

Abstract
Detection of microbial components by immune cells via Toll-like receptors (TLRs) with subsequent induction of inflammation is essential for host defense. However, an overactive immune response can cause tissue damage and sepsis. The endogenous molecule hemoglobin and its derivative heme are often released into tissue compartments where there is infection in the presence of degrading blood. We found that hemoglobin synergizes with multiple TLR agonists to induce high levels of tumor necrosis factor and interleukin-6 from macrophages and that this synergy is independent of TLR4 and MyD88. In contrast, heme synergized with some but not all TLR agonists studied. Furthermore, the synergy of both hemoglobin and heme with lipo-polysaccharide was suppressed by hemopexin, a plasma heme-binding protein. These studies suggest that hemoglobin and heme may substantially contribute to microbe-induced inflammation when bacterial or viral infection coexists with blood degradation and that hemopexin may play a role in controlling inflammation in such settings.
Much of the pathophysiology that occurs early and during microbial infection is believed to be due to the induction of inflammation in tissues that has evolved as an essential part of the defense against early microbial challenge. Integral to this concept is the early identification of microbes in tissues by specialized cells, such as macrophages, with the production of secondary mediators, such as cytokines, that amplify the signal and communicate with other local and distant tissues. It is now appreciated that certain molecules on microorganisms known as microbial-associated molecular pattern molecules (MAMPs) interact with a limited number of pattern recognition receptors called Toll-like receptors (TLRs), to initiate a cascade of events that ultimately result in a signal being transmitted to the nucleus to produce cytokines. Each TLR has receptor-specific ligands, such as lipopolysaccharide (LPS), which signals through TLR4; lipoteichoic acid and peptidoglycan, which signal through TLR2; viral nucleic acid structures, such as double-stranded RNA poly I: C, which signals through TLR3; the guanosine analogue loxoribine, which signals through TLR7; and bacterial DNA (CpG), which signals through TLR9 [1, 2].
In some clinical situations, a hyperactive immune response can cause tissue damage. Sepsis syndrome, which is defined by certain parameters of systemic inflammation in the setting of infection [3], is characterized at early stages by high levels of proinflammatory cytokines [4]. Although the precise mechanisms underlying sepsis syndrome are not well understood, it is widely believed that excessive production of cytokines may be a driving force, a situation sometimes referred to as a “cytokine storm.” A better understanding of the mechanisms responsible for the liberation of these cytokines may permit the development of effective control strategies.
Exogenous microbial TLR ligands synergize with one another to activate signaling pathways with subsequent induction of proinflammatory cytokines from macrophages and other immune cells [5–10]. Data from our laboratory suggest that the outcome of stimulation with different microbial TLR ligands is dependent on differential engagement of MyD88-dependent and MyD88-independent pathways [11]. Recent studies suggest that endogenous host molecules also can act as TLR ligands and that molecules released or induced during tissue damage may contribute to the induction of inflammatory cytokines in sepsis [4]. For example, such oxidants as hydrogen peroxide [12], heat-shock proteins (HSPs; ie, HSP-60, HSP-70, and Gp-96), and self-messenger RNA have been proposed to synergize with exogenous TLR agonists [13].
Visible or microscopic blood is often present in tissues where there is infection and necrosis, so that hemoglobin and microorganisms coexist in infected microenvironments. This situation is particularly common when there is invasive bacterial or viral infection with tissue necrosis; after trauma, burns, or surgery; or in any infection where there is breach of capillaries. Older studies revealed that blood and hemoglobin enhanced growth of bacteria by providing heme as a nutrient source [14]. However, hemoglobin is also known to synergize with LPS to augment macrophage induction of tumor necrosis factor (TNF) [15–18]. It has been proposed that hemoglobin preparations increase the biological activity of LPS through physically interacting with LPS [19, 20]. Many of the studies involving the interactions of hemoglobin and LPS have focused on the development of artificially cross-linked hemoglobin for use as a cell-free blood transfusion substitute, where such an interaction has major potential implications [15, 16, 21, 22].
Given the ubiquity of blood in infected tissues and the broad array of bacterial and viral infections in which blood might play a role, we studied the activation of macrophages in the presence of hemoglobin and multiple different TLR ligand agonists (ie, ligands for TLR2, TLR3, TLR4, TLR7, and TLR9). We observed extensive synergy with all of these TLR agonists. We found that this synergy is not TLR4 dependent and not MyD88 dependent and that the degraded hemoglobin product, hemin, synergizes with some (but not all) TLR agonists. Finally, we observed that hemopexin (Hx), a plasma protein that binds heme with an extremely high affinity, blocks the synergy of both hemoglobin and hemin with LPS, raising the possibility that Hx may be involved in local regulation of this synergy. Our findings also suggest that exogenously administered Hx might be a candidate for treating inflammation induced by microorganisms in tissues that contain blood breakdown products through a novel mechanism of blocking this synergistic activation of inflammation.
MATERIALS AND METHODS
Materials
The following TLR agonists were purchased: smooth LPS from Escherichia coli O55:B5 (List Biologicals), Pam3Cys (EMC Microcollections), poly I:C, loxoribine, and CpG DNA (Invivogen). All these TLR agonists were dissolved in pyrogen-free H2O and saved as aliquots at −80°C. E. coli O4 was the kind gift of Alan Cross (University of Maryland), and it was heat killed and washed before use, as described elsewhere [23]. Heat-killed Staphylococcus aureus was purchased from Invivogen. Hemin chloride was purchased from Frontier Scientific. Hemin solutions were made immediately before use in the dark, as described elsewhere [24]. In brief, the powder was dissolved in pyrogen-free 0.1 N NaOH at 10 mg/mL and was further diluted in serum-free medium as desired; these steps were followed by filtration through 0.22-μm Millipore membranes. C57BL/6, C3H/HeN, and C3H/HeJ mice were obtained from Charles River Laboratories. MyD88 knockout (MyD88−/−) mice were generated by Kawai and colleagues [25] and had been backcrossed >10 generations into the C57BL/6J strain.
Purification of mouse hemoglobin
Hemoglobin was purified as previously described elsewhere [26], with modifications, under pyrogen-free conditions. In brief, mouse blood was collected from C57BL/6 mice by means of cardiac puncture, and it was washed with an equal weight of isotonic saline solution (0.9% NaCl, wt/vol) three times by centrifugation at 1000 g, to remove serum proteins. Equal volumes of saline were added to the pellet containing red blood cells, and this solution was sonicated for 5 × 10 s at 40% amplitude with 1-min laps between pulses in a Branson 450 sonicator from Branson Ultrasonics. The hemoglobin solution was diluted with an equal volume of saline and subjected to a second centrifugation at 2000 g for 1 h. The resulting hemoglobin solution, removed from the center layer, was filtered through 0.22-μm Millipore membranes and saved at −20°C in the dark. The concentration of purified mouse hemoglobin was measured using Micro-BCA. The purity of the hemoglobin was confirmed to be >99%, by means of nondenaturing polyacrylamide gel electrophoresis and high-pressure liquid chromatography.
Purification of Hx from mouse or human serum
Mouse serum Hx (mHx) or human serum Hx (hHx) was purified using heme affinity chromatography essentially as we have described elsewhere [27]. In brief, after filtration through 0.22- μm Millipore membranes, the abundant albumin in the serum was precipitated and removed by use of cold 1.68% rivanol solution (pH 8.0). The sample obtained after rivanol precipitation was dialyzed against pyrogen-free phosphate-buffered saline. Protease inhibitors (0.5 mmol/L 4-[2-aminoethyl] ben-zenesulfonyl fluoride, 10 μmol/L E-64, 2 μg/mL aprotinin, and 1 μmol/L pepstatin A) were added, and they interacted with the dialyzed sample obtained after rivanol precipitation for 15 min by gentle agitation at 4°C. The mixture was applied to 6 mL of heminagarose column (Sigma) 3 times, followed by extensive washing with 1200 mL of phosphate-buffered saline containing 0.5 mol/L NaCl overnight at 4°C to remove unbound proteins. Hx bound to the column was eluted by 0.2 mol/L citric acid (pH 2.0), followed by immediate neutralization with 10 mol/L NaOH. Proteins in the buffer were exchanged, concentrated in PBS at 4°C by use of Centriprep YM- 30 (Millipore), and saved in aliquots at −80°C.
Limulus amebocyte lysate assay
The limulus amebocyte lysate assay was performed as previously described elsewhere [28].
Preparation of macrophages
Bone marrow–derived macrophages (BMDMs) were prepared from mice, as described elsewhere [29], with minor modifications [11, 27]. BMDMs were seeded at 1.28 × 105 cells/well in 96-well tissue culture plates and were allowed to adhere overnight before use in assays.
Macrophage assays and cytokine production
BMDMs were washed 3 times in serum-free medium, followed by incubation overnight with different TLR agonists with or without hemoglobin or hemin chloride at indicated concentrations. Purified mHx was added to the culture in some experiments, as noted. Levels of TNF and interleukin (IL)–6 in the supernatants were quantitated by enzyme-linked immunosorbent assay (ELISA; R & D Systems), in accordance with the manufacturer’s instructions.
Human whole-blood assay
Whole-blood stimulations were performed as previously described elsewhere [30]. Heparinized whole blood, collected aseptically from healthy human volunteers, was diluted 1:4 in pyrogen-free RPMI 1640 (Cellgro; Mediatech) and placed in a 96-well plate at 100 μL/well. The diluted blood was incubated with desired concentrations of stimuli at 37°C, followed by centrifugation at 500 g for 5 min. The supernatants were saved for quantification of cytokines by ELISA.
Statistics
Except where indicated, representative data from at least three experiments are presented in the figures. Data are expressed as means, and error bars denote standard errors. The data were analyzed using Prism 5 software (GraphPad). One-way analysis of variance followed by Dunnett’s post hoc test was used to assess cytokine levels produced by macrophages treated with different TLR agonists in the absence or presence of hemin or hemoglobin at desired concentrations. Student’s t test was used to compare the cytokine levels produced by macrophages from C3H/HeN with C3H/HeJ or C57BL/6 with MyD88−/− mice in the presence of TLR agonists and hemin or hemoglobin at desired concentrations. Two-way analysis of variance followed by Bonferroni’s post hoc test was used to assess cytokine levels from macrophages treated with LPS with or without hemin or hemoglobin at different concentrations in the presence of Hx or phosphate-buffered saline. P <.05 was considered to denote statistical significance.
RESULTS
Strong synergy of hemoglobin with TLR2, TLR3, TLR4, TLR7, and TLR9 agonists and with bacteria to induce TNF and IL-6 from macrophages
Differing concentrations of hemoglobin were incubated with a predetermined optimized concentration of each TLR agonist in cell culture with BMDMs, as described in Materials and Methods. Levels of the proinflammatory cytokines TNF and IL-6 induced in the supernatant were measured by ELISA. Hemoglobin significantly enhanced the induction of TNF and IL-6 from BMDMs by all TLR agonist ligands studied (LPS, Pam3Cys, Poly I:C, loxoribine, and CpG) in a dose-dependent manner (Figure 1). Concentrations of hemoglobin as low as 30 μg/mL led to synergistic induction of the cytokines with LPS and Pam3Cys (Figure 1A–D), whereas higher concentrations (~300 μg/mL) of hemoglobin were required for loxoribine (Figure 1G, H) and CpG DNA (Figure 1I and 1J). Similar results were obtained with different concentrations of the TLR agonists (data not shown) as well as with killed E. coli and S. aureus (Figure 2A, 2B, 2E, and 2F). Synergy was observed as early as 3 h after the cells were stimulated (Figure 2C, 2D, 2G, and 2H).
Figure 1.

Synergy of hemoglobin (Hb) with lipopolysaccharide (LPS), Pam3Cys, Poly I:C, loxoribine, and bacterial DNA (CpG). Bone marrow–derived macrophages from C57BL/6 mice were washed 3 times with serum-free medium and then were cultured overnight with either Hb alone or Hb with various Toll-like receptor agonists: LPS (1 ng/mL) (A and B ), Pam3Cys (P3C; 10 ng/mL) (C and D ), Poly I:C (150 μg/mL) (E and F ), loxoribine (250 μmol/L) (G and H ), and CpG (0.1 μmol/L) (I and J ). Concentrations of tumor necrosis factor (TNF) (A, C, E, G, and I ) and interleukin (IL)–6 (B, D, F, H, and J ) in the supernatants were determined by enzyme-linked immunosorbent assay. The results denote the mean ± standard error and are representative of 4 independent experiments. *P <.05 and **P <.01, compared between cells treated with and without Hb.
Figure 2.

Synergy of hemoglobin (Hb) with killed Escherichia coli and Staphylococcus aureus. Bone marrow–derived macrophages from C57BL/6 mice were washed 3 times with serum-free medium and then cultured overnight (A, B, E, and F ) or at different times (C, D, G, and H ) with heat-killed E. coli at indicated concentrations (A and B ) or at 105 cfu/mL (C and D ) or with S. aureus at indicated concentrations (E and F ) or at 107 cfu/mL (G and H ) in the absence or presence of Hb at 300 μg/mL. Concentrations of tumor necrosis factor (TNF) (A, C, E, and G ) and interleukin (IL)–6 (B, D, F, and H ) in the supernatants were determined by enzyme-linked immunosorbent assay. The results denote the mean ± standard error and are representative of 4 independent experiments. *P <.05, **P <.01, ***P <.001, compared between cells treated with and without Hb.
TLR4- and MyD88-independent synergistic induction of proinflammatory cytokines by hemoglobin and various TLR agonists
Because heme has been described as a TLR4 ligand agonist [24], we next addressed the question of whether hemoglobin acts through TLR4-signaling pathways to induce the synergy that we observed. For these experiments, the synergistic induction of cytokines induced in BMDMs from C3H/HeJ mice, which are deficient in TLR4, was compared with induction of cytokines from control BMDMs from C3H/HeN mice, by use of the same TLR agonists shown in Figure 1. As expected, neither LPS nor LPS with hemoglobin induced TNF and IL-6 from C3H/HeJ macrophages (data not shown). The synergy for non-LPS ligands with hemoglobin was present despite the deficiency of TLR4 (Figure 3), suggesting that heme ligation of TLR4 [24] was not a prominent mechanism in the synergy. To be able to compare between cell preparations from the different strains, these results are presented as the percentage change.
Figure 3.
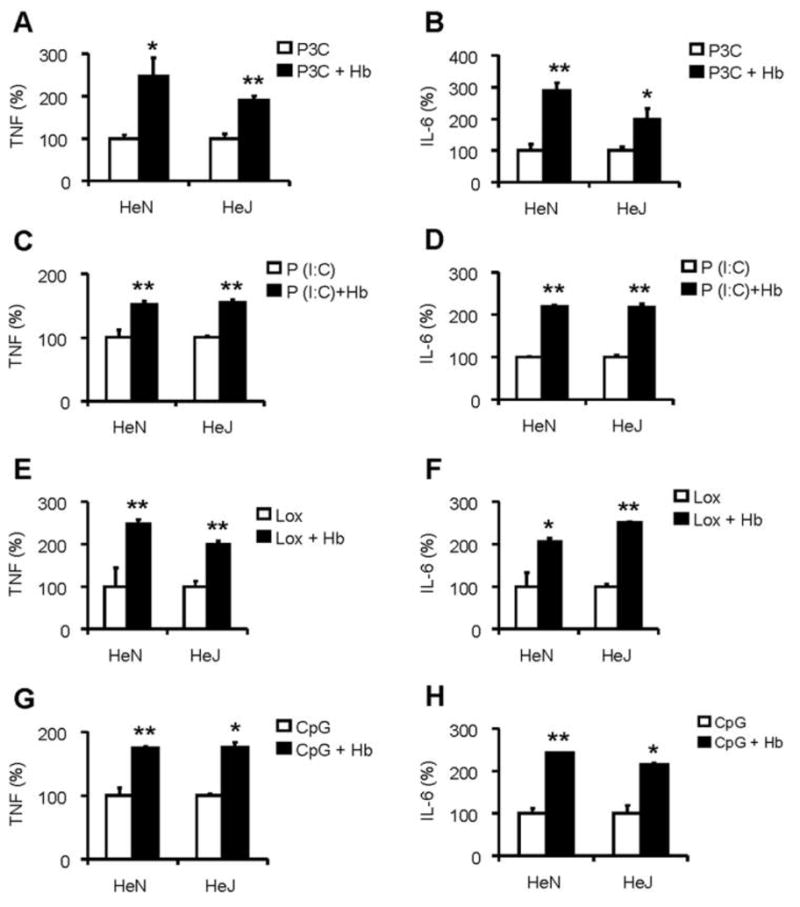
Synergy of hemoglobin (Hb) with Toll-like receptor (TLR) agonists is not TLR4 dependent. Bone marrow–derived macrophages from HeN and HeJ mice were washed 3 times with serum-free medium and then cultured overnight with TLR agonists alone, including Pam3Cys (P3C; 10 ng/mL) (A and B ), Poly I:C (P[I:C]; 150 μg/mL) (C and D ), loxoribine (Lox; 250 μmol/L) (E and F ), and bacterial DNA (CpG; 0.1 μmol/L) (G and H ), or with Hb at 100 μg/mL (A–D ) or 300 μg/mL (E and F ) or 1000 μg/mL (G and H ) indicated concentrations. Concentrations of tumor necrosis factor (TNF) (A, C, E, and G ) and interleukin (IL)–6 (B, D, F, and H ) in the supernatants were determined by enzyme-linked immunosorbent assay. Each graph denotes the percentage of cytokine production when cytokine production by TLR agonist alone is assigned to 100%. Results denote the mean ± the standard error and are representative of 4 independent experiments. *P <.05 and **P <.01, compared between cells treated with and without Hb.
We previously noted that the synergy between different microbial TLR agonists is altered depending on whether the agonists function through the MyD88-dependent signaling pathway [11]. MyD88 is an essential adaptor molecule in the signaling pathway of many TLRs, with the exception that TLR3 is exclusively and TLR4 is partially, but not predominantly, MyD88 independent [11]. Accordingly, we assessed the synergy of hemoglobin with ligands of TLR3 and TLR4 in BMDMs from MyD88−/− mice. As shown in Figure 4, hemoglobin induced a similar percentage increase in TNF from both wild-type (C57BL/6) and MyD88-deficient BMDMs, when stimulated with TLR4 agonist LPS and TLR3 agonist poly I:C. There was no significant IL-6 produced from MyD88-deficient BMDMs (not shown). As expected, other TLR agonists (ie, Pam3Cys, loxoribine, and CpG DNA) did not induce TNF and IL-6 from MyD88-deficient BMDMs, with or without hemoglobin (data not shown).
Figure 4.
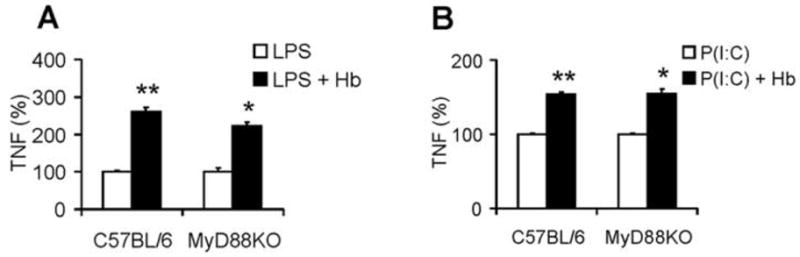
Synergy of hemoglobin (Hb) with lipopolysaccharide (LPS) and Poly I:C is not MyD88 dependent. Bone marrow–derived macrophages from C57BL/6 and MyD88 knockout (MyD88KO) mice were washed 3 times with serum-free medium and then cultured with Toll-like receptor (TLR) agonists LPS (1 ng/mL) (A) or Poly I:C (P[I:C]; 150 μg/mL) (B ) and Hb at 100 μg/mL. Concentrations of tumor necrosis factor (TNF) in the supernatants were determined by enzyme-linked immunosorbent assay. The data denote the percentage of cytokine production, when cytokine production by TLR agonist alone is assigned to 100%. The results denote the mean ± the standard error and are representative of 3 independent experiments. *P <.05 and **P <.01, compared between cells treated with and without Hb.
Synergistic induction of proinflammatory cytokines from macrophages with hemin and agonists of TLR3, TLR4, and TLR7—but not with TLR2 and TLR9—and the role of TLR4 and MyD88
Hemoglobin consists of 2 moieties: hemin and globin. It has been hypothesized but not directly shown that hemin is the active moiety that synergizes with LPS [31]. To directly study this question, we evaluated the synergy of hemin chloride in the presence and absence of different TLR agonists. As previously reported, we found that hemin alone weakly but significantly enhanced production of TNF (data not shown) [24]. Hemin synergized with LPS (Figure 5A), poly I:C (Figure 5B), and loxoribine (Figure 5C). We also found that the synergy of hemin with poly I:C and loxoribine was not TLR4 dependent (Figure 5B and 5C) and that synergy with LPS was partially but not completely MyD88 dependent (Figure 5D and 5E). Of note, we did not find any hemin synergy with Pam3Cys and CpG DNA, although a large range of hemin chloride concentrations (0.01 to ~30 μmol/L) was tested (data not shown).
Figure 5.

Synergy of hemin with lipopolysaccharide (LPS), Poly I:C, and loxoribine, and the role of Toll-like receptor (TLR)–4 and MyD88. Bone marrow–derived macrophages from C57BL/6, C3H/HeN, C3H/HeJ, or MyD88 knockout (MyD88KO) mice were washed 3 times with serum-free medium and then cultured with TLR agonists. A, C57BL/6 cells, hemin concentrations as shown (the open bar denotes no hemin; vertical lines, 1 μmol/L hemin; and the solid bar, 3 μmol/L hemin) and, where indicated, LPS (1 ng/mL). The tumor necrosis factor (TNF) concentrations in the culture are shown. B and C, C3H/HeN (HeN) and C3H/HeJ (HeJ) cells cultured with Poly I:C (P[I:C], 150 μg/mL) (B ) or loxoribine (Lox; 250 μmol/L) (C ) in the absence or presence of hemin (3 μmol/L). D and E, C57BL/6 or MyD88 KO cells cultured with Poly I:C (P[I:C]; 150 μg/mL) (D ) or LPS (1 ng/mL) in the absence or presence of hemin (3 μmol/L) (E ). B–E, The percentage of TNF production when TNF by the same type of cells stimulated by TLR agonist alone is assigned to 100%. The results denote the mean ± the standard error and are representative of four independent experiments. *P <.05, **P <.01, and ***P <.001, compared between cells treated with and without hemin.
Suppressive effect of Hx on the synergistic induction of pro-inflammatory cytokines from macrophages by hemin and hemoglobin with LPS
Hx is the major heme scavenger in plasma and has been described to bind free heme in a 1:1 ratio with remarkably high affinity [32, 33]. In addition, Hx has some immunomodulatory activities in that it modestly down-regulates proinflammatory cytokines from macrophages [27] and functions as an anti-inflammatory component of serum high-density lipoprotein in atherosclerosis [34]. It was therefore of interest to assess whether Hx would alter the induction of pro-inflammatory cytokines that were synergistically induced by hemin with LPS. We found that Hx abrogated the synergistic induction of TNF (Figure 6A). Similar results were obtained with IL-6 (data not shown). Unexpectedly, Hx also significantly down-regulated the synergistic induction of TNF by hemoglobin with LPS (Figure 6B). Similar results were obtained with IL-6 (data not shown). As previously described elsewhere [27], in both situations, Hx decreased LPS-induced TNF when studied in the absence of hemin or hemoglobin (as designated by the zero point in Figures 6A and 6B).
Figure 6.
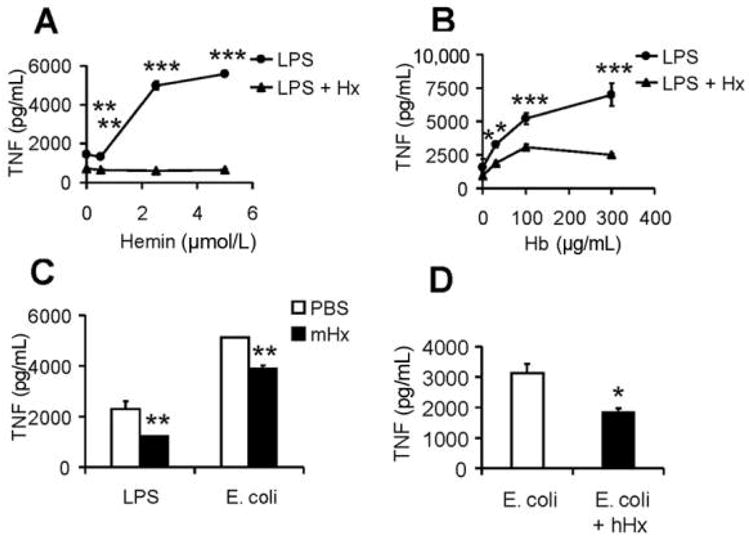
Effect of mouse or human hemopexin (mHx or hHx) on hemin or hemoglobin (Hb) synergy with lipopolysaccharide (LPS) or Escherichia coli on bone marrow–derived macrophages (BMDMs) or human whole blood. BMDMs from C57BL/6 mice were washed 3 times with serum-free medium and then cultured overnight with 1 ng/mL of LPS in the absence (●) or presence (▲) of 300 μg/mL mHx and indicated concentrations of hemin (A) or Hb (B ). Concentrations of tumor necrosis factor (TNF) in the supernatants were determined by enzyme-linked immunosorbent assay. Fresh human whole blood was diluted 1:4 in serum-free medium and cultured for 1 h and then were coincubated overnight with 2.5 ng/mL LPS or 105 cfu/mL E. coli in the absence or presence of mHx (300 μg/mL) (C ), or 105 cfu/mL of E. coli with or without hHx (400 μg/mL) (D ). The results denote the mean ± the standard error and are representative of more than 4 independent experiments. *P <.05, **P <.01, ***P <.001, compared between samples treated with and without Hx.
Suppressive effect of the induction of proinflammatory cytokines by LPS and E. coli in whole human blood
A human whole-blood stimulation assay has been proposed as a more physiological system in which to assess anti-inflammatory drugs [35]. We found that induction of TNF by LPS and E. coli was significantly reduced by the addition of mHx and hHx in this system (Figure 6C and 6D).
DISCUSSION
Inflammation induced by microbes through activation of TLRs is believed to play a critical role in host defense; however, in some circumstances, this same inflammation is also believed to cause sepsis and tissue injury. We previously studied the synergy between different exogenous microbial TLR agonists, in an effort to dissect the initiation process of this inflammation [11]. In the present study, we report that multiple TLR agonists synergize remarkably with 2 endogenous molecules, hemoglobin and its derivative heme. This finding likely has broad importance in the clinic, because microscopic blood and blood degradation products are present in many infected local microenvironments. Furthermore, we found that this synergy is blocked by Hx, suggesting that Hx may be important in controlling the inflammation in which even small amounts of degraded blood are present.
Older studies revealed that blood and hemoglobin enhance growth of bacteria by providing a source of heme [14]. Our findings extend prior available information related to the induction of cytokines by microbial cell walls in the setting of blood. Much of this information comes from studies to evaluate the safety of using soluble cross-linked hemoglobin preparations as a potential substitute for red blood cells, although enhancement of LPS-induced inflammation by native hemoglobin has also been reported in multiple previous studies [16, 18, 31]. In addition to the synergy with LPS, hemoglobin has been reported to increase IL-6, when present in combination with lipoteichoic acid [36], and several cytokines, when present in combination with the cell wall of Streptococcus suis [37]. To our knowledge, this is the first study to systematically evaluate the synergy of native hemoglobin on inflammation induced by multiple TLR agonists, including ligands for TLR3, TLR7, and TLR9, which are involved in inducing inflammation after viral infections [38, 39], to report synergy of TLR agonists with heme and to report that the synergy is suppressed with Hx.
The mechanisms responsible for the synergy of hemoglobin with different TLR agonists remain unclear. It has been proposed that one mechanism for hemoglobin synergy with LPS is through direct binding of hemoglobin to LPS leading to better presentation of LPS to TLR4 or is by inducing the conformational changes in the lipid A moiety of LPS [19, 20]. Other studies suggest that hemoglobin may produce cell hypersensitivity to LPS by increasing the intracellular load of heme iron, which catalyze cellular redox changes and oxidant damage without direct interacting with LPS [40]. In a separate study, it was reported that hemoglobin synergy with lipotechoic acid was partially dependent on TLR4 [36]. Although we cannot exclude the possibility that each of these different TLR agonist molecules bound to hemoglobin or heme and that presentation was enhanced to the TLR, this seems unlikely given their markedly different molecular properties. We also found that TLR4 is not required for hemoglobin synergy with the synthetic TLR2 agonist Pam3Cys as well as with TLR 3, TLR7, and TLR9 agonists and that MyD88 signaling is not required for synergy with LPS and poly I:C, which activate cells via partial and exclusive MyD88-independent pathways, respectively. The reason for the partial dependence on MyD88 for the heme synergy with LPS is unclear. Taken together, these findings suggest that the role of hemoglobin itself in the synergy may be independent of TLR activation. One possible mechanism may be through quenching of the anti-inflammatory action of nitric oxide [41].
Blood and/or blood degradation products in infected body fluids are common. Sites at which this occurs include but are not limited to pleural infection, peritoneal infection, central nervous system infection, surgical wounds, burn wounds, trauma wounds, pancreatitis, and necrotizing infections of any tissue, including lung tissue. Numerous bacterial, fungal, parasitic, and viral infections also directly lead to hemorrhagic lesions or are present together with bloody secretions. In addition, microbial TLR agonists are present with hemoglobin in circulating blood in blood vessels if there is even minor hemolysis during bacteremia, parasitemia, fungemia, or viremia. It is difficult to estimate the concentrations of available free hemoglobin or heme in such infected tissues, drainage fluids, or blood. The total concentration of hemoglobin in red blood cells in whole blood is ~150 mg/mL. We found significant synergy with several TLR agonists (LPS, Pam3Cys, and Poly I:C) at 30 μg/mL, which is 1/5000th of this concentration. Estimation of the effective concentration of heme is even more difficult. The approximate total heme concentration in red blood cells in whole blood is approximately 9 mmol/L. We found significant synergy with 1 and 3 μmol/L, which is approximately 1/3000th of this concentration. However, much of the free hemoglobin and heme that is released from damaged red blood cells is likely to be bound and blocked by haptoglobin [42, 43] or by Hx [32] (Figure 6).
One of our most striking results was the suppressive effect of the Hx on both heme and hemoglobin-LPS synergy. The suppression of the synergy of hemoglobin with LPS was unexpected. There may be several potential mechanisms involved. First, Hx might simply sequester free heme from interaction with macrophages. Indeed, this might also explain the suppression of synergy with hemoglobin as well, because heme can be directly transferred from hemoglobin to Hx [44]. Alternatively, heme-Hx complexes might induce hemoxygenase-1, which has anti-inflammatory effects through its enzymatic products carbon monoxide and biliverdin [45–47]. Finally, some other unknown mechanisms could be involved. Elucidation of the mechanisms for the suppression of hemoglobin synergy with LPS by Hx will need more studies.
A next step in evaluating the relevance of the findings will be to study the synergy of hemoglobin and heme in some sort of in vivo system. Unfortunately, choosing an appropriate model system that is reflective of human disease may be challenging [48]. Intrinsically, mice are multiple orders of magnitude more resilient to TLR4 agonist challenge than humans, and the same relationship likely is true for other TLR agonists [23]. Therefore, assessment of the synergistic inflammation caused by blood products and microbes might be better performed in a species that is similar to humans in sensitivity, such as rabbits [23].
In summary, our studies suggest that hemoglobin and/or heme could contribute substantially to the initiation of the inflammatory response induced by different microbial TLR agonists in settings where trace amounts of blood coexist with bacterial or viral infection. The latter is of special relevance given the current epidemic of H1N1 influenza and the knowledge that severe disease can be accompanied by blood in the alveolar spaces [49]. Further work will be needed to evaluate whether local concentrations of Hx are depleted in infected tissue microenvironments and whether infusion of Hx to bind, neutralize, or clear hemoglobin or hemin could be beneficial in some settings.
Acknowledgments
Financial support: National Institutes of Health (grants AI059010 and GM59694) and the Shriners Hospital for Crippled Children (grant 8720).
Footnotes
Potential conflicts of interest: in accordance with institutional policy, H.S.W. has reported the use of hemopexin as a potential anti-inflammatory agent, and an application for patent protection has been filed. All other authors: no conflicts.
References
- 1.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 4.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–87. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lombardi V, Van Overtvelt L, Horiot S, Moingeon P. Human dendritic cells stimulated via TLR7 and/or TLR8 induce the sequential production of Il-10, IFN-γ, and IL-17A by naive CD4+ T cells. J Immunol. 2009;182:3372–9. doi: 10.4049/jimmunol.0801969. [DOI] [PubMed] [Google Scholar]
- 6.Strandskog G, Skjaeveland I, Ellingsen T, Jorgensen JB. Double-stranded RNA- and CpG DNA-induced immune responses in Atlantic salmon: comparison and synergies. Vaccine. 2008;26:4704–15. doi: 10.1016/j.vaccine.2008.06.054. [DOI] [PubMed] [Google Scholar]
- 7.Vanhoutte F, Paget C, Breuilh L, et al. Toll-like receptor (TLR)2 and TLR3 synergy and cross-inhibition in murine myeloid dendritic cells. Immunol Lett. 2008;116:86–94. doi: 10.1016/j.imlet.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 8.He H, Genovese KJ, Nisbet DJ, Kogut MH. Synergy of CpG oligo-deoxynucleotide and double-stranded RNA (poly I:C) on nitric oxide induction in chicken peripheral blood monocytes. Mol Immunol. 2007;44:3234–42. doi: 10.1016/j.molimm.2007.01.034. [DOI] [PubMed] [Google Scholar]
- 9.Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, et al. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of Toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005;52:2313–22. doi: 10.1002/art.21278. [DOI] [PubMed] [Google Scholar]
- 10.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–76. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bagchi A, Herrup EA, Warren HS, et al. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J Immunol. 2007;178:1164–71. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]
- 12.Paul-Clark MJ, Sorrentino R, Bailey LK, Sriskandan S, Mitchell JA. Gram-positive and gram-negative bacteria synergize with oxidants to release CXCL8 from innate immune cells. Mol Med. 2008;14:238–46. doi: 10.2119/2007-00098.Paul-Clark. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H. Role of Toll-like receptors in the development of sepsis. Shock. 2008;29:315–21. doi: 10.1097/SHK.0b013e318157ee55. [DOI] [PubMed] [Google Scholar]
- 14.Bornside GH, Bouis PJ, Jr, Cohn I., Jr Hemoglobin and Escherichia coli, a lethal intraperitoneal combination. J Bacteriol. 1968;95:1567–71. doi: 10.1128/jb.95.5.1567-1571.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roth RI, Levin FC, Levin J. Distribution of bacterial endotoxin in human and rabbit blood and effects of stroma-free hemoglobin. Infect Immun. 1993;61:3209–15. doi: 10.1128/iai.61.8.3209-3215.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su D, Roth RI, Yoshida M, Levin J. Hemoglobin increases mortality from bacterial endotoxin. Infect Immun. 1997;65:1258–66. doi: 10.1128/iai.65.4.1258-1266.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su D, Roth RI, Levin J. Hemoglobin infusion augments the tumor necrosis factor response to bacterial endotoxin (lipopolysaccharide) in mice. Crit Care Med. 1999;27:771–8. doi: 10.1097/00003246-199904000-00034. [DOI] [PubMed] [Google Scholar]
- 18.Bodet C, Chandad F, Grenier D. Hemoglobin and LPS act in synergy to amplify the inflammatory response. J Dent Res. 2007;86:878–82. doi: 10.1177/154405910708600914. [DOI] [PubMed] [Google Scholar]
- 19.Kaca W, Roth RI, Levin J. Hemoglobin, a newly recognized lipopolysaccharide (LPS)-binding protein that enhances LPS biological activity. J Biol Chem. 1994;269:25078–84. [PubMed] [Google Scholar]
- 20.Brandenburg K, Garidel P, Andra J, et al. Cross-linked hemoglobin converts endotoxically inactive pentaacyl endotoxins into a physiologically active conformation. J Biol Chem. 2003;278:47660–9. doi: 10.1074/jbc.M304743200. [DOI] [PubMed] [Google Scholar]
- 21.Bornside GH, Bouis PJ, Jr, Cohn I., Jr Enhancement of Escherichia coli infection and endotoxic activity by hemoglobin and ferric ammonium citrate. Surgery. 1970;68:350–5. [PubMed] [Google Scholar]
- 22.Su D, Roth RI, Levin J. Hemoglobin infusion augments the tumor necrosis factor response to bacterial endotoxin (lipopolysaccharide) in mice. Crit Care Med. 1999;27:771–8. doi: 10.1097/00003246-199904000-00034. [DOI] [PubMed] [Google Scholar]
- 23.Warren HS, Fitting C, Hoff E, et al. Resilience to bacterial infection: difference between species could be due to proteins in serum. J Infect Dis. 2010;201:223–32. doi: 10.1086/649557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Figueiredo RT, Fernandez PL, Mourao-Sa DS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282:20221–9. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- 25.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–22. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 26.Andrade CT, Barros LA, Lima MC, Azero EG. Purification and characterization of human hemoglobin: effect of the hemolysis conditions. Int J Biol Macromol. 2004;34:233–40. doi: 10.1016/j.ijbiomac.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 27.Liang X, Lin T, Sun G, Beasley-Topliffe L, Cavaillon JM, Warren HS. Hemopexin down-regulates LPS-induced proinflammatory cytokines from macrophages. J Leukoc Biol. 2009;86:229–35. doi: 10.1189/jlb.1208742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Novitsky TJ, Roslansky PF, Siber GR, Warren HS. Turbidometric method for quantifying serum inhibition of limulus amoebocyte lysate response. J Clin Micro. 1985;20:211–6. doi: 10.1128/jcm.21.2.211-216.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schilling D, Thomas K, Nixdorff K, Vogel SN, Fenton MJ. Toll-like receptor 4 and Toll–IL-1 receptor domain-containing adapter protein (TIRAP)/myeloid differentiation protein 88 adapter-like (Mal) contribute to maximal IL-6 expression in macrophages. J Immunol. 2002;169:5874–80. doi: 10.4049/jimmunol.169.10.5874. [DOI] [PubMed] [Google Scholar]
- 30.Liang MD, Bagchi A, Warren HS, et al. Bacterial peptidoglycan-associated lipoprotein: a naturally occurring Toll-like receptor 2 agonist that is shed into serum and has synergy with lipopolysaccharide. J Infect Dis. 2005;191:939–48. doi: 10.1086/427815. [DOI] [PubMed] [Google Scholar]
- 31.Yang H, Wang H, Bernik TR, et al. Globin attenuates the innate immune response to endotoxin. Shock. 2002;17:485–90. doi: 10.1097/00024382-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 32.Tolosano E, Altruda F. Hemopexin: structure, function, and regulation. DNA Cell Biol. 2002;21:297–306. doi: 10.1089/104454902753759717. [DOI] [PubMed] [Google Scholar]
- 33.Paoli M, Anderson BF, Baker HM, Morgan WT, Smith A, Baker EN. Crystal structure of hemopexin reveals a novel high-affinity heme site formed between two beta-propeller domains. Nat Struct Biol. 1999;6:926–31. doi: 10.1038/13294. [DOI] [PubMed] [Google Scholar]
- 34.Watanabe J, Grijalva V, Hama S, et al. Hemoglobin and its scavenger protein haptoglobin associate with ApoA-1–containing particles and influence the inflammatory properties and function of high density lipoprotein. J Biol Chem. 2009;284:18292–301. doi: 10.1074/jbc.M109.017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson BMG, Severn A, Rapson NT, Chana J, Hopkins P. A convenient human whole blood culture system for studying the regulation of tumour necrosis factor release by bacterial lipopolysaccharide. J Immunol Methods. 1991;139:233–40. doi: 10.1016/0022-1759(91)90193-j. [DOI] [PubMed] [Google Scholar]
- 36.Cox KH, Ofek I, Hasty DL. Enhancement of macrophage stimulation by lipoteichoic acid and the costimulant hemoglobin is dependent on Toll-like receptors 2 and 4. Infect Immun. 2007;75:2638–41. doi: 10.1128/IAI.01320-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanabe S, Gottschalk M, Grenier D. Hemoglobin and Streptococcus suis cell wall act in synergy to potentiate the inflammatory response of monocyte-derived macrophages. Innate Immun. 2008;14:357–63. doi: 10.1177/1753425908098388. [DOI] [PubMed] [Google Scholar]
- 38.Takeda K, Akira S. Toll-like receptors. In: Coligan JE, et al., editors. Current Protocols in Immunology. unit 14.12. chapter 14. 2007. [DOI] [PubMed] [Google Scholar]
- 39.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 40.Crowley JP, Metzger J, Gray A, Pivacek LE, Cassidy G, Valeri CR. Infusion of stroma-free cross-linked hemoglobin during acute gram-negative bacteremia. Circ Shock. 1993;41:144–9. [PubMed] [Google Scholar]
- 41.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–16. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 42.Schaer DJ, Alayash AI, Buehler PW. Gating the radical hemoglobin to macrophages: the anti-inflammatory role of CD163, a scavenger receptor. Antioxid Redox Signal. 2007;9:991–9. doi: 10.1089/ars.2007.1576. [DOI] [PubMed] [Google Scholar]
- 43.Polticelli F, Bocedi A, Minervini G, Ascenzi P. Human haptoglobin structure and function—a molecular modelling study. FEBS J. 2008;275:5648–56. doi: 10.1111/j.1742-4658.2008.06690.x. [DOI] [PubMed] [Google Scholar]
- 44.Hrkal Z, Vodrazka Z, Kalousek I. Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur J Biochem. 1974;43:73–8. doi: 10.1111/j.1432-1033.1974.tb03386.x. [DOI] [PubMed] [Google Scholar]
- 45.Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest. 2008;118:239–47. doi: 10.1172/JCI32730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–55. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 47.Pamplona A, Ferreira A, Balla J, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med. 2007;13:703–10. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- 48.Warren HS. Editorial: Mouse models to study sepsis syndrome in humans. J Leukoc Biol. 2009;86:199–201. doi: 10.1189/jlb.0309210. [DOI] [PubMed] [Google Scholar]
- 49.Perez-Padilla R, Rosa-Zamboni D, Ponce DL, et al. Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N Engl J Med. 2009;361:680–9. doi: 10.1056/NEJMoa0904252. [DOI] [PubMed] [Google Scholar]