

Abstract
The kinase Akt is a key signaling node in regulating cellular growth and survival. It is implicated in cancer by mutation and its role in the downstream transmission of aberrant PI3K signaling. For these reasons, Akt has become an increasingly important target of drug development efforts and several inhibitors are now reaching clinical trials. Paradoxically it has been observed that active site kinase inhibitors of Akt lead to hyperphosphorylation of Akt itself. To investigate this phenomenon we here describe the application of a chemical genetics strategy that replaces native Akt with a mutant version containing an active site substitution that allows for the binding of an engineered inhibitor. This analog sensitive strategy allows for the selective inhibition of a single kinase. In order to create the inhibitor selective for the analog sensitive kinase, a diversity of synthetic approaches was required, finally resulting in the compound PrINZ, a 7-substiututed version of the Abbott Labs Akt inhibitor A-443654.
Introduction
The serine/threonine kinase Akt (PKB) is a signaling node in many cellular processes including cell growth and survival. It is a key downstream effector of phosphoinositide 3-kinase (PI3K) and sends signals to a wide range of apoptotic and metabolic regulators including GSK3, FOXO and TSC2.1 Because the PI3K/Akt signaling pathway is dysregulated in a number of human diseases such as cancer and diabetes, modulation of the pathway is a significant therapeutic goal. Three Akt inhibitors have now advanced into late stage development or early clinical trials: MK-2206 (Merck),2 GSK-690693 (GlaxoSmithKline),3 and A-674563 (Abbott).4 As a result of the large size of the human kinome and the close structural relationship between kinases, small molecule inhibitors of kinases are rarely perfectly selective for their intended targets and often inhibit other related kinases as well. To address the challenge of specific kinase inhibition we have developed an approach which exploits the power of genetics to sensitize a single kinase to inhibition by a pyrazolopyrimidine based series of inhbitors. We have termed this approach chemical genetics and applied it to over 40 different protein kinases.5-9 In attempting to apply the approach to Akt we realized that the pyrazolopyrimidine based scaffold was not optimal for potent and selective inhibition of each Akt isoform. Here, we describe the development of a class of indazole based chemical genetic inhibitors against each isoform of Akt to overcome this limitation.
Mammalian cells contain three genes that encode three closely related and highly conserved isoforms of Akt, termed Akt1/2/3. Mouse knockout studies have uncovered distinct physiological functions for the three Akt isoforms: Akt1-deficient mice display developmental defects, Akt2-deficient mice have defects in glucose homeostasis, and Akt3-deficient mice show defects in neuronal development.10 Akt1 has also been shown to be required for ErbB2 induced mammary oncogenesis and governs breast cancer progression in vivo.11 The development of isoform selective Akt inhibitors will ultimately allow dissection of the complex biology controlled by each of these critical kinases.
Abbott labs reported the first selective Akt inhibitors based on an indazole scaffold (Fig. 1a). Our own profiling of this compound against a panel of protein kinases revealed this inhibitor also inhibits a wide variety of kinases closely related to Akt. Similarly, GSK reported GSK690693 as an ATP competitive Akt inhibitor.3 In contrast to the ATP competitive inhibitors A-443654 and GSK690693, the Merck Akt inhibitor series based on a quinoxaline scaffold binds outside of the ATP binding pocket and exhibits unusual Akt selectivity compared to other kinases.12 Merck has further refined this compound to obtain MK-2206, which is now being tested in clinical trials.2 These compounds show a remarkable ability to inhibit subsets of Akt isoforms, but absolute isoform specificity remains elusive.
Fig. 1.

(a) Structures of disclosed Akt inhibitors in clinical development. (b) Analog sensitive design strategy. Crystal structure of Akt2 with A443654 shows the native gatekeeper methionine and its proximity to the 7 position of indazole ring. Structures of PP1 derivatives commonly used to inhibit analog sensitive kinases.
We have recently reported strikingly different cellular responses to ATP competitive Akt inhibitors compared to the non-ATP site class of inhibitors.13 Our group and others have observed that ATP competitive inhibitors induce a striking and counterintuitive hyperphosphorylation of Akt itself.14 To investigate this paradoxical result we required a highly selective ATP competitive Akt inhibitor to perform a mechanistic analysis of this unusual signaling effect. Thus we decided to employ a chemical genetics strategy using Akt isoforms that have been engineered to enlarge the ATP pocket, resulting in sensitization to inactive derivatives of kinase inhibitors.15 This approach exploits a conserved, large hydrophobic residue in the kinase active site (termed the gatekeeper), which is in direct contact with the N6 amino group of ATP. By mutating the gatekeeper residue to a small residue such as glycine or alanine, mutant kinases have been engineered to accept inhibitors that cannot interact with the wild-type (wt) counterpart (Fig. 1b). Knocking-in an as-kinase (analog-sensitive) by substituting for the endogenous wt kinase in cells or organisms followed by treatment with an as-kinase specific inhibitor allows for the real-time pharmacological investigation of the role of a single kinase.16 In addition to the use of pyrazolopyrimidine1 (PP1) analogues, a versatile starting point for the development of many analog sensitive kinase inhibitors,6,17 we report here the development of inhibitors based on a completely different scaffold, that of the Abbott Akt inhibitor, A-443654.18 These molecules are more potent against Akt-as isoforms than PP1 analogues either in vitro or in vivo without disrupting Akt-wt and will allow for the detailed investigation of the role of individual Akt isoforms in normal and disease physiology.
Results and discussion
Chemical synthesis
A-443654 analogues with bulky substituents at the C-7 position predicted to be oriented toward the Akt gatekeeper residue (M227 in Akt1, M225 in Akt2, and M229 in Akt3) were synthesized by Stille coupling of stannyl pyridine Y218 with bromoindazole Y1 substituted at the C7 position followed by deprotection of the Boc group on the tryptophanol moiety18 (Scheme 1). Syntheses of bromoindazole building blocks (Y1a, Y1b and Y1i) is described in Scheme 2. Commercially available 2-alkyl-4-bromo-6-methylanilines (in the case of Y6a and Y6i), which can be prepared by bromination of commercially available 2-alkyl-6-methylaniline (in a case of Y5b), were diazotized and quenched with t-butylthiol to give diazosulfides (Y7). Basic treatment of Y7 lead to an intramolecular cyclization affording bromoindazole intermediate Y1.19 However, due to poor commercial availability of Y5 and Y6, a procedure to introduce an alkyl group at the C7 position on indazole ring was required.
Scheme 1.

(a) Pd2(dba)2, P(o-tol)3, Et3N, DMF, 90 °C, over night, (b) TFA, CH2Cl2, rt, 1 h, yield: 8–29% for 2 steps.
Scheme 2.

(a) Br2, AcOH, 0 °C, 2.5 h, yield: 88% for Y5b, (b) NaNO2, HCl, H2O, −20 °C, 30 min then tBuSH, 0 °C, 1 h, (c) tBuOK, DMSO, rt, over night, yield: 54–71% for 2 steps.
We next attempted a direct substitution of the chloride group on the indazole ring of Y3i with an alkyl group by Suzuki coupling using alkylboronic acid under Hartwig palladium catalyst conditions20 (Scheme 3). Treatment of Y3i with n-butylboronic acid in presence of Pd(dba)2 and Q-phos followed by deprotection of Boc group afforded the desired Y4d with 2% yield. Although the simplicity of this route makes it an attractive alternative for derivatizing the C7 position, because of the low yield, we decided to introduce the C7 substituent prior to the Stille coupling.
Scheme 3.

(a) nBuB(OH)2, Pd(dba)2,K3PO4, Q-Phos, toluene, 90 °C, over night, (b) TFA, CH2Cl2, rt, 1 h, yield: 2% for 2 steps.
We thus used Suzuki coupling of 2-bromo-6-methylaniline with vinylboronic acids to prepare C7-alkylsubstituted bromoindazole building blocks (Scheme 4). Suzuki coupling of vinylboronic acids with 2-bromo-6-methylaniline (Y8) followed by hydrogenation of the double-bond to give 2-alkyl-6-methylaniline (Y9), led to corresponding bromoindazoles Y1c and Y1e. However, since the commercially availability of vinylboronic acids was also poor, alternative methods were desired to prepare A-443654 analogs with more diverse substituents at the C7 position. Thus we investigated a synthetic route as shown in Scheme 5 using direct alkyl-substitution of 2-bromo-6-methylnitorobenzene (Y12) by Suzuki coupling under a Hartwig catalyst.20 Suzuki coupling of Y12 with alkylboronic acids under the catalysis of Pd(dba)2 and Q-phos, followed by reduction of the nitro group successfully afforded 2-alkyl-6-methylaniline (Y13). In this reaction, it was necessary to use 2-bromo-6-methylnitorobenzene (Y12) as a starting material instead of 2-bromo-6-methylaniline (Y8) because the nitro group activates the arylbromide and under the condition of palladium-Q-phos, the aniline group would likely be reactive. The resulting Y13 was converted to Y1 with the same procedure as shown above.
Scheme 4.

(a) R1CH=CHB(OH)2, Pd(PPh3)4, Na2CO3, DME, 85 °C, over night, (b) H2, Pd/C, EtOH, rt, over night, yield: 62–76% for 2 steps, (c) Br2, AcOH, 0 °C, 1 h, yield: 60–68%, (d) NaNO2, HCl, H2O, −20 °C, 30 min, then tBuSH, 0 °C, 1 h, (e) tBuOK, DMSO, rt, over night, yield: 71–77% for 2 steps.
Scheme 5.

(a) R2B(OH)2, Pd(dba)2, K3PO4, Q-Phos, toluene, 100 °C, 2.5 h, (b) H2, Pd/C, EtOH, rt, over night, (c) Br2, AcOH, 0 °C, 1 h, yield: 24–63% for 3 steps, (d) NaNO2, HCl, H2O, −20 °C, 30 min, then tBuSH, 0 °C, 1 h, (e) tBuOK, DMSO, rt, over night, yield: 48–81% for 2 steps.
Analogues of A-443654 having a methyl group at the C3 position on the indazole ring were synthesized as depicted in Scheme 6 and Scheme 7. An intermediate with an ethyl substituent at C7 position (Y1j) was synthesized with the same procedure as shown above owing to its symmetrical structure (Scheme 6). Due to a lack of ring closure selectivity in an asymmetrical azosulfide, an alternative synthesis for analogues with a larger alkyl group substitution at C7 position was investigated (Scheme 7). Wittig reaction of commercially available aldehyde Y18 afforded a styrene intermediate which was reduced to linear (Y19k, R5 = H) or branched (Y19l, R5 = Me) alkyl chain with hydrogenation or diimide reduction, respectively.21 In the hydrogenation to produce Y19k, use of THF as a solvent was essential and reaction in either ethyl acetate or ethanol did not give the product. In the case of tri-substituted styrene reduction to give Y19l, palladium-carbon hydrogenation in any solvent did not yield the desired product. To avoid the use of a more reactive catalyst for hydrogenation in order not to lose the bromide functional group at 4-position, diimide reduction was tested, affording the saturated alkylbenzene (Y19l) without loss of the bromide. Fluorine group-directed ortho lithiation22 followed by treatment with DMF gave formylbenzene (Y20), which was converted to methylketone (Y21) by Grignard reaction followed by oxidation of the resulting alcohol. Ring closure using excess amount of hydrazine afforded desired 3-methylindazole (Y1k and Y1l) but the yield of the reaction was poor because of the sterically hindered nature of the fluorine center.
Scheme 6.

(a) NaNO2, HCl, H2O, −20 °C, 30 min, then tBuSH, 0 °C, 1 h, (b) tBuOK, DMSO, rt, over night, yield: 86% for 2 steps.
Scheme 7.

a) [Ph P-CHMeR5]+ 3 Br−, tBuOK / THF, 2–3 h, (b) H2, Pd/C, THF, rt, over night (R5 = H) or TsNH2NH2, THF, 100 °C, 3 h (R5 = Me), (c) LDA, THF, −78 °C, 1 h then DMF–THF, 78 °C, 1 h, yield: 47–53% for 3 steps, (d) MeMgBr, Et2O, 0 °C, 30 min, (e) MnO2, dioxane, 90 °C, 9 h, yield: 63–88% for 2 steps, (f) NH2NH2, DMF, 120 °C, 24 h, yield: 7–11%.
Analogue sensitive Akt generation and evaluation
To establish the analogue sensitive system for all Akt isoforms, mutations enlarging the size of the ATP-binding pocket were introduced by substituting the gatekeeper methionine (M) with glycine (G) or alanine (A) (i.e. M227A/M225A/M229A for Akt1/2/3-as1 and M227G/M225G/M229G for Akt1/2/3-as2). In vitro immunoprecipitation kinase assays revealed that both Ala and Gly mutants of all three isoforms of Akt-as retained approximately 30% of the activity of the corresponding Akt-wt isoforms (Fig. 2a). The level of Akt1/2/3-as1/2 activity in cells was also determined by measurement of GSK-3β Ser-9 phosphorylation. Akt constructs containing a c-Src myristoylation recognition sequence (myr-HA-Akt-as) are constitutively membrane localized and thus constitutively active without growth factor stimulation. As expected, expression of myr-HA-Akt1/2/3-as1/2 or myr-HA-Akt1/2/3-wt in HEK293 cells resulted in elevated phosphorylation of the Akt substrate GSK3β at Ser9 (Fig. 2b). Elevation of GSK3β phosphorylation by myr-HA-Akt1/2/3-as1/2 transfection was comparable to that by myr-HA-Akt1/2/3-wt transfection, confirming the cellular activity of each Akt-as isoform is similar to the corresponding activity of Akt-wt isoforms despite the significantly lower measured in vitro activity.
Fig. 2.

(a) In vitro kinase activity of wt and analog sensitive (as) Akt kinases. (b) in vivo kinase activity of wt and as kinase activity. Myristoylated Akt was transfected into HEK293T cells and blotted for Akt substrates.
Development of Akt-as selective inhibitors
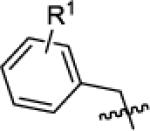
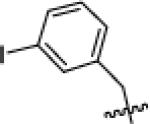
With suffciently active Akt-as isoforms in hand, we next sought to develop Akt-as specific inhibitors that do not perturb endogenous Akt-wt and related kinases. The pyrazolopyrimidine1 (PP1) scaffold has proven to be a versatile starting point for the development of many analog sensitive kinase inhibitors.15 Over 60% of the analog sensitive kinases we have characterized are potently (<25 nM IC50 @ 1 μM ATP) inhibited by one or more of the following pyrazolopyrimidine analogs (1-NM-PP1, 1-NaPP1, or 3-MB-PP1) (C.Z. and K.M.S., unpublished data). In the case of Akt1-as2 however, none of these three widely used inhibitors were potent ligands for the engineered kinase although 1-NM-PP1 exhibited the most promising level of inhibition (IC50 = 61 nM). A more diverse series of benzyl substituted pyrazolopyrimidine analogues were screened against Akt1/2/3-as1/2 at a single concentration (1 uM), leading to the discovery of some modest hits. Most of the hits from the screening have a substituent at the meta position on the benzyl group as often seen for SAR against other as-kinases. Determination of IC50 values of these hits (Table 1) led to the identification of the most potent analogue: 3-iodobenzyl PP1 (X1b, 3-IB-PP1). This molecule inhibits Akt1/2/3-as1/2 with good potency, without inhibiting Akt1/2/3-wt (Table 1). The in vitro potency and selectivity of 3-IB-PP1 for Akt1-as1/2 (IC50 = 18 nM for as1, 28 nM for as2) vs. Akt1-wt (IC50 > 10 000 nM) provides a valuable tool for cellular studies of Akt1 specific functions. In contrast, the potency of 3-IB-PP1 for Akt2-as1/2 (IC50 = 1100 nM for as1, 240 nM for as2) and Akt3-as1/2 (IC50 = 76 nM for as1, 120 nM for as2) is insuffcient for an ATP-competitive kinase inhibitor.23 Although 3IB-PP1 exhibited suitable potency against Akt1-as, the 10-fold drop in potency upon application to Akt2 or Akt3-as alleles precludes their use to study these important Akt isoforms.
Table 1.
IC50 values of pyrazolopyrimidine series X1 against wt and as Akt1/2/3
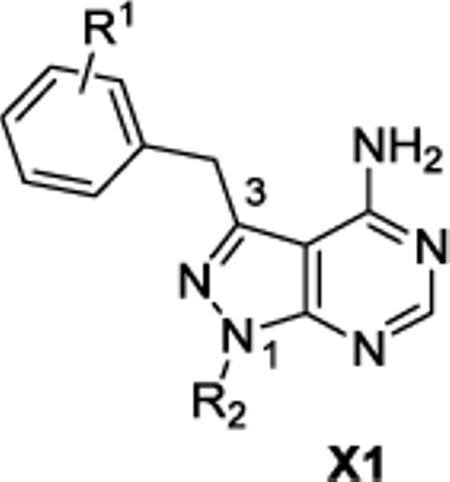
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Akt variants IC50 (nM) |
||||||||||
| Compd | R2 | Akt1-as1 | Akt1-as2 | wtAkt1 | Akt2-as1 | Akt2-as2 | wtAkt2 | Akt3-as1 | Akt3-as2 | wtAkt3 | |
| X1a |
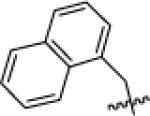
|
tBu | 71 | 61 | >10 000 | 1200 | 280 | >10 000 | 150 | 110 | >10 000 |
| X1b(3-IB-PP1) |
|
tBu | 18 | 28 | >10 000 | 1100 | 240 | >10 000 | 76 | 120 | >10 000 |
| X1c |
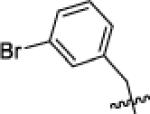
|
tBu | 32 | 60 | >10 000 | 520 | 380 | >10 000 | 78 | 90 | >10 000 |
| X1d |
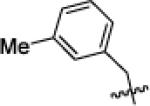
|
tBu | 67 | 84 | >10 000 | N.D. | 360 | >10 000 | 63 | 150 | >10 000 |
| X1e |
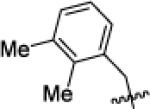
|
tBu | 71 | 100 | >10 000 | 1000 | 320 | >10 000 | 72 | 84 | >10 000 |
| X1f |
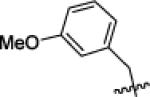
|
tBu | 61 | 180 | >10 000 | 1100 | 860 | >10 000 | 130 | 340 | >10 000 |
| X1g |
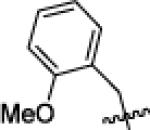
|
tBu | 290 | 660 | >10 000 | 1900 | 4900 | >10 000 | 260 | 800 | >10 000 |
| X1h |
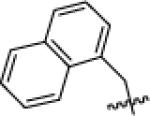
|
tPr | 120 | 140 | >10 000 | N.D. | 1200 | >10 000 | 250 | 110 | >10 000 |
| X1i |
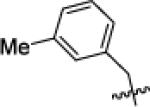
|
Cyclo Pen | 81 | 110 | >10 000 | N.D. | 470 | >10 000 | 82 | 150 | >10 000 |
To obtain a more effective Akt-as1/2 specific inhibitor, we turned to a scaffold based on the known pan-Akt inhibitor A-443654. The parent compound exhibits unusual potency against Akt (Ki = 160 pM for Akt1) and a co-crystal structure was available to guide derivative design. Evaluation of the crystal structure24 of Akt2 with A-443654 suggested the C7 position on the indazole ring of A-443654 to be a promising position for introducing large substituents that would clash with the gatekeeper methionine of Akt-wt (Fig. 1b). SAR studies of various C7 substituted A-443654 analogues were carried out. Since the distance between the methylthio group of the gatekeeper methionine and C7-carbon on A-443654 is 3.7 Å (resolution: 2.30 Å), a small alkyl group was substituted for hydrogen on the C7 position on the indazole ring. We hypothesized a neutral group such as a hydrocarbon chain was a suitable substitution in order not to disrupt already existing H-bonds. Substitution at the C6 position was also considered but we were concerned about changing the dihedral angle between the indazole ring and the pyridine ring.
We first synthesized analogues without a methyl group at the C3 position of the indazole moiety because of synthetic accessibility of this series (R2 = H). A-443654 analogues having a methyl group at C3 position are previously reported to be ~5-fold more potent against Akt than the corresponding 3-hydrogen-derivartives.18 We thus planned to introduce a 3-methyl substituent on 3-hydrogen-analogues having an optimized C7 substituent. As shown in Table 2, introduction of a methyl group (Y4a) to the C7 position does not inhibit Akt-wt, suggesting a small substituent such as a methyl group is suffcient to exhibit orthogonality against Akt-wt presumably by clashing with the gatekeeper methionine. Moreover Y4a weakly inhibited Akt-as isoforms.
Table 2.
IC50 values of indazole series Y4 against wt and as Akt1/2/3
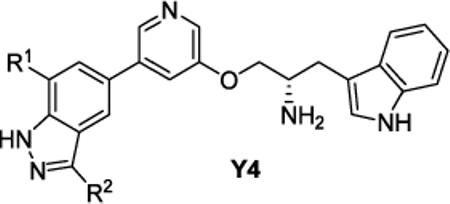
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Akt variants IC50 (nM) |
||||||||||||
| Compd | Scheme | R1 | R2 | Akt1-as1 | Akt1-as2 | wtAkt1 | Akt2-as1 | Akt2-as2 | wtAkt2 | Akt3-as1 | Akt3-as2 | wtAkt3 |
| Z(A-443654) | 1 | H– | Me– | ND | ND | 3 | ND | ND | 30 | 30 | ND | 51 |
| Y4a | 2 | Me– | H– | 240 | 100 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| Y4b | 2 | Et– | H– | 860 | 25 | 570 | >1000 | 930 | >1000 | >1000 | 770 | >1000 |
| Y4c | 4 | nPr– | H– | 330 | 6 | >1000 | >1000 | 110 | >1000 | >1000 | 63 | >1000 |
| Y4d | 3 | nBu– | H– | 350 | 4 | >1000 | >1000 | 43 | >1000 | 530 | 33 | >1000 |
| Y4e | 5 | iBu– | H– | >1000 | 6 | >1000 | >1000 | 19 | >1000 | >1000 | 28 | >1000 |
| Y4f | 4 | nPen– | H– | 380 | 26 | >1000 | >1000 | 58 | >1000 | 750 | 34 | >1000 |
| Y4g | 5 | iPen– | H– | 270 | 17 | >1000 | >1000 | 35 | >1000 | 300 | 25 | >1000 |
| Y4h | 5 | PhCH2CH2– | H– | 560 | 34 | >1000 | >1000 | 100 | >1000 | 630 | 50 | >1000 |
| Y4i | 2 | Cl– | H– | 320 | 87 | 720 | >1000 | 700 | >1000 | >1000 | >1000 | >1000 |
| Y4j | 6 | Et– | Me– | 270 | 17 | 740 | >1000 | 200 | >1000 | 900 | 110 | >1000 |
| Y4k (PrlDZ) | 7 | nPr– | Me– | 56 | 2.8 | >1000 | >1000 | 31 | >1000 | 360 | 13 | >10 000 |
| Y4l | 7 | iBu– | Me– | 720 | 6.6 | >1000 | >1000 | 6.7 | >1000 | >1000 | 25 | >10 000 |
Encouraged by these results, we next synthesized 3-hydrogen analogues with a larger alkyl substituent at C7 position on the indazole ring (Y4b-Y4i). Assays against Akt-as isoforms revealed that these analogues potently inhibit Akt-as2. Interestingly the derivatives do not show strong inhibition against Akt-as1 isoform. It remains unclear why this reduced potency exists for the Akt-as1 isoform. However, the methyl side chain of the mutated alanine residue at gatekeeper position (as1) may clash with the substituent at the C7 position on the inhibitors. Compounds having relatively larger alkyl groups at C7 position potently inhibit Akt-as2 and surprisingly, even compounds with a very large substituent such as phenethyl (Y4h) potently inhibit Akt-as2, suggesting existence of a large pocket behind the gatekeeper. Maximum potency occurred with C3–C5 alkyl substituents at C7 (Y4c-Y4g). As anticipated based on the close proximity of these substituents to the gatekeeper sidechain, these analogues did not inhibit Akt-wt.
Finally we attempted to gain additional potency by exploiting the known enhancement of Akt potency by derivatization at the C3 position. Due to the preference for rigid substituents, we chose X4b (Et), X4c (nPr) and X4e (iBu) for further derivatization at C3 position. As anticipated based on SAR studies by the Abbott group, introduction of a C3-methyl substituent dramatically improved (ca. 10-fold) the binding affnity to afford compounds showing excellent potency against all isoforms of Akt-as2 (low-nanomolar-range IC50 against all isoforms of Akt-as2) (Y4k and Y4l). We chose Y4k as the optimal inhibitor for each analog specific Akt isoform and named it PrIDZ (Propylindazole).
Specificity and cellular effects of Akt-as selective inhibitors
With inhibitors PrIDZ and 3-IB-PP1 optimized against Akt1/2/3-as, we sought to confirm their orthogonality against the human kinome both in vivo and In vitro. To assess their specificity for Akt-as, we screened both compounds against 191 human kinases and disease mutants using the SelectScreen® kinase profiling service from Invitrogen (Supplementary Table 1). Compared to the parent compound, A-443654, which we previously reported inhibited 47/220 kinases at 1 μM,13 the PrIDZ analog was far more selective. Only 10 kinases were inhibited at greater than 80% at 5 μM and none were found in the canonical PI3K → Akt → mTOR signaling pathway. 3-IB-PP1 was similarly selective, only inhibiting 9 kinases at 5 μM and only one of these kinases was also inhibited by PrIDZ (Casein Kinase 1d, CK1d). Together these results validate our analog sensitive strategy and confirm that any results observed with the two inhibitors are likely due to on-target effects against Akt-as.
We next validated the ability of compounds PrIDZ and 3-IB-PP1 to effectively and potently inhibit Akt-as2 kinases expressed in cells. We previously validated the cellular orthogonality of our inhibitors against Akt-wt, by measuring in vivo inhibition of downstream Akt targets by our engineered compounds and the parent A-443654 and observed no cross-reactivity.13 The data confirm that these as-specific inhibitors are suffciently selective against Akt-wt, and that as predicted from the In vitro screening, off-target effects of these compounds do not have observable effects on upstream or downstream of Akt.
Cellular effects of these inhibitors (PrIDZ and 3-IB-PP1) against Akt-as2 isoforms were assessed in HEK 293 cells transfected with the constitutively activated myr-HA-Akt1/2/3-as2. Treatment of cells transfected with myr-HA-Akt1/2/3-as2 with various concentrations of PrIDZ and 3-IB-PP1 was performed to analyze the phosphorylation status of GSK3β (Fig. 3) (Akt1 previously reported in ref. 13). Both inhibitors decreased the phosphorylation level of Ser9 on GSK3β in a dose-dependent manner and PrIDZ was particularly effective at inhibiting Akt-as2, validating In vitro inhibitory activities (IC50 values could not be independently determined from western blots). The data suggested that the treatment of cells expressing Akt-as2 isoforms with PrIDZ is an effective means of potently inhibiting each of the three Akt isoform functions in cells.
Fig. 3.

in vivo activity of engineered inhibitors 3-IB-PP1 and PrIDZ in HEK 293 cells transfected with myristoylated analog sensitive Akt2 (a) or Akt3 (b).
Conclusion
In an effort to develop highly specific inhibitors of each individual human kinase we have developed a widely used chemical genetic method that exploits the conserved nature of the gatekeeper residue across the kinome. This method has been applied widely using a pyrazolopyrimidine based scaffold for inhibition of kinases engineered to contain a Gly or Ala gatekeeper residue. It is perhaps not surprising that a single scaffold would be unable to potently bind all engineered kinases, since the PP1 scaffold was first identified by Pfizer as an inhibitor of the Src tyrosine kinase family.15 In attempting to develop a potent inhibitor of the serine threonine kinase Akt, we found that the pyrazolopyrimidine scaffold could be tuned to inhibit analog sensitive Akt1, but we were unable to potently inhibit the Akt2 or Akt3 isoforms with this inhibitor class. In order to achieve more potent inhibition of all Akt isoforms we turned to the Akt inhibitor A-443654 from Abbott based on the indazole scaffold. Modifications at the C-7 position rendered the inhibitor unable to bind to wt Akt isoforms yet while retaining the ability to potently inhibit each of the three Akt-as isoforms.
These studies provide an additional avenue to development of chemical genetic inhibitors of a wide variety of kinases. The extensive expansion of drug discovery efforts focused on protein kinases have afforded many new scaffolds that as our work shows can be relatively easily repurposed for the development of mutant specific kinase inhibitors. In addition to the development of potent inhibitors, there is an additional benefit to extension beyond the pyrazolopyrimidine scaffold. As with any small molecule used in complex biological systems, one must always be concerned with off-target effects due to binding of the particular molecule to unintended targets. This concern is greatly ameliorated by the use of a chemical genetic system, which allows side-by-side comparison of cells containing a single point mutation in a single target within an isogenic cell line. Nonetheless, another type of control based on changing the inhibitor structure is beneficial. The use of a second chemical series with comparable In vitro specificity for the target of interest (Akt-as) can be used in cellular experiments. If the cellular effects of the two structurally distinct compounds are similar, then one can gain increasing confidence in effects being on-target. The key assumption is that compounds with vastly different structure may share one target in common but are largely unlikely to share a common off target effect.
The development of this chemical genetics approach complements traditional genetic techniques.25,26 An analog sensitive kinase and inhibitor can phenocopy the selectivity of genetic knockouts while being reversible and temporally controllable. Heat sensitive or drug inducible knockout systems do allow for control of gene expression but both are binary in nature and neither offer the simple and rapid titration of kinase activity possible with chemical genetics. Generation of analog sensitive model organisms can allow for the deconvolution of kinase function required in development versus adult homeostasis.16 Many of the isoform specific effects in Akt knockout mice were gross developmental defects that might differ significantly from knocking out the kinase in adults. Generation of Akt-as mice would allow for the elucidation of the role of Akt isoforms in adult biology and may more directly inform upon their contributions to cancer where Akt dysregulation is clinically implicated.
The availability of potent and specific Akt inhibitors that bind in the ATP site allowed recent elucidation of a new form of kinase inhibition. Kinase dead Akt mutants treated with selective inhibitors became hyperphosphorylated independent of downstream kinase activity.13 This demonstrated a new mode of kinase activation whereby conformational changes induced by inhibitor binding altered the kinase's accessibility to being phosphorylated by upstream kinases. This observation illustrates the complexity inherent in signaling networks and the frequent unexpected feedback loops present in these systems.
Experimental
Buffer solutions
Buffer A: 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% (v/v) Triton X, 2.5 mM sodium pyrophosphate, 1 mM β-glycerphosphate, Complete protease inhibitor cocktail (Roche Applied Sciences), phosphatase inhibitor cocktail 1 (Sigma-Aldrich), phosphatase inhibitor cocktail 2 (Sigma-Aldrich), and 20 nM microcystin LR (Calbiochem). Buffer B: 25 mM Tris (pH 7.5), 10 mM MgCl2, 5 mM β-glycerphosphate, 0.1 mM sodium orthovanadate and 2 mM DTT.
In vitro kinase assays
HEK293T cells were plated in six-well dishes and transfected at approximately 90% confluence with the appropriate pcDNA3-myr-HA-Akt1/2/3-as1/2 plasmid using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's instructions. After 48 h cells were washed and detached with Ca2+, Mg2+ free ice cold PBS and pelleted by centrifugation at 14 000g for 20 min at 4 °C. Pellets were lysed using Buffer A then immunoprecipated overnight at 4 °C by using anti-HA Affnity Matrix (Roche Applied Science) pre-blocked with 1% BSA in PBS. Immunoprecipitates were washed twice with Buffer A and then twice with Buffer B before performing the In vitro kinase assay. Serial dilutions of individual inhibitors were incubated with immunoprecipitated myr-HA-Akt constructs, 30 μM synthetic peptide GRPRTSSFAEG (Crosstide, Upstate), 1.7% DMSO, 28 nM (2.5 μCi) [γ-32P]ATP (3000 Ci/mmol, Perkin Elmer Life Science) and 50 μM ATP n Buffer B for 45 min. Reaction mixtures were spotted into phosphocellulose paper (Whatman) and washed 5× with 1% H3PO4. Membranes were exposed to a phosphoimager and scanned on Typhoon analyzer (Amersham Biosciences) to quantify the radioactivity with SPOT software.27
Immunoblots
After appropriate stimulation and/ or drug treatment HEK 293 cells were pelleted and lysed as described for the In vitro kinase assays. Cell lysates were subjected to SDS/PAGE and transferred to nitrocellulose membranes (Bio-Rad Laboratories). Blots were blocked with 5% (w/v) skim milk in 0.1% (v/v) Tween-20/Tris-buffered Saline (TBST). They were then probed with various primary antibodies in 5% BSA (w/v) in TBST according to manufacture's instructions (All primary antibodies from Cell Signaling Technology). Primary antibodies were detected by appropriate peroxidase-conjugated IgGs (Pierce Biotechnology or Santa Cruz Biotechnology) in 5% (w/v) BSA/TBST and protein signal was visualized using enhanced chemoluminescence (Pierce Biotechnology) by exposure to CL-X Posure film (Pierce Biotechnology).
Plasmid constructs
pcDNA3-myr-HA-Akt1/2/3 were kind gifts of W. Sellers via Addgene (Akt1/2/3)-Addgene plasmid 9008/9016/9017). Point mutations in Akt for the gatekeeper were introduced by a modified method of Stratgene QuikChange® site-directed mutagenesis (SQC).28 Generation of Akt1/2/3-as2 (M → G gatekeeper mutation) was described previously in the supplemental methods to ref. 13. Akt1/2/3-as1 (M227A, M229A and M225A respectively) were generated using the same methods with the following Quikchange primers. M227A in Akt1 (fwd: 5′ TT GTC gcc GAG TAC GCC AAC GGG GGC GAG CTG TTC, rev: 5′ TA CTC ggc GAC AAA GCA GAG GCG GTC GTG GGT CTG); M229A in Akt2 (fwd: 5′ TT GTG gcc GAG TAT GCC AAC GGG GGT GAG CTG TTC, rev: 5′ TA CTC ggc CAC AAA GCA CAG GCG GTC GTG GGT CTG); M225A in Akt3 (fwd, 5′ GT TTT GTG gcc GAA TAT GTT AAT GGG GGC GAG CTG, rev 5′ C ATA TTC ggc CAC AAA ACA CAA ACG GTC TTT TGT C).
Accession code
Protein Data Bank: The cocrystal structure of Akt2 with A-443654 was generated in a previous study and deposited under accession code 2JDR.
Chemical synthesis
Unless otherwise noted, all reactions were performed under argon and stirred magnetically in oven-dried glassware fitted with rubber septa. All 1H and 13C NMR were recorded on a Varian Innova 400 spectrometer at 400 MHz and 100 MHz, respectively. 1H chemical shifts are reported as chemical shifts (δ) in ppm, multiplicity (s: singlet; d: doublet; t: triplet; dd: doublet of doublets; m: multiplet; br: broad), number of protons and coupling constant (J) in hertz (Hz). 13C NMR data are reported as chemical shifts (δ) in ppm and coupling constant (J) in hertz (Hz) for C-F coupling. Low resolution electrospray ionization LCMS (LR ESI-LCMS) and liquid chromatography retention times were recorded on a Waters Micromass ZQ equipped with a Waters 2695 Separations Module using an XTerra MS C18 3.5 μm column (Waters). Retention times were obtained using an initial flow rate of 1 mL/min of 95% A (H2O with 0.1% formic acid) and 5% B (CH3CN with 0.1% formic acid) for 0.5 min followed by a linear gradient with a flow rate of 0.5 mL/min of 95% A and 5% B to 5% A and B in 4.5 min followed by 0.5 min at of 5% A and 95% B. High-resolution electron impact mass spectra were recorded on a MicoMass VG70E spectrometer at the University of California-San Francisco center for Mass Spectrometry. Preparative HPLC was performed on a Varian ProStar solvent delivery system equipped with a Zorbax 300-SB C18 column using a CH3CN (0.1% TFA)/H2O (0.1% TFA) gradient (0-100%) as the mobile phase and monitored by UV at λ = 254 nm. Automated silica gel chromatography was performed on an AnaLogix IntelliFlash 280 system using Analogix SuperFlash Sepra Si 50 columns and monitored by UV at λ = 254 nm. Unless otherwise noted, all reagents and solvents were obtained from commercial suppliers and were used without further purification.
A-443654 derived analogs
5-bromo-7-methyl-1H-indazole (Y1a)
To a suspension of aniline Y6a (2.0 g, 10.0 mmol) in 24% hydrochloric acid (5.5 ml) was added a solution of sodium nitrite (730 mg, 10.5 mmol) in water (1.6 ml) dropwise at −20 °C. After stirring for 30 min at −20 °C, the mixture was buffered to ca. pH 5 with solid sodium acetate. The mixture was added to a solution of tert-butylthiol (1.10 ml, 10.0 mmol) in ethanol at 0 °C and stirred at rt for 1 h. After dilution with ethyl acetate, the organic layer was and then dried and concentrated to give diazosulfide Y7a (3.6 g). A solution of Y7a (3.6 g) in DMSO was added potassium tert-butoxide (9.9 g, 88.2 mmol) at 0 °C and the mixture was stirred over night at rt. The reaction was quenched with 1 M HCl aq. at 0 °C and diluted with ethyl acetate. The organic layer was washed with water and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-ethyl acetate gradient) to afford Y1a (1.5 g, 7.11 mmol, 71% for 2 steps). 1H NMR (400 MHz, CDCl3) λ 2.60 (s, 3H), 7.34 (s, 1H), 7.78 (s, 1), 8.09 (s, 1H). LR ESI-LCMS m/z calculated 211, 213, found 211, 213 [M+H]+.
5-bromo-7-ethyl-1H-indazole (Y1b)
To a suspension of aniline Y5b (2.0 g, 14.8 mmol) in acetic acid (8.0 ml) was added a solution of Br2 (800 μl, 15.5 mmol) in acetic acid (8 ml) dropwise at 0 °C. After stirring for 2.5 h at 0 °C, the mixture was diluted with ethyl acetate and water, and then neutralized with 6 M NaOHaq. The organic layer was washed with sat. Na2S2O3aq. and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-ethyl acetate gradient) to afford Y6b (2.8 g, 13.1 mmol, 88%) of suffcient purity to carry on to the next step. Compound Y1b was synthesized from Y6b (2.8 g, 13.1 mmol) according to the procedure for Y1a by substituting Y6b for Y6a. Yield: 1.9 g (8.4 mmol, 64% for 2 steps). 1H NMR (400 MHz, CDCl3) λ 1.34 (t, J = 5.8 Hz, 3H), 2.88 (t, J =7.6Hz,2H),7.26(s,1H),7.71(s,1H),7.99(s,1H).LR ESI-LCMS m/z calculated 225, 227, found 225, 227 [M+H]+.
5-bromo-7-chloro-1H-indazole (Y1i)
Compound Y1i was synthesized from Y6i (3.2 g, 14.4 mmol) according to the procedure for Y1a by substituting Y6i for Y6a. Yield: 1.8 g (7.8 mmol, 54% for 2 steps). 1H NMR (400 MHz, DMSO-d6) λ 7.65 (s, 1H), 8.02 (s, 1H), 8.19 (s, 1H). LR ESI-LCMS m/z calculated 231, 233, found 231, 233 [M+H]+.
4-bromo-2-methyl-6-propylbenzenamine (Y10c)
To a solution of bromobenzene Y8 (3.5 g, 18.8 mmol) in DME (94 ml) and water (23 ml) were added Pd(PPh3)4 (1.2 g, 1.0 mmol), Na2CO3 (2.0 g, 18.8 mmol) and trans-1-propen-1-ylboronic acid (2.03 g, 23.6 mmol). After stirring over night at 85 °C, the mixture was diluted with ethyl acetate. The organic layer was washed with water and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-ethyl acetate gradient) to afford the styrene analogue (2.5 g) of suffcient purity to carry on to the next step. To a solution of the styrene analogue (2.5 g) in ethanol (40 ml) was added 20% Pd-C wet (500 mg) and the mixture was stirred over night at rt under H2 atmosphere. After filtration over Celite, the solvent was removed in vacuo to give crude Y9c (2.1 g, 14.3 mmol, 76% for 2 steps). The crude product Y9c was used for the next step without further purification. To a solution of Y9c (2.6 g, 17.4 mmol) in acetic acid (13 ml) was added a solution of Br2 (940 μl, 18.3 mmol) in acetic acid (13 ml) dropwise at 0 °C. After stirring for 1 h at 0 °C, the mixture was diluted with ethyl acetate and water, and then neutralized with 6 M NaOH aq. The organic layer was washed with sat. Na2S2O3 aq. and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-ethyl acetate gradient) to afford Y10c (2.7 g, 11.9 mmol, 68%). 1H NMR (400 MHz, CDCl3) δ 0.98 (t, J = 7.4 Hz, 3H), 1.59–1.65 (m, 2H), 2.16 (s, 3H), 2.44 (t, J = 7.6 Hz, 2H), 7.04 (d, J = 4.4 Hz, 2H). LR ESI-LCMS m/z calculated 228, 230, found 228, 230 [M+H]+.
5-bromo-7-propyl-1H-indazole (Y1c)
Compound Y1c was synthesized from Y10c (2.7 g, 11.8 mmol) according to the procedure for Y1a by substituting Y10c for Y6a. Yield: 2.0 g (8.37 mmol, 71% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 1.04 (t, J = 7.4 Hz, 3H), 1.77-1.85 (m, 2H), 2.91 (t, J =7.6 Hz, 2H), 7.35 (s, 1H), 7.79 (s, 1H), 8.10 (s, 1H). LR ESI-LCMS m/z calculated 239, 241, found 239, 241 [M+H]+.
4-bromo-2-methyl-6-pentylbenzenamine (Y10f)
Compound Y10f was synthesized from Y8 (5.5 g, 28.1 mmol) according to the procedure for Y10c by substituting 1-penten-1-ylboronic acid for trans-1-propen-1-ylboronic acid. Yield: 2.7 g (10.4 mmol, 37% for 3 steps). 1H NMR (400MHz, CDCl3) δ 0.91 (d, J = 6.4 Hz, 3H), 1.36 (m, 4H), 1.60 (m, 2H), 2.15 (s, 3H), 2.44 (t, J = 7.8 Hz, 2H), 7.05 (s, 2H). LR ESI-LCMS m/z calculated 256, 258, found 256, 258 [M+H]+.
5-bromo-7-pentyl-1H-indazole (Y1f)
Compound Y1f was synthesized from Y10f (500 mg, 1.95 mmol) according to the procedure for Y1a by substituting Y10f for Y6a. Yield: 400 mg (1.50 mmol, 77% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 0.91 (t, J = 7.0 Hz, 3H), 1.39 (m, 4H), 1.76 (m, 2H), 2.85 (t, J = 7.8 Hz, 2H), 7.28 (s, 1H), 7.75 (s, 1H), 8.01 (s, 1H). LR ESI-LCMS m/z calculated 267, 269, found 267, 269 [M+H]+.
4-bromo-2-isobutyl-6-methylbenzenamine (Y14e)
To a solution of nitrobromobenzene Y12 (4.0 g, 18.5 mmol) in toluene (80 ml) were added bis(dibenzylideneacetone)palladium(0) (106 mg, 0.185 mmol), 1,2,3,4,5-pentaphenyl-1′-(di-tert-butylphosphino)ferrocene (285 mg, 0.37 mmol), K3PO4 (7.8 g, 37.0 mmol) and isobutylboronic acid (2.26 g, 22.2 mmol). After stirring for 2.5 h at 100 °C, the mixture was diluted with ethyl acetate. The organic layer was washed with 1 M HCl aq. and 1 M NaOH aq., and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-ethyl acetate gradient) to afford the dialkylnitrobenzene analogue (3.5 g) of suffcient purity to carry on to the next step. To a solution of the dialkylnitrobenzene analogue (3.5 g) in ethanol (40 ml) was added 20% Pd-C wet (600 mg) and the mixture was stirred over night at rt under H2 atmosphere. After filtration over Celite, the solvent was removed in vacuo to give crude Y13e (2.1 g). The crude product Y13e was used for the next step without further purification. To a solution of Y13e (2.1 g, 14.1 mmol) in acetic acid (8 ml) was added a solution of Br2 (800 μl, 15.5 mmol) in acetic acid (8 ml) dropwise at 0 °C. After stirring for 1 h at 0 °C, the mixture was diluted with ethyl acetate and water, and then neutralized with 6 M NaOH aq. The organic layer was washed with sat. Na2S2O3 aq. and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-ethyl acetate gradient) to afford Y14e (1.06 g, 4.38 mmol, 24% for 3 steps). 1H NMR (400 MHz, CDCl3) δ 0.95 (d, J = 6.4 Hz, 6H), 1.92 (m, 1H), 2.14 (s, 3H), 2.34 (d, J = 7.2 Hz, 2H), 7.00 (s, 1H), 7.06 (s, 1H). LR ESI-LCMS m/z calculated 242, 244, found 242, 244 [M+H]+.
5-bromo-7-isobutyl-1H-indazole (Y1e)
Compound Y1e was synthesized from Y14e (1.06 g, 4.38 mmol) according to the procedure for Y1a by substituting Y14e for Y6a. Yield: 900 mg (3.56 mmol, 81% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 0.98 (d, J = 6.8 Hz, 6H), 2.06 (m, 1H), 2.72 (d, J = 7.6 Hz, 2H), 7.25 (s, 1H), 7.76 (s, 1H), 8.01 (s, 1H). LR ESI-LCMS m/z calculated 253, 255, found 253, 255 [M+H]+.
4-bromo-2-isopentyl-6-methylbenzenamine (Y14g)
Compound Y14g was synthesized from Y12 (4.0 g, 18.5 mmol) according to the procedure for Y14e by substituting isopentylboronic acid for isobutylboronic acid. Yield: 3.0 g (11.7 mmol, 63% for 3 steps). 1H NMR (400 MHz, CDCl3) δ 0.90 (d, J = 6.4 Hz, 6H), 1.50 (m, 2H), 1.65 (m, 1H), 2.52 (s, 3H), 2.81 (t, J = 8.2 Hz, 2H), 7.20 (s, 2H). LR ESI-LCMS m/z calculated 256, 258, found 256, 258 [M+H]+.
5-bromo-7-isopentyl-1H-indazole (Y14g)
Compound Y1g was synthesized from Y14g (3.0 g, 11.7 mmol) according to the procedure for Y1a by substituting Y14g for Y6a. Yield: 2.0 g (7.49 mmol, 64% for 2 steps). 1H NMR (400 MHz, CDCl3) d 1.01 (d, J = 6.4 Hz, 6H), 1.71 (m, 3H), 3.02 (m, 2H), 7.42 (s, 1H), 7.82 (s, 1H), 8.19 (s, 1H). LR ESI-LCMS m/z calculated 267, 269, found 267, 269 [M+H]+.
4-bromo-2-methyl-6-phenethylbenzenamine (Y14h)
Compound Y14h was synthesized from Y12 (4.0 g, 18.5 mmol) according to the procedure for Y14h by substituting phenethylboronic acid for isobutylboronic acid. Yield: 2.0 g (6.90 mmol, 37% for 3 steps). 1H NMR (400 MHz, CDCl3) δ 2.15 (s, 3H), 2.73–2.77 (m, 2H), 2.89–2.94 (m, 2H), 7.08 (s, 2H), 7.19–7.33 (m, 5H).
5-bromo-7-phenethyl-1H-indazole (Y1h)
Compound Y1h was synthesized from Y14h (2.0 g, 6.90 mmol) according to the procedure for Y1a by substituting Y14h for Y6a. Yield: 1.0 g (3.32 mmol, 48% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 3.03–3.07 (m, 2H), 3.14–3.18 (m, 2H), 7.14–7.15 (m, 2H), 7.24–7.29 (m, 4H), 7.75 (s, 1H), 7.95 (s, 1H). LR ESI-LCMS m/z calculated 301, 303, found 301, 303 [M+H]+.
5-bromo-7-ethyl-3-methyl-1H-indazole (Y1j)
Compound Y1j was synthesized Y16 (2.0 g, 8.77 mmol) according to the procedure for Y1a by substituting Y16 for Y6a. Yield: 1.8 g (7.53 mmol, 86% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 1.35 (t, J = 7.4 Hz, 3H), 2.54 (s, 3H), 2.86 (q, J = 7.6 Hz, 2H), 7.26 (s, 1H), 7.65 (s, 1). LR ESI-LCMS m/z calculated 239, 241, found 239, 241 [M+H]+.
5-bromo-2-fluoro-3-propylbenzaldehyde (Y20k)
To a solution of ethyltriphenylphosphonium bromide (16.6 g, 44.8 mmol) in THF (230 ml) was added potassium tert-butoxide (5.0 g, 44.8 mmol) at 0 °C. After stirring for 10 min at rt, Y18 (7.0 g, 34.4 mmol) was added, then the mixture was stirred at rt for 3 h. After removal of the solvent in vacuo, the residue was dissolved in EtOAc and filterated through celite. After concentration of the filtrate, the residue was purified by filtration through a pad of silica gel in a large sintered glass funnel eluting with hexanes to afford styrene analogue (12.5 g). To a solution of the styrene analogue (8.4 g) in THF (84 ml) was added 20% Pd–C (wet) (1.7 g), and the resulting mixture was stirred at rt under H2 atmosphere for 20 h. After filteration on celite, the solvent was removed in vacuo to give crude Y19k (7.0 g). To a solution of Y19k (7.0 g) in dry THF was added 2M solution of LDA in heptanes, THF and ethylbenzene (16.1 ml, 32.2 mmol) at −78 °C and stirred for 1 h at the same temperature. DMF (3.3 ml, 41.9 mmol) was added and the mixture was stirred for additional 1 h at −78 °C. The reaction was quenched with ice chips and diluted with EtOAc. The organic layer was washed with 1N HClaq. and water, and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-EtOAc gradient) to afford Y20k (4.2 g, 17.1 mmol, 57% for 3 steps). 1H NMR (400 MHz, CDCl3) δ 0.98 (t, J = 7.4 Hz, 3H), 1.68 (m, 2H), 2.66 (t, J = 7.4 Hz, 2H), 7.56 (dd, J = 6.4, 2.8 Hz, 1H), 7.80 (dd, J = 5.8, 2.6 Hz, 1H), 10.29 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 13.8, 23.2, 30.6 (d, JC–F = 2 Hz), 117.4 (d, JC–F = 4 Hz), 125.4 (d, JC–F = 10 Hz), 128.9 (d, JC–F = 2 Hz), 133.4 (d, JC–F = 18 Hz), 139.5 (d, JC–F = 6 Hz), 162.3 (d, JC–F = 256 Hz), 186.4 (d, JC–F = 8 Hz). LR ESI-LCMS m/z calculated 245, 247, found 245, 247 [M+H]+.
1-(5-bromo-2-fluoro-3-propylphenyl)ethanone (21k)
To a solution of Y20k (4.2 g, 17.1 mmol) in ether (50 ml) was added 3 M solution of CH3MgBr in ether (11.4 ml, 34.3 mmol) at 0 °C. After stirring for 30 min at 0 °C, the reaction was quenched with ice chips and diluted with EtOAc. The organic layer was washed with 1 M HCl aq., sat. NaHCO3 aq. and brine, and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-EtOAc gradient) to afford alcohol analogue (4.1 g) of suffcient purity to carry on to the next step. To a solution of the alcohol (4.1 g, 15.7 mmol) in dioxane (50 ml) was added MnO2 (6.8 g, 78.5 mmol). After stirring for 9 h at 90 °C, the reaction mixture was filtered through celite and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-EtOAc gradient) to afford 21k as yellow oil (3.9 g, 15.1 mmol, 88% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 0.94 (t, J = 7.4 Hz, 3H), 1.62 (m, 2H), 2.58-2.62 (m, 4H), 7.44 (dd, J = 6.2, 2.6 Hz, 1H), 7.74 (dd, J = 6.0, 2.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 13.7, 23.1, 30.8 (d, JC–F = 3 Hz), 31.4 (d, JC–F = 8 Hz), 116.8 (d, JC–F = 4 Hz), 127.2 (d, JC–F = 15 Hz), 130.7 (d, JC–F = 3 Hz), 133.2 (d, JC–F = 20 Hz), 137.6 (d, JC–F 6 Hz), 159.6 (d, JC–F = 253 Hz), 195.0 (d, JC–F = 3 Hz). LR ESI-LCMS m/z calculated 259, 261, [M+H]+.
5-bromo-3-methyl-7-propyl-1H-indazole (Y1k)
To a solution of Y21k (2.0 g, 7.72 mmol) in DMF (10 ml) was added hydrazine monohydrate (7.5 ml, 154 mmol). After stirring for 24 h at 120 °C, the reaction was diluted with EtOAc. The organic layer was washed with 1N HCl aq. and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-EtOAc gradient) to afford Y1k (210 mg, 0.83 mmol, 11%). 1H NMR (400 MHz, CDCl3) δ 1.01 (t, J = 7.4 Hz, 3H), 1.78 (m, 2H), 2.55 (s, 3H), 2.80 (t, J = 7.8 Hz, 3H), 7.26 (s, 1H), 7.67 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 12.3, 14.0, 22.5, 33.3, 113.7, 120.7, 124.3, 126.4, 129.1, 139.6, 143.3. LR ESI-LCMS m/z calculated 253, 255, found 253, 255 [M+H]+.
5-bromo-2-fluoro-3-isobutylbenzaldehyde (Y20l)
To a solution of isopropyltriphenylphosphonium iodide (25.5 g, 59.1 mmol) in THF (300 ml) was added a 2.5 M solution of n-butyllithium in hexane (23.6 ml, 59.1 mmol) at 0 °C. After stirring for 30 min at 0 °C, Y18 (10.0 g, 49.3 mmol) was added, then the mixture was stirred for 2 h at rt. After removal of the solvent in vacuo, the residue was dissolved in EtOAc and filterated on Celite. After concentration of the filtrate, the residue was purified by filtration through a pad of silica gel in a large sintered glass funnel eluting with hexanes to afford styrene analogue (11.7 g). To a solution of the styrene analogue (11.7 g, 51.1 mmol) in THF (450 ml) were added p-toluenesulfonyl hydrazide (95.0 g, 511 mmol) and sodium acetate (41.9 g, 511 mmol), and the resulting mixture was stirred at 100 °C for 2 h. After the solvent was removed in vacuo, the residue was dissolved in hexane and filtered through a pad of silica gel in a large sintered glass funnel eluting with hexanes. Four times continues treatments with p-toluenesulfonyl hydrazide and sodium acetate followed by filtration through silica gel were carried out for reducing styrene completely to yield Y19l (8 g). To a solution of Y19l (8.0 g) in dry THF was added 2 M solution of LDA in heptanes, THF and ethylbenzene (24.0 ml, 48.5 mmol) at −78 °C and stirred for 1 h at the same temperature. DMF (4.0 ml, 51.9 mmol) was added and the mixture was stirred for additional 1 h at −78 °C. The reaction was quenched with ice chips and diluted with EtOAc. The organic layer was washed with 1 M HClaq. and water, and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (hexane-EtOAc gradient) to afford Y20l (6.0 g, 23.2 mmol, 47% for 3 steps). 1H NMR (400 MHz, CDCl3) δ 0.95 (d, J = 6.8 Hz, 6H), 1.92 (m, 1H), 2.55 (dd, J = 7.2, 1.2 Hz, 2H), 7.53 (dd, J = 6.4, 2.4 Hz, 1H), 7.81 (dd, J = 6.2, 2.2 Hz, 1H), 10.29 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 22.4, 29.4, 37.7 (d, JC–F = 2 Hz), 117.3 (d, JC–F = 4 Hz), 125.4 (d, JC–F = 10 Hz), 129.0 (d, JC–F = 2 Hz), 132.5 (d, JC–F = 17 Hz), 140.1 (d, JC–F =7Hz), 162.4 (d, JC–F = 256 Hz), 186.3 (d, JC–F = 8 Hz). LR ESI-LCMS m/z calculated 259, 261, found 259, 261 [M+H]+.
1-(5-bromo-2-fluoro-3-isobutylphenyl)ethanone (Y21l)
Compound Y21l was synthesized from Y20l (6.0 g, 23.2 mmol) according to the procedure for Y21k by substituting Y20l for Y20k. Yield: 4.0 g (14.7 mmol, 63% for 2 steps). 1H NMR (400 MHz, CDCl3) δ 0.94 (d, J = 6.8 Hz, 6H), 1.91 (m, 1H), 2.53 (dd, J = 7.2, 1.6 Hz, 2H), 2.62 (d, J = 5.2 Hz, 3H), 7.44 (m, 1H), 7.78 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 22.3, 29.4, 31.5 (d, JC–F = 5 Hz), 38.0 (d, JC–F = 2 Hz), 116.1 (d, JC–F = 4 Hz), 127.3 (d, JC–F = 16 Hz), 130.9 (d, JC–F =3Hz), 132.4 (d, JC–F = 20 Hz), 138.4 (d, JC–F = 6 Hz), 159.7 (d, JC–F = 253 Hz), 195.2 (d, JC–F = 3 Hz). LR ESI-LCMS m/z calculated 273, 275, found 273, 275 [M+H]+.
5-bromo-7-iso-butyl-3-methyl-1H-indazole (Y21l)
Compound Y1l was synthesized from Y21l (4.0 g, 14.7 mmol) according to the procedure for Y1k by substituting Y21l for Y21k. Yield: 160 mg (0.60 mmol, 7%). 1H NMR (400 MHz, CDCl3) δ 0.96 (d, J = 6.8 Hz, 6H), 2.05 (m,1H), 2.55 (s,3H), 2.69 (d, J = 7.2Hz, 2H), 7.23 (s, 1H), 7.67 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 12.2, 22.8, 29.1, 40.8, 113.6, 120.5, 124.3, 125.6, 129.8, 139.9, 143.3. LR ESI-LCMS m/z calculated 267, 269, found 267, 269 [M+H]+.
(S)-1-(5-(7-methyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4a)
To a solution of stannyl compound Y2 (200 mg, 0.38 mmol) in DMF (5 ml) were added Y1a (120 mg, 0.57 mmol), Pd2(dba)3 (52 mg, 0.057 mmol), P(o-tol)3 (35 mg, 0.11 mmol) and triethylamine (83 μl, 0.57 mmol). After stirring over night at 90 °C, the mixture was diluted with ethyl acetate. The organic layer was washed with water and then dried and concentrated. The resulting residue was purified by column chromatography on silica gel (CHCl3-methanol gradient) to afford Y3a (92 mg) of sufficient purity to carry on to the next step. A solution of crude Y3a (92 mg) in CH2Cl2 (2 ml) and TFA (2 ml) was stirred for 1 h and concentrated. Purification by reversed phase HPLC (gradient of water containing 0.1% TFA-acetonitrile containing 0.1% TFA) followed by lyophilization afforded 2X TFA salt of Y4a (40 mg, 0.064 mmol, 17% based on Y2 for 2 steps). LC Retention 3 : 24. 1H NMR (400 MHz, DMSO-d6) δ 2.58 (s, 3H), 3.09–3.17 (m, 3H), 3.84 (brs, 1H), 4.17 (m, 1H), 4.34 (m, 1H), 7.00 (t, J = 7.6 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 7.29 (s, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.48 (s, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.69 (s, 1H), 7.91 (s, 1H), 8.14 (s, 1H), 8.19 (brs, 2H), 8.31 (s, 1H), 8.58 (s, 1H), 11.03 (s, 1H). HR ESI m/z calculated 398.1975, found 398.1987 [M+H]+.
(S)-1-(5-(7-ethyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4b)
Compound Y4b (2X TFA salt) was synthesized from Y1b (64 mg, 0.29 mmol) and Y2 (100 mg, 0.19 mmol) according to the procedure for Y4a by substituting Y1b for Y1a. Yield: 23 mg (0.036 mmol, 19% based on Y2 for 2 steps). LC Retention 3 : 30. 1H NMR (400 MHz, DMSO-d6) δ 1.33 (t, J = 7.6 Hz, 3H), 2.97 (q, J = 7.6 Hz, 2H), 3.09–3.17 (m, 3H), 3.84 (brs, 1H), 4.17 (dd, J = 10.4, 6.0 Hz, 1H), 4.34 (dd, J = 10.6, 3.0 Hz, 1H), 7.00 (t, J = 7.2 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 7.29 (s, 1H), 7.37 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.67 (s, 1H), 7.91 (s, 1H), 8.14 (s, 1H), 8.20 (brs, 2H), 8.30 (s, 1H), 8.58 (s, 1H), 11.03 (s, 1H). HR ESI m/z calculated 412.2131, found 412.2117 [M+H]+.
(S)-1-(5-(7-propyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4c)
Compound Y4c (2X TFA salt) was synthesized from Y1c (110 mg, 0.46 mmol) and Y2 (200 mg, 0.38 mmol) according to the procedure for Y4a by substituting Y1c for Y1a. Yield: 19 mg (0.029 mmol, 7.6% based on Y2 for 2 steps). LC Retention 3 : 46. 1H NMR (400 MHz, DMSO-d6) δ 0.96 (t, J = 7.2 Hz, 3H), 1.74 (m, 2H), 2.92 (t, J = 7.4 Hz, 1H), 3.09–3.17 (m, 3H), 3.84 (brs, 1H), 4.17 (dd, J = 10.6, 5.8 Hz, 1H), 4.34 (dd, J = 10.8, 3.2 Hz, 1H), 7.00 (t, J = 7.4 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 7.29 (s, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.46 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.66 (s, 1H), 7.91 (s, 1H), 8.13 (s, 1H), 8.20 (brs, 2H), 8.30 (s, 1H), 8.57 (s, 1H), 11.03 (s, 1H). HR ESI m/z calculated 426.2288, found 426.2272 [M+H]+.
(S)-1-(5-(7-isobutyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4e)
Compound Y4e (2X TFA salt) was synthesized from Y1e (191 mg, 0.75 mmol) and Y2 (200 mg, 0.38 mmol) according to the procedure for Y4a by substituting Y1e for Y1a. Yield: 28 mg (0.042 mmol, 11% based on Y2 for 2 steps). LC Retention 3 : 56. 1H NMR (400 MHz, DMSO-d6) δ 0.93 (d, J = 6.4 Hz, 6H), 2.06 (m, 1H), 2.83 (d, J = 7.2 Hz, 2H), 3.10–3.18 (m, 3H), 3.81 (brs, 1H), 4.25 (dd, J = 10.8, 6.0 Hz, 1H), 4.36 (dd, J = 10.4, 2.8 Hz, 1H), 7.01 (t, J = 7.2 Hz, 1H), 7.10 (t, J = 7.0 Hz, 1H), 7.30 (s, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.44 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.70 (s, 1H), 7.93 (s, 1H), 8.14 (s, 1H), 8.20 (brs, 2H), 8.32 (s, 1H), 8.60 (s, 1H), 11.04 (s, 1H). HR ESI m/z calculated 440.2444, found 440.2464 [M+H]+.
(S)-1-(5-(7-n-pentyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4f)
Compound Y4f (2X TFA salt) was synthesized from Y1f (201 mg, 0.75 mmol) and Y2 (200 mg, 0.38 mmol) according to the procedure for Y4a by substituting Y1f for Y1a. Yield: 32 mg (0.047 mmol, 12% based on Y2 for 2 steps). LC Retention 3 : 72. 1H NMR (400 MHz, DMSO-d6) δ 0.91 (t, J = 7.0 Hz, 3H), 1.39 (m, 4H), 1.77 (m, 2H), 2.99 (t, J = 7.6 Hz, 2H), 3.12–3.22 (m, 3H), 4.23 (dd, J = 10.4, 6.0 Hz, 1H), 4.40 (dd, J = 10.6, 3.0 Hz, 1H), 7.05 (t, J = 7.4 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 7.34 (s, 1H), 7.42 (d, J = 8.4 Hz, 1H), 7.51 (s, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.74 (s, 1H), 7.96 (s, 1H), 8.18 (s, 1H), 8.24 (brs, 2H), 8.36 (s, 1H), 8.64 (s, 1H), 11.08 (s, 1H). HR ESI m/z calculated 454.2601, found 454.2595 [M+H]+.
(S)-1-(5-(7-iso-pentyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4g)
Compound Y4g (2X TFA salt) was synthesized from Y1g (201 mg, 0.75 mmol) and Y2 (200 mg, 0.38 mmol) according to the procedure for Y4a by substituting Y1g for Y1a. Yield: 52 mg (0.076 mmol, 20% based on Y2 for 2 steps). LC Retention 3 : 70. 1H NMR (400 MHz, DMSO-d6) δ 0.97 (d, J = 6.0 Hz, 6H), 1.63 (m, 3H), 2.96 (t, J = 7.4 Hz, 2H), 3.10–3.18 (m, 3H), 3.85 (brs, 1H), 4.19 (dd, J = 10.8, 6.0 Hz, 1H), 4.36 (dd, J = 10.8, 3.2 Hz, 1H), 7.01 (t, J = 7.0 Hz, 1H), 7.10 (t, J = 7.2 Hz, 1H), 7.30 (s, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.69 (s, 1H), 7.91 (s, 1H), 8.15 (s, 1H), 8.20 (brs, 2H), 8.32 (s, 1H), 8.59 (s, 1H), 11.04 (s, 1H). HR ESI m/z calculated 454.2601, found 454.2620 [M+H]+.
(S)-1-(5-(7-phenethyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4h)
Compound Y4h (2X TFA salt) was synthesized from Y1h (230 mg, 0.75 mmol) and Y2 (200 mg, 0.38 mmol) according to the procedure for Y4a by substituting Y1h for Y1a. Yield: 53 mg (0.074 mmol, 19% based on Y2 for 2 steps). LC Retention 3 : 70. 1H NMR (400 MHz, DMSO-d6) δ 3.04 (t, J = 8.2 Hz, 2H), 3.09–3.19 (m, 3H), 3.25 (t, J = 8.2 Hz, 2H), 3.86 (brs, 1H), 4.18 (dd, J = 10.6, 5.8 Hz, 1H), 4.35 (dd, J = 10.6, 3.0 Hz, 1H), 7.01 (t, J = 7.4 Hz, 1H), 7.10 (t, J = 7.2 Hz, 1H), 7.18 (t, J = 7.2 Hz, 1H), 7.27–7.39 (m, 6H), 7.48 (s, 1H), 7.63 (d, J = 7.6 Hz, 1H), 7.66 (s, H), 7.94 (s, 1H), 8.17 (s, 1H), 8.21 (brs, 2H), 8.33 (s, 1H), 8.56 (s, 1H), 11.04 (s, 1H). HR ESI m/z calculated 488.2444, found 488.2438 [M+H]+.
(S)-1-(5-(7-chloro-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4i)
Compound Y4i (2X TFA salt) was synthesized from Y1i (104 mg, 0.45 mmol) and Y2 (119 mg, 0.22 mmol) according to the procedure for Y4a by substituting Y1h for Y1a. Yield: 39 mg (0.061 mmol, 28% based on Y2 for 2 steps). LC Retention 3 : 34. 1H NMR (400 MHz, DMSO-d6) δ 3.08–3.18 (m, 3H), 3.86 (brs, 1H), 4.19 (dd, J = 10.8, 6.0 Hz, 1H), 4.36 (dd, J = 10.6, 3.0 Hz, 1H), 7.01 (t, J = 7.0 Hz, 1H), 7.10 (t, J = 7.2 Hz, 1H), 7.30 (s, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.41 (s, 1H), 7.84 (s, 1H), 8.12 (s, 1H), 8.20 (brs, 2H), 8.29 (s, 1H), 8.35 (s, 1H), 8.62 (s, 1H), 11.03 (s, 1H). HR ESI m/z calculated 418.1429, found 418.1426 [M+H]+.
(S)-1-(5-(7-ethyl-3-methyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4j)
Compound Y4j (2X TFA salt) was synthesized from Y1j (94 mg, 0.39 mmol) and Y2 (200 mg, 0.38 mmol) according to the procedure for Y4a by substituting Y1j for Y1a. Yield: 45 mg (0.069 mmol, 18% based on Y2 for 2 steps). LC Retention 3 : 42. 1H NMR (400 MHz, DMSO-d6) δ 1.31 (t, J = 7.6 Hz, 3H), 2.53 (s, 3H), 2.93 (q, J = 7.5 Hz, 2H), 3.06–3.18 (m, 3H), 3.83 (brs, 1H), 4.18 (dd, J = 10.6, 5.8 Hz, 1H), 4.35 (dd, J = 10.6, 3.0 Hz, 1H), 7.00 (t, J = 7.4 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 7.29 (s,1H),7.37(d, J = 8.0 Hz, 1H), 7.46 (s, 1H), 7.62 (d, J = 7.6Hz, 1H), 7.13 (s, 1H), 7.87 (s, 1H), 8.20 (brs, 2H), 8.31 (s, 1H), 8.62 (s, 1H), 11.03 (s, 1H). HR ESI m/z calculated 426.2288, found 426.2298 [M+H]+.
(S)-1-(5-(3-methyl-7-n-propyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4k)
Compound Y4k (2X TFA salt) was synthesized from Y1k (100 mg, 0.40 mmol) and Y2 (116 mg, 0.22 mmol) according to the procedure for Y4a by substituting Y1k for Y1a. Yield: 43 mg (0.064 mmol, 29% based on Y2 for 2 steps). LC Retention 3 : 48. 1H NMR (400 MHz, DMSO-d6) δ 1.00 (t, J = 7.4 Hz, 3H), 1.77 (m, 2H), 2.58 (s, 3H), 2.93 (t, J = 7.6 Hz, 2H), 3.16–3.22 (m, 3H), 3.89 (brs, 1H), 4.24 (dd, J = 10.6, 5.8 Hz, 1H), 4.40 (dd, J = 10.4, 2.8 Hz, 1H), 7.05 (1H, t, J = 7.4 Hz, 1H), 7.14 (1H, t, J = 7.2 Hz, 1H), 7.34 (d, J = 2.4 Hz, 1H), 7.42 (d, J =8.0 Hz, 1H), 7.50 (d, J = 1.2 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.79 (t, J = 2.0 Hz, 1H), 7.93 (d, J = 1.6 Hz, 1H), 8.25 (brd, J = 3.2 Hz, 3H), 8.37 (d, J = 2.8 Hz, 1H), 8.37 (d, J = 2.8 Hz, 1H), 8.68 (d, J = 1.6 Hz, 1H), 11.08 (d, J = 2.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 11.7, 13.7, 22.7, 25.0, 32.9, 50.8, 67.6, 107.8, 111.9, 116.1, 117.9, 118.6, 120.0, 120.5, 122.8, 124.8, 125.5, 127.0, 128.0, 135.0, 136.3, 137.8, 140.4, 142.1, 154.7, 158.0, 158.3. HR ESI m/z calculated 440.2444, found 440.2442 [M+H]+.
(S)-1-(5-(7-iso-butyl-3-methyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4l)
Compound Y4l (2X TFA salt) was synthesized from Y1l (160 mg, 0.60 mmol) and Y2 (176 mg, 0.33 mmol) according to the procedure for Y4a by substituting Y1l for Y1a. Yield: 55 mg (0.080 mmol, 24% based on Y2 for 2 steps). LC Retention 3 : 62. 1H NMR (400 MHz, DMSO-d6) δ 0.87 (d, J = 6.8 Hz, 6H), 2.00 (m, 1H), 2.49 (s, 3H), 2.73 (d, J = 7.2 Hz, 2H), 3.02–3.15 (m, 3H), 3.80 (brs, 1H), 4.14 (m, 1H), 4.29 (dd, J = 22.2, 15 Hz, 1H), 6.96 (t, J = 7.0 Hz, 1H), 7.05 (t, J = 7.6 Hz, 1H), 7.25 (d, J = 2.4 Hz, 1H), 7.33 (d, J = 8.4 Hz, 1H), 7.37 (d, J = 0.8 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.64 (s, 1H), 7.83 (d, J = 1.6 Hz, 1H), 8.18 (brs, 3H), 8.26 (d, J = 2.4 Hz, 1H), 8.56 (d, J = 1.2 Hz, 1H), 9.08 (brs, 1H), 11.00 (s, 1H). 13C NMR (100 MHz, DMSO-d6) d11.8, 22.3, 25.0, 28.5, 50.7, 67.5, 107.8, 111.6, 116.2, 118.1, 118.6, 119.6, 121.2, 122.8, 124.6, 125.0, 127.0, 128.1, 135.7, 136.3, 137.4, 140.5, 142.0, 154.5, 157.8, 158.1. HR ESI m/z calculated 454.2601, found 454.2579 [M+H]+.
(S)-1-(5-(7-butyl-1H-indazol-5-yl)pyridin-3-yloxy)-3-(1H-indol-3-yl)propan-2-amine (Y4d)
To a solution of Y3i (170 mg, 0.33 mmol) in toluene (5.5 ml) were added n-butylboronic acid (135 mg, 1.33 mmol), Pd(dba)2 (19 mg, 0.033 mmol), 1,2,3,4,5-pentaphenyl-1′-(di-tert-butylphosphino)ferrocene (51 mg, 0.066 mmol) and K3PO4 (211 mg , 1.0 mmol). After stirring over night at 90 °C, the mixture was diluted with ethyl acetate. The organic layer was washed with water and then dried and concentrated. The resulting residue was purified by reversed phase HPLC (gradient of water containing 0.1% TFA-acetonitrile containing 0.1% TFA) followed by lyophilization afforded the coupling product (10 mg). A solution of the coupling product (10 mg) in CH2Cl2 (0.5 ml) and TFA (0.5 ml) was stirred for 1 h and concentrated. Purification by reversed phase HPLC (gradient of water containing 0.1% TFA-acetonitrile containing 0.1% TFA) followed by lyophilization afforded 2X TFA salt of Y4d (3.7 mg, 0.0055 mmol, 2% from Y3i for 2 steps). LC Retention 3:58. 1H NMR (400 MHz, DMSO-d6) δ 0.93 (t, J = 7.4 Hz, 3H), 1.39 (m, 2H), 1.71 (m, 2H), 2.96 (t, J = 7.6 Hz, 1H), 3.09–3.18 (m, 3H), 3.85 (brs, 1H), 4.19 (dd, J = 10.4, 6.0 Hz, 1H), 4.36 (dd, J = 10.6, 3.0 Hz, 1H), 7.01 (t, J = 7.4 Hz, 1H), 7.10 (t, J = 7.4 Hz, 1H), 7.30 (s, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.47 (s, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.70 (s, 1H), 7.92 (s, 1H), 8.14 (s, 1H), 8.20 (brs, 2H), 8.33 (s, 1H), 8.60 (s, 1H), 11.04 (s, 1H). HR ESI m/z calculated 440.2444, found 440.2430 [M+H]+.
Supplementary Material
Footnotes
Electronic supplementary information (ESI) available: Supplementary Table 1.
This article is part of a Molecular BioSystems themed issue on Chemical Genomics.
References
- 1.Manning BD, Cantley LC. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tolcher AW, Yap TA, Fearen I, Taylor A, Carpenter C, Brunetto AT, Beeram M, Papadopoulos K, Yan L, de Bono J. J. Clin. Oncol. (Meeting Abstracts) 2009;27:3503. [Google Scholar]
- 3.Heerding DA, Rhodes N, Leber JD, Clark TJ, Keenan RM, Lafrance LV, Li M, Safonov IG, Takata DT, Venslavsky JW, Yamashita DS, Choudhry AE, Copeland RA, Lai Z, Schaber MD, Tummino PJ, Strum SL, Wood ER, Duckett DR, Eberwein D, Knick VB, Lansing TJ, McConnell RT, Zhang S, Minthorn EA, Concha NO, Warren GL, Kumar R. J. Med. Chem. 2008;51:5663–5679. doi: 10.1021/jm8004527. [DOI] [PubMed] [Google Scholar]
- 4.Luo Y, Shoemaker AR, Liu X, Woods KW, Thomas SA, de Jong R, Han EK, Li T, Stoll VS, Powlas JA, Oleksijew A, Mitten MJ, Shi Y, Guan R, McGonigal TP, Klinghofer V, Johnson EF, Leverson JD, Bouska JJ, Mamo M, Smith RA, Gramling-Evans EE, Zinker BA, Mika AK, Nguyen PT, Oltersdorf T, Rosenberg SH, Li Q, Giranda VL. Mol. Cancer Ther. 2005;4:977–986. doi: 10.1158/1535-7163.MCT-05-0005. [DOI] [PubMed] [Google Scholar]
- 5.Abeliovich H, Zhang C, Dunn WA, Jr., Shokat KM, Klionsky DJ. Mol. Biol. Cell. 2003;14:477–490. doi: 10.1091/mbc.E02-07-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop AC, Kung C-Y, Shah K, Witucki L, Shokat KM, Liu Y. J. Am. Chem. Soc. 1999;121:627–631. [Google Scholar]
- 7.Eblen ST, Kumar NV, Shah K, Henderson MJ, Watts CK, Shokat KM, Weber MJ. J. Biol. Chem. 2003;278:14926–14935. doi: 10.1074/jbc.M300485200. [DOI] [PubMed] [Google Scholar]
- 8.Hindley AD, Park S, Wang L, Shah K, Wang Y, Hu X, Shokat KM, Kolch W, Sedivy JM, Yeung KC. FEBS Lett. 2004;556:26–34. doi: 10.1016/s0014-5793(03)01352-8. [DOI] [PubMed] [Google Scholar]
- 9.Ulrich SM, Kenski DM, Shokat KM. Biochemistry. 2003;42:7915–7921. doi: 10.1021/bi030042a. [DOI] [PubMed] [Google Scholar]
- 10.Dummler B, Hemmings BA. Biochem. Soc. Trans. 2007;35:231–235. doi: 10.1042/BST0350231. [DOI] [PubMed] [Google Scholar]
- 11.Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, Zhou J, Turner J, Lisanti MP, Russell RG, Mueller SC, Ojeifo J, Chen WS, Hay N, Pestell RG. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7438–7443. doi: 10.1073/pnas.0605874104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. Bioorg. Med. Chem. Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Okuzumi T, Fiedler D, Zhang C, Gray DC, Aizenstein B, Hoffman R, Shokat KM. Nat. Chem. Biol. 2009;5:484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han EK, Leverson JD, McGonigal T, Shah OJ, Woods KW, Hunter T, Giranda VL, Luo Y. Oncogene. 2007;26:5655–5661. doi: 10.1038/sj.onc.1210343. [DOI] [PubMed] [Google Scholar]
- 15.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Ye H, Kuruvilla R, Ramanan N, Scangos KW, Zhang C, Johnson NM, England PM, Shokat KM, Ginty DD. Neuron. 2005;46:13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 17.Tamguney T, Zhang C, Fiedler D, Shokat K, Stokoe D. Exp. Cell Res. 2008;314:2299–2312. doi: 10.1016/j.yexcr.2008.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woods KW, Fischer JP, Claiborne A, Li T, Thomas SA, Zhu GD, Diebold RB, Liu X, Shi Y, Klinghofer V, Han EK, Guan R, Magnone SR, Johnson EF, Bouska JJ, Olson AM, de Jong R, Oltersdorf T, Luo Y, Rosenberg SH, Giranda VL, Li Q. Bioorg. Med. Chem. 2006;14:6832–6846. doi: 10.1016/j.bmc.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 19.Dell'Erba C, Novi M, Petrillo G, Tavani C. Tetrahedron. 1994;50:3529–3536. [Google Scholar]
- 20.Kataoka N, Shelby Q, Stambuli JP, Hartwig JF. J. Org. Chem. 2002;67:5553–5566. doi: 10.1021/jo025732j. [DOI] [PubMed] [Google Scholar]
- 21.Dubost C, MarkŬ IE, Bryans J. Tetrahedron Lett. 2005;46:4005–4009. [Google Scholar]
- 22.Antonysamy S, Hirst G, Park F, Sprengeler P, Stappenbeck F, Steensma R, Wilson M, Wong M. Bioorg. Med. Chem. Lett. 2009;19:279–282. doi: 10.1016/j.bmcl.2008.08.064. [DOI] [PubMed] [Google Scholar]
- 23.Knight ZA, Shokat KM. Chem. Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Davies TG, Verdonk ML, Graham B, Saalau-Bethell S, Hamlett CC, McHardy T, Collins I, Garrett MD, Workman P, Woodhead SJ, Jhoti H, Barford D. J. Mol. Biol. 2007;367:882–894. doi: 10.1016/j.jmb.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Specht KM, Shokat KM. Curr. Opin. Cell Biol. 2002;14:155–159. doi: 10.1016/s0955-0674(02)00317-4. [DOI] [PubMed] [Google Scholar]
- 26.Weiss WA, Taylor SS, Shokat KM. Nat. Chem. Biol. 2007;3:739–744. doi: 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knight ZA, Feldman ME, Balla A, Balla T, Shokat KM. Nat. Protoc. 2007;2:2459–2466. doi: 10.1038/nprot.2007.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng L, Baumann U, Reymond JL. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.