 1.4 kg (3.1lb) per month of treatment. Specific tricyclics that have been associated with weight gain include nortriptyline (Weber et al., 2000), doxepin (Feighner et al., 1986), and amitriptyline (Montgomery et al., 1998). Within the class of selective serotonin reuptake inhibitors (SSRIs), paroxetine appears to be more likely to cause weight gain than other agents (Sussman et al., 2001).
Weight changes in patients undergoing treatment for depression are complicated by several factors. First, weight gain can represent an improvement in those who may have lost weight as a result of their depression. Second, ongoing weight gain may be a residual symptoms of depression. Last, it can be a side effect of treatment (Aronne and Segal, 2003). This last possibility may be most likely when substantial weight gain occurs during the acute phase of treatment or when it continues following complete remission of depressive symptoms. Weight gain associated with use of antidepresssant medications is an important source of treatment noncompliance and may also contribute to the increased health risks associated with overweight and obesity. The relative risk for weight gain associated with antidepressant therapy has been reviewed by Fava (2000). That analysis suggests that tricyclic antidepressants and monammine oxidase inhibitors are more likely to cause weight gain than are the SSRIs or some of the newer antidepressants. However, data on SSRIs are confounded by the fact that in some patients weight is lost initially, only to be regained, with steady weight gain over the long term (Aronne and Segal, 2003).
The atypical antidepressants buproprion and nefazodone appear to cause less weight gain in the long term. Nefazodone is reportedly weight neutral (Sussman et al., 2001), and bupropion may be associated with weight loss (Croft et al., 2002).
Fava et al. (2000) reported that more patients treated with paroxetine than with either fluoxetine or sertraline gained at least 7% of their baseline weight during 26 to 32 weeks of treatment. Sussman et al. (2001) noted that published data on newer antidepressants and treatment-emergent changes in weight are somewhat limited and that many reports of unexpected weight gain with SSRIs were anecdotal or from small uncontrolled trials. These investigators conducted a retrospective analysis of data from trials comparing nefazodone with various SSRIs and with the tricyclic antidepressant imipramine. Medications were considered to cause clinically significant weight change if the patients gained or lost at least 7% of their baseline weight. Acute effects were assessed after 6 or 8 weeks of treatment. Long-term effects of treatment were evaluated from 16 to 46 weeks. SSRIs were associated with greater initial weight loss than nefazodone during acute treatment: 4.3% of SSRI-treated patients lost weight compared with 1.7% of nefazodone-treated patients (p = .017) (Sussman et al., 2001). In contrast, during long-term treatment, 17.9% of SSRI-treated patients gained weight compared with 8.3% of nefazodone-treated patients (p = .003). Imipramine produced greater weight gain than did nefazodone during both the acute treatment phase (4.9% vs. 0.9%, respectively; p = .027) and long-term treatment (24.5% vs. 9.5%, respectively).
Lithium has been widely used since 1970 for treating bipolar disorder despite a number of side effects, a narrow therapeutic window, and limited effectiveness in some patients. Weight gain is a common side effect of lithium therapy, occurring in one third to two thirds of patients (Garland et al., 1988; Baptista et al., 1995). The weight gain associated with the use of lithium can range from 4 kg (8.8 lbs) within 1 to 2 years to 4.5 to 15.6 kg (10.0 to 34. 6 lb) over 2 years (Coxhead et al., 1992; Baptista et al., 1995).
Although better tolerated than the older antipsychotic agents because of the lack of extrapyridimal side effects, many of the new atypical antipsychotic agents have weight gain as a common side effect (Allison and Casey, 2001). This weight gain is of clinical concern because it impedes patient compliance and has deleterious health consequences (Allison and Casey, 2001; Kurzthaler and Fleischhacker, 2001). The differential effect of atypical antipsychotics on histamine (H1) receptors, anticholinergic effects, and serotonin 5-HT2C antagonistic effects may explain differences in weight gain among the drugs. Moreover, the prevalence of overweight and obesity in patients with schizophrenia—without regard to therapy—may be compared to that of the nonschizophrenic population, whereas the complications of obesity (i.e., diabetes, dyslipidemias, and hypertension) may be underdiagnosed and undertreated in these patients (Allison and Casey, 2001). Recent studies document the risks of obesity: Henderson and colleagues demonstrated that weight gain associated with clozapine treatment continued for as long as 46 months was accompanied by a significant increase in triglyceride levels and a 37% increase in the incidence of type 2 diabetes over the 5-year period of observation (Henderson et al., 2000). Allison and Casey (2001) noted the effects of switching patients from olanzapine to ziprasidone: patients lost weight when switched to ziprasidone, and this weight loss was associated with improvements in their serum lipid profile and glucose tolerance.
Antidiabetic medications such as sulfonylureas, thiazolidinediones, and insulin can cause significant weight gain. In the United Kingdom Prospective Diabetes Study, weight gain of 10 kg was seen in obese diet-failure subjects randomly assigned to insulin, compared with a weight gain of 5.3 kg with sulfonylureas and a weight loss of 1.3 kg with metformin (U.K. Prospective Diabetes Study Group, 1998). Studies comparing sulfonylureas to placebo (Simonson et al., 1997) or another hypoglycemic agent (Segal et al., 1997) have shown weight gains ranging from 1.4 kg (3.1lb) to 2.3 kg (5.1lb). Thiazolidinediones can cause weight gain and a redistribution of body fat from visceral to subcutaneous stores as well as edema. A study by Wallace et al. (2004) showed weight gain of 0.70 kg (1.5 lb) in patients with type 2 diabetes taking pioglitazone after a period of 12 weeks.
Weight gain is also associated with the use of antihypertensive medications, specifically β-adrenergic blockers, but this is not a consistent finding (Kumpusalo and Takala, 2001). A recent study showed a mean weight gain of 1.19 kg (2.6 lb) in patients taking metoprolol tartrate compared with those taking carvedilol for hypertension, suggesting that weight gain is not a class effect (Messerli et al., 2007). A meta-analysis (Sharma et al., 2001) of body weight changes in a series of randomized controlled hypertension trials of at least 6 months duration showed that body weight was higher in the β-blocker group than in the control group at the end of the study, with a median difference of 1.2 kg between the beta blocker group and the control group.
There is conflicting research on weight gain and oral contraceptive use. Many studies are unable to distinguish oral contraceptive-related weight changes from weight changes associated with changes in diet or physical activity. One study showed that after 4 cycles, 31% of the women gained weight, but only 5% gained more than 5% of their baseline body weight (Rosenberg, 1998). A greater magnitude of weight gain is associated with the use of progestins such as depomedroxyprogesterone. A study by Espey and colleagues (Epsey et al., 2000) indicated an average excess weight gain of 6 pounds at 1 year and 11 pounds at 2 years compared to a control group. A study of 19 women with polycystic ovarian syndrome measured the effects of contraceptive treatment on weight and lipids (Vrbikova et al., 2006). The results indicated a significant increase in body weight (p < 0.05), total cholesterol (p < 0.05), and triglycerides (p<0.004) in patients before and after contraceptive use. The link between weight gain and hormone replacement therapy is difficult to measure given that menopause itself is associated with changes in body composition and energy metabolism.
Antihistamine users may also experience weight gain. Research is inconclusive regarding differences in the weight gain potential of sedating vs. nonsedating antihistamines, but it appears that the more potent the antihistamine, the greater the potential for weight gain (Aronne, 2002a). Corticosteroids have also been shown to cause weight gain. One study showed a 2.0 kg (4.4 lb) weight gain after 24 weeks in patients taking prednisone (Prummel et al., 1993).
Treatments for human immunodeficiency disease include administration of antiretroviral therapy and protease inhibitors. Although effective for suppressing HIV viral activity and improving health, such treatments are associated with weight gain and increased deposition of abdominal adipose tissue (Stricker and Goldberg, 1998). One study of 10 HIV patients treated with protease inhibitor-containing regimens found that patients gained an average of 19 lbs (p=0.006) after a period of 6 months (Stricker and Goldberg, 1998).
Conclusion
The potential contribution of commonly prescribed medications to the growing obesity epidemic is highlighted by the fact that several million people take the above-mentioned drugs on a chronic basis. Furthermore, weight, once gained, is not easily lost, accumulating an even larger group of people who have taken the drugs for shorter periods of time. This source of weight gain is preventable, and highlights the need for increased education among clinicians. In many cases, there are alternative medications with little or no potential for weight gain. A lack of acceptable alternatives warrants prescribing the minimal dose to produce clinical efficacy and careful monitoring of weight, with early institution of a program of diet and lifestyle that may minimize drug-induced weight gain.
Ambient Temperature and Obesity
General Statement of the Putative Cause and Hypothesized Mechanisms of Action
The thermal environment affects both energy expenditure and energy intake to maintain homeostasis (Garrow et al 1978), and a thermal neutral zone (TNZ) can be defined as a range of ambient temperatures across which energy expenditure is not allocated towards maintaining a constant body temperature. At temperatures above and below the TNZ, energy intake and expenditure are adjusted to maintain thermal homeostasis. We postulate that an increased usage of climate control permits humans to spend more time in the TNZ which results in a positive energy balance and is manifested as weight gain. Furthermore, we propose that recent trends in the use of climate control may be a modest, yet significant contributor to the recent increase in the prevalence of obesity.
Basic science evidence
The relationship between ambient temperature and body weight has been addressed by several animal studies. Rowe et al (Rowe et al 1982) showed that the body weight and fat stores of rats housed at thermal neutral conditions (25°C) were greater than rats housed under cooler conditions (18°C). Presumably, rats housed below thermal neutral temperatures would be allocating their energy intake towards increasing thermoregulation, whereas rats housed within thermal neutral conditions would be storing this excess energy intake as adipose tissue. Conversely, exposure to ambient temperatures above thermal neutral conditions are associated with reduced body weight. The effect of high ambient temperature on body weight has been extensively studied in the agricultural field, where studies in cattle (Mader et al 2003), pigs (Collin et al 2001) and chickens (Yahav et al 1996) have shown that weight gain is reduced when animals are housed at high ambient temperatures. These studies suggest that the reduction in weight gain is primarily due to decreased food intake at high ambient temperatures. However, the increased metabolic cost of maintaining a constant body temperature (via postural adjustments, evaporative cooling) is likely to exacerbate the effect of high ambient temperatures on body weight.
Ecological correlation evidence
In humans, the TNZ is approximately 25–30°C, and there is substantial evidence that the average ambient temperature has increased in recent history. Between 1970 and 2000, the average internal home temperature in the United Kingdom has increased from 13°C to 18°C (EHCS 2000). Perceptions of thermal comfort in the United States have also increased, with the thermal standard for winter comfort increasing from 18°C in 1923 to 24.6°C in 1986 (Space Heating Technology Atlas 2004). In addition to increased use of climate control for heating, there is evidence that individuals are less likely to be exposed to temperatures above the TNZ. For example, the percentage of homes in the United States with central air conditioning has increased from 23.0% to 47.1% between 1978 and 1997 (Figure 10).
Figure 10.

Figure Percentage of homes with air conditioning. From US DOE, <http://www.eia.doe.gov/emeu/consumptionbriefs/recs/actrends/recs_ac_trends_table2.html>.
Epidemiologic evidence
It is tempting to speculate that historical modifications in the thermal environment have contributed to the obesity epidemic by influencing postnatal weight gain. There is a growing appreciation for the effect of the environment during infancy on the risk of late-life disease, especially with regards to the prevalence of obesity and diabetes (Strauss et al 1997) and several studies suggest that the perinatal thermal environment is related to the risk of obesity. For example, one study of adolescents aged 15–19 years old revealed that females with a BMI > 85th percentile tended to be born during warmer ambient temperatures (>13.2°C), although the relationship was only statistically significant for African American females (van Hanswijck de Jonge et al 2002). Another study showed that one week-old infants placed in an incubator at 36.5°C for two weeks had greater increases in body weight and length compared to a group of infants placed in an incubator maintained at 35°C despite being identical food intake (Glass et al 1968). Additionally, weight gain during the first four months of life is positively correlated with childhood overweight status (Stettler 2002), and weight gain during this period is greater for infants born in the spring (and presumably reared in warmer ambient temperatures) than infants born in the fall, although this was only significant for Puerto Rican and African Americans (van Hanswijck de Jonge et al 2003).
Experimental evidence from human studies
The increase in ambient temperature is likely to result in decreased metabolic rate as shown in several studies in humans: energy expenditure was 167 kcal day−1 lower in males exposed to 22°C compared to 16°C (Westerterp-Plantenga et al 2002), and Buemann et al (Buemann et al 1992) reported a nearly identical reduction (168 kcal day−1) in women exposed to similar thermal conditions (24°C versus 16°C). Other studies report smaller (23 kcal day−1) (Dauncey 1981) or larger (239 kcal day−1) (van Marken Lichtenbelt et al 2001), 185 kcal day−1 (Westerterp-Plantenga et al 2002) reductions in metabolic rate in the TNZ compared to cooler temperatures. Nonetheless, it is clear that metabolic rate in humans is lower within the TNZ compared to temperatures that were historically the norm.
Increased weight gain is a logical consequence of the decreased metabolic rate observed in the TNZ. Although the absolute decrease in metabolic rate in the human studies was quantitatively small (23–239 kcal day−1), other studies have demonstrated that similar differences in rates of energy expenditure alone are predictive of future weight gain (Ravussin et al 1988; Tataranni et al 2003). Therefore, the historical increase in ambient temperature may have had subtle, yet significant effects on metabolic rate and consequently, weight gain. This relationship assumes that humans have reduced their food intake in parallel with the historical increase in ambient temperature, and we feel that this is unlikely.
Increased utilization of air conditioning may exert a more substantial effect on weight gain by attenuating the negative influence of hot environments on food intake. Food intake is generally lower in warm environments (Stroebele et al 2004) and studies of food selection in cafeterias show that there is a trend for selection of lower-calorie food items the late summer (Zifferblatt et al 1980). Furthermore, Johnson et al (Johnson et al 1947) report a strong negative relationship between voluntary food intake and environmental temperature in military troops. In addition to decreasing energy intake, exposure to high ambient temperatures also increases metabolic rate: when exposed to hot environments, core body temperature is elevated and metabolic rate increases by 7–17% per °C rise in body temperature (Saxton et al 1981). Thus, an increased use of air conditioning is likely to lead to a positive energy balance by increasing energy intake while reducing energy expenditure.
Intrauterine and intergenerational effects
General Statement of the Putative Cause and Hypothesized Mechanisms of Action
Genotype and phenotype are not identical, and a key linkage is via the processes of developmental plasticity through which environmental signals acting during early development influence functional and organ development (West-Eberhard 2003); epigenetic processes are thought to be central (Burdge et al 2007). Developmental plasticity is a normative process by which change is made in an attempt to better adapt the organism to its anticipated environment. Other developmental responses, generally under more severe conditions, are made as a result of tradeoffs to allow either the fetus or mother to survive concurrent environmental stress. These two classes of response have an adaptive origin although both may have maladaptive consequences. A further set of developmental responses induced by an extreme environment, toxins or infection may disrupt the developmental program – this cannot be considered adaptive in origin (Gluckman et al 2005b). As a key part of an organism’s life-course strategy depends on matching nutritional demand and energy stores to the environment, these developmental considerations deserve a greater focus in understanding the etiology of excess adiposity. A further consideration is the particular role of fat in infant survival – humans have been described as the fattest species at birth and it has been strongly argued that this fat is an important buffer against the risk of malnutrition following weaning (Kuzawa 1998).
There is growing evidence to suggest that these various mechanisms play a significant role in explaining the emergent patterns of metabolic compromise and obesity. A harder issue is to quantify the relative importance of these effects, as they do not act in isolation; indeed the experimental evidence would suggest that the primary effect of the developmental phase is to alter the risk of an individual developing obesity and insulin resistance in particular energetic environments later in life (Gluckman et al 2004).
There are at least two developmental pathways to an increased risk of obesity that have their origin in prenatal or early infant life. These are likely to involve quite distinct processes and they will therefore be considered separately. Together, they are likely to account for the U- or J-shaped relationship between birth weight and later obesity or insulin resistance (McCance et al 1994; Curhan et al 1996).
The mismatch and related pathways
It is clear that an impaired embryonic, fetal or infant environment can lead to a greater risk of visceral obesity and metabolic compromise in later life. Indeed, there is growing evidence that relative visceral obesity may be present from birth in small-for-gestational-age infants (Harrington et al 2004). This pathway has been the focus of a growing field of research known as the “developmental origins of health and disease”. A tendency to pathogenic adiposity and its associated diseases are the potential maladaptive consequences of developmentally plastic responses made in early life that either lead to immediate responses requiring fetal adaptations for survival (e.g. growth impairment) or which through anticipatory processes induce a phenotype matched for a more thrifty adult environment (Gluckman et al 2005b). The distinction between these processes is not clear; they may be a continuum and involve overlapping proximate mechanisms. In either case, they lead to mature individuals who have followed a developmental trajectory appropriate for a lower energy environment but who find themselves in a high energy environment and with physiological and structural adaptations that are now inappropriate and associated with a greater propensity to lay down fat and develop insulin resistance – this is manifest as obesity and its complications (Gluckman et al 2004b). At least in its anticipatory form, this is a normative physiological process (Gluckman et al 2007b) and it can be considered as the outcome of a mismatch between the phenotype defined by evolutionary and developmental processes and the energy environment the individual now lives in (Gluckman et al 2006).
Epidemiological evidence
In the 1970s there were data that showed that experimentally induced fetal growth retardation in rats led to offspring with a greater risk of insulin resistance and abnormal pancreatic function (Aerts et al 1979; De Prins et al 1981) but these provocative observations were ignored until a series of epidemiological observations by Barker and colleagues showed that impaired human fetal development as reflected in reduced birth size had later cardiovascular and metabolic effects (Barker et al 1986; Hales et al 1991). Subsequently the same group returned to one of these cohorts and demonstrated that those born smaller had as 65 year olds greater body fat, less lean body mass and greater visceral adiposity (Kensara et al 2005). These data were based on a comparison of low birth weight versus normal/high birth weight. Although the data relating birth size to metabolic outcomes was initially controversial, there are now a large number of studies relating birth size to later insulin resistance (Godfrey 2006) and a smaller number relating birth size to adult adiposity (Martorell et al 2001). From such studies one important point stands out: later clinical outcomes such as cardiovascular disease are not related to a low birth size per se, but rather there is a continuous relationship between birth size and later disease risk which continues across the entire birth size range although at the highest birth sizes the risk may again rise as within this group lies the offspring of the gestational diabetic mother (Barker et al 1989; Osmond et al 1993; Curhan et al 1996).
Birth size is not envisaged as the causative factor, but is rather seen as a surrogate for maternal, environmental and placental factors that impair fetal nutrition – the actual delivery of nutrients across the placenta to the fetus. A variety of factors can impair fetal nutrition either directly or indirectly (Harding et al 1995; Gluckman et al 2004a). Rather than focus on the extremes caused by overt maternal and placental disease, there is a growing consideration of the non-pathophysiological factors that impact on fetal nutrition or fetal growth. For example, fetal growth is impaired in those who are born to smaller mothers, to first-time mothers, or to very young mothers. Thus, first-born children may be at particular risk (Stettler et al 2000) and the percentage of first born children in the population has risen as family size has fallen.
These are all processes which lead to reduced nutrition and are generally considered under the rubric “maternal constraint”. There is also evidence that variation in maternal nutrition within the unremarkable range of intakes of Western women can affect fetal physiology, size at birth, and subsequent endothelial function in childhood (Godfrey et al 1996a; Godfrey et al 1996b; Javaid et al 2004; Gale et al 2006).
Clinical evidence
More recently, researchers have begun a number of prospective cohort studies of children from birth or before. These have shown that children of lower birth size present the pattern of excess adipose gain (Ong et al 2006). This is supported by studies in detail of more particular subgroups. For example, a study of children born with intrauterine growth retardation shows specific gain of visceral adipose tissue by 6 years of age (Ibáñez et al 2008).
There is also a growing number of MRI studies of body composition at birth (Harrington et al 2004; Modi et al 2006). These show that while smaller babies have reduced subcutaneous fat with the reduction proportionate to the fall in birth size, visceral fat is not lost to the same extent so that the smaller babies have relative visceral adiposity. Given the metabolic properties of visceral fat (Ostman et al 1979), this provides a plausible route to later metabolic compromise.
Experimental and mechanistic evidence
There is a very large body of evidence, reviewed elsewhere (McMillen et al 2005), relating compromised fetal development to later obesity and metabolic compromise. While the bulk of studies are in the rat, there are also studies in sheep, in primates, mice and pigs. In general these studies challenge fetal development by reducing overall maternal nutrition or by specifically reducing maternal protein intake or by administering glucocorticoids to the mother. Several key points arise from such studies. First, there is an interaction between the prenatal and postnatal environments – such that the combination of a prenatal low nutrient diet and a post-weaning high fat diet has a far greater effect on adiposity than does a postnatal high fat diet alone (Vickers et al 2000). Secondly the studies suggest multiple components of the adipose phenotype are affected. These include such central effects as appetite control and food preference – offspring of nutritionally impaired pregnancies have a preference for higher fat foods and are hyperphagic (Vickers et al 2000; Bellinger et al 2004). Provocatively, these rats have reduced activity in open field testing – it is not yet clear whether this reflects mood alterations or reduced voluntary exercise (Vickers et al 2003). They have neuroendocrine alterations in the hypothalamus (Ikenasio-Thorpe et al 2007). They are also sarcopenic, develop endothelial dysfunction, visceral, hepatic and subcutaneous obesity and have disordered expression of metabolic genes in peripheral tissues. These findings suggest that there is a coordinated central and peripheral response to an impaired fetal experience. Intriguingly the phenotype is remarkably similar irrespective of its mode of induction, suggesting common underlying mechanisms. There are data in which the effect of nutrition on later outcomes has extended from the periconceptional period to the lactation period (McMillen et al 2005), but as yet it is unclear whether the effects are very specific to different developmental phases within this window.
The likely proximate mechanisms are reviewed elsewhere (Burdge et al 2007; Waterland et al 2007), with much interest focusing on the epigenetic processes reviewed below. There is growing evidence that the alterations in gene expression in metabolic pathways are in turn underpinned by upstream methylation changes in specific regulatory genes such as for the peroxisome proliferator activated receptor α and the glucocorticoid receptor, which are central to control of carbohydrate and lipid metabolism (Lillycrop et al 2005; Gluckman et al 2007c). These are not parentally imprinted genes and it has been suggested that this reflects the role of non-imprinted epigenetic change in the regulation of developmental plasticity.
Several studies have now shown in the rodent that administration of leptin in the neonatal period, either directly or via the mother, prevents the development of the metabolic phenotype and in particular the development of adiposity (Stocker et al 2004; Vickers et al 2005). This in turn is associated with a reversal of the changes in gene expression and methylation of these genes in liver tissue when examined as adults (Gluckman et al 2007c). The mode of action of leptin is subject of speculation. It may be acting by effects on neonatal food intake but this seems unlikely given that large changes in infant food intake only cause minor modification of the adult phenotype. There is evidence that leptin has effects on hypothalamic maturation (Bouret et al 2004) and a key research question is whether this effect has a critical period which is accessible in other species, such as the human, more mature at birth.
Mechanisms
While the proximate mechanisms have been discussed above, if there are physiological mechanisms it is necessary to consider the ultimate mechanisms that have protected these processes through evolution. Life history theory points to the importance of trade-offs in development (Stearns 1992). One trade-off is if the intrauterine environment is nutritionally restrictive, growth is impaired initially to conserve nutrients for the fetal brain and under more severe conditions to protect the mother. This may become irreversible, and the offspring is left to cope with the various adaptations made in the growth impaired fetus. This is the origin of the thrifty phenotype model proposed by Hales and Barker (1992).
However the continuous association between birth weight and outcome extends across the normative range of birth size and such severe tradeoffs seem unlikely to be the explanation for the outcomes for the majority of infants born in this range. Bateson (2001) and Gluckman & Hanson (2004c) independently proposed an anticipatory model based on the considerable comparative evidence that organisms in early development may make adaptive responses which are of no advantage at the time they are made but are able to better match the organism to the later predicted environment. These have been termed ‘predictive adaptive responses’ (Gluckman et al 2005a). If the developing organism anticipates a future lower nutritional environment then the appropriate response is to induce a suite of coordinated responses which will maximize reproductive fitness in a nutritionally compromised environment. These would include a tendency to prefer a high fat diet, hyperphagia, less investment in muscle mass, and a tendency to visceral adipose stores when excess energy was available (Gluckman et al 2004b; Gluckman et al 2007b). These would be of no consequence in a nutritionally limited environment, as was likely to have been the case for humans in preagricultural history, but is likely to lead to mismatch in modern environments. As the likelihood of high energy environments grows in modern times, the risk of mismatch and thus obesity grows.
Importantly, maternal constraint limits the maximal environment the fetus can anticipate by putting a cap on nutrient transfer, and this in turn limits the range of postnatal environments which the offspring can manage without risk of mismatch (Gluckman et al 2004a). Such a mechanism could well explain differential relationships across ethnic groups; for example, South Asians, who have much lower birth weights, have a greater risk than Europeans of metabolic compromise for a given body mass (Whincup et al 2002).
Ecological context
The biggest research challenge is to identify the prevalence of this pathway. Elsewhere it has been argued that it is a major contributor to the risks of heart disease and diabetes (Barker et al 2002). However, we currently lack a useful biomarker that can detect alterations in the trajectory of metabolic development. The most likely candidate is probably some measure of specific epigenetic change, but until such a biomarker exists, the prevalence and importance of the mismatch pathway to the risk of obesity can only be estimated from the substantial and experimental data. The model can explain a number of features of the demography of obesity, such as the rapid appearance in populations in transition (Prentice 2006) and the higher incidence in first-born children (Stettler et al 2000). One concern is recent evidence from the UK that shows that the incidence of imprudent diets in pregnancy is very high. Up to 50% of women in the lowest educational groups had a diet inappropriate to support an optimal fetal outcome (Robinson et al 2004). In Japan birth weight is falling and this is paralleled by a reduction in maternal weight gain in pregnancy which is secondary to a number of cultural changes including a unsubstantiated belief that dietary restriction of weight improves pregnancy outcome (Gluckman et al 2007a). The recent Foresight exercise in the UK suggested that of all the putative strategies to attack obesity, the focus on early development was the most likely to be productive (http://www.foresight.gov.uk/Obesity/obesity_final/17.pdf).
Maternal overnutrition, perinatal hyperinsulinemia, and related processes
Insulin is adipogenic in late fetal and infant life, probably increasing both fat cell number and fat cell content (Wu et al 1999). The presence of more fat cells from birth thus sets a template on which it is easier later to develop obesity in an obesogenic environment. This pathway is most well defined in relation to the offspring of mothers who have type 2 diabetes or who develop gestational diabetes, which itself reflects the insulin resistance arising from altered metabolic partitioning during pregnancy. The pathophysiological pathway is believed to be that maternal hyperglycemia leads to greater transfer of glucose across the placenta, which occurs via a process of facilitated diffusion and is therefore partially concentration dependent. In turn, increased delivery of glucose to the fetus in late gestation leads to increased fetal insulin release and this in turn leads to greater adipogenesis. This is supported by experiments in which fetal monkeys have been infused with insulin (Susa et al 1984) and by clinical observations (Eidelman et al 2002). Generally infants of gestational diabetics are macrosomic, with the degree of macrosomia partially dependent on the degree of diabetic control. While fetal hyperinsulinemia leads to a small increase in lean body mass, the greater effect is on fat mass. Irrespective of size at birth, however, postnatal weight gain curves show, by four years of age, a faster gain of body mass relative to offspring of non-diabetic mothers (Silverman et al 1998).
The prevalence of gestational diabetes in the US is now about 9% (Lawrence et al 2008); while this may represent only a relatively small pathway to obesity, there is growing concern about the role of maternal obesity itself independent of clinically apparent diabetes. There is a growing body of evidence that fat mothers give birth to babies who become fat children. For example, in a US cohort maternal first-trimester obesity led to a 2- to 3-fold increase in the risk of childhood obesity in their progeny, such that 24% of children of obese mothers were themselves obese at age 4 compared with 9% of children of normal-weight mothers (Whitaker 2004). Conversely, in a UK cohort the strength of association between parental BMI and offspring BMI at age 7.5 was similar for both parents (Davey Smith et al 2007), suggesting a cultural rather than biological basis for the phenomenon. Nevertheless, there is growing experimental evidence that high fat diets in pregnant rodents lead to obese offspring (Samuelsson et al 2008). Given the increasing prevalence of maternal obesity, this may be a growing and compounding pathway by which sensitivity to obesogenic diets is enhanced in the perinatal period.
Formula feeding in infancy is associated with an increased risk of obesity. Some (Gillman et al 2001; Owen et al 2005a; Harder et al 2005) but not all (Owen et al 2005b) observational studies and meta-analyses have found that breast feeding is protective against obesity in later life. If indicative of causation, there are several possible mechanisms. Formula has a very different protein content and composition (Ziegler 2006). Formula-fed children drink higher volumes, which may entrain satiety differently (Taveras et al 2004). Breast-fed infants have a different gut biota because of the oligosaccharide content of human milk (Parracho et al 2007; Boehm et al 2007) and thus the biotic conversion of food may be different, leading to different absorption (Turnbaugh et al 2006). Alternatively it may be that bottle feeding after a period of relative constraint in fetal life merely exaggerates a mismatch. All these may induce short-term effects on adipose tissue development which, as they are occurring through a phase of metabolic and neuronal plasticity, may later manifest in greater sensitivity to fat in the diet.
Epidemiological and clinical evidence
There is strong epidemiological evidence that a maternal hypernutritional state, whether due to pre-existing type 2 diabetes, gestational diabetes or obesity, generates long-term risk of obesity for the child. Pre-existing type 2 diabetes or gestational diabetes affect 8–9% of pregnancies in the US (Lawrence et al 2008), whereas overweight or obesity affect nearly 50% of pregnancies (Ehrenberg et al 2004). Correspondingly, high birth weight, whether defined as macrosomia (birth weight >4000g) or large for gestational age (> 90th centile), has increased in developed countries, now occurring in 20% of infants in Denmark and increasing by 23% in Sweden and by 24% in Canada during the 1990s (Catalano et al 2007). Increasing hyperglycemia in pregnancy is associated with an increased risk of childhood obesity (Hillier et al 2007), Obesity itself increases the risk of gestational diabetes by 5-fold (Baeten et al 2001), but even obese women with normal glucose tolerance have a two-fold risk of having a macrosomic baby (Kleigman et al 1985). Importantly, adiposity patterns may be altered in the children of women with gestational diabetes even in the absence of macrosomia, with relative increases in fat mass and proportion of body fat (Catalano et al 2007). Maternal weight loss by pre-pregnancy bariatric surgery seems to prevent transmission of obesity to children compared with the offspring of mothers who did not undergo the surgery and remained obese (Kral et al 2006). Gestational weight gain irrespective of pre-pregnancy body mass is positively associated with obesity at 3 years (Oken et al 2007). In turn, higher birth weight is linked by strong epidemiological evidence to childhood, adolescent and adult obesity, as demonstrated by the large cohort studies showing a greater prevalence of overweight in individuals born large (Martorell et al 2001) as well as more recent clinical studies showing that mothers with a higher prepregnant BMI or a larger mid-upper arm circumference during pregnancy tend to have children with greater adiposity at age 9 years (Gale et al 2007).
The effect of a hypernutritional state may extend into postnatal life. Numerous studies, summarized in systematic reviews and meta-analyses (Baird et al 2005; Monteiro et al 2005; Ong et al 2006; Stettler 2007), have shown that rapid weight gain during early infancy is associated with susceptibility to obesity in adulthood. Such rapid weight gain is often associated with formula feeding, and the protective effect of breastfeeding against later obesity (see above) may reflect this. Indeed, studies that have manipulated the composition of formula feeds have shown protective effects of slower growth against cardiovascular risk factors in adolescence (Singhal et al 2003; Singhal et al 2004) and of low-protein formula against infancy weight gain.
Experimental evidence
As reviewed elsewhere (Nathanielsz et al 2007), the epidemiological and clinical observations on maternal obesity and gestational diabetes are supported by experimental studies showing similar effects in animal models. For example, feeding an obesogenic diet to female mice from before mating through lactation leads to maternal obesity as well as hyperphagia, increased adiposity, decreased muscle mass, and reduced locomotor activity in the offspring (Samuelsson et al 2008). Models of postnatal hypernutrition also exist. The overfeeding caused by suckling rats in small litters leads to later appetite dysregulation and obesity, even if a normal diet is fed after weaning (Plagemann et al 1992). An artificial high-carbohydrate but low-fat diet in the pre-weaning period causes obesity in rats (Srinivasan et al 2003), as does maternal gestational diabetes (Franke et al 2005), an effect that can be mimicked by neonatal administration of insulin (Harder et al 1998). This last observation, together with our observation of the protective effects of neonatal leptin treatment (Vickers et al 2005), confirms the importance of the neonatal period in the programming of obesity, at least in rodents.
Mechanisms
There are a number of mechanisms by which maternal hyperglycemia and/or obesity, or early-life overnutrition, may translate into offspring obesity. One relates to adipogenesis and adipocyte metabolism. The increase in the mass of adipose tissue associated with obesity involves both hyperplasia via differentiation of preadipocytes and hypertrophy of existing adipocytes (Prins et al 1997). Adipogenesis begins in fetal life, and there is evidence for maternal nutritional programming of fetal adipogenesis in both undernutrition (Budge et al 2005) and overnutrition (Taylor et al 2006; Samuelsson et al 2008) models. For example, in sheep, increased maternal nutrition results in increased in adipogenic, lipogenic, and adipokine gene expression in fetal adipose tissue (Mühlhäusler et al 2007). Experimental evidence suggests this is mediated at least in part by insulin (Susa et al 1984).
A further possible mechanism relates to ‘programming’ of hypothalamic appetite control systems in the offspring. The hypothalamus, particularly the arcuate and paraventricular nuclei, integrates signals of nutrient availability to control feeding behavior via secretion of orexigenic and anorexigenic neuropeptides (Mühlhäusler 2007). These appetite control circuits are plastic in early life, although the precise timing of the critical windows during which their development can be affected by nutritional factors depends on the maturation stage at birth of the particular species. There is abundant experimental evidence suggesting that factors such as maternal or fetal hyperglycemia, maternal overfeeding during pregnancy or postnatal overfeeding of the offspring can affect the maturation and functioning of the hypothalamic neural networks controlling appetite, both in species such as the sheep where (as in humans) the regulatory circuits are relatively mature at birth (Mühlhäusler et al 2006) and in species such as the rat where the circuits are not fully developed until shortly before weaning (Davidowa et al 2004).
A third mechanism relates to cultural or familial effects causing intergenerational transmission of dietary or exercise habits. For example, patterns of diet and exercise can affect risk across more than one generation (Brook et al 1999).
Integration
These two classes of developmental pathway, broadly defined by undernutrition and hypernutrition in early life, are not mutually exclusive and may co-exist in a single individual For example, women who themselves were born small may be more likely to become insulin resistant in pregnancy and thus the fetus is both constrained through the mismatch pathway, presuming maternal glucose is not the sole signal of maternal nutritional state to the fetus, and has excess adiposity secondary to maternal gestational diabetes (Gluckman and Hanson 2005). So-called ‘catch-up’ or ‘compensatory’ growth, defined as a period of accelerated growth after early nutritional restriction, may pose particular problems for later metabolic health. The deleterious effects of catch-up growth are apparent from theoretical models (Mangel et al 2005), animal studies (Hales et al 2003), clinical evidence (Ibáñez et al 2006; Ibáñez et al 2008) and epidemiological studies (Bhargava et al 2004; Barker et al 2005). The relative contributions of impaired prenatal growth and accelerated postnatal growth to later metabolic compromise remain to be disentangled, although experimental studies (Jimenez-Chillaron et al 2007) suggest that attenuation of catch-up growth after low birth weight is protective against later obesity. The key point is that developmental cues create sensitivity to later conditions, impacting on behaviors such as appetite and on the routes to utilization of energy consumed.
The clinical and societal implications of these developmental pathways to obesity are several-fold. Increasing evidence for the effects of accelerated early growth on later obesity has led to debate on the optimal management of the small-for-gestational-age infant (Ong 2007) and on the design of supplementary feeding programs in developing countries (Uauy et al 2002). More widely, adoption of new child growth standards recommending slower early weight gain may help to prevent later obesity. Although, with some exceptions (Gluckman et al 2007a), low birth weight is a decreasing problem in developed countries, evidence that a suboptimal intrauterine environment may lead to metabolic compromise without decrease in birth weight (Barker et al 1989; Osmond et al 1993; Catalano et al 2007), together with demonstration of the later effects on the offspring of even subtle changes in macronutrient (Gale et al 2006) or micronutrient (Yajnik et al 2008) status during pregnancy, underlines the importance of a balanced maternal diet before and during pregnancy. Worldwide, the metabolic mismatch resulting from the ‘nutrition transition’ in developing countries, with its characteristic suboptimal fetal development coupled with later energy-dense nutrition, presents a major public health challenge (Monteiro et al 2004; Prentice 2006). Breaking these intergenerational cycles of obesity requires a focus on optimal nutrition through the life course.
Overall Summary & Conclusions
Overall it is clear that multiple plausible causes of obesity exist outside of the big two and that as of yet these alternatives have had little if any influence on mainstream discussions of clinical and public health approaches to obesity. This is unfortunate because some of these putative causes could lead directly to practical intervention and prevention approaches. An obvious example building on the idea of sleep debt would be to try to get people to sleep more and then to see if doing so produced more weight loss or less weight gain. According to the ClinicalTrials.gov website, at least one such study is underway in adults (ClinicalTrials.gov Identifier: NCT00261898) and another in infants (ClinicalTrials.gov Identifier: NCT00125580). Perhaps instead of additional large scale studies investigating increasing PE in schools as an obesity reduction strategy, when such programs have often been shown to be ineffective, we should be promoting napping in children during school hours and then assessing the results. It is certainly possible that such a program would work no better then PE, but given the available data it seems worth trying.
How well supported are the putative causes we have reviewed? Figure 11 illustrates the increasing prevalence of some of these putative contributing factors over time, and Table 4 provides an overall summary, permitting one to obtain a sense of a reasonable ranking of each factor in terms of the strength of the evidence that there is a nonzero effect. In some cases, e.g. drug-induced weight gain, data are clear and consistent in every area of evidence including relatively long-term randomized controlled trials in humans that this is indeed an influence. It would obviously be useful to rank the factors in terms of the magnitude of their effects, but at present, we have insufficient information to do so. In other such cases as, for example, increased fecundity among certain segments of the BMI distribution, the evidence is too inchoate to support firm conclusions.
Figure 11.

Prevalence of putative contributors against the increasing prevalence of obesity. a) prevalence of obesity (CDC), b) mean age of mothers at first birth (NCHS, 2003), c) percentage of homes using air conditioning (Energy Information Administration), d) Patient visits involving Antipsychotic prescriptions (Olfsen et al, 2006), e) Glitazone prescriptions (IMS Health), f) PBDE in breastmilk (Noren et al, 2000; Guvenius et al, 2003), g) Average hours of sleep per night (data points from Terman and Hocking 1913; Hammond 1964; McGhie and Russel 1962; Tune 1968; Hicks et al 2001, Palmer et al 1980; NSF 2000, 2001, 2002, and 2008; Punjabi et al 2003).
For all of these putative causes, we believe further research is warranted. For some such as sleep debt, research is warranted to determine whether the factor is indeed contributing to obesity levels. For some such as obesity-promoting drugs, research investigating mechanisms that may lead to an understanding of how to modulate the effects or design alternatives that will not have such deleterious effects is warranted. For some, such as sleep debt or ambient temperature effects, studies involving manipulation of these factors in human randomized controlled trials to evaluate effects (if any) on weight seem warranted, and indeed some such studies are underway (the sleep studies were mentioned above; for ambient temperature, see ClinicalTrials.gov Identifier: NCT00521729).
Finally, we believe that the factors we identify above do not exhaust the list of plausible contributors. Rather they serve as a clarion call for scientists to remain skeptical of simplistic conclusions about complex phenomena which public health advocates in the obesity field in particular seem prone to accept as complete and adequate explanations. In this unusually public discussion it is imperative that scientists remain open to alternative ideas, insist that claims about the causes of the obesity epidemic be grounded in the best available data, and recognize that any one explanatory theory must be viewed in the context of the constellation of plausible, interrelated, and in many cases still unproven hypotheses.
Figure 7.

Four proposed mechanisms for endocrine disruptor function. ER, estrogen receptor; E2, estradiol; T, testosterone, P450s, cytochrome P450 aromatases which convert testosterone to estradiol; ED, endocrine disruptors; NR, nuclear receptors; 1–4, four possible mechanisms for endocrine disruptor function. Mechanism 1 corresponds to enhanced gene transcription by estradiol via endocrine disruptors; mechanism 2 to supressed transcription by estradiol via endocrine disruptors, mechanism 3 to suppresion of converstion of testosterone to estradiol by endocrine disruptors, and mechanism 4 to the enhancement of testosterone to estradiol by endocrine disruptors.
Figure 8. Percent of adults 18 years of age and over reporting antidepressant use in the past month by age and sex: United States, 1988–94 and 1999–2000.

National Center for Health Statistics (2004). Health, United States, 2004 with chartbook on trends in the health of Americans. Hyattsville, MD, p 59.
Table 2.
Allison, DB, Mentore JL, Heo, M, et al. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry 1999:156:1686–1696.
| Drug or Study Condition and Number of Studieda | Weight Change (kg): Fixed Effects Model |
Test For Heterogeneity in Fixed Effects Model |
Weight Change (kg): Random Effects Model |
Estimated Weight Change (kg) at 10 Weeks: Fined Effects Modelb |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | 95% Cl | χ2 | df | p | Mean | 95% Cl | Mean | 95% CI | |
| Chlorpromazine (N=25; 13) | 6.19 | 5.84 to 6.54 | 746.2 | 24 | <0.0005 | 4.19 | 2.94 to 5.44 | 2.10 | 0.85 to 3.35 |
| Clozapine (N=14; 12) | 4.37 | 4.00 to 4.74 | 148.2 | 13 | <0.0005 | 5.67 | 4.34 to 7.00 | 3.99 | 2.72 to 5.26 |
| Fluphenazine (N=11; 10) | 0.95 | 0.73 to 1.17 | 142.0 | 10 | <0.0005 | 1.13 | 0.09 to 2.17 | 0.43 | −0.65 to 1.51 |
| Haloperidol (N=25; 19] | 0.18 | 0.02 to 0.34 | 78.5 | 24 | <0.0005 | 0.51 | 0.20 to 0.82 | 0.48 | 0.07 to 1.03 |
| Loxapine (N=5; 3) | 0.75 | 0.06 to 1.44 | 71.4 | 4 | <0.0005 | 0.65 | −2.56 to 3.86 | — | — |
| Molindone (N=17; 10) | −1.06 | −1.51 to −0.61 | 154.0 | 16 | <0.0005 | −0.10 | −1.39 to 1.19 | −0.81 | −2.16 to 0.54 |
| Nonpharmacologic control (N=7; 4) | 0.79 | 0.46 to 1.12 | 21.0 | 6 | 0.002 | 0.82 | 0.08 to 1.56 | 1.33 | 0.84 to 1.82 |
| Olanzapine (N=157; 7) | 1.53 | 1.49 to 1.57 | 4009.8 | 156 | <0.0005 | 4.17 | 3.70 to 4.64 | 3.51 | 3.29 to 3.73 |
| Perphenazine (N=4; 4) | 2.79 | 1.63 to 3.95 | 19.4 | 3 | <0.0005 | 5.77 | 0.44 to 11.10 | — | — |
| Pimozide (N=2; 2) | −3.53 | −7.65 to 0.59 | 21.1 | 1 | 0.15 | −2.69 | −9.30 to 3.92 | — | — |
| Placebo (N=25; 22) | −0.50 | −0.70 to −0.30 | 238.7 | 24 | <0.0005 | −0.97 | −1.79 to −0.15 | −0.41 | −1.29 to 0.47 |
| Polypharmacy (N=26; 13) | 0.47 | 0.25 to 0.69 | 89.9 | 25 | <0.0005 | 0.46 | 0.24 to 0.68 | 1.22 | 0.36 to 2.08 |
| Quetiapine (N=8; 3)d | 2.61 | 2.07 to 3.14 | 28.8 | 7 | <0.0005 | 2.49 | 1.51 to 3.47 | — | — |
| Risperidone (N=38; 26) | 1.38 | 1.26 to 1.48 | 289.6 | 37 | <0.0005 | 1.67 | 1.38 to 1.96 | 2.00 | 1.61 to 2.39 |
| Sertindole (N=7; 4) | 2.94 | 2.70 to 3.18 | 6.2 | 6 | 0.39 | 2.94 | 2.70 to 3.18 | 2.92 | 1.76 to 4.08 |
| Thioridazine/mesoridazine (N= 16; 12) | 1.97 | 1.58 to 2.36 | 129.1 | 15 | <0.0005 | 2.81 | 1.59 to 4.03 | 3.49 | 1.75 to 5.23 |
| Thiothixene (N=4; 3) | 2.31 | 1.45 to 3.17 | 5.2 | 3 | 0.16 | 2.89 | 1.01 to 4.77 | — | — |
| Trifluoperazine (N=2; 2) | 0.34 | −0.86 to 1.54 | 0.1 | 1 | 0.75 | 0.34 | −0.86 to 1.54 | — | — |
| Ziprasidone (N=25; 22) | 0.64 | 0.40 to 0.88 | 69.2 | 24 | <0.0005 | 0.28 | −0.27 to 0.83 | 0.04 | −0.49 to 0.57 |
Some of the observations entering into the calculations are not independent (i.e., they may be from the same subjects measured at multiple points in time). This was not taken into account in calculation of the standard errors. The Ns shown are total number of means and number of independent cohorts the means came from. The number of means will always be greater than or equal to the number of independent means, because soma cohorts may have been measured at multiple points in time. However, the number of independent means can exceed the number of trials, because some trials contained more than one independent cohort. For example, six trials provided data on ziprasidone, but because the data for men end women were provided separately and several different dose conditions were used with multiple groups, the six trials yield 22 independent cohorts.
Estimated from the fixed effects fitted regression (see text).
Acknowledgments
Supported in part By: NIH Grant P30DK056336. to DBA and NIH1R01DK066164 awarded to NVD. The opinions expressed herein are those of the authors and not necessarily the NIH or any other organization with which they are affiliated.
Footnotes
Author Contribution: David Allison conceived and oversaw the project, drafted the introduction and discussion, and edited all sections. Emily McAllister, Nikhil Dhurandhar, and Scott Keith contributed to specific sections, edited the entire document, and contributed to the overall development of the manuscript. All other authors had the opportunity to edit the complete manuscript and wrote first drafts of specific sections as follows: Eisenmann contributed to the introduction; Boggiano, Hanlon and Benca to Sleep Debt; Elobeid and Ruden to Endocrine Disruptors; Pietrobelli and Barger to Ambient Temperature; Dhurandhar and McAllister to Infections; Aronne and Wright to Pharmaceutical Iatrogenesis; Baskin and Biggio to Increasing Gravida Age; Gluckman to Intrauterine and Intergenerational Effects; Waterland to Epigenetic Effects; Keith, Wang, and Fontaine to Increased Fecundity; and Redden and Katzmarzyk to Assortative mating.
References
- Postponed childbearing--United States, 1970–1987. MMWR Morb Mortal Wkly Rep. 1989;38:810–812. [PubMed] [Google Scholar]
- UKPDS 28: a randomized trial of efficacy of early addition of metformin in sulfonylurea-treated type 2 diabetes. U.K. Prospective Diabetes Study Group. Diabetes care. 1998b;21:87–92. doi: 10.2337/diacare.21.1.87. [DOI] [PubMed] [Google Scholar]
- Abraham SF, Beumont PJV. How patients describe bulimia or binge eating. Psychological Med. 1982;12:625–635. doi: 10.1017/s0033291700055732. [DOI] [PubMed] [Google Scholar]
- Aerts L, Van Assche FA. Is gestational diabetes an acquired condition? Journal of Developmental Physiology. 1979;1:219–225. [PubMed] [Google Scholar]
- Agras WS, Hammer LD, McNicholas F, Kraemer HC. Risk factors for childhood overweight: a prospective study from birth to 9.5 years. J Pediatr. 2004;145:20–25. doi: 10.1016/j.jpeds.2004.03.023. [DOI] [PubMed] [Google Scholar]
- Allison DB, Neale MC, Kezis MI, Alfonso VC, Heshka S, Heymsfield SB. Assortative mating for relative weight: Genetic implications. Behav Genet. 1996;26:103–111. doi: 10.1007/BF02359888. [DOI] [PubMed] [Google Scholar]
- Allison DB, Mentore JL, Heo M, Chandler LP, Cappelleri JC, Infante MC, Weiden PJ. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry. 1999;156:1686–1696. doi: 10.1176/ajp.156.11.1686. [DOI] [PubMed] [Google Scholar]
- Allison DB, Casey DE. Antipsychotic-induced weight gain: a review of the literature. J Clin Psychiatry. 2001;62(Suppl 7):22–31. [PubMed] [Google Scholar]
- Allison DB, Pietrobelli A, Faith MS, Fontaine KR, Gropp E, Fernandez JR. Obesity: Mechanisms & Clinical Management. Elsevier; New York: 2003. Genetic influences on obesity; pp. 1–74. [Google Scholar]
- Allison DBPA, Faith MS, Fontaine KR, Gropp E, Fernández JR. Genetic Influences on Obesity. In: Eckel R, editor. Obesity: Mechanisms & Clinical Management. Elsevier; New York: 2003. pp. 31–74. [Google Scholar]
- Alvarez GG, Ayas NT. The impact of daily sleep duration on health: a review of the literature. Prog Cardiovasc Nurs. 2004;19:56–59. doi: 10.1111/j.0889-7204.2004.02422.x. Review. [DOI] [PubMed] [Google Scholar]
- American Medical Association. Report 3 of the Council on Science and Public Health (A-08) [Accessed: 6/29/2008];The Health Effects of High Fructose Syrup. 2008 http://www.ama-assn.org/ama/pub/category/18641.html.
- Andersen RE, Crespo CJ, Bartlett SJ, Cheskin LJ, Pratt M. Relationship of physical activity and television watching with body weight and level of fatness among children. JAMA. 1998;279:938–942. doi: 10.1001/jama.279.12.938. [DOI] [PubMed] [Google Scholar]
- Anderson M, Matsa DA. [Accessed 6/29/2008];Are restaurants really super-sizing America. 2007 http://are.berkeley.edu/Papers/anderson08.pdf.
- Aronne L. A practical guide to drug-induced weight gain. Minneapolis: McGraw-Hill; 2002a. Drug-Induced Weight Gain: Non-CNS Medications; pp. 77–91. [Google Scholar]
- Aronne LJ. A practical guide to drug-induced weight gain. McGraw-Hill; Minneapolis: 2002b. Drug-Induced Weight Gain: Non-CNS Medications; pp. 77–91. [Google Scholar]
- Aronne LJ, Segal KR. Weight gain in the treatment of mood disorders. J Clin Psychiatry. 2003;64(Suppl 8):22–29. [PubMed] [Google Scholar]
- Artiga AI, Viana JB, Maldonado CR, Chandler-Laney PC, Oswald KD, Boggiano MM. Body composition and endocrine status of long-term stress-induced binge-eating rats. Physiol Behav. 2007:424–431. doi: 10.1016/j.physbeh.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrup AV, Rossner S, Sorensen TI. Alternative causes of obesity. Ugeskr Laeger. 2006;168:135–137. [PubMed] [Google Scholar]
- Atkinson RL, Dhurandhar NV, Allison DB, Bowen RL, Israel BA, Albu JB, Augustus AS. Human adenovirus-36 is associated with increased body weight and paradoxical reduction of serum lipids. Int J Obes (Lond) 2005;29:281–286. doi: 10.1038/sj.ijo.0802830. [DOI] [PubMed] [Google Scholar]
- Ayas NT, White DP, Al-Delaimy WK, Manson JE, Stampfer MJ, Speizer FE, Patel S, Hu FB. A prospective study of self-reported sleep duration and incident diabetes in women. Diabetes Care. 2003;26:380–384. doi: 10.2337/diacare.26.2.380. [DOI] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeten JM, Bukusi EA, Lambe M. Pregnancy complications and outcomes among overweight and obese nulliparous women. American Journal of Public Health. 2001;91:436–440. doi: 10.2105/ajph.91.3.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8:185–192. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- Baird J, Fisher D, Lucas P, Kleijnen J, Roberts H, Law C. Being big or growing fast: Systematic review of size and growth in infancy and later obesity. British Medical Journal. 2005;331:929–931. doi: 10.1136/bmj.38586.411273.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird PA, Sadovnick AD, Yee IM. Maternal age and birth defects: a population study. Lancet. 1991;337:527–530. doi: 10.1016/0140-6736(91)91306-f. [DOI] [PubMed] [Google Scholar]
- Baptista T, Teneud L, Contreras Q, Alastre T, Burguera JL, de Burguera M, de Baptista E, Weiss S, Hernandez L. Lithium and body weight gain. Pharmacopsychiatry. 1995;28:35–44. doi: 10.1055/s-2007-979586. [DOI] [PubMed] [Google Scholar]
- Barcelo A, Barbe F, Llompart E, de la Pena M, Duran-Cantolla J, Ladaria A, Bosch M, Guerra L, Agusti AG. Neuropeptide Y and leptin in patients with obstructive sleep apnea syndrome: Role of obesity. Am J Respir Crit Care Med. 2004 doi: 10.1164/rccm.200405-579OC. in press. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077–1081. doi: 10.1016/s0140-6736(86)91340-1. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Forsen TJ, Kajantie E, Eriksson JG. Trajectories of growth among children who have coronary events as adults. New England Journal of Medicine. 2005;353:1802–1809. doi: 10.1056/NEJMoa044160. [DOI] [PubMed] [Google Scholar]
- Barker DJP, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- Barker DJP, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–1239. doi: 10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- Baskin ML, Ard J, Franklin F, Allison DB. Prevalence of obesity in the United States. Obes Rev. 2005;6:5–7. doi: 10.1111/j.1467-789X.2005.00165.x. [DOI] [PubMed] [Google Scholar]
- Bastian LA, West NA, Corcoran C, Munger RG. Number of children and the risk of obesity in older women. Prev Med. 2005;40:99–104. doi: 10.1016/j.ypmed.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Bateson P. Fetal experience and good adult design. Int J Epidemiol. 2001;30:928–934. doi: 10.1093/ije/30.5.928. [DOI] [PubMed] [Google Scholar]
- Baum CL. The effects of cigarette costs on BMI and obesity. Health Econ. 2008;10:1002. doi: 10.1002/hec.1340. [DOI] [PubMed] [Google Scholar]
- Bellinger L, Lilley C, Langley-Evans SC. Prenatal exposure to a maternal low-protein diet programmes a preference for high-fat foods in the young adult rat. Br J Nutr. 2004;92:513–520. doi: 10.1079/bjn20041224. [DOI] [PubMed] [Google Scholar]
- Bernard A, Akaoka H, Giraudon P, Belin M. Viruses and the neuroendocrine system: model of murine obesity induced by cerebral infection by canine distemper virus. Ann Biol Clin (Paris) 1999;57:291–299. [PubMed] [Google Scholar]
- Bhanot JL, Chhina GS, Singh B, Sachdeva U, Kumar VM. REM sleep deprivation and food intake. Indian J Physiol Pharmacol. 1989;33:139–145. [PubMed] [Google Scholar]
- Bhargava SK, Sachdev HS, Fall CHD, Osmond C, Lakshmy R, Barker DJP, Biswas SK, Ramji S, Prabhakaran D, Reddy KS. Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. New England Journal of Medicine. 2004;350:865–875. doi: 10.1056/NEJMoa035698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bistrian BR, Khaodhiar L. Chronic systemic inflammation in overweight and obese adults. JAMA. 2000;283:2235. author reply 2236. [PubMed] [Google Scholar]
- Bjorvatn B, Sagen IM, Oyane N, Waage S, Fetveit A, Pallesen S, Ursin R. The association between sleep duration, body mass index and metabolic measures in the Hordaland Health Study. J Sleep Res. 2007;16:66–76. doi: 10.1111/j.1365-2869.2007.00569.x. [DOI] [PubMed] [Google Scholar]
- Blair NJ, Thompson JM, Black PN, Becroft DM, Clark PM, Han DY, Robinson E, Waldie KE, Wild CJ, Mitchell EA. Risk factors for obesity in 7-year-old European children: the Auckland Birthweight Collaborative Study. Arch Dis Child. 2007;92:866–871. doi: 10.1136/adc.2007.116855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc P, Corsi AM, Gabbuti A, Peduzzi C, Meacci F, Olivieri F, Lauretani F, Francesco M, Ferrucci L. Chlamydia pneumoniae seropositivity and cardiovascular risk factors: The InCHIANTI Study. J Am Geriatr Soc. 2004;52:1626–1631. doi: 10.1111/j.1532-5415.2004.52453.x. [DOI] [PubMed] [Google Scholar]
- Bliwise DL. Historical change in the report of daytime fatigue. Sleep. 1996;19:462–464. doi: 10.1093/sleep/19.6.462. [DOI] [PubMed] [Google Scholar]
- Bobrowski RA, Bottoms SF. Underappreciated risks of the elderly multipara. Am J Obstet Gynecol. 1995;172:1764–1767. doi: 10.1016/0002-9378(95)91409-9. discussion 1767–1770. [DOI] [PubMed] [Google Scholar]
- Bodosi B, Gardi J, Hajdu I, Szentirmai E, Obal F, Jr, Krueger JM. Rhythms of ghrelin, leptin, and sleep in rats: effects of the normal diurnal cycle, restricted feeding, and sleep deprivation. Am J Physiol Regul Integr Comp Physiol. 2004a;287:R1071–1079. doi: 10.1152/ajpregu.00294.2004. [DOI] [PubMed] [Google Scholar]
- Bodosi B, Gardi J, Hajdu I, Szentirmai E, Obal FJ, Krueger JM. Rhythms of ghrelin, leptin, and sleep in rats: effects of the normal diurnal cycle, restricted feeding, and sleep deprivation. Am J Physiol Regul Integr Comp Physiol. 2004b;287:R1071–R1079. doi: 10.1152/ajpregu.00294.2004. [DOI] [PubMed] [Google Scholar]
- Boehm G, Stahl B. Oligosaccharides from milk. Journal of Nutrition. 2007;137:847S–849S. doi: 10.1093/jn/137.3.847S. [DOI] [PubMed] [Google Scholar]
- Bolumar F, Olsen J, Rebagliato M, Saez-Lloret I, Bisanti L. Body mass index and delayed conception: a European Multicenter Study on Infertility and Subfecundity. Am J Epidemiol. 2000;151:1072–1079. doi: 10.1093/oxfordjournals.aje.a010150. [DOI] [PubMed] [Google Scholar]
- Bonnet MH, Arand DL. We are chronically sleep deprived. Sleep. 1995;18:908–911. doi: 10.1093/sleep/18.10.908. [DOI] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- Bray GA, Champagne CM. Beyond energy balance: there is more to obesity than kilocalories. J Am Diet Assoc. 2005;105:S17–23. doi: 10.1016/j.jada.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. 2004;79:537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- Broman JE, Lundh LG, Hetta J. Insufficient sleep in the general population. Neurophysiol Clin. 1996;26:30–39. doi: 10.1016/0987-7053(96)81532-2. [DOI] [PubMed] [Google Scholar]
- Brook JS, Whiteman M, Brook DW. Transmission of risk factors across three generations. Psychol Rep. 1999;85:227–241. doi: 10.2466/pr0.1999.85.1.227. [DOI] [PubMed] [Google Scholar]
- Budge H, Gnanalingham MG, Gardner DS, Mostyn A, Stephenson T, Symonds ME. Maternal nutritional programming of fetal adipose tissue development: long-term consequences for later obesity. Birth Defects Research Part C - Embryo Today: Reviews. 2005;75:193–199. doi: 10.1002/bdrc.20044. [DOI] [PubMed] [Google Scholar]
- Buemann B, Astrup A, Christensen NJ, Madsen J. Effect of moderate cold exposure on 24-h energy expenditure: similar response in postobese and nonobese women. Am J Physiol. 1992;263:E1040–1045. doi: 10.1152/ajpendo.2006.263.6.E1040. [DOI] [PubMed] [Google Scholar]
- Burdge GC, Hanson MA, Slater-Jeffries JL, Lillycrop KA. Epigenetic regulation of transcription: a mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? Br J Nutr. 2007;97:1036–1046. doi: 10.1017/S0007114507682920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero B. The global epidemic of obesity: an overview. Epidemiol Rev. 2007;29:1–5. doi: 10.1093/epirev/mxm012. [DOI] [PubMed] [Google Scholar]
- Calegari L, Gorenstein C, Gentil V, Planeta CS, Nunes-de-Souza RL. Effect of chronic treatment with clomipramine on food intake, macronutrient selection and body weight gain in rats. Biol Pharm Bull. 2007;30:1541–1546. doi: 10.1248/bpb.30.1541. [DOI] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- Carp RI, Meeker H, Sersen E, Kozlowski P. Analysis of the incubation periods, induction of obesity and histopathological changes in senescence-prone and senescence-resistant mice infected with various scrapie strains. J Gen Virol. 1998;79 (Pt 11):2863–2869. doi: 10.1099/0022-1317-79-11-2863. [DOI] [PubMed] [Google Scholar]
- Carter JK, Garlich JD, Donaldson WE, Smith RE. Influence of diet on a retrovirus-induced obesity and stunting syndrome. Avian Dis. 1983a;27:317–322. [PubMed] [Google Scholar]
- Carter JK, Ow CL, Smith RE. Rous-associated virus type 7 induces a syndrome in chickens characterized by stunting and obesity. Infect Immun. 1983b;39:410–422. doi: 10.1128/iai.39.1.410-422.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano PM, Thomas A, Huston-Presley L, Amini SB. Phenotype of infants of mothers with gestational diabetes. Diabetes Care. 2007;30:S156–S160. doi: 10.2337/dc07-s209. [DOI] [PubMed] [Google Scholar]
- Cawley J, Meyerhoefer C, Newhouse D. The impact of state physical education requirements on youth physical activity and overweight. Health economics. 2007;16:1287–1301. doi: 10.1002/hec.1218. [DOI] [PubMed] [Google Scholar]
- Charriere G, Cousin B, Arnaud E, Andre M, Bacou F, Penicaud L, Casteilla L. Preadipocyte conversion to macrophage. Evidence of plasticity. J Biol Chem. 2003;278:9850–9855. doi: 10.1074/jbc.M210811200. [DOI] [PubMed] [Google Scholar]
- Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- Chou SY, Grossman M, Saffer H. An economic analysis of adult obesity: results from the Behavioral Risk Factor Surveillance System. J Health Econ. 2004;23:565–587. doi: 10.1016/j.jhealeco.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Christakis NA, Fowler JH. The spread of obesity in a large social network over 32 years. N Engl J Med. 2007;357:370–379. doi: 10.1056/NEJMsa066082. [DOI] [PubMed] [Google Scholar]
- Chua JL, Touyz S, Hill AJ. Negative mood-induced overeating in obese binge eaters: an experimental study. Int J Obes Relat Metab Disord. 2004;28:606–610. doi: 10.1038/sj.ijo.0802595. [DOI] [PubMed] [Google Scholar]
- Cnattingius S, Forman MR, Berendes HW, Isotalo L. Delayed childbearing and risk of adverse perinatal outcome. A population-based study. JAMA. 1992;268:886–890. [PubMed] [Google Scholar]
- Collin A, van Milgen J, Dubois S, Noblet J. Effect of high temperature on feeding behaviour and heat production in group-housed young pigs. Br J Nutr. 2001;86:63–70. doi: 10.1079/bjn2001356. [DOI] [PubMed] [Google Scholar]
- Collins S. The limit of human adaptation to starvation. Nature Med. 1995;1:810–814. doi: 10.1038/nm0895-810. [DOI] [PubMed] [Google Scholar]
- Contoyannis P, Wildman J. Using relative distributions to investigate the body mass index in England and Canada. Health economics. 2007;16:929–944. doi: 10.1002/hec.1240. [DOI] [PubMed] [Google Scholar]
- Cooper GD, Pickavance LC, Wilding JP, Harrold JA, Halford JC, Goudie AJ. Effects of olanzapine in male rats: enhanced adiposity in the absence of hyperphagia, weight gain or metabolic abnormalities. J Psychopharmacol. 2007;21:405–413. doi: 10.1177/0269881106069637. [DOI] [PubMed] [Google Scholar]
- Cope MB, Nagy TR, Fernandez JR, Geary N, Casey DE, Allison DB. Antipsychotic drug-induced weight gain: development of an animal model. Int J Obes (Lond) 2005;29:607–614. doi: 10.1038/sj.ijo.0802928. [DOI] [PubMed] [Google Scholar]
- Cournot M, Ruidavets JB, Marquie JC, Esquirol Y, Baracat B, Ferrieres J. Environmental factors associated with body mass index in a population of Southern France. Eur J Cardiovasc Prev Rehabil. 2004;11:291–297. doi: 10.1097/01.hjr.0000129738.22970.62. [DOI] [PubMed] [Google Scholar]
- Coxhead N, Silverstone T, Cookson J. Carbamazepine versus lithium in the prophylaxis of bipolar affective disorder. Acta Psychiatr Scand. 1992;85:114–118. doi: 10.1111/j.1600-0447.1992.tb01453.x. [DOI] [PubMed] [Google Scholar]
- Croft H, Houser TL, Jamerson BD, Leadbetter R, Bolden-Watson C, Donahue R, Metz A. Effect on body weight of bupropion sustained-release in patients with major depression treated for 52 weeks. Clin Ther. 2002;24:662–672. doi: 10.1016/s0149-2918(02)85141-4. [DOI] [PubMed] [Google Scholar]
- Crossley N. Global anti-corporate struggle: a preliminary analysis. Br J Sociol. 2002;53:667–691. doi: 10.1080/0007131022000021542. [DOI] [PubMed] [Google Scholar]
- Curhan GC, Chertow GM, Willett WC, Spiegelman D, Colditz GA, Manson JE, Speizer FE, Stampfer MJ. Birth weight and adult hypertension and obesity in women. Circulation. 1996;94:1310–1315. doi: 10.1161/01.cir.94.6.1310. [DOI] [PubMed] [Google Scholar]
- Dahmen N, Bierbrauer J, Kasten M. Increased prevalence of obesity in narcoleptic patients and relatives. Eur Arch Psychiatry Clin Neurosci. 2001;251:85–89. doi: 10.1007/s004060170057. [DOI] [PubMed] [Google Scholar]
- Dallman MF, Pecoraro N, Akana SF, La Fleur SE, Gomez F, Houshyar H, Bell ME, Bhatnagar S, Laugero KD, Manalo S. Chronic stress and obesity: a new view of “comfort food”. Proc Natl Acad Sci U S A. 2003;100:11696–11701. doi: 10.1073/pnas.1934666100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damstra T. Potential effects of certain persistent organic pollutants and endocrine disrupting chemicals on the health of children. J Toxicol Clin Toxicol. 2002;40:457–465. doi: 10.1081/clt-120006748. [DOI] [PubMed] [Google Scholar]
- Dart AM, Martin JL, Kay S. Association between past infection with Chlamydia pneumoniae and body mass index, low-density lipoprotein particle size and fasting insulin. Int J Obes Relat Metab Disord. 2002;26:464–468. doi: 10.1038/sj.ijo.0801890. [DOI] [PubMed] [Google Scholar]
- Dauncey MJ. Influence of mild cold on 24 h energy expenditure, resting metabolism and diet-induced thermogenesis. Br J Nutr. 1981;45:257–267. doi: 10.1079/bjn19810102. [DOI] [PubMed] [Google Scholar]
- Davey Smith G, Steer C, Leary S, Ness A. Is there an intrauterine influence on obesity? Evidence from parent-child associations in the Avon Longitudinal Study of Parents and Children (ALSPAC) Archives of Disease in Childhood. 2007;92:876–880. doi: 10.1136/adc.2006.104869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidowa H, Plagemann A. Hypothalamic neurons of postnatally overfed, overweight rats respond differentially to corticotropin-releasing hormones. Neuroscience Letters. 2004;371:64–68. doi: 10.1016/j.neulet.2004.08.042. [DOI] [PubMed] [Google Scholar]
- De Prins FA, Van Assche FA. Fetal growth retardation. Churchill Livingstone; Edinburgh: 1981. Intrauterine growth retardation and development of endocrine pancreas in the experimental rat; pp. 188–196. [DOI] [PubMed] [Google Scholar]
- DeVore NE. Parenthood postponed. Am J Nurs. 1983;83:1160–1163. [PubMed] [Google Scholar]
- Dhurandhar NV, Kulkarni P, Ajinkya SM, Sherikar A. Effect of adenovirus infection on adiposity in chicken. Vet Microbiol. 1992;31:101–107. doi: 10.1016/0378-1135(92)90068-5. [DOI] [PubMed] [Google Scholar]
- Dhurandhar NV, Kulkarni PR, Ajinkya SM, Sherikar AA, Atkinson RL. Association of adenovirus infection with human obesity. Obes Res. 1997;5:464–469. doi: 10.1002/j.1550-8528.1997.tb00672.x. [DOI] [PubMed] [Google Scholar]
- Dhurandhar NV, Israel BA, Kolesar JM, Mayhew GF, Cook ME, Atkinson RL. Increased adiposity in animals due to a human virus. Int J Obes Relat Metab Disord. 2000;24:989–996. doi: 10.1038/sj.ijo.0801319. [DOI] [PubMed] [Google Scholar]
- Dhurandhar NV. Infectobesity: obesity of infectious origin. J Nutr. 2001;131:2794S–2797S. doi: 10.1093/jn/131.10.2794S. [DOI] [PubMed] [Google Scholar]
- Dhurandhar NV, Whigham LD, Abbott DH, Schultz-Darken NJ, Israel BA, Bradley SM, Kemnitz JW, Allison DB, Atkinson RL. Human adenovirus Ad-36 promotes weight gain in male rhesus and marmoset monkeys. J Nutr. 2002;132:3155–3160. doi: 10.1093/jn/131.10.3155. [DOI] [PubMed] [Google Scholar]
- Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104:13056–13061. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff RG, Vogt PK. Characteristics of two new avian tumor virus subgroups. Virology. 1969;39:18–30. doi: 10.1016/0042-6822(69)90344-4. [DOI] [PubMed] [Google Scholar]
- Duncan BB, Schmidt MI, Chambless LE, Folsom AR, Heiss G. Inflammation markers predict increased weight gain in smoking quitters. Obes Res. 2003;11:1339–1344. doi: 10.1038/oby.2003.181. [DOI] [PubMed] [Google Scholar]
- Dunn BE, Cohen H, Blaser MJ. Helicobacter pylori. Clin Microbiol Rev. 1997;10:720–741. doi: 10.1128/cmr.10.4.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Tauleria E, Rona RJ, Chinn S. Factors associated with weight for height and skinfold thickness in British children. J Epidemiol Community Health. 1995;49:466–473. doi: 10.1136/jech.49.5.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzaja A, Dalal MA, Himmerich H, Uhr M, Pollmacher T, Schuld A. Sleep enhances nocturnal plasma ghrelin levels in healthy subjects. Am J Physiol Endocrinol Metab. 2004;286:E963–967. doi: 10.1152/ajpendo.00527.2003. [DOI] [PubMed] [Google Scholar]
- Ehrenberg HM, Mercer BM, Catalano PM. The influence of obesity and diabetes on the prevalence of macrosomia. American Journal of Obstetrics & Gynecology. 2004;191:964–968. doi: 10.1016/j.ajog.2004.05.052. [DOI] [PubMed] [Google Scholar]
- Eidelman AI, Samueloff A. The pathophysiology of the fetus of the diabetic mother. Seminars in Perinatology. 2002;26:232–236. doi: 10.1053/sper.2002.34215. [DOI] [PubMed] [Google Scholar]
- Eisenmann JC. Insight into the causes of the recent secular trend in pediatric obesity: Common sense does not always prevail for complex, multi-factorial phenotypes. Prev Med. 2006;42:329–335. doi: 10.1016/j.ypmed.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Ekesbo R, Nilsson PM, Lindholm LH, Persson K, Wadstrom T. Combined seropositivity for H. pylori and C. pneumoniae is associated with age, obesity and social factors. J Cardiovasc Risk. 2000;7:191–195. doi: 10.1177/204748730000700305. [DOI] [PubMed] [Google Scholar]
- Ellis L, Haman D. Population increases in obesity appear to be partly due to genetics. J Biosoc Sci. 2004;36:547–559. doi: 10.1017/s0021932003006357. [DOI] [PubMed] [Google Scholar]
- Eriksson UJ, Borg LA. Diabetes and embryonic malformations. Role of substrate-induced free-oxygen radical production for dysmorphogenesis in cultured rat embryos. Diabetes. 1993;42:411–419. doi: 10.2337/diab.42.3.411. [DOI] [PubMed] [Google Scholar]
- Espey E, Steinhart J, Ogburn T, Qualls C. Depo-provera associated with weight gain in Navajo women. Contraception. 2000;62:55–58. doi: 10.1016/s0010-7824(00)00144-x. [DOI] [PubMed] [Google Scholar]
- Everson CA, Crowley WR. Reductions in circulating anabolic hormones induced by sustained sleep deprivation in rats. Am J Physiol Endocrinol Metab. 2004;286:E1060–1070. doi: 10.1152/ajpendo.00553.2003. [DOI] [PubMed] [Google Scholar]
- Faith MS, Fontaine KR, Baskin ML, Allison DB. Toward the reduction of population obesity: macrolevel environmental approaches to the problems of food, eating, and obesity. Psychol Bull. 2007;133:205–226. doi: 10.1037/0033-2909.133.2.205. [DOI] [PubMed] [Google Scholar]
- Falck G, Gnarpe J, Hansson LO, Svardsudd K, Gnarpe H. Comparison of individuals with and without specific IgA antibodies to Chlamydia pneumoniae: respiratory morbidity and the metabolic syndrome. Chest. 2002;122:1587–1593. doi: 10.1378/chest.122.5.1587. [DOI] [PubMed] [Google Scholar]
- Fava M. Weight gain and antidepressants. The Journal of clinical psychiatry. 2000;61(Suppl 11):37–41. [PubMed] [Google Scholar]
- Fava M, Judge R, Hoog SL, Nilsson ME, Koke SC. Fluoxetine versus sertraline and paroxetine in major depressive disorder: changes in weight with long-term treatment. J Clin Psychiatry. 2000;61:863–867. doi: 10.4088/jcp.v61n1109. [DOI] [PubMed] [Google Scholar]
- Feighner J, Hendrickson G, Miller L, Stern W. Double-blind comparison of doxepin versus bupropion in outpatients with a major depressive disorder. J Clin Psychopharmacol. 1986;6:27–32. [PubMed] [Google Scholar]
- Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- Fernandez-Real JM, Lopez-Bermejo A, Vendrell J, Ferri MJ, Recasens M, Ricart W. Burden of infection and insulin resistance in healthy middle-aged men. Diabetes Care. 2006;29:1058–1064. doi: 10.2337/diacare.2951058. [DOI] [PubMed] [Google Scholar]
- Fernandez-Real JM, Ferri MJ, Vendrell J, Ricart W. Burden of infection and fat mass in healthy middle-aged men. Obesity (Silver Spring) 2007;15:245–252. doi: 10.1038/oby.2007.541. [DOI] [PubMed] [Google Scholar]
- Fields LE, Burt VL, Cutler JA, Hughes J, Roccella EJ, Sorlie P. The burden of adult hypertension in the United States 1999 to 2000: a rising tide. Hypertension. 2004;44:398–404. doi: 10.1161/01.HYP.0000142248.54761.56. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Troiano RP. Changes in the distribution of body mass index of adults and children in the US population. Int J Obes Relat Metab Disord. 2000;24:807–818. doi: 10.1038/sj.ijo.0801232. [DOI] [PubMed] [Google Scholar]
- Flegal KM. The effects of changes in smoking prevalence on obesity prevalence in the United States. Am J Public Health. 2007;97:1510–1514. doi: 10.2105/AJPH.2005.084343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Kothare SV, Zihlif M, Suarez E, Adams R, Legido A, De Luca F. Association between inadequate sleep and insulin resistance in obese children. J Pediatr. 2007;150:364–369. doi: 10.1016/j.jpeds.2006.08.063. [DOI] [PubMed] [Google Scholar]
- Fogelholm M, Kronholm E, Kukkonen-Harjula K, Partonen T, Partinen M, Härmä M. Sleep-related disturbances and physical inactivity are independently associated with obesity in adults. Int J Obesity. 2007 doi: 10.1038/sj.ijo.0803663. Jun 19 Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Forshee RA, Storey ML, Allison DB, Glinsmann WH, Hein GL, Lineback DR, Miller SA, Nicklas TA, Weaver GA, White JS. A critical examination of the evidence relating high fructose corn syrup and weight gain. Crit Rev Food Sci Nutr. 2007;47:561–582. doi: 10.1080/10408390600846457. [DOI] [PubMed] [Google Scholar]
- Franke K, Harder T, Aerts L, Melchior K, Fahrenkrog S, Rodekamp E, Ziska T, Van Assche FA, Dudenhausen JW, Plagemann A. ‘Programming’ of orexigenic and anorexigenic hypothalamic neurons in offspring of treated and untreated diabetic mother rats. Brain Research. 2005;1031:276–283. doi: 10.1016/j.brainres.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Fregly MJ, Marshall NB, Mayer J. Effect of changes in ambient temperature on spontaneous activity, food intake and body weight of goldthioglucose-obese and nonobese mice. Am J Physiol. 1957;188:435–438. doi: 10.1152/ajplegacy.1957.188.3.435. [DOI] [PubMed] [Google Scholar]
- Frese KK, Lee SS, Thomas DL, Latorre IJ, Weiss RS, Glaunsinger BA, Javier RT. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene. 2003;22:710–721. doi: 10.1038/sj.onc.1206151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch RE. Body fat, menarche, fitness and fertility. Hum Reprod. 1987;2:521–533. doi: 10.1093/oxfordjournals.humrep.a136582. [DOI] [PubMed] [Google Scholar]
- Fujihara H, Serino R, Ueta Y, Sei H, Morita Y. Six-hour selective REM sleep deprivation increases the expression of the galanin gene in the hypothalamus of rats. Brain Res Mol Brain Res. 2003;119:152–159. doi: 10.1016/j.molbrainres.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Gale CR, Jiang B, Robinson SM, Godfrey KM, Law CM, Martyn CN. Maternal diet during pregnancy and carotid intima-media thickness in children. Arterioscler Thromb Vasc Biol. 2006;26:1877–1882. doi: 10.1161/01.ATV.0000228819.13039.b8. [DOI] [PubMed] [Google Scholar]
- Gale CR, Javaid MK, Robinson SM, Law CM, Godfrey KM, Cooper C. Maternal size in pregnancy and body composition in children. Journal of Clinical Endocrinology and Metabolism. 2007;92:3904–3911. doi: 10.1210/jc.2007-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangwisch J, Heymsfield S. Sleep deprivation as a risk factor for obesity: results based on the NHANES I. North American Association for the Study of Obesity (NAASO) 2004 Abstract no. 42-OR:A11. [Google Scholar]
- Garland EJ, Remick RA, Zis AP. Weight gain with antidepressants and lithium. J Clin Psychopharmacol. 1988;8:323–330. [PubMed] [Google Scholar]
- Garrow J. Energy Balance and Obesity in Man. Elsevier/North-Holland Biomedical Press; Amsterdam: 1978. [Google Scholar]
- Gillman MW, Rifas-Shiman SL, Camargo CA, Berkey CS, Frazier AL, Rockett HRH, Field AE, Colditz GA. Risk of overweight among adolescents who were breastfed as infants. Journal of the American Medical Association. 2001;285:2461–2467. doi: 10.1001/jama.285.19.2461. [DOI] [PubMed] [Google Scholar]
- Glass L, Silverman WA, Sinclair JC. Effect of the thermal environment on cold resistance and growth of small infants after the first week of life. Pediatrics. 1968;41:1033–1046. [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. Maternal constraint of fetal growth and its consequences. Semin Fetal Neonatal Med. 2004a;9:419–425. doi: 10.1016/j.siny.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004b;305:1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab. 2004c;15:183–187. doi: 10.1016/j.tem.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. The Fetal Matrix: Evolution, Development, and Disease. Cambridge University Press; Cambridge: 2005a. [Google Scholar]
- Gluckman PD, Hanson MA, Spencer HG. Predictive adaptive responses and human evolution. Trends Ecol Evol. 2005b;20:527–533. doi: 10.1016/j.tree.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Spencer HG, Bateson P. Environmental influences during development and their later consequences for health and disease: implications for the interpretation of empirical studies. Proc Royal Soc Lond B. 2005c;272:671–677. doi: 10.1098/rspb.2004.3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. Mismatch - why our world no longer fits our bodies. Oxford University Press; Oxford: 2006. [Google Scholar]
- Gluckman PD, Chong YS, Fukuoka H, Beedle AS, Hanson MA. Low birthweight and subsequent obesity in Japan. Lancet. 2007a;369:1081–1082. doi: 10.1016/S0140-6736(07)60524-8. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol. 2007b;19:1–19. doi: 10.1002/ajhb.20590. [DOI] [PubMed] [Google Scholar]
- Gluckman PD, Lillycrop KA, Vickers MH, Pleasants AB, Phillips ES, Beedle AS, Burdge GC, Hanson MA. Metabolic plasticity during mammalian development is directionally dependent on early nutritional status. Proc Natl Acad Sci U S A. 2007c;104:12796–12800. doi: 10.1073/pnas.0705667104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey K, Robinson S, Barker DJ, Osmond C, Cox V. Maternal nutrition in early and late pregnancy in relation to placental and fetal growth. British Medical Journal. 1996a;312:410–414. doi: 10.1136/bmj.312.7028.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey K. Developmental Origins of Health and Disease. Cambridge University Press; Cambridge: 2006. The ‘developmental origins’ hypothesis: epidemiology; pp. 6–32. [Google Scholar]
- Godfrey KM, Robinson S, Hales CN, Barker DJ, Osmond C, Taylor KP. Nutrition in pregnancy and the concentrations of proinsulin, 32–33 split proinsulin, insulin, and C-peptide in cord plasma. Diabetic Medicine. 1996b;13:868–873. doi: 10.1002/(SICI)1096-9136(199610)13:10<868::AID-DIA261>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Goldstone AP. Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends Endocrinol Metab. 2004;15:12–20. doi: 10.1016/j.tem.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Gomez-Merino D, Chennaoui M, Drogou C, Bonneau D, Guezennec CY. Decrease in serum leptin after prolonged physical activity in men. Med Sci Sports Exerc. 2002;34:1594–1599. doi: 10.1097/00005768-200210000-00010. [DOI] [PubMed] [Google Scholar]
- Gosztonyi G, Ludwig H. Borna disease--neuropathology and pathogenesis. Curr Top Microbiol Immunol. 1995;190:39–73. [PubMed] [Google Scholar]
- Gottlieb DJ, Punjabi NM, Newman AB, Resnick HE, Redline S, Baldwin CM, Nieto FJ. Association of sleep time with diabetes mellitus and impaired glucose tolerance. Arch Intern Med. 2005;165:863–867. doi: 10.1001/archinte.165.8.863. [DOI] [PubMed] [Google Scholar]
- Grossman M, Sindelar JL, Mullahy J, Anderson R. Alcohol and cigarette taxes. Journal of Economic Perspectives. 1993;7(4):211–222. [Google Scholar]
- Gu Q, Paulose-Ram R, Dillon C, Burt V. Antihypertensive medication use among US adults with hypertension. Circulation. 2006;113:213–221. doi: 10.1161/CIRCULATIONAHA.105.542290. [DOI] [PubMed] [Google Scholar]
- Guvenius DM, Aronsson A, Ekman-Ordeberg G, Bergman A, Noren K. Human prenatal and postnatal exposure to polybrominated diphenyl ethers, polychlorinated biphenyls, polychlorobiphenylols, and pentachlorophenol. Environ Health Perspect. 2003;111(9):1235–1241. doi: 10.1289/ehp.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan MM, Shuman ES, Oswald KD, Corcoran KJ, Profitt JH, Blackburn K, Schwiebert MW, Chandler PC, Birbaum C. Incidence of chaotic eating behaviors in binge-eating disorder: Contributing factors. Behavioral Medicine. 2002;28:99–105. doi: 10.1080/08964280209596048. [DOI] [PubMed] [Google Scholar]
- Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- Hale L, Do DP. Racial differences in self-reports of sleep duration in a population-based study. Sleep. 2007a;30:1096–1103. doi: 10.1093/sleep/30.9.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale L, Do DP. Racial differences in self-reports of sleep duration in a population-based study. Sleep. 2007b;30:1096–1103. doi: 10.1093/sleep/30.9.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. British Medical Journal. 1991;303:1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35:595–601. doi: 10.1007/BF00400248. [DOI] [PubMed] [Google Scholar]
- Hales CN, Ozanne SE. The dangerous road of catch-up growth. Journal of Physiology. 2003;547.1:5–10. doi: 10.1113/jphysiol.2002.024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond EC. Some preliminary findings on physical complaints from a prospective study of 1,064,004 men and women. Am J Public Health Nations Health. 1964;54:11–23. doi: 10.2105/ajph.54.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen JB, Kristiansen K. Regulatory circuits controlling white versus brown adipocyte differentiation. Biochem J. 2006;398:153–168. doi: 10.1042/BJ20060402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder T, Plagemann A, Rohde W, Dörner G. Syndrome X-like alterations in adult female rats due to neonatal insulin treatment. Metabolism. 1998;47:855–862. doi: 10.1016/s0026-0495(98)90126-3. [DOI] [PubMed] [Google Scholar]
- Harder T, Bergmann R, Kallischnigg G, Plagemann A. Duration of breastfeeding and risk of overweight: a meta-analysis. American Journal of Epidemiology. 2005;162:397–403. doi: 10.1093/aje/kwi222. [DOI] [PubMed] [Google Scholar]
- Harding JE, Johnston BM. Nutrition and fetal growth. Reproduction, Fertility, & Development. 1995;7:539–547. doi: 10.1071/rd9950539. [DOI] [PubMed] [Google Scholar]
- Hardy R, Kuh D, Langenberg C, Wadsworth ME. Birthweight, childhood social class, and change in adult blood pressure in the 1946 British birth cohort. Lancet. 2003;362:1178–1183. doi: 10.1016/S0140-6736(03)14539-4. [DOI] [PubMed] [Google Scholar]
- Harrington TAM, Thomas EL, Frost G, Modi N, Bell JD. Distribution of adipose tissue in the newborn. Pediatric Research. 2004;55:437–441. doi: 10.1203/01.PDR.0000111202.29433.2D. [DOI] [PubMed] [Google Scholar]
- Hasler G, Buysse DJ, Klaghofer R, Gamma A, Ajdacic V, Eich D, Rossler W, Angst J. The association between short sleep duration and obesity in young adults: a 13-year prospective study. Sleep. 2004;27:661–666. doi: 10.1093/sleep/27.4.661. [DOI] [PubMed] [Google Scholar]
- Hebebrand J, Wulftange H, Goerg T, Ziegler A, Hinney A, Barth N, Mayer H, Remschmidt H. Epidemic obesity: Are genetic factors involved via increased rates of assortative mating? Int J Obes Relat Metab Disord. 2000;24:345–353. doi: 10.1038/sj.ijo.0801135. [DOI] [PubMed] [Google Scholar]
- Heck KE, Schoendorf KC, Ventura SJ, Kiely JL. Delayed childbearing by education level in the United States, 1969–1994. Matern Child Health J. 1997;1:81–88. doi: 10.1023/a:1026218322723. [DOI] [PubMed] [Google Scholar]
- Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-alpha knockout mice. Proc Natl Acad Sci U S A. 2000;97:12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen LA, Henderson RM. Changes in the distribution of body mass index of white US men, 1890–2000. Ann Hum Biol. 2004;31:174–181. doi: 10.1080/03014460410001663434. [DOI] [PubMed] [Google Scholar]
- Helmink SK, Shanks RD, Leighton EA. Investigation of breeding strategies to increase the probability that German shephard dog and Labrador retreiver dog guides would attain optimal size. J Anim Sci. 2003;81:2950–2958. doi: 10.2527/2003.81122950x. [DOI] [PubMed] [Google Scholar]
- Henderson DC, Cagliero E, Gray C, Nasrallah RA, Hayden DL, Schoenfeld DA, Goff DC. Clozapine, diabetes mellitus, weight gain, and lipid abnormalities: A five-year naturalistic study. The American journal of psychiatry. 2000;157:975–981. doi: 10.1176/appi.ajp.157.6.975. [DOI] [PubMed] [Google Scholar]
- Henry CJ. Body mass index and the limits of human survival. European journal of clinical nutrition. 1990;44:329–335. [PubMed] [Google Scholar]
- Henry CJK. Variability in adult body size: Uses in defining the limits of human survival. In: Ulijaszek SJ, Masci-Taylor CGN, editors. Anthropometry: The individual and the population. Cambridge University Press; New York: 1994. pp. 117–129. [Google Scholar]
- Herden C, Herzog S, Richt JA, Nesseler A, Christ M, Failing K, Frese K. Distribution of Borna disease virus in the brain of rats infected with an obesity-inducing virus strain. Brain Pathol. 2000;10:39–48. doi: 10.1111/j.1750-3639.2000.tb00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman-Giddens ME, Slora EJ, Wasserman RC, Bourdony CJ, Bhapkar MV, Koch GG, Hasemeier CM. Secondary sexual characteristics and menses in young girls seen in office practice: a study from the Pediatric Research in Office Settings network. Pediatrics. 1997;99:505–512. doi: 10.1542/peds.99.4.505. [DOI] [PubMed] [Google Scholar]
- Herman CP. Effects of heat on appetite. In: Marriot BM, editor. Nutritional Needs in Hot Environments: Applications for MIlitary Personnel in Field Operations. National Academy Press; Washington D.C: 1993. pp. 187–214. [PubMed] [Google Scholar]
- Heslop P, Smith GD, Metcalfe C, Macleod J, Hart C. Sleep duration and mortality: The effect of short or long sleep duration on cardiovascular and all-cause mortality in working men and women. Sleep Med. 2002;3:305–314. doi: 10.1016/s1389-9457(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Hicks RA, Fernandez C, Pellegrini RJ. Self-reported sleep durations of college students: normative data for 1978–79, 1988–89, and 2000–01. Percept Mot Skills. 2001;93:139–140. doi: 10.2466/pms.2001.93.1.139. [DOI] [PubMed] [Google Scholar]
- Hillier TA, Pedula KL, Schmidt MM, Mullen JA, Charles MA, Pettitt DJ. Childhood obesity and metabolic imprinting: The ongoing effects of maternal hyperglycemia. Diabetes Care. 2007;30:2287–2292. doi: 10.2337/dc06-2361. [DOI] [PubMed] [Google Scholar]
- Hitchins MP, Wong JJ, Suthers G, Suter CM, Martin DI, Hawkins NJ, Ward RL. Inheritance of a cancer-associated MLH1 germ-line epimutation. N Engl J Med. 2007;356:697–705. doi: 10.1056/NEJMoa064522. [DOI] [PubMed] [Google Scholar]
- Holt VL, Scholes D, Wicklund KG, Cushing-Haugen KL, Daling JR. Body mass index, weight, and oral contraceptive failure risk. Obstet Gynecol. 2005;105:46–52. doi: 10.1097/01.AOG.0000149155.11912.52. [DOI] [PubMed] [Google Scholar]
- Ibáñez L, Ong K, Dunger DB, de Zegher F. Early development of adiposity and insulin resistance after catch-up weight gain in small-for-gestational-age children. Journal of Clinical Endocrinology and Metabolism. 2006;91:2153–2158. doi: 10.1210/jc.2005-2778. [DOI] [PubMed] [Google Scholar]
- Ibáñez L, Suarez L, Lopez-Bermejo A, Diaz M, Valls C, de Zegher F. Early development of visceral fat excess following spontaneous catch-up growth in children with low birthweight. Journal of Clinical Endocrinology and Metabolism. 2008 doi: 10.1210/jc.2007-1618. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Iglowstein I, Jenni OG, Molinari L, Largo RH. Sleep duration from infancy to adolescence: reference values and generational trends. Pediatrics. 2003;111:302–307. doi: 10.1542/peds.111.2.302. [DOI] [PubMed] [Google Scholar]
- Ikenasio-Thorpe BA, Breier BH, Vickers MH, Fraser M. Prenatal influences on susceptibility to diet-induced obesity are mediated by altered neuroendocrine gene expression. Journal of Endocrinology. 2007;193:31–37. doi: 10.1677/joe.1.07017. [DOI] [PubMed] [Google Scholar]
- Irwin M, Thompson J, Miller C, Gillin JC, Ziegler M. Effects of sleep and sleep deprivation on catecholamine and interleukin-2 levels in humans: clinical implications. J Clin Endocrinol Metab. 1999;84:1979–1985. doi: 10.1210/jcem.84.6.5788. [DOI] [PubMed] [Google Scholar]
- Ishii Y, Blundell JE, Halford JC, Upton N, Porter R, Johns A, Rodgers RJ. Satiety enhancement by selective orexin-1 receptor antagonist SB-334867: influence of test context and profile comparison with CCK-8S. Behav Brain Res. 2005;160:11–24. doi: 10.1016/j.bbr.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Jacobson P, Torgerson JS, Sjostrom L, Bouchard C. Spouse resemblance in body mass index: effects on adult obesity prevalence in the offspring generation. Am J Epidemiol. 2007;165:101–108. doi: 10.1093/aje/kwj342. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Javaid MK, Godfrey KM, Taylor P, Shore SR, Breier B, Arden NK, Cooper C. Umbilical venous IGF-1 concentration, neonatal bone mass, and body composition. J Bone Miner Res. 2004;19:56–63. doi: 10.1359/JBMR.0301211. [DOI] [PubMed] [Google Scholar]
- Jeanrenaud B, Rohner-Jeanrenaud F. Effects of neuropeptides and leptin on nutrient partitioning: dysregulations in obesity. Annu Rev Med. 2001;52:339–351. doi: 10.1146/annurev.med.52.1.339. Review. [DOI] [PubMed] [Google Scholar]
- Jensen TK, Scheike T, Keiding N, Schaumburg I, Grandjean P. Fecundability in relation to body mass and menstrual cycle patterns. Epidemiology. 1999;10:422–428. doi: 10.1097/00001648-199907000-00011. [DOI] [PubMed] [Google Scholar]
- Jensen TK, Andersson AM, Jorgensen N, Andersen AG, Carlsen E, Petersen JH, Skakkebaek NE. Body mass index in relation to semen quality and reproductive hormones among 1,558 Danish men. Fertil Steril. 2004;82:863–870. doi: 10.1016/j.fertnstert.2004.03.056. [DOI] [PubMed] [Google Scholar]
- Jimenez-Chillaron JC, Patti ME. To catch up or not to catch up: Is this the question? Lessons from animal models. Curr Opin Endocrinol Diabetes Obes. 2007;14:23–29. doi: 10.1097/MED.0b013e328013da8e. [DOI] [PubMed] [Google Scholar]
- Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RE, Kark RM. Environment and food intake in man. Science. 1947;105:378–379. doi: 10.1126/science.105.2728.378. [DOI] [PubMed] [Google Scholar]
- Jokela M, Kivimaki M, Elovainio M, Viikari J, Raitakari OT, Keltikangas-Jarvinen L. Body mass index in adolescence and number of children in adulthood. Epidemiology. 2007;18:599–606. doi: 10.1097/EDE.0b013e3181257158. [DOI] [PubMed] [Google Scholar]
- Kanayama T, Kobayashi N, Mamiya S, Nakanishi T, Nishikawa J. Organotin compounds promote adipocyte differentiation as agonists of the peroxisome proliferator-activated receptor gamma/retinoid X receptor pathway. Mol Pharmacol. 2005;67:766–774. doi: 10.1124/mol.104.008409. [DOI] [PubMed] [Google Scholar]
- Katzmarzyk PT, Pérusse L, Rao DC, Bouchard C. Spousal resemblance and risk of 7-year increases in obesity and central adiposity in the Canadian population. Obes Res. 1999a;7:545–551. doi: 10.1002/j.1550-8528.1999.tb00712.x. [DOI] [PubMed] [Google Scholar]
- Katzmarzyk PT, Perusse L, Rao DC, Bouchard C. Familial risk of obesity and central adipose tissue distribution in the general Canadian population. Am J Epidemiol. 1999b;149:933–942. doi: 10.1093/oxfordjournals.aje.a009737. [DOI] [PubMed] [Google Scholar]
- Katzmarzyk PT, Hebebrand J, Bouchard C. Spousal resemblance in the Canadian population: implications for the obesity epidemic. Int J Obes Relat Metab Disord. 2002;26:241–246. doi: 10.1038/sj.ijo.0801870. [DOI] [PubMed] [Google Scholar]
- Katzmarzyk PT, Bouchard C. Genetic influences on human body composition. In: Heymsfield S, Lohman T, Wang Z, Going S, editors. Human Body Composition. 2. Human Kinetics; Champaign, IL: 2005. pp. 243–257. [Google Scholar]
- Keith SW, Redden DT, Katzmarzyk PT, Boggiano MM, Hanlon EC, Benca RM, Ruden D, Pietrobelli A, Barger JL, Fontaine KR, Wang C, Aronne LJ, Wright SM, Baskin M, Dhurandhar NV, Lijoi MC, Grilo CM, DeLuca M, Westfall AO, Allison DB. Putative contributors to the secular increase in obesity: exploring the roads less traveled. Int J Obes (Lond) 2006;30:1585–1594. doi: 10.1038/sj.ijo.0803326. [DOI] [PubMed] [Google Scholar]
- Kensara OA, Wootton SA, Phillips DI, Patel M, Jackson AA, Elia M The Hertfordshire Study, G. Fetal programming of body composition: relation between birth weight and body composition measured with dual-energy X-ray absorptiometry and anthropometric methods in older Englishmen. American Journal of Clinical Nutrition. 2005;82:980–987. doi: 10.1093/ajcn/82.5.980. [DOI] [PubMed] [Google Scholar]
- Kim SA, Stein AD, Martorell R. Country development and the association between parity and overweight. Int J Obes (Lond) 2007;31:805–812. doi: 10.1038/sj.ijo.0803478. [DOI] [PubMed] [Google Scholar]
- Kim YS, Carp RI, Callahan SM, Wisniewski HM. Scrapie-induced obesity in mice. J Infect Dis. 1987;156:402–405. doi: 10.1093/infdis/156.2.402. [DOI] [PubMed] [Google Scholar]
- Kleigman RM, Gross T. Prenatal problems of the obese mother and her infant. Obstetrics & Gynecology. 1985;66:299–306. [PubMed] [Google Scholar]
- Knuiman MW, Divitini ML, Bartholomew HC, Welborn TA. Spouse correlations in cardiovascular risk factors and the effect of marriage duration. Am J Epidemiol. 1996;143:48–53. doi: 10.1093/oxfordjournals.aje.a008656. [DOI] [PubMed] [Google Scholar]
- Knutson KL, Spiegel K, Penev P, Van Cauter E. The metabolic consequences of sleep deprivation. Sleep Med Rev. 2007;11:163–178. doi: 10.1016/j.smrv.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko GT, Chan JC, Chan AW, Wong PT, Hui SS, Tong SD, Ng SM, Chow F, Chan CL. Association between sleeping hours, working hours and obesity in Hong Kong Chinese: the ‘better health for better Hong Kong’ health promotion campaign. Int J Obes. 2007;31:254–260. doi: 10.1038/sj.ijo.0803389. [DOI] [PubMed] [Google Scholar]
- Kok SW, Meinders AE, Overeem S, Lammers GJ, Roelfsema F, Frolich M, Pijl H. Reduction of plasma leptin levels and loss of its circadian rhythmicity in hypocretin (orexin)-deficient narcoleptic humans. J Clin Endocrinol Metab. 2002;87:805–809. doi: 10.1210/jcem.87.2.8246. [DOI] [PubMed] [Google Scholar]
- Konkle AT, Sreter KB, Baker SL, Bielajew C. Chronic paroxetine infusion influences macronutrient selection in male Sprague-Dawley rats. Pharmacol Biochem Behav. 2003;74:883–890. doi: 10.1016/s0091-3057(03)00021-2. [DOI] [PubMed] [Google Scholar]
- Kotagal S, Krahn LE, Slocumb N. A putative link between childhood narcolepsy and obesity. Sleep Med Rev. 2004;5:147–150. doi: 10.1016/j.sleep.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau M, Marceau P. Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics. 2006a;118:e1644–e1649. doi: 10.1542/peds.2006-1379. [DOI] [PubMed] [Google Scholar]
- Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, Marceau P. Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics. 2006b;118:e1644–1649. doi: 10.1542/peds.2006-1379. [DOI] [PubMed] [Google Scholar]
- Kripke DF, Garfinkel L, Wingard DL, Klauber MR, Marler MR. Mortality associated with sleep duration and insomnia. Arch Gen Psychiatry. 2002;59:131–136. doi: 10.1001/archpsyc.59.2.131. [DOI] [PubMed] [Google Scholar]
- Kuczmarski RJ, Carroll MD, Flegal KM, Troiano RP. Varying body mass index cutoff points to describe overweight prevalence among U.S. adults: NHANES III (1988 to 1994) Obes Res. 1997;5:542–548. doi: 10.1002/j.1550-8528.1997.tb00575.x. [DOI] [PubMed] [Google Scholar]
- Kumpusalo EA, Takala JK. Do beta-blockers put on weight? Hypertension. 2001;38:E4–5. doi: 10.1161/01.hyp.38.1.e4. [DOI] [PubMed] [Google Scholar]
- Kurzthaler I, Fleischhacker WW. The clinical implications of weight gain in schizophrenia. J Clin Psychiatry. 2001;62(Suppl 7):32–37. [PubMed] [Google Scholar]
- Kuzawa CW. Adipose tissue in human infancy and childhood: an evolutionary perspective. Yearbook of Physical Anthropology. 1998;41:177–209. doi: 10.1002/(sici)1096-8644(1998)107:27+<177::aid-ajpa7>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Kyrkouli SE, Stanley BG, Leibowitz SF. Galanin: stimulation of feeding induced by medial hypothalamic injection of this novel peptide. Eur J Pharmacol. 1986;122:159–160. doi: 10.1016/0014-2999(86)90175-5. [DOI] [PubMed] [Google Scholar]
- Lake JK, Power C, Cole TJ. Women’s reproductive health: the role of body mass index in early and adult life. Int J Obes Relat Metab Disord. 1997;21:432–438. doi: 10.1038/sj.ijo.0800424. [DOI] [PubMed] [Google Scholar]
- Lawlor DA, Najman JM, Sterne J, Williams GM, Ebrahim S, Davey Smith G. Associations of parental, birth, and early life characteristics with systolic blood pressure at 5 years of age: findings from the Mater-University study of pregnancy and its outcomes. Circulation. 2004;110:2417–2423. doi: 10.1161/01.CIR.0000145165.80130.B5. [DOI] [PubMed] [Google Scholar]
- Lawrence JM, Contreras R, Chen W, Sacks DA. Trends in the prevalence of pre-existing diabetes and gestational diabetes mellitus among a racially/ethnically diverse population of pregnant women, 1999–2005. Diabetes Care. 2008 doi: 10.2337/dc2307-2345. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Le Stunff C, Fallin D, Bougneres P. Paternal transmission of the very common class I INS VNTR alleles predisposes to childhood obesity. Nat Genet. 2001;29:96–99. doi: 10.1038/ng707. [DOI] [PubMed] [Google Scholar]
- Leslie WS, Hankey CR, Lean ME. Weight gain as an adverse effect of some commonly prescribed drugs: a systematic review. QJM. 2007;100:395–404. doi: 10.1093/qjmed/hcm044. [DOI] [PubMed] [Google Scholar]
- Levin BE. The obesity epidemic: metabolic imprinting on genetically susceptible neural circuits. Obes Res. 2000;8:342–347. doi: 10.1038/oby.2000.41. [DOI] [PubMed] [Google Scholar]
- Levine JA, Jensen MD, Eberhardt NL, O’Brien T. Adipocyte macrophage colony-stimulating factor is a mediator of adipose tissue growth. J Clin Invest. 1998;101:1557–1564. doi: 10.1172/JCI2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J Nutr. 2005;135:1382–1386. doi: 10.1093/jn/135.6.1382. [DOI] [PubMed] [Google Scholar]
- Lipowicz A. Effect of husbands’ education on fatness of wives. Am J Hum Biol. 2003;15:1–7. doi: 10.1002/ajhb.10119. [DOI] [PubMed] [Google Scholar]
- Locard E, Mamelle N, Billette A, Miginiac M, Munoz F, Rey S. Risk factors of obesity in a five year old population. Parental versus environmental factors. Int J Obes Relat Metab Disord. 1992;16:721–729. [PubMed] [Google Scholar]
- Lowell BB, VSS, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, Kozak LP, Flier JS. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993;366:740–742. doi: 10.1038/366740a0. [DOI] [PubMed] [Google Scholar]
- Lu X, Barr AM, Kinney JW, Sanna P, Conti B, Behrens MM, Bartfai T. A role for galanin in antidepressant actions with a focus on the dorsal raphe nucleus. Proc Natl Acad Sci U S A. 2005;102:874–879. doi: 10.1073/pnas.0408891102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MJ, Faust IM, Hemmes RB, Buskirk DR, Hirsch J, Zabriskie JB. A virally induced obesity syndrome in mice. Science. 1982;216:82–85. doi: 10.1126/science.7038878. [DOI] [PubMed] [Google Scholar]
- Maachi M, Pieroni L, Bruckert E, Jardel C, Fellahi S, Hainque B, Capeau J, Bastard JP. Systemic low-grade inflammation is related to both circulating and adipose tissue TNFalpha, leptin and IL-6 levels in obese women. Int J Obes Relat Metab Disord. 2004;28:993–997. doi: 10.1038/sj.ijo.0802718. [DOI] [PubMed] [Google Scholar]
- Macari M, Zuim SM, Secato ER, Guerreiro JR. Effects of ambient temperature and thyroid hormones on food intake by pigs. Physiol Behav. 1986;36:1035–1039. doi: 10.1016/0031-9384(86)90476-2. [DOI] [PubMed] [Google Scholar]
- Mader TL. Environmental stress in confined beef cattle. J Anim Sci. 2003;81:E110–E119. [Google Scholar]
- Madrigal L, Relethford JH, Crawford MH. Heritability and anthropometric influences on human fertility. Am J Hum Biol. 2003;15:16–22. doi: 10.1002/ajhb.10109. [DOI] [PubMed] [Google Scholar]
- Maes HHM, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27:325–351. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- Mangel M, Munch SB. A life-history perspective on short- and long-term consequences of compensatory growth. Am Nat. 2005;166:E155–E176. doi: 10.1086/444439. [DOI] [PubMed] [Google Scholar]
- Marti A, Marcos A, Martinez JA. Obesity and immune function relationships. Obes Rev. 2001;2:131–140. doi: 10.1046/j.1467-789x.2001.00025.x. [DOI] [PubMed] [Google Scholar]
- Martin JA, Hamilton BE, Sutton PD, Ventura SJ, Menacker F, Kirmeyer S. Births: final data for 2004. Natl Vital Stat Rep. 2006;55:1–101. [PubMed] [Google Scholar]
- Martorell R, Stein AD, Schroeder DG. Early nutrition and later adiposity. Journal of Nutrition. 2001;131:874S–880S. doi: 10.1093/jn/131.3.874S. [DOI] [PubMed] [Google Scholar]
- Masand PS. Weight gain associated with psychotropic drugs. Expert opinion on pharmacotherapy. 2000;1:377–389. doi: 10.1517/14656566.1.3.377. [DOI] [PubMed] [Google Scholar]
- Mathews TJ, Hamilton BE. Mean age of mother, 1970–2000. Natl Vital Stat Rep. 2002;51:1–13. [PubMed] [Google Scholar]
- McCance DR, Pettitt DJ, Hanson RL, Jacobsson LT, Bennett PH. Birth weight and non-insulin dependent diabetes: thrifty genotype, thrifty phenotype, or surviving small baby genotype? British Medical Journal. 1994;308:942–945. doi: 10.1136/bmj.308.6934.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy SL, Frye MA, Suppes T, Dhavale D, Keck PE, Jr, Leverich GS, Altshuler L, Denicoff KD, Nolen WA, Kupka R, Grunze H, Walden J, Post RM. Correlates of overweight and obesity in 644 patients with bipolar disorder. J Clin Psychiatry. 2002;63:207–213. doi: 10.4088/jcp.v63n0306. [DOI] [PubMed] [Google Scholar]
- McGhie A, Russell SM. The Subjective Assessment of Normal Sleep Patterns. J Ment Sci. 1962;108:642–654. [Google Scholar]
- McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiological Reviews. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- Mead MN. Origins of obesity. Environ Health Perspect. 2004;112:A344. doi: 10.1289/ehp.112-a344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzner I, Scott V, Dorsch K, Fischer P, Wabitsch M, Bruderlein S, Hasel C, Moller P. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J Biol Chem. 2002;277:45420–45427. doi: 10.1074/jbc.M208511200. [DOI] [PubMed] [Google Scholar]
- Mendelson, S. D., un;158(6):963–4. Related Articles, L., and . (2001). Treatment of anorexia nervosa with tramadol. Am J Psychiatry 158
- Messerli FH, Bell DS, Fonseca V, Katholi RE, McGill JB, Phillips RA, Raskin P, Wright JT, Jr, Bangalore S, Holdbrook FK, Lukas MA, Anderson KM, Bakris GL. Body weight changes with beta-blocker use: results from GEMINI. Am J Med. 2007;120:610–615. doi: 10.1016/j.amjmed.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Middleton K, HE, Xu J. National Hospital Ambulatory Medical Care Survey: 2005 outpatient department summary. Adv Data. 2007;39:1–37. [PubMed] [Google Scholar]
- Mieda M, Williams SC, Sinton CM, Richardson JA, Sakurai T, Yanagisawa M. Orexin neurons function in an efferent pathway of a food-entrainable circadian oscillator in eliciting food-anticipatory activity and wakefulness. J Neurosci. 2004;24:10493–10501. doi: 10.1523/JNEUROSCI.3171-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikamo E, Harada S, Nishikawa J, Nishihara T. Endocrine disruptors induce cytochrome P450 by affecting transcriptional regulation via pregnane X receptor. Toxicol Appl Pharmacol. 2003;193:66–72. doi: 10.1016/j.taap.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Miles R, Panton LB, Jang M, Haymes EM. Residential context, walking and obesity: two African-American neighborhoods compared. Health Place. 2008;14:275–286. doi: 10.1016/j.healthplace.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Minet-Ringuet J, Even PC, Lacroix M, Tome D, de Beaurepaire R. A model for antipsychotic-induced obesity in the male rat. Psychopharmacology (Berl) 2006;187:447–454. doi: 10.1007/s00213-006-0433-0. [DOI] [PubMed] [Google Scholar]
- Minet-Ringuet J, Even PC, Valet P, Carpéné C, Visentin V, Prévot D, Daviaud D, Quignard-Boulange A, Tomé D, de Beaurepaire R. Alterations of lipid metabolism and gene expression in rat adipocytes during chronic olanzapine treatment. Mol Psychiatry. 2007;12(6):562–571. doi: 10.1038/sj.mp.4001948. [DOI] [PubMed] [Google Scholar]
- Modi N, Thomas EL, Harrington TAM, Uthaya S, Doré CJ, Bell JD. Determinants of adiposity during preweaning postnatal growth in appropriately grown and growth-retricted term infants. Pediatric Research. 2006;60:345–348. doi: 10.1203/01.pdr.0000232732.93000.52. [DOI] [PubMed] [Google Scholar]
- Monteiro CA, Moura EC, Conde WL, Popkin BM. Socioeconomic status and obesity in adult populations of developing countries: a review. Bull World Health Organ. 2004;82:940–946. [PMC free article] [PubMed] [Google Scholar]
- Monteiro PO, Victora CG. Rapid growth in infancy and childhood and obesity in later life -- a systematic review. Obes Rev. 2005;6:143–154. doi: 10.1111/j.1467-789X.2005.00183.x. [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Reimitz PE, Zivkov M. Mirtazapine versus amitriptyline in the long-term treatment of depression: a double-blind placebo-controlled study. Int Clin Psychopharmacol. 1998;13:63–73. doi: 10.1097/00004850-199803000-00002. [DOI] [PubMed] [Google Scholar]
- Moreno CR, Louzada FM, Teixeira LR, Borges F, Lorenzi-Filho G. Short sleep is associated with obesity among truck drivers. Chronobiol Int. 2006;23:1295–1303. doi: 10.1080/07420520601089521. [DOI] [PubMed] [Google Scholar]
- Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- Muhlhausler BS, Adam-Dagger PA, Findlay CL, Duffield JA, McMillen IC. Increased maternal nutrition alters development of the appetite regulating network in the brain. FASEB Journal. 2006;20:1257–1259. doi: 10.1096/fj.05-5241fje. [DOI] [PubMed] [Google Scholar]
- Muhlhausler BS. Programming of the appetite-regulating neural network: A link between maternal overnutrition and the programming of obesity? Journal of Neuroendocrinology. 2007;19:67–72. doi: 10.1111/j.1365-2826.2006.01505.x. [DOI] [PubMed] [Google Scholar]
- Muhlhausler BS, Duffield JA, McMillen IC. Increased maternal nutrition stimulates peroxisome proliferator activated receptor-?, adiponectin, and leptin messenger ribonucleic acid expression in adipose tissue before birth. Endocrinology. 2007;148:878–885. doi: 10.1210/en.2006-1115. [DOI] [PubMed] [Google Scholar]
- Muller J, Moller DS, Kjaer M, Nyvad O, Larsen NA, Pedersen EB. Chlamydia pneumoniae DNA in peripheral blood mononuclear cells in healthy control subjects and patients with diabetes mellitus, acute coronary syndrome, stroke, and arterial hypertension. Scand J Infect Dis. 2003;35:704–712. doi: 10.1080/00365540310016538. [DOI] [PubMed] [Google Scholar]
- Mullington JM, Chan JL, Van Dongen HP, Szuba MP, Samaras J, Price NJ, Meier-Ewert HK, Dinges DF, Mantzoros CS. Sleep loss reduces diurnal rhythm amplitude of leptin in healthy men. J Neuroendocrinol. 2003;15:851–854. doi: 10.1046/j.1365-2826.2003.01069.x. [DOI] [PubMed] [Google Scholar]
- Nabukera S, Wingate MS, Alexander GR, Salihu HM. First-time births among women 30 years and older in the United States: patterns and risk of adverse outcomes. J Reprod Med. 2006;51:676–682. [PubMed] [Google Scholar]
- Nathanielsz PW, Poston L, Taylor PD. In Utero exposure to maternal obesity and diabetes: animal models that identify and characterize implications for future health. Clinics in Perinatology. 2007;34:515–526. doi: 10.1016/j.clp.2007.09.005. [DOI] [PubMed] [Google Scholar]
- National Center for Health Statistics. Mean age of mother by live-birth order, according to race and Hispanic origin of mother: United States, 1968–2003. Center for Disease Control; 2003. http://www.cdc.gov/nchs/datawh/statab/unpubd/natality/natab2003.htm. [Google Scholar]
- Nevels M, Tauber B, Spruss T, Wolf H, Dobner T. “Hit-and-run” transformation by adenovirus oncogenes. J Virol. 2001;75:3089–3094. doi: 10.1128/JVI.75.7.3089-3094.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold RR, Jefferson WN, Padilla-Banks E, Haseman J. Developmental exposure to diethylstilbestrol (DES) alters uterine response to estrogens in prepubescent mice: low versus high dose effects. Reprod Toxicol. 2004;18:399–406. doi: 10.1016/j.reprotox.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Newell-Price J, King P, Clark AJ. The CpG island promoter of the human proopiomelanocortin gene is methylated in nonexpressing normal tissue and tumors and represses expression. Mol Endocrinol. 2001;15:338–348. doi: 10.1210/mend.15.2.0599. [DOI] [PubMed] [Google Scholar]
- NIDDK. Chronic Sleep Deprivation as a Risk Factor for Metabolic Syndrome and Obesity. National Institutes of Health Clinical Center (CC); 2007. Identifier # NCT00261898. [Google Scholar]
- Nilsson R. Endocrine modulators in the food chain and environment. Toxicol Pathol. 2000;28:420–431. doi: 10.1177/019262330002800311. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355:39–40. doi: 10.1016/S0140-6736(99)05582-8. [DOI] [PubMed] [Google Scholar]
- Noren K, Meironyte D. Certain organochlorine and organobromine contaminants in Swedish human milk in perspective of past 20–30 years. Chemosphere. 2000;40:1111–1123. doi: 10.1016/s0045-6535(99)00360-4. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Fryar CD, Carroll MD, Flegal KM. Mean body weight, height, and body mass index, United States 1960–2002. Adv Data. 2004:1–17. [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007;132:2087–2102. doi: 10.1053/j.gastro.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Flegal KM. High body mass index for age among US children and adolescents, 2003–2006. JAMA. 2008;299:2401–2405. doi: 10.1001/jama.299.20.2401. [DOI] [PubMed] [Google Scholar]
- Ohayon MM. Interactions between sleep normative data and sociocultural characteristics in the elderly. J Psychosom Res. 2004a;56:479–486. doi: 10.1016/j.psychores.2004.04.365. [DOI] [PubMed] [Google Scholar]
- Ohayon MM. Interactions between sleep normative data and sociocultural characteristics in the elderly. J Psychosom Res. 2004b;56:479–486. doi: 10.1016/j.psychores.2004.04.365. [DOI] [PubMed] [Google Scholar]
- Oken E, Taveras EM, Kleinman KP, Rich-Edwards JW, Gillman MW. Gestational weight gain and child adiposity at age 3 years. American Journal of Obstetrics & Gynecology. 2007:196. doi: 10.1016/j.ajog.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olfson M, Blanco C, Liu L, Moreno C, Laje G. National trends in the outpatient treatment of children and adolescents with antipsychotic drugs. Arch Gen Psychiatry. 2006;63(6):679–685. doi: 10.1001/archpsyc.63.6.679. [DOI] [PubMed] [Google Scholar]
- Ong K, Loos R. Rapid infancy weight gain and subsequent obesity: Systematic reviews and hopeful suggestions. Acta Paediatrica, International Journal of Paediatrics. 2006;95:904–908. doi: 10.1080/08035250600719754. [DOI] [PubMed] [Google Scholar]
- Ong KK. Catch-up growth in small for gestational age babies: Good or bad? Current Opinion in Endocrinology, Diabetes and Obesity. 2007;14:30–34. doi: 10.1097/MED.0b013e328013da6c. [DOI] [PubMed] [Google Scholar]
- Osmond C, Barker DJP, Winter PD, Fall CHD, Simmonds SJ. Early growth and death from cardiovascular disease in women. British Medical Journal. 1993;307:1519–1524. doi: 10.1136/bmj.307.6918.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostman J, Amer P, Engfeldt P, Kager L. Regional differences in the control of lipolysis in human adipose tissue. Metabolism. 1979;28:1198–1205. doi: 10.1016/0026-0495(79)90131-8. [DOI] [PubMed] [Google Scholar]
- Owen CG, Martin RM, Whincup PH, Davey-Smith G, Gillman MW, Cook DG. The effect of breastfeeding on mean body mass index throughout life: A quantitative review of published and unpublished observational evidence. American Journal of Clinical Nutrition. 2005a;82:1298–1307. doi: 10.1093/ajcn/82.6.1298. [DOI] [PubMed] [Google Scholar]
- Owen CG, Martin RM, Whincup PH, Davey Smith G, Cook DG. Effect of infant feeding on the risk of obesity across the life course: a quantitative review of published evidence. Pediatrics. 2005b;115:1367–1377. doi: 10.1542/peds.2004-1176. [DOI] [PubMed] [Google Scholar]
- Owens J, Maxim R, McGuinn M, Nobile C, Msall M, Alario A. Television-viewing habits and sleep disturbance in school children. Pediatrics. 1999;104:e27. doi: 10.1542/peds.104.3.e27. [DOI] [PubMed] [Google Scholar]
- Palmer CD, Harrison GA, Hiorns RW. Sleep patterns and life style in Oxfordshire villages. J Biosoc Sci. 1980;12:437–467. doi: 10.1017/s0021932000013043. [DOI] [PubMed] [Google Scholar]
- Parracho H, McCartney AL, Gibson GR. Probiotics and prebiotics in infant nutrition. Proceedings of the Nutrition Society. 2007;66:405–411. doi: 10.1017/S0029665107005678. [DOI] [PubMed] [Google Scholar]
- Pasarica M, Mashtalir N, McAllister EJ, Kilroy GE, Koska J, Permana P, de Courten B, Yu M, Ravussin E, Gimble JM, Dhurandhar NV. Adipogenic humanadenovirus Ad-36 induces commitment, differentiation, and lipid accumulation in human adipose-derived stem cells. Stem Cells. 2008;26:969–978. doi: 10.1634/stemcells.2007-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M, Loiler S, Dhurandhar NV. Acute effect of infection by adipogenic human adenovirus Ad36. Arch Virol. 2008;153:2097–2102. doi: 10.1007/s00705-008-0219-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M, Dhurandhar NV. Infectobesity: obesity of infectious origin. Adv Food Nutr Res. 2007;52:61–102. doi: 10.1016/S1043-4526(06)52002-9. [DOI] [PubMed] [Google Scholar]
- Pasarica M, Shin AC, Yu M, Ou Yang HM, Rathod M, Jen KL, MohanKumar S, MohanKumar PS, Markward N, Dhurandhar NV. Human adenovirus 36 induces adiposity, increases insulin sensitivity, and alters hypothalamic monoamines in rats. Obesity (Silver Spring) 2006;14:1905–1913. doi: 10.1038/oby.2006.222. [DOI] [PubMed] [Google Scholar]
- Patel SR, Malhotra A, White DP, Gottlieb DJ, Hu FB. Association between reduced sleep and weight gain in women. Am J Epidemiol. 2006;164:947–954. doi: 10.1093/aje/kwj280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson ML, Stern S, Crawford PB, McMahon RP, Similo SL, Schreiber GB, Morrison JA, Waclawiw MA. Sociodemographic factors and obesity in preadolescent black and white girls: NHLBI’s Growth and Health Study. J Natl Med Assoc. 1997;89:594–600. [PMC free article] [PubMed] [Google Scholar]
- Paulose-Ram R, Safran MA, Jonas BS, Gu Q, Orwig D. Trends in psychotropic medication use among U.S. adults. Pharmacoepidemiol Drug Saf. 2007;16:560–570. doi: 10.1002/pds.1367. [DOI] [PubMed] [Google Scholar]
- Pecoraro N, Reyes F, Gomez F, Bhargava A, Dallman MF. Chronic stress promotes palatable feeding, which reduces signs of stress: feedforward and feedback effects of chronic stress. Endocrinology. 2004;145:3754–3762. doi: 10.1210/en.2004-0305. [DOI] [PubMed] [Google Scholar]
- Pedrazzoli M, D’Almeida V, Martins PJ, Machado RB, Ling L, Nishino S, Tufik S, Mignot E. Increased hypocretin-1 levels in cerebrospinal fluid after REM sleep deprivation. Brain Res. 2004;995:1–6. doi: 10.1016/j.brainres.2003.09.032. [DOI] [PubMed] [Google Scholar]
- Pembrey ME, Bygren LO, Kaati G, Edvinsson S, Northstone K, Sjostrom M, Golding J. Sex-specific, male-line transgenerational responses in humans. Eur J Hum Genet. 2006;14:159–166. doi: 10.1038/sj.ejhg.5201538. [DOI] [PubMed] [Google Scholar]
- Perez-Iglesias R, Crespo-Facorro B, Amado JA, Garcia-Unzueta MT, Ramirez-Bonilla ML, Gonzalez-Blanch C, Martinez-Garcia O, Vazquez-Barquero JL. A 12-week randomized clinical trial to evaluate metabolic changes in drug-naive, first-episode psychosis patients treated with haloperidol, olanzapine, or risperidone. J Clin Psychiatry. 2007;68:1733–1740. doi: 10.4088/jcp.v68n1113. [DOI] [PubMed] [Google Scholar]
- Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–1292. doi: 10.1007/s001250050822. [DOI] [PubMed] [Google Scholar]
- Pickup JC, Crook MA. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia. 1998;41:1241–1248. doi: 10.1007/s001250051058. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Heidrich I, Gotz F, Rohde W, Dorner G. Obesity and enhanced diabetes and cardiovascular risk in adult rats due to early postnatal overfeeding. Experimental and Clinical Endocrinology. 1992;99:154–158. doi: 10.1055/s-0029-1211159. [DOI] [PubMed] [Google Scholar]
- Prentice AM. The emerging epidemic of obesity in developing countries. International Journal of Epidemiology. 2006;35:93–99. doi: 10.1093/ije/dyi272. [DOI] [PubMed] [Google Scholar]
- Prins JB, O’Rahilly S. Regulation of adipose cell number in man. Clinical Science. 1997;92:3–11. doi: 10.1042/cs0920003. [DOI] [PubMed] [Google Scholar]
- Prummel MF, Mourits MP, Blank L, Berghout A, Koornneef L, Wiersinga WM. Randomized double-blind trial of prednisone versus radiotherapy in Graves’ ophthalmopathy. Lancet. 1993;342:949–954. doi: 10.1016/0140-6736(93)92001-a. [DOI] [PubMed] [Google Scholar]
- Prysak M, Lorenz RP, Kisly A. Pregnancy outcome in nulliparous women 35 years and older. Obstet Gynecol. 1995;85:65–70. doi: 10.1016/0029-7844(94)00330-g. [DOI] [PubMed] [Google Scholar]
- Punjabi NM, Bandeen-Roche K, Young T. Predictors of objective sleep tendency in the general population. Sleep. 2003;26:678–683. doi: 10.1093/sleep/26.6.678. [DOI] [PubMed] [Google Scholar]
- Ramlau-Hansen CH, Thulstrup AM, Nohr EA, Bonde JP, Sorensen TI, Olsen J. Subfecundity in overweight and obese couples. Hum Reprod. 2007;22:1634–1637. doi: 10.1093/humrep/dem035. [DOI] [PubMed] [Google Scholar]
- Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, Argyropoulos G, Walts B, Perusse L, Bouchard C. The human obesity gene map: the 2005 update. Obesity (Silver Spring) 2006;14:529–644. doi: 10.1038/oby.2006.71. [DOI] [PubMed] [Google Scholar]
- Rathod M, Vangipuram SD, Krishnan B, Heydari AR, Holland TC, Dhurandhar NV. Viral mRNA expression but not DNA replication is required for lipogenic effect of human adenovirus Ad-36 in preadipocytes. Int J Obes (Lond) 2007;31:78–86. doi: 10.1038/sj.ijo.0803358. [DOI] [PubMed] [Google Scholar]
- Rathod MA, Rogers PM, Vangipuram SD, Vangipuram McAllister EJ, Dhurandhar NV. Adipogenic Cascade Can Be Induced Without Adipogenic Media by a Human Adenovirus. Obesity (Silver Spring) 2009 January; doi: 10.1038/oby.2008.630. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravussin E, Lillioja S, Knowler WC, Christin L, Freymond D, Abbott WG, Boyce V, Howard BV, Bogardus C. Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med. 1988;318:467–472. doi: 10.1056/NEJM198802253180802. [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Bergmann BM, Everson CA, Kushida CA, Gilliland MA. Sleep deprivation in the rat: X. Integration and discussion of the findings. 1989. Sleep. 2002;25:68–87. [PubMed] [Google Scholar]
- Redgrave TG, Wallace P, Jandacek RJ, Tso P. Treatment with a dietary fat substitute decreased Arochlor 1254 contamination in an obese diabetic male. J Nutr Biochem. 2005;16:383–384. doi: 10.1016/j.jnutbio.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Reichborn-Kjennerud T, Bulik CM, Sullivan PF, Tambs K, Harris JR. Psychiatric and medical symptoms in binge eating in the absence of compensatory behaviors. Obes Res. 2004;12:1445–1454. doi: 10.1038/oby.2004.181. [DOI] [PubMed] [Google Scholar]
- Reilly JJ, Armstrong J, Dorosty AR, Emmett PM, Ness A, Rogers I, Steer C, Sherriff A Team., A. L. S. o. P. a. C. S. Early life risk factors for obesity in childhood: cohort study. 2005;330:1357. doi: 10.1136/bmj.38470.670903.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricquier D. Respiration uncoupling and metabolism in the control of energy expenditure. Proc Nutr Soc. 2005;64:47–52. doi: 10.1079/pns2004408. [DOI] [PubMed] [Google Scholar]
- Robinson SM, Crozier SR, Borland SE, Hammond J, Barker DJ, Inskip HM. Impact of educational attainment on the quality of young women’s diets. European journal of clinical nutrition. 2004;58:1174–1180. doi: 10.1038/sj.ejcn.1601946. [DOI] [PubMed] [Google Scholar]
- Rogers PM, Fusinski KA, Rathod MA, Loiler SA, Pasarica M, Shaw MK, Kilroy G, Sutton GM, McAllister EJ, Mashtalir N, Gimble JM, Holland TC, Dhurandhar NV. Human adenovirus Ad-36 induces adipogenesis via its E4 orf-1 gene. Int J Obes (Lond) 2007;32:397–406. doi: 10.1038/sj.ijo.0803748. [DOI] [PubMed] [Google Scholar]
- Rogers PM, Mashtalir N, Rathod MA, Dubuisson O, Wang Z, Dasuri K, Babin S, Gupta A, Markward N, Cefalu WT, Dhurandhar NV. Metabolically Favorable Remodeling of Human Adipose Tissue by Human Adenovirus Ad-36. Diabetes. 2008 doi: 10.2337/db07-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls BJ, Rowe EA. Exercise and the development and persistence of dietary obesity in male and female rats. Physiol Behav. 1979;23:241–247. doi: 10.1016/0031-9384(79)90361-5. [DOI] [PubMed] [Google Scholar]
- Rolls BJ, Rowe EA, Turner RC. Persistent obesity in rats following a period of consumption of a mixed, high energy diet. J Physiol. 1980;298:415–427. doi: 10.1113/jphysiol.1980.sp013091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls BJ, Rowe EA. Pregnancy and lactation in the obese rat: effects on maternal and pup weights. Physiol Behav. 1982;28:393–400. doi: 10.1016/0031-9384(82)90130-5. [DOI] [PubMed] [Google Scholar]
- Rolls BJ, van Duijvenvoorde PM, Rowe EA. Effects of diet and obesity on body weight regulation during pregnancy and lactation in the rat. Physiol Behav. 1984;32:161–168. doi: 10.1016/0031-9384(84)90124-0. [DOI] [PubMed] [Google Scholar]
- Rosenberg L, Palmer JR, Wise LA, Horton NJ, Kumanyika SK, Adams-Campbell LL. A prospective study of the effect of childbearing on weight gain in African-American women. Obes Res. 2003;11:1526–1535. doi: 10.1038/oby.2003.204. [DOI] [PubMed] [Google Scholar]
- Rosenberg M. Weight change with oral contraceptive use and during the menstrual cycle. Results of daily measurements. Contraception. 1998;58:345–349. doi: 10.1016/s0010-7824(98)00127-9. [DOI] [PubMed] [Google Scholar]
- Rowe EA, Rolls BJ. Effects of environmental temperature on dietary obesity and growth in rats. Physiol Behav. 1982;28:219–226. doi: 10.1016/0031-9384(82)90065-8. [DOI] [PubMed] [Google Scholar]
- Sakaue H, Ogawa W, Matsumoto M, Kuroda S, Takata M, Sugimoto T, Spiegelman BM, Kasuga M. Posttranscriptional control of adipocyte differentiation through activation of phosphoinositide 3-kinase. J Biol Chem. 1998;273:28945–28952. doi: 10.1074/jbc.273.44.28945. [DOI] [PubMed] [Google Scholar]
- Salihu HM, Kinniburgh BA, Aliyu MH, Kirby RS, Alexander GR. Racial disparity in stillbirth among singleton, twin, and triplet gestations in the United States. Obstet Gynecol. 2004;104:734–740. doi: 10.1097/01.AOG.0000139944.15133.e3. [DOI] [PubMed] [Google Scholar]
- Sallmen M, Sandler DP, Hoppin JA, Blair A, Baird DD. Reduced fertility among overweight and obese men. Epidemiology. 2006;17:520–523. doi: 10.1097/01.ede.0000229953.76862.e5. [DOI] [PubMed] [Google Scholar]
- Samuelsson AJ, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EHJM, Piersma AH, Ozanne SE, Fernandez-Twinn D, Remacle C, Rowlerson A, Poston L, Taylor PD. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51:383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- Saxton C. Effects of severe heat stress on respiration and metabolic rate in resting man. Aviat Space Environ Med. 1981;52:281–286. [PubMed] [Google Scholar]
- Schilder RJ, Marden JH. Metabolic syndrome and obesity in an insect. Proc Natl Acad Sci U S A. 2006;103:18805–18809. doi: 10.1073/pnas.0603156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuld A, Hebebrand J, Geller F, Pollmacher T. Increased body mass index in patients with narcolepsy. Lancet. 2000;355:1274–1275. doi: 10.1016/S0140-6736(05)74704-8. [DOI] [PubMed] [Google Scholar]
- Segal NL, Allison DB. Twins and virtual twins: bases of relative body weight revisited. Int J Obes Relat Metab Disord. 2002;26:437–441. doi: 10.1038/sj.ijo.0801941. [DOI] [PubMed] [Google Scholar]
- Segal P, Feig PU, Schernthaner G, Ratzmann KP, Rybka J, Petzinna D, Berlin C. The efficacy and safety of miglitol therapy compared with glibenclamide in patients with NIDDM inadequately controlled by diet alone. Diabetes Care. 1997;20:687–691. doi: 10.2337/diacare.20.5.687. [DOI] [PubMed] [Google Scholar]
- Sekine M, Yamagami T, Handa K, Saito T, Nanri S, Kawaminami K, Tokui N, Yoshida K, Kagamimori S. A dose-response relationship between short sleeping hours and childhood obesity: results of the Toyama Birth Cohort Study. Child Care Health Dev. 2002;28:163–170. doi: 10.1046/j.1365-2214.2002.00260.x. [DOI] [PubMed] [Google Scholar]
- Sgoifo A, Buwalda B, Roos M, Costoli T, Merati G, Meerlo P. Effects of sleep deprivation on cardiac autonomic and pituitary-adrenocortical stress reactivity in rats. Psychoneuroendocrinology. 2006;31:197–208. doi: 10.1016/j.psyneuen.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Sharma AM, Pischon T, Hardt S, Kunz I, Luft FC. Hypothesis: Beta-adrenergic receptor blockers and weight gain: A systematic analysis. Hypertension. 2001;37:250–254. doi: 10.1161/01.hyp.37.2.250. [DOI] [PubMed] [Google Scholar]
- Shen L, Kondo Y, Guo Y, Zhang J, Zhang L, Ahmed S, Shu J, Chen X, Waterland RA, Issa JP. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007;3:2023–2036. doi: 10.1371/journal.pgen.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinomiya N, Shinomiya M. Dichlorodiphenyltrichloroethane suppresses neurite outgrowth and induces apoptosis in PC12 pheochromocytoma cells. Toxicol Lett. 2003;137:175–183. doi: 10.1016/s0378-4274(02)00401-0. [DOI] [PubMed] [Google Scholar]
- Silva MJ, Barr DB, Reidy JA, Malek NA, Hodge CC, Caudill SP, Brock JW, Needham LL, Calafat AM. Urinary levels of seven phthalate metabolites in the U.S. population from the National Health and Nutrition Examination Survey (NHANES) 1999–2000. Environ Health Perspect. 2004;112:331–338. doi: 10.1289/ehp.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman BL, Cho NH, Rizzo TA, Metzger BE. Long-term effects of the intrauterine environment. Diabetes Care. 1998;21:B142–B149. [PubMed] [Google Scholar]
- Simon C, Gronfier C, Schlienger JL, Brandenberger G. Circadian and ultradian variations of leptin in normal man under continuous enteral nutrition: relationship to sleep and body temperature. J Clin Endocrinol Metab. 1998;83:1893–1899. doi: 10.1210/jcem.83.6.4864. [DOI] [PubMed] [Google Scholar]
- Simonson DC, Kourides IA, Feinglos M, Shamoon H, Fischette CT. Efficacy, safety, and dose-response characteristics of glipizide gastrointestinal therapeutic system on glycemic control and insulin secretion in NIDDM. Results of two multicenter, randomized, placebo-controlled clinical trials. The Glipizide Gastrointestinal Therapeutic System Study Group. Diabetes Care. 1997;20:597–606. doi: 10.2337/diacare.20.4.597. [DOI] [PubMed] [Google Scholar]
- Singh M, Drake CL, Roehrs T, Hudgel DW, Roth T. The association between obesity and short sleep duration: a population-based study. J Clin Sleep Med. 2005;1:357–363. [PubMed] [Google Scholar]
- Singhal A, Fewtrell M, Cole TJ, Lucas A. Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. Lancet. 2003;361:1089–1097. doi: 10.1016/S0140-6736(03)12895-4. [DOI] [PubMed] [Google Scholar]
- Singhal A, Cole TJ, Fewtrell M, Deanfield J, Lucas A. Is slower early growth beneficial for long-term cardiovascular health? Circulation. 2004;109:1108–1113. doi: 10.1161/01.CIR.0000118500.23649.DF. [DOI] [PubMed] [Google Scholar]
- Skaer TL, Sclar DA, Robison LM, Galin RS. Trends in the rate of depressive illness and use of antidepressant pharmacotherapy by ethnicity/race: an assessment of office-based visits in the United States, 1992–1997. Clin Ther. 2000;22:1575–1589. doi: 10.1016/s0149-2918(00)83055-6. [DOI] [PubMed] [Google Scholar]
- So PW, Herlihy AH, Bell JD. Adiposity induced by adenovirus 5 inoculation. Int J Obes (Lond) 2005;29:603–606. doi: 10.1038/sj.ijo.0802917. [DOI] [PubMed] [Google Scholar]
- Sohoni P, Sumpter JP. Several environmental oestrogens are also anti-androgens. J Endocrinol. 1998;158:327–339. doi: 10.1677/joe.0.1580327. [DOI] [PubMed] [Google Scholar]
- Sothern MS. Obesity prevention in children: physical activity and nutrition. Nutrition. 2004;20:704–708. doi: 10.1016/j.nut.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–1439. doi: 10.1016/S0140-6736(99)01376-8. [DOI] [PubMed] [Google Scholar]
- Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on physiological rhythms. Rev Neurol (Paris) 2001;159:6S11–20. French. [PubMed] [Google Scholar]
- Spiegel K, Leproult R, L’hermite-Baleriaux M, Copinschi G, Penev PD, Van Cauter E. Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004a;89:5762–5771. doi: 10.1210/jc.2004-1003. [DOI] [PubMed] [Google Scholar]
- Spiegel K, Tasali E, Penev P, Van Cauter E. Brief communication: Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med. 2004b;141:846–850. doi: 10.7326/0003-4819-141-11-200412070-00008. [DOI] [PubMed] [Google Scholar]
- Spiegel K, Knutson K, Leproult R, Tasali E, Van Cauter E. Sleep loss: a novel risk factor for insulin resistance and Type 2 diabetes. J Appl Physiol. 2005;99:2008–2019. doi: 10.1152/japplphysiol.00660.2005. [DOI] [PubMed] [Google Scholar]
- Spraul M, Ravussin E, Fontvieille AM, Rising R, Larson DE, Anderson EA. Reduced sympathetic nervous activity. A potential mechanism predisposing to body weight gain. J Clin Invest. 1993;92:1730–1735. doi: 10.1172/JCI116760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spuhler JN. Assortative mating with respect to physical characteristics. Eugenics Quarterly. 1968;15:128–140. doi: 10.1080/19485565.1968.9987763. [DOI] [PubMed] [Google Scholar]
- Srinivasan M, Laychock SG, Hill DJ, Patel MS. Neonatal nutrition: metabolic programming of pancreatic islets and obesity. Exp Biol Med. 2003;228:15–23. doi: 10.1177/153537020322800102. [DOI] [PubMed] [Google Scholar]
- Stahlhut RW, van Wijngaarden E, Dye TD, Cook S, Swan SH. Concentrations of urinary phthalate metabolites are associated with increased waist circumference and insulin resistance in adult U.S. males. Environ Health Perspect. 2007;115:876–882. doi: 10.1289/ehp.9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamford BA, Matter S, Fell RD, Papanek P. Effects of smoking cessation on weight gain, metabolic rate, caloric consumption, and blood lipids. Am J Clin Nutr. 1986;43:486–494. doi: 10.1093/ajcn/43.4.486. [DOI] [PubMed] [Google Scholar]
- Stearns SC. The evolution of life histories. Oxford University Press; Oxford: 1992. [Google Scholar]
- Stettler N, Tershakovec AM, Zemel BS, Leonard MB, Boston RC, Katz SH, Stallings VA. Early risk factors for increased adiposity: a cohort study of African American subjects followed from birth to young adulthood. American Journal of Clinical Nutrition. 2000;72:378–383. doi: 10.1093/ajcn/72.2.378. [DOI] [PubMed] [Google Scholar]
- Stettler N, Zemel BS, Kumanyika S, Stallings VA. Infant weight gain and childhood overweight status in a multicenter, cohort study. Pediatrics. 2002;109:194–199. doi: 10.1542/peds.109.2.194. [DOI] [PubMed] [Google Scholar]
- Stettler N. Nature and strength of epidemiological evidence for origins of childhood and adulthood obesity in the first year of life. International Journal of Obesity. 2007;31:1035–1043. doi: 10.1038/sj.ijo.0803659. [DOI] [PubMed] [Google Scholar]
- Stocker C, O’Dowd J, Morton NM, Wargent E, Sennitt MV, Hislop D, Glund S, Seckl JR, Arch JR, Cawthorne MA. Modulation of susceptibility to weight gain and insulin resistance in low birthweight rats by treatment of their mothers with leptin during pregnancy and lactation. Int J Obes Relat Metab Disord. 2004;28:129–136. doi: 10.1038/sj.ijo.0802476. [DOI] [PubMed] [Google Scholar]
- Stoger R. In vivo methylation patterns of the leptin promoter in human and mouse. Epigenetics. 2006;1:155–162. doi: 10.4161/epi.1.4.3400. [DOI] [PubMed] [Google Scholar]
- Strauss RS. Effects of the intrauterine environment on childhood growth. Br Med Bull. 1997;53:81–95. doi: 10.1093/oxfordjournals.bmb.a011608. [DOI] [PubMed] [Google Scholar]
- Stricker RB, Goldberg B. Weight gain associated with protease inhibitor therapy in HIV-infected patients. Res Virol. 1998;149:123–126. doi: 10.1016/s0923-2516(98)80088-5. [DOI] [PubMed] [Google Scholar]
- Stroebele N, De Castro JM. Effect of ambience on food intake and food choice. Nutrition. 2004;20:821–838. doi: 10.1016/j.nut.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Sturm R. Childhood obesity - what we can learn from existing data on societal trends, part 1. Prev Chronic Dis. 2005;2:A12. [PMC free article] [PubMed] [Google Scholar]
- Sugimori H, Yoshida K, Izuno T, Miyakawa M, Suka M, Sekine M, Yamagami T, Kagamimori S. Analysis of factors that influence body mass index from ages 3 to 6 years: A study based on the Toyama cohort study. Pediatr Int. 2004;46:302–310. doi: 10.1111/j.1442-200x.2004.01895.x. [DOI] [PubMed] [Google Scholar]
- Surkan PJ, Hsieh CC, Johansson AL, Dickman PW, Cnattingius S. Reasons for increasing trends in large for gestational age births. Obstet Gynecol. 2004;104:720–726. doi: 10.1097/01.AOG.0000141442.59573.cd. [DOI] [PubMed] [Google Scholar]
- Susa JB, Widness JA, Hintz R, Liu FR, Sehgal P, Schwartz R. Somatomedins and insulin in diabetic pregnancies: effects on fetal macrosomia in the human and rhesus monkey. Journal of Clinical Endocrinology and Metabolism. 1984;58:1099–1105. doi: 10.1210/jcem-58-6-1099. [DOI] [PubMed] [Google Scholar]
- Sussman N, Ginsberg DL, Bikoff J. Effects of nefazodone on body weight: a pooled analysis of selective serotonin reuptake inhibitor- and imipramine-controlled trials. J Clin Psychiatry. 2001;62:256–260. [PubMed] [Google Scholar]
- Symonds ME, Pearce S, Bispham J, Gardner DS, Stephenson T. Timing of nutrient restriction and programming of fetal adipose tissue development. Proc Nutr Soc. 2004;63:397–403. doi: 10.1079/pns2004366. [DOI] [PubMed] [Google Scholar]
- Szentirmai E, Krueger JM. Obestatin alters sleep in rats. Neurosci Lett. 2006;404:222–226. doi: 10.1016/j.neulet.2006.05.053. [DOI] [PubMed] [Google Scholar]
- Szentirmai E, Kapas L, Krueger JM. Ghrelin microinjection into forebrain sites induces wakefulness and feeding in rats. Am J Physiol Regul Integr Comp Physiol. 2007;292:R575–585. doi: 10.1152/ajpregu.00448.2006. [DOI] [PubMed] [Google Scholar]
- Tabb MM, BB New modes of action for endocrine-disrupting chemicals. Mol Endocrinol. 2007;20:475–482. doi: 10.1210/me.2004-0513. [DOI] [PubMed] [Google Scholar]
- Taheri S, Lin L, Austin D, Young T, Mignot E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004;1:e62. doi: 10.1371/journal.pmed.0010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri S. The link between short sleep duration and obesity: we should recommend more sleep to prevent obesity. Arch Dis Child. 2006a;91:881–884. doi: 10.1136/adc.2005.093013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri S. The link between short sleep duration and obesity: we should recommend more sleep to prevent obesity. T. Arch Dis Child. 2006b;91:881–884. doi: 10.1136/adc.2005.093013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taheri S. Sleep and metabolism: bringing pieces of the jigsaw together. Sleep Med Rev. 2007;11:159–162. doi: 10.1016/j.smrv.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Tamashiro KL, Wakayama T, Akutsu H, Yamazaki Y, Lachey JL, Wortman MD, Seeley RJ, D’Alessio DA, Woods SC, Yanagimachi R, Sakai RR. Cloned mice have an obese phenotype not transmitted to their offspring. Nat Med. 2002;8:262–267. doi: 10.1038/nm0302-262. [DOI] [PubMed] [Google Scholar]
- Tanneeru M. Health. CNN; 2006. Obesity: A looming national threat? [Google Scholar]
- Tasali E, Van Cauter E. Sleep-disordered breathing and the current epidemic of obesity: consequence or contributing factor? Am J Respir Crit Care Med. 2002;165:562–563. doi: 10.1164/ajrccm.165.5.2201001b. [DOI] [PubMed] [Google Scholar]
- Tataranni PA, Harper IT, Snitker S, Del Parigi A, Vozarova B, Bunt J, Bogardus C, Ravussin E. Body weight gain in free-living Pima Indians: effect of energy intake vs expenditure. Int J Obes Relat Metab Disord. 2003;27:1578–1583. doi: 10.1038/sj.ijo.0802469. [DOI] [PubMed] [Google Scholar]
- Taveras EM, Scanlon KS, Birch L, Rifas-Shiman SL, Rich-Edwards JW, Gillman MW. Association of breastfeeding with maternal control of infant feeding at age 1 year. Pediatrics. 2004;114:e577–e583. doi: 10.1542/peds.2004-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PD, Poston L. Developmental programming of obesity in mammals. Experimental Physiology. 2006;92:287–298. doi: 10.1113/expphysiol.2005.032854. [DOI] [PubMed] [Google Scholar]
- Tchernof A, Poehlman ET. Effects of the menopause transition on body fatness and body fat distribution. Obes Res. 1998;6:246–254. doi: 10.1002/j.1550-8528.1998.tb00344.x. [DOI] [PubMed] [Google Scholar]
- Terman L, Hocking A. The sleep of school children, its distribution according to age, and its relationship to physical and mental efficiency. J Educ Psychol. 1913a;4:269–282. [Google Scholar]
- Terman L, Hocking A. The sleep of school children: It’s distribution according to age, and its relation to physical and mental efficiency. J Ed Psych. 1913b;4:138–147. [Google Scholar]
- Terman L, HA The sleep of school children, its distribution according to age, and its relationship to physical and mental efficiency. J Educ Psychol. 1913;4:269–282. [Google Scholar]
- Thomas GCR, Tilkorn D, Keramidaris E, Morrison W, Penington A, Blick T. Effect of human adenovirus -36 in a localized in vivo model of neoadipogenesis. Obesity. 2008;16:S74. [Google Scholar]
- Toppila J, Stenberg D, Alanko L, Asikainen M, Urban JH, Turek FW, Porkka-Heiskanen T. REM sleep deprivation induces galanin gene expression in the rat brain. Neurosci Lett. 1995;183:171–174. doi: 10.1016/0304-3940(94)11143-7. [DOI] [PubMed] [Google Scholar]
- Trayhurn P, Beattie JH. Physiological role of adipose tissue: white adipose tissue as an endocrine and secretory organ. Proc Nutr Soc. 2001;60:329–339. doi: 10.1079/pns200194. [DOI] [PubMed] [Google Scholar]
- Trenell MI, Marshall NS, Rogers NL. Sleep and metabolic control: waking to a problem? Clin Exp Pharmacol Physiol. 2007;34:1–9. doi: 10.1111/j.1440-1681.2007.04541.x. [DOI] [PubMed] [Google Scholar]
- Tune G. Sleep and wakefulness in normal human adults. British Med J. 1968a;2:269–271. doi: 10.1136/bmj.2.5600.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tune G. Sleep and wakefulness in 509 normal human adults. Br J Med Psychol. 1969;42:75–80. doi: 10.1111/j.2044-8341.1969.tb02060.x. [DOI] [PubMed] [Google Scholar]
- Tune GS. Sleep and wakefulness in normal human adults. Br Med J. 1968b;2:269–271. doi: 10.1136/bmj.2.5600.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Uauy R, Kain J. The epidemiological transition: need to incorporate obesity prevention into nutrition programmes. Pub Health Nutr. 2002;5:223–229. doi: 10.1079/PHN2001297. [DOI] [PubMed] [Google Scholar]
- Understanding Comfort, Behavior and Productivity. Space Heating Technology Atlas. 2004 [ http://www.esource.com/esource/getpub/public/pdf/Heating.pdf]
- Uzumcu M, Suzuki H, Skinner MK. Effect of the anti-androgenic endocrine disruptor vinclozolin on embryonic testis cord formation and postnatal testis development and function. Reprod Toxicol. 2004;18:765–774. doi: 10.1016/j.reprotox.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Van Cauter E, Spiegel K. Sleep as a mediator of the relationship between socioeconomic status and health: a hypothesis. Ann N Y Acad Sci. 1999;896:254–261. doi: 10.1111/j.1749-6632.1999.tb08120.x. [DOI] [PubMed] [Google Scholar]
- Van Cauter E, Holmback U, Knutson K, Leproult R, Miller A, Nedeltcheva A, Pannain S, Penev P, Tasali E, Spiegel K. Impact of sleep and sleep loss on neuroendocrine and metabolic function. Horm Res. 2007;67(Suppl 1):2–9. doi: 10.1159/000097543. [DOI] [PubMed] [Google Scholar]
- van der Lely AJ, Tschöp M, Heiman ML, Ghigo E. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004a;25:426–457. doi: 10.1210/er.2002-0029. [DOI] [PubMed] [Google Scholar]
- van der Lely AJ, Tschop M, Heiman ML, Ghigo E. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004b;25:426–457. doi: 10.1210/er.2002-0029. [DOI] [PubMed] [Google Scholar]
- van Hanswijck de Jonge L, Stettler N, Kumanyika S, Stoa Birketvedt G, Waller G. Environmental temperature during gestation and body mass index in adolescence: new etiologic clues? Int J Obes Relat Metab Disord. 2002;26:765–769. doi: 10.1038/sj.ijo.0802011. [DOI] [PubMed] [Google Scholar]
- van Hanswijck de Jonge L, Waller G, Stettler N. Ethnicity modifies seasonal variations in birth weight and weight gain of infants. J Nutr. 2003;133:1415–1418. doi: 10.1093/jn/133.5.1415. [DOI] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Westerterp-Plantenga MS, van Hoydonck P. Individual variation in the relation between body temperature and energy expenditure in response to elevated ambient temperature. Physiol Behav. 2001;73:235–242. doi: 10.1016/s0031-9384(01)00477-2. [DOI] [PubMed] [Google Scholar]
- Vangipuram SD, Sheele J, Atkinson RL, Holland TC, Dhurandhar NV. A human adenovirus enhances preadipocyte differentiation. Obes Res. 2004;12:770–777. doi: 10.1038/oby.2004.93. [DOI] [PubMed] [Google Scholar]
- Vangipuram SD, Yu M, Tian J, Stanhope KL, Pasarica M, Havel PJ, Heydari AR, Dhurandhar NV. Adipogenic human adenovirus-36 reduces leptin expression and secretion and increases glucose uptake by fat cells. Int J Obes (Lond) 2007;31:87–96. doi: 10.1038/sj.ijo.0803366. [DOI] [PubMed] [Google Scholar]
- Vanitallie TB. Sleep and energy balance: Interactive homeostatic systems. Metabolism. 2006;55:S30–S35. doi: 10.1016/j.metabol.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Ventura SJ. Trends and variations in first births to older women, United States, 1970–86. Vital Health Stat. 1989;21:1–27. [PubMed] [Google Scholar]
- Verlaeten O, Griffond B, Khuth ST, Giraudon P, Akaoka H, Belin MF, Fellmann D, Bernard A. Down regulation of melanin concentrating hormone in virally induced obesity. Mol Cell Endocrinol. 2001;181:207–219. doi: 10.1016/s0303-7207(01)00488-9. [DOI] [PubMed] [Google Scholar]
- Vestri HS, Maianu L, Moellering DR, Garvey WT. Atypical antipsychotic drugs directly impair insulin action in adipocytes: effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology. 2007;32(4):765–772. doi: 10.1038/sj.npp.1301142. [DOI] [PubMed] [Google Scholar]
- Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD. Fetal origins of hyperphagia, obesity and hypertension and its postnatal amplification by hypercaloric nutrition. American Journal of Physiology. 2000;279:E83–E87. doi: 10.1152/ajpendo.2000.279.1.E83. [DOI] [PubMed] [Google Scholar]
- Vickers MH, Breier BH, McCarthy D, Gluckman PD. Sedentary behaviour during postnatal life is determined by the prenatal environment and exacerbated by postnatal hypercaloric nutrition. American Journal of Physiology. 2003;285:R271–R273. doi: 10.1152/ajpregu.00051.2003. [DOI] [PubMed] [Google Scholar]
- Vickers MH, Gluckman PD, Coveny AH, Hofman PL, Cutfield WS, Gertler A, Breier BH, Harris M. Neonatal leptin treatment reverses developmental programming. Endocrinology. 2005;146:4211–4216. doi: 10.1210/en.2005-0581. [DOI] [PubMed] [Google Scholar]
- Vioque J, Torres A, Quiles J. Time spent watching television, sleep duration and obesity in adults living in Valencia, Spain. Int J Obes Relat Metab Disord. 2000a;24:1683–1688. doi: 10.1038/sj.ijo.0801434. [DOI] [PubMed] [Google Scholar]
- Vioque J, Torres A, Quiles J. Time spent watching television, sleep duration and obesity in adults in Valencia, Spain. Int J Eat Obes Relat Metab Disord. 2000b;24:1683–1688. doi: 10.1038/sj.ijo.0801434. [DOI] [PubMed] [Google Scholar]
- von Kries R, Toschke AM, Wurmser H, Sauerwald T, Koletzko B. Reduced risk for overweight and obesity in 5- and 6-y-old children by duration of sleep--a cross-sectional study. Int J Obes Relat Metab Disord. 2002;26:710–716. doi: 10.1038/sj.ijo.0801980. [DOI] [PubMed] [Google Scholar]
- Vorbrodt AW, Dobrogowska DH, Tarnawski M, Meeker HC, Carp RI. Quantitative immunogold study of glucose transporter (GLUT-1) in five brain regions of scrapie-infected mice showing obesity and reduced glucose tolerance. Acta Neuropathol. 2001;102:278–284. doi: 10.1007/s004010100382. [DOI] [PubMed] [Google Scholar]
- Vorona RD, Winn MP, Babineau TW, BPE, Feldman HR, Ware JC. Overweight and obese patients in a primary care population report less sleep than patients with a normal body mass index. Arch Intern Med. 2005a;165:25–30. doi: 10.1001/archinte.165.1.25. [DOI] [PubMed] [Google Scholar]
- Vorona RD, Winn MP, Babineau TW, Eng BP, Feldman HR, Ware JC. Overweight and obese patients in a primary care population report less sleep than patients with a normal body mass index. Arch Intern Med. 2005b;165:25–30. doi: 10.1001/archinte.165.1.25. [DOI] [PubMed] [Google Scholar]
- Vrbikova J, Dvorakova K, Hill M, Starka L. Weight change and androgen levels during contraceptive treatment of women affected by polycystic ovary. Endocr Regul. 2006;40:119–123. [PubMed] [Google Scholar]
- Wade GN, Gray JM, Bartness TJ. Gonadal influences on adiposity. Int J Obes. 1985;9(Suppl 1):83–92. [PubMed] [Google Scholar]
- Wadhera S. Trends in birth and fertility rates, Canada, 1921–1987. Health Rep. 1989;1:211–223. [PubMed] [Google Scholar]
- Wadhera S, Millar WJ. Patterns and change in Canadian fertility 1971–1988: first births after age 30. Health Rep. 1991;3:149–162. [PubMed] [Google Scholar]
- Wallace TM, Levy JC, Matthews DR. An increase in insulin sensitivity and basal beta-cell function in diabetic subjects treated with pioglitazone in a placebo-controlled randomized study. Diabet Med. 2004;21:568–576. doi: 10.1111/j.1464-5491.2004.01218.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Lobstein T. Worldwide trends in childhood overweight and obesity. Int J Pediatric Obesity. 2006;1:11–25. doi: 10.1080/17477160600586747. [DOI] [PubMed] [Google Scholar]
- Wang JX, Davies M, Norman RJ. Body mass and probability of pregnancy during assisted reproduction treatment: retrospective study. BMJ. 2000;321:1320–1321. doi: 10.1136/bmj.321.7272.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Beydoun MA. The obesity epidemic in the United States--gender, age, socioeconomic, racial/ethnic, and geographic characteristics: a systematic review and meta-regression analysis. Epidemiol Rev. 2007;29:6–28. doi: 10.1093/epirev/mxm007. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Cefalu WT, Zhang XH, Yu Y, Qin J, Son L, Rogers PM, Mashtalir N, Bordelon JR, Ye J, Dhurandhar NV. Human adenovirus type 36 enhances glucose uptake in diabetic and nondiabetic human skeletal muscle cells independent of insulin signaling. Diabetes. 2008;57:1805–1813. doi: 10.2337/db07-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland RA, Garza C. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr. 1999;69:179–197. doi: 10.1093/ajcn/69.2.179. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland RA. Does nutrition during infancy and early childhood contribute to later obesity via metabolic imprinting of epigenetic gene regulatory mechanisms? In: Hernell OS, editor. Feeding during late infancy and early childhood: Impact on health. Nestec Ltd; Vevey: 2005. pp. 157–174. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Dolinoy DC, Lin JR, Smith CA, Shi X, Tahiliani KG. Maternal methyl supplements increase offspring DNA methylation at Axin Fused. Genesis. 2006;44:401–406. doi: 10.1002/dvg.20230. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–388. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obes (Lond) 2008;32:1373–9. doi: 10.1038/ijo.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson NF, Goldberg J, Arguelles L, Buchwald D. Genetic and environmental influences on insomnia, daytime sleepiness, and obesity in twins. Sleep. 2006;29:645–649. doi: 10.1093/sleep/29.5.645. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Webb WB, Agnew HW. Are we chronically sleep deprived? Bull Psychon Soc. 1975;6:47–48. [Google Scholar]
- Weber E, Stack J, Pollock BG, Mulsant B, Begley A, Mazumdar S, Reynolds CFr. Weight change in older depressed patients during acute pharmacotherapy with paroxetine and nortriptyline: a double-blind randomized trial. Am J Geriatr Psychiatry. 2000;8:245–250. [PubMed] [Google Scholar]
- Welsh JA, Cogswell ME, Rogers S, Rockett H, Mei Z, Grummer-Strawn LM. Overweight among low-income preschool children associated with the consumption of sweet drinks: Missouri, 1999–2002. Pediatrics. 2005;115:e223–229. doi: 10.1542/peds.2004-1148. [DOI] [PubMed] [Google Scholar]
- Weng HH, Bastian LA, Taylor DH, Jr, Moser BK, Ostbye T. Number of children associated with obesity in middle-aged women and men: results from the health and retirement study. J Womens Health (Larchmt) 2004;13:85–91. doi: 10.1089/154099904322836492. [DOI] [PubMed] [Google Scholar]
- West-Eberhard MJ. Developmental Plasticity and Evolution. Oxford University Press; New York: 2003. [Google Scholar]
- Westerterp-Plantenga MS, van Marken Lichtenbelt WD, Cilissen C, Top S. Energy metabolism in women during short exposure to the thermoneutral zone. Physiol Behav. 2002a;75:227–235. doi: 10.1016/s0031-9384(01)00649-7. [DOI] [PubMed] [Google Scholar]
- Westerterp-Plantenga MS, van Marken Lichtenbelt WD, Strobbe H, Schrauwen P. Energy metabolism in humans at a lowered ambient temperature. European journal of clinical nutrition. 2002b;56:288–296. doi: 10.1038/sj.ejcn.1601308. [DOI] [PubMed] [Google Scholar]
- Westoff CF. Fertility in the United States. Science. 1986;234:554–559. doi: 10.1126/science.3532324. [DOI] [PubMed] [Google Scholar]
- Whigham LD, Israel BA, Atkinson RL. Adipogenic potential of multiple human adenoviruses in vivo and in vitro in animals. Am J Physiol Regul Integr Comp Physiol. 2006;290:R190–194. doi: 10.1152/ajpregu.00479.2005. [DOI] [PubMed] [Google Scholar]
- Whincup PH, Gilg JA, Papacosta O, Seymour C, Miller GJ, Alberti KG, Cook DG. Early evidence of ethnic differences in cardiovascular risk: cross sectional comparison of British South Asian and white children. British Medical Journal. 2002;324:635. doi: 10.1136/bmj.324.7338.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker RC. Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics. 2004;114:e29–e36. doi: 10.1542/peds.114.1.e29. [DOI] [PubMed] [Google Scholar]
- Wilkinson PW, Parkin JM, Pearlson J, Philips PR, Sykes P. Obesity in childhood: A community study in Newcastle upon Tyne. Lancet. 1977;1:350–352. doi: 10.1016/s0140-6736(77)91147-3. [DOI] [PubMed] [Google Scholar]
- Williamson DF, Madans J, Anda RF, Kleinman JC, Giovino GA, Byers T. Smoking cessation and severity of weight gain in a national cohort. N Engl J Med. 1991;324:739–745. doi: 10.1056/NEJM199103143241106. [DOI] [PubMed] [Google Scholar]
- Wittassek M, Wiesmuller GA, Koch HM, Eckard R, Dobler L, Muller J, Angerer J, Schluter C. Internal phthalate exposure over the last two decades--a retrospective human biomonitoring study. Int J Hyg Environ Health. 2007;210:319–333. doi: 10.1016/j.ijheh.2007.01.037. [DOI] [PubMed] [Google Scholar]
- Wolff GL, Roberts DW, Mountjoy KG. Physiological consequences of ectopic agouti gene expression: the yellow obese mouse syndrome. Physiol Genomics. 1999;1:151–163. doi: 10.1152/physiolgenomics.1999.1.3.151. [DOI] [PubMed] [Google Scholar]
- Wolfson AR, Carskadon MA. Sleep schedules and daytime functioning in adolescents. Child Dev. 1998;69:875–887. [PubMed] [Google Scholar]
- Woodhouse AJ, Cooke GM. Suppression of aromatase activity in vitro by PCBs 28 and 105 and Aroclor 1221. Toxicol Lett. 2004;152:91–100. doi: 10.1016/j.toxlet.2004.04.009. [DOI] [PubMed] [Google Scholar]
- World HO. Obesity: Preventing and Managing the Global Epidemic. Report of a WHO Consultation on Obesity; Geneva. 3–5 June, 1997; Geneva: World Health Organization; 1998. [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Spiegelman BM. Transcriptional activation of adipogenesis. Current Opinion in Cell Biology. 1999;11:689–694. doi: 10.1016/s0955-0674(99)00037-x. [DOI] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahav S, Straschnow A, Plavnik I, Hurwitz S. Effects of diurnally cycling versus constant temperatures on chicken growth and food intake. Br Poult Sci. 1996;37:43–54. doi: 10.1080/00071669608417835. [DOI] [PubMed] [Google Scholar]
- Yajnik CS, Deshpande SS, Jackson AA, Refsum H, Rao S, Fisher DJ, Bhat DS, Naik SS, Coyaji KJ, Joglekar CV, Joshi N, Lubree HG, Deshpande VU, Rege SS, Fall CH. Vitamin B(12) and folate concentrations during pregnancy and insulin resistance in the offspring: the Pune Maternal Nutrition Study. Diabetologia. 2008;51:29–38. doi: 10.1007/s00125-007-0793-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LH, Chen TM, Yu ST, Chen YH. Olanzapine induces SREBP-1-related adipogenesis in 3T3-L1 cells. Pharmacol Res. 2007;56(3):202–208. doi: 10.1016/j.phrs.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Yang Q, Greenland S, Flanders WD. Associations of maternal age- and parity-related factors with trends in low-birthweight rates: United States, 1980 through 2000. Am J Public Health. 2006;96:856–861. doi: 10.2105/AJPH.2004.049312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N Engl J Med. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- Youngstedt SD, Kripke DF. Long sleep and mortality: rationale for sleep restriction. Sleep Med Rev. 2004;8:159–174. doi: 10.1016/j.smrv.2003.10.002. Review. [DOI] [PubMed] [Google Scholar]
- Zhang JV, Ren PG, Avsian-Kretchmer O, Luo CW, Rauch R, Klein C, Hsueh AJ. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science. 2005a;310:996–999. doi: 10.1126/science.1117255. [DOI] [PubMed] [Google Scholar]
- Zhang JV, Ren PG, vsian-Kretchmer O, Luo CW, Rauch R, Klein C, Hsueh AJ. Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science. 2005b;310:996–999. doi: 10.1126/science.1117255. [DOI] [PubMed] [Google Scholar]
- Ziegler EE. Growth of breast-fed and formula-fed infants. Nestle Nutr Workshop Ser Pediatr Program. 2006;58:59. doi: 10.1159/000095010. [DOI] [PubMed] [Google Scholar]
- Zifferblatt SM, Wilbur CS, Pinsky JL. Influence of ecologic events on cafeteria food selections: understanding food habits. J Am Diet Assoc. 1980;76:9–14. [PubMed] [Google Scholar]