

Abstract
Prolonged cardiac overexpression of the mitochondrial biogenesis regulatory transcriptional coactivator PGC-1α disrupts cardiac contractile function and its genetic ablation limits cardiac capacity to enhance work-load. In contrast, transient induction of PGC-1α alleviates neuronal cell oxidative stress and enhances skeletal myotube antioxidant defenses. We explored whether transient upregulation of PGC-1α in the heart protects against ischemia-reperfusion injury. The transient induction of PGC-1α in the cardiac-restricted inducible PGC-1α transgenic mouse, increased PGC-1α protein levels 5-fold. Following 25 minutes of ischemia and 2 hours of reperfusion on a Langendorff perfusion apparatus, contractile recovery and the rate pressure product was significantly blunted in mice overexpressing PGC-1α vs. controls. Affymetrix gene array analysis showed a 3-fold PGC-1α-mediated upregulation of adenine nucleotide translocase 1 (ANT1). As ANT1 upregulation induces cardiomyocyte cell death we investigated whether the induction of ANT1 by PGC-1α contributes to this enhanced ischemia-stress susceptibility. Infection with adenovirus harboring PGC-1α into cardiac-derived H9c2 cells significantly upregulates ANT1 without changing basal cell viability. In response to anoxia-reoxygenation injury cell death is significantly increased following PGC-1α overexpression. This detrimental effect is abolished following siRNA knockdown of ANT1. Similarly, the attenuation of ANT-1 in the presence of PGC-1α overexpression preserves the mitochondrial membrane potential in response to hydrogen-peroxide stress. Interestingly, the isolated knockdown of ANT1 also protects H9c2 cells from anoxia-reoxygenation injury. Taken together these data suggest that transient induction of PGC-1α in the murine heart decreases ischemia-reperfusion contractile recovery and diminishes anoxia-reoxygenation tolerance in H9c2 cells. These adverse phenotypes appear to be mediated, in part, by PGC-1α induced upregulation of ANT1.
Keywords: PGC-1α, Cardiac ischemia-reperfusion, ANT1
Mitochondrial homeostasis and regulatory adaptations are recognized as central to the myocardial capacity to tolerate both ischemic and oxidative stress [1, 2]. In light of our increased understanding of the molecular programs governing mitochondrial homeostasis it is postulated that modulation of mitochondrial biology may be a feasible strategy to enhance tolerance to mitochondrial stress with concomitant cellular survival advantage [3, 4]. The most extensively investigated mitochondrial regulator is the mitochondrial biogenesis master regulator peroxisome proliferator activated receptor gamma coactivator 1 alpha (PGC-1α). The genetic deletion of PGC-1α results in diminished cardiac mitochondrial enzyme activities, diminished ATP production, blunted cardiac postnatal growth, diminished chronotropic capacity and an inability to appropriately augment cardiac workload in response to exercise or to β-adrenergic stimulation [5, 6]. In contrast, unfettered cardiac overexpression of PGC-1α results in a progressive and uncontrolled increase in mitochondrial number with resultant disruption in cardiomyocyte sarcomeric structure and myocardial contractile function [7]. Collectively these data show that robust bidirectional modulation in PGC-1α levels in the heart severely disrupts mitochondrial homeostasis resulting in adverse myocardial function and stress-susceptibility. In contrast, transient overexpression of PGC-1α has been shown to enhance anti-oxidant enzymes in skeletal myotubes and to protect neuronal cells against oxidative stress [8, 9]. Moreover, in delayed cardiac ischemic preconditioning PGC-1α is temporarily induced during the transient ischemic stress which is then associated with subsequent enhanced myocardial ischemia-reperfusion tolerance [10]. We therefore investigated whether transient upregulation of PGC-1α in the heart confers protection against ischemia-perfusion injury.
Methods
Animal Studies
Double transgenic, TRE-PGC1α MHC-rtTA mice were kindly provided by Dr. Daniel Kelly. Briefly, TRE-PGC1α mice were crossed with mice harboring the reverse tetracycline transactivator downstream of the α-myosin heavy chain promoter (MHC-rtTA) on a FVB/N background [11]. To transiently induce cardiac-specific PGC-1α expression doxycycline supplemented drinking water (2g/L Doxycycline, 5% sucrose) was provided to the TRE-PGC-1α MHC-rtTA mice. Control mice (MHC-rtTA) were similarly given 2g/L doxycycline and 5% sucrose in their drinking water. Mice were sacrificed after three days and their hearts removed for subsequent studies. All animal procedures were approved by the Animal Care and Use Committee of the National Heart, Lung, and Blood Institute, National Institutes of Health.
RNA isolation and expression levels
H9c2 RNA was prepared using RNeasy RNA isolation kit (Qiagen). RNA was quantified and used for RT-PCR following first strand synthesis and for gene array analyses. H9c2 RNA amplification and gene array analysis were performed using mouse genome microarrays (Affymetrix). Real-time quantitative PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) with a MJ Research DNA Engine Opticon 2 fluorescence detection system. Ribosomal 18s and Tbp1 primers were used as internal standards.
Protein analysis
Primary antibodies: goat ANT (Santa Cruz); mouse actin (Ambion); mouse ANT1 (MitoSciences); rabbit PGC1α (gift from D. Kelly) and mouse NDUFa9 (Abcam). H9c2 cells were harvested and lysed in RIPA buffer containing protease inhibitors. Mouse cardiac mitochondria protein was prepared as described above. Samples were used for SDS PAGE and Western immunoblotting.
Cardiac response to ischemia-reperfusion injury
TRE-PGC-1α MHC-rtTA and control mice were anesthetized with an intraperitoneal injection of 0.10 cc pentobarbital sodium diluted 1:5 in perfusate. The abdominal cavity was exposed with a transverse incision and 0.05 cc heparin was administered to the inferior vena cava. The heart was quickly isolated and placed in ice-cold Krebs-Heinseleit (KH) buffer (25 mM NaHCO3, 120 mM NaCl, 11 mM glucose, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, and 1.75 mM CaCl2) to arrest the heart. The heart was perfused via the aorta. A water-filled latex balloon was inserted into the left ventricle to measure hemodynamic parameters using a PowerLab 2/25 and Chart v5.5 software (AD Instruments, Colorado Springs, CO). All hearts were perfused with KH buffer gassed with 95% O2 and 5% CO2 and maintained at 37°C for 30 min before being subjected to 25 min of no-flow ischemia followed by 2 h of reperfusion. Recovery of left ventricular developed pressure (LVDP) and rate pressure product (RPP; LVDP × heart rate), expressed as a percentage of the initial values prior to ischemia, were measured at 2 h of reflow. Infarct size was determined with triphenyltetrazolium chloride stain (TTC) as previously described [12].
Mitochondria isolation and respiration
In brief, all mitochondria isolation procedures and buffers were kept cold on ice. Mouse cardiac ventricles were homogenized in KE buffer (0.18 M KCl, 0.01M EDTA, pH 7.4) using a Dounce homegenizer. Samples were then centrifuged at 2380rpm for 5 minutes to pellet cellular debris. Supernatants were centrifuged for 5 minutes at 4500rpm and the mitochondria pellet resuspended in KE buffer and used immediately for experiments. Mitochondria respiration was measured using fiber-optic oxygen spectrophotometry (Instech Laboratories). Mitochondria (0.5 mg/mL) were resuspended in respiration buffer (0.25M sucrose, 25mM Tris-HCl, 8.5mM K2HPO4, pH 7.4) and basal state 3 respiration determined in the presence of 5 mM glutamate, 5 mM malate, and 500 μM ADP.
To evaluate mitochondria copy number the ratio of mitochondrial gene expression to nuclear gene expression was calculated. Genomic DNA was isolated from mouse heart using a DNeasy tissue DNA isolation kit (Qiagen). Real time quantitative PCR for mitochondrial genes ND1, cyt.b, and the nuclear gene 18s was performed as described above.
Cell culture and transfection
H9c2 rat myoblasts (ATCC) were cultured in 10% FBS in DMEM with 1% penicillin/streptomycin at 37 °C, 5% CO2. Cells were transfected with control siRNA (Invitrogen) or ANT1 siRNA (Qiagen) using Lipofectamine RNAiMAX (Invitrogen) and were cultured 48 hours prior to experiments. Transient transfection in H9c2 with a control vector pcDNA3.1 (Invitrogen) or pcDNA3.1-ANT1 was performed using Lipofectamine 2000 (Invitrogen) 48 hours prior to experiments. For flow cytometry experiments, transient transfection of a flag tagged PGC1α construct [13] was accomplished with Fugene HD (Roche, Indianapolis, IN).
Adenoviral vectors
H9c2 cells were infected with adenoviral vectors encoding for full-length mouse PGC-1α-Ad-Track-FLAG-HA-PGC-1α, kindly provided by P. Puigserver (Harvard Medical School, Boston, MA). Adeno-GFP used for infection controls was kindly provided by K. Walsh (Boston University School of Medicine, Boston, MA).
Mitochondrial membrane potential and flow cytometry
H9c2 cells were provided with siRNA and DNA plasmids as described. After 72 hours, cells were given 1 mM H2O2 for 30 minutes, trypsinized, and incubated with 50 nM Tetramethyl Rhodamine Methyl Ester (TMRM; Invitrogen, Molecular Probes) in DMEM cell media for 15 minutes at 37°C. The cells were then washed with 1X PBS and resuspended in 1X PBS with 1% BSA. The suspended cells were then analyzed on a FACScalibur flow cytometer (BD Biosciences). FCS Express V3 software (De Novo Software) was used to analyze FACS data.
ATP assay
ATP assays were performed using the ATP determination kit (Invitrogen) per manufacturer’s instructions. Data was recorded with a Tecan GENios Plus plate reader.
Cell viabilit
H9c2 were subject to anoxia-reoxygenation injury as previously described [14]. In brief, transfected cells were exposed to 17-h anoxia in DMEM media (no glucose, pyruvate, or antibiotics), using a GasPak EZ system (Becton–Dickenson). Following anoxia, normal growth medium was added and cells incubated for an additional 2 hours. Cellular injury was determined using a LDH cell viability assay (CytoTox 96, Promega) with subsequent analysis on a Tecan microtiter plate reader.
Statistical Analysis
Gene array expression levels were analyzed after segregation to exclude gene probesets employing a false discovery rate (FDR) of <20% and a fold change >1.2. Differences between groups were evaluated for significance using the Student t-test. Analyses to assess cell death were evaluated for significance using ANOVA. P < 0.05 was considered statistically significant. Data are expressed as mean ± S.E.M.
Results
PGC-1α was upregulated in the cardiac-restricted inducible double transgenic mice following the addition of doxycycline to the drinking water. Three-days of doxycycline treatment were found to be the shortest time period necessary to obtain a consistent increase in PGC-1α protein levels (Figure 1A). At three days of PGC-1α induction mitochondrial genomic copy number was unchanged (Figure 1B), mitochondrial respiration in response to glutamate/malate and to palmitoylcarnitine was unchanged (data not shown) and the gene transcript levels of numerous PGC-1α target genes were upregulated less than 2 fold in the double-transgene versus the vehicle treated control mice (Figure 1C). To assess ischemia-reperfusion tolerance, isolated mouse hearts were interrogated using the Langendorff perfusion apparatus and contractile recovery and infarct size were assessed at numerous time points of ischemia followed by two hours of reperfusion. Following 25 minutes of ischemia and 2 hours of reperfusion (experimental protocol shown in Figure 2A), the recovery of left ventricular developed pressure (LVDP, Figure 2B) and rate pressure product (RPP, Figure 2C) were significantly more depressed in the PGC-1α transgene (LVDP 33.57 ± 7.07%; RPP 27.4 ± 5.2%) compared to their littermate controls (LVDP 52.4 ± 6.45 %; RPP 45.0 ± 4.6) (n=12 per group, p < 0.03 for both parameters). Despite these significant changes, a clear difference in ventricular infarct size was not evident comparing the PGC-1α transgenic mice to littermate controls in this same study sample (Figure 2D).
Figure 1.
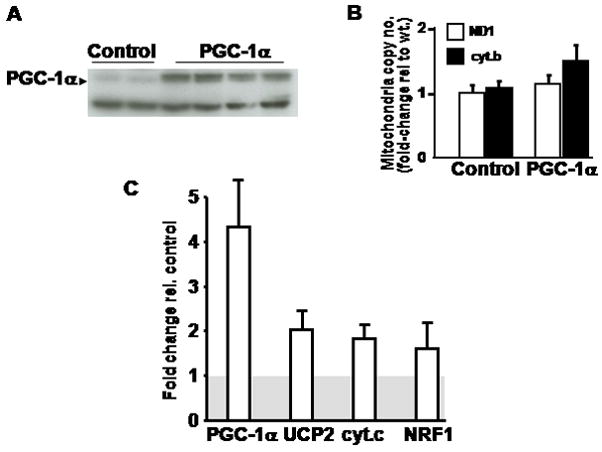
A) Western blot analysis shows upregulation of the PGC-1α after 72 hours of doxycycline administration to the mice. The lower non-specific band shows equal loading. B) Mitochondrial copy number as measured by the ratio of mitochondrial genomic transcripts (ND1 or cyt B) to genomic DNA (18s). ND1 – NADH dehydrogenase subunit 1, cyt B – cytochrome B. C) Relative transcript induction following doxycycline administration to mice. The grey shaded box represents the controls normalized to = 1. UCP2 – uncoupling protein 2, cyt c – cytochrome c, NRF1 – nuclear respiratory factor 1.
Figure 2.
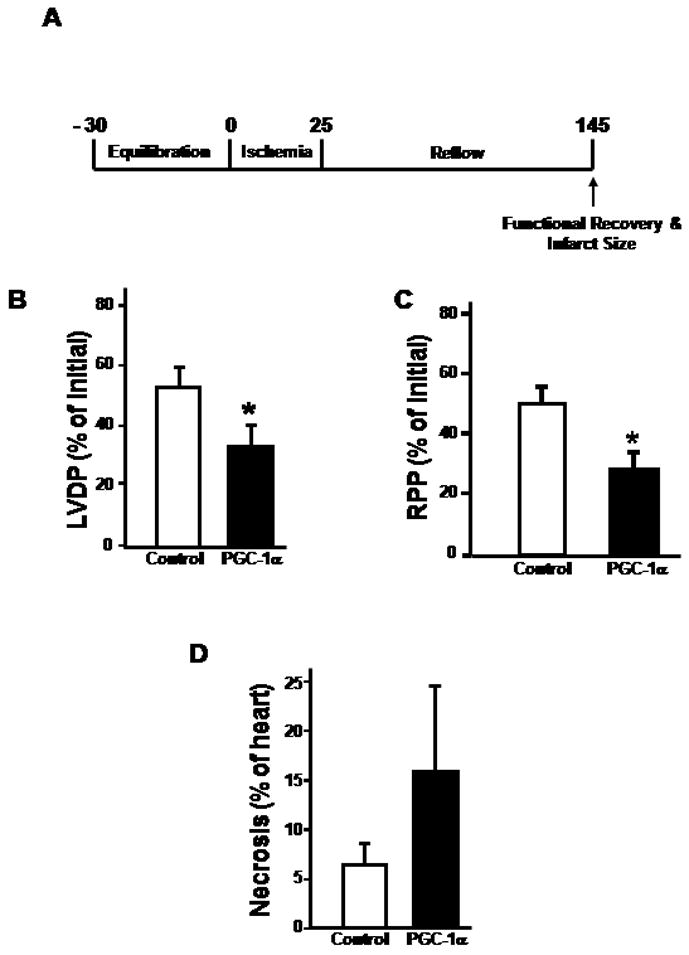
A) Schematic of time frame of study to investigate cardiac contractile recovery in the isolated perfused mouse hearts. B) Recovery of LVDP after a 25-minute ischemic period and 2-hour reperfusion. C) Recovery of rate pressure product following a 25-minute ischemic period and 2-hour reperfusion. Asterisk represents a p value of < 0.03 in both histograms versus their respective controls. D) Infarct size determination following the same ischemic-reperfusion insult. Twelve mice were used in each group for these experiments.
In light of the adverse effect of acute upregulation of PGC-1α in the heart, which contrasts to salutary effects in other tissues, we employed Affymetrix gene array analysis to identify putative targets that could mediate this effect. The heat map and gene array data are shown as supplemental figures 1 and table 1 respectively. As would be expected the gene expression profiles of numerous mitochondrial regulatory proteins and mitochondrial metabolic genes were modestly induced in response to upregulation of PGC-1α. Two findings that were intriguing with respect to the possible adverse response to PGC-1α upregulation included the downregulation of the uncoupling protein 3 (UCP3) and the upregulation of adenine nucleotide translocator - ANT1. As we have previously shown the requirement of UCP3 in protecting both cardiac cells and skeletal muscle against oxidative stress [15, 16] we investigated whether ANT1 induction contributes to PGC-1α-mediated reduction in oxidative stress tolerance.
We initially employed RT-PCR to confirm the induction of ANT1 in the doxycycline inducible mice. Following seventy-two hours of doxycycline administration ANT1 was shown to be upregulated approximately 5-fold in the PGC-1α inducible mice compared to the doxycycline treated control mice (Figure 3A). In order to manipulate ANT1 and PGC-1α in concert, subsequent studies were performed in cardiac derived H9c2 cells. These cardiac cells have previously been employed to study mechanisms modulating anoxia-reoxygenation tolerance [16]. Infection of H9c2 cells with an adenoviral construct harboring PGC-1α resulted in a significant upregulation of the transcript encoding for ANT1 (Figure 3B) with a parallel and robust induction of the steady-state protein levels of ANT1 (Figure 3C). siRNA targeting of ANT1 was employed to evaluate whether the knockdown of this mitochondrial protein would modulate PGC-1α-induced effects on H9c2 cell viability. In parallel with the overexpression of PGC-1α, the knockdown of ANT1 reduces ANT1 levels by ≈ 15 percent, p = 0.001 (Figure 3D) which functionally results in a similar percent reduction in cellular ATP levels compared to the overexpression of PGC-1α and scrambled siRNA, p = 0.013 (Figure 3E).
Figure 3.
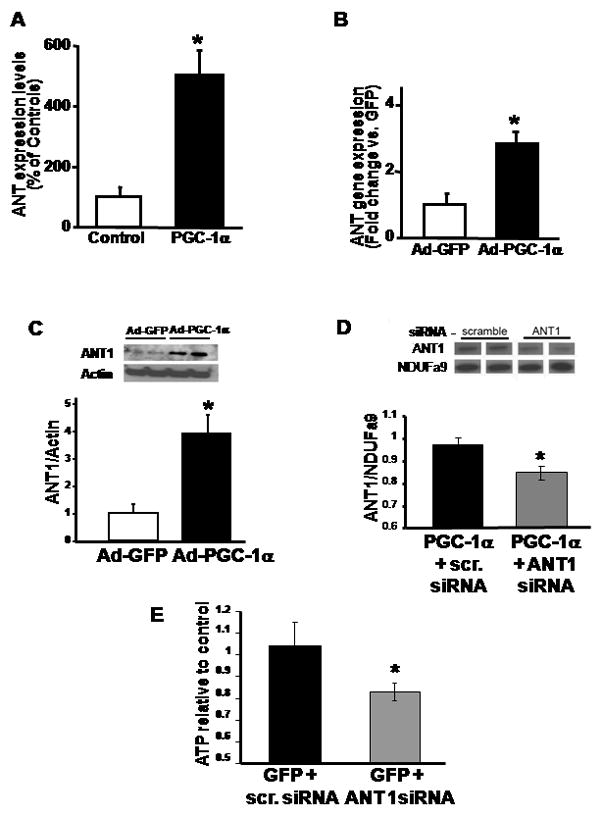
A) RT-PCR analysis of ANT1 expression in mouse hearts following 3-day doxycycline-induced PGC1α expression. ANT1 expression is normalized to 18s and expressed as percentage of controls. N=6, * p < 0.05 versus control mice. B) RT-PCR analysis of ANT1 in H9c2 after 48 h infection with either adenoviral PGC-1α (Ad-PGC) or adenoviral GFP (Ad-GFP) control. Results were normalized versus 18s and expressed as fold-change versus Ad-GFP. N = 6, * p < 0.05 versus Ad-GFP. C) Densitometric quantification of ANT1 protein in H9c2 after 48 h infection by Ad-PGC-1α or Ad-GFP, measured by Western immunoblot analysis. Actin was used to evaluate protein loading. Upper panel, representative Western immunoblot. N = 3 experiments in duplicate, * p < 0.05 versus Ad-GFP. D) Western blot of ANT1 expression in PGC-1α transfected H9c2 cells with or without ANT1 siRNA after 72 hours. Densitometric quantification of ANT1 protein in H9c2 mitochondria. NDUFa9 expression shows mitochondrial protein levels (n = 4 per group, * p < 0.01 versus scrambled siRNA) E) ATP levels in H9c2 cells used in experiments show decreased ATP with knockdown of ANT1. n = 4–5 per group, * p < 0.05 versus scrambled siRNA.
To delineate the functional effect of ANT1 induction in response to PGC-1α, we explored the differential effects of manipulating these proteins on H9c2 cell viability in response to anoxia-reoxygenation and to the ability to maintain the mitochondrial membrane potential in response to H2O2 induced oxidative stress. In parallel with the murine findings, the infection of H9c2 cells with PGC-1α resulted in increased cell injury versus adenoviral control vector treated H9c2 cells in response to an anoxia-reoxygenation insult (Figure 4A). Interestingly, the knockdown of ANT1 alone enhanced resilience to anoxia-reoxygenation injury and the combined overexpression of PGC-1α with the depletion of ANT1 restores anoxia-reoxygenation tolerance to control levels (Figure 4A). As the knockdown of ANT1 has recently been shown to desensitize the mitochondrial permeability transition in the brain [17], we investigated whether the knockdown of ANT1 would blunt the loss of the mitochondrial membrane potential in PGC-1α-overexpressing cells in response to oxidative stress. In response to H2O2, the reduction in ANT1 levels in PGC-1α infected cells maintained their relative mitochondrial membrane potential to a greater degree compared to PGC-1α infected cells in the presence of scrambled siRNA (Figure 4B). Taken together, these data implicate the induction of ANT1 levels as a mediator of the adverse stress-tolerant phenotype in cardiac derived H9c2 cells in response to transient robust upregulation of PGC-1α and would support a similar action in the intact heart, although this was not directly assessed.
Figure 4.
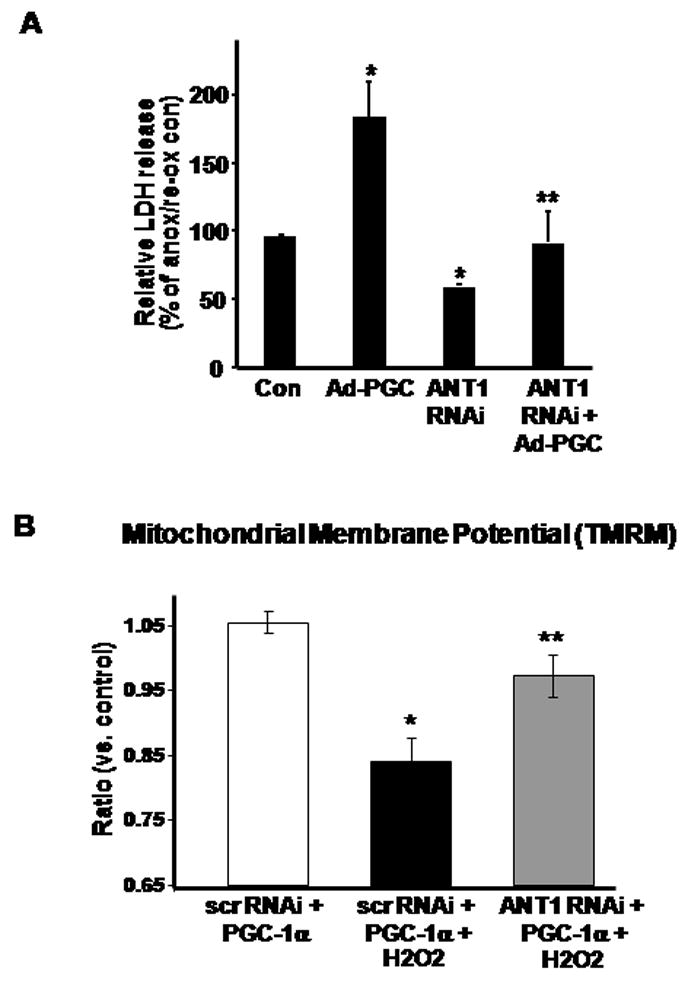
A) Relative LDH release in H9c2 treated with Ad-PGC-1α or ANT1 siRNA, or both in combination. After 48 h treatment, H9c2 were subject to 17 h anoxia – 2 h reperfusion with subsequent determination of LDH release. N = 6, * p < 0.05 versus control (Con), ** p < 0.05 versus Ad-PGC. B) Mitochondrial membrane potential in H9c2 cells as measured by flow cytometery with TMRM. At 72 hours post-transfection, cells were treated with 1 mM H2O2 for 30 minutes. The graph is a comparison of geometric means between 3–5 experiments relative to the control geometric mean. * p <0.05 versus control and ** p<0.05 versus PGC1α with H2O2.
Discussion
The major findings in this paper show that despite salutary effects in other tissues, transient induction of PGC-1α in the heart diminishes cardiac contractile recovery in response to an ischemia-reperfusion insult. Furthermore, these data implicate the upregulation of ANT-1 in response to PGC-1α induction as a component of this adverse phenotype. The role of ANT-1 in this stress-tolerance program is validated in cardiac derived H9c2 cells as ANT-1 knockdown abolishes the detrimental response to PGC-1α following anoxia-reoxygenation cell injury and in response to H2O2 stress.
As PGC-1α is a transcriptional coactivator, its induction is associated with a plethora of gene regulatory events that modulate mitochondrial biogenesis [7, 18], metabolic substrate utilization [19] and antioxidant defense programs [9]. Robust or chronic induction of this transcriptional coactivator has been shown to be detrimental in the heart, as it promotes the excessive proliferation of mitochondria that are thought to structurally impede the cardiomyocyte contractile machinery [7]. However, transient induction has been shown to evoke ameliorative effects against neuronal cell oxidative stress [8], in improving skeletal muscle insulin resistance [13, 19] and in upregulating anti-oxidant enzymes in skeletal muscle [9]. Moreover, in delayed ischemic preconditioning in the heart, the transcript of PGC-1α and cognate target protein levels are transiently induced by the sublethal transient ischemic preconditioning stress [10]. Based on these collective data, we hypothesized that the isolated and transient induction of PGC-1α in the heart would have salutary effects against ischemia-reperfusion injury. This hypothesis was tested in transgenic mice with cardiac restricted doxycycline inducible induction of PGC-1α for a 72 hour period. Despite no robust effect on mitochondrial biogenesis, this regulatory perturbation worsened post-ischemic contractile function in the isolated perfused mouse heart. This detrimental effect was probably not related to the model system in that infection of cardiac derived H9c2 cells also show diminished tolerance to anoxia-reoxygenation and oxidative stress. Hence, despite the advantageous effect of transient induction of this regulatory coactivator in other cell types, these salutary effects are not operational in the heart or in cardiac-derived transformed myoblasts. Although the role of PGC-1α has not been functionally validated, the difference in these data with the transient induction of PGC-1α in response to delayed ischemic preconditioning may hypothetically be explained by the myriad of additional ‘triggers’ induced by delayed preconditioning (reviewed [20, 21]) that are likely not present with the isolated induction of PGC-1α.
As the murine results invalidated our initial hypothesis, we employed unbiased techniques to identify potential candidates that could confer stress susceptibility. In addition to transcriptional regulation PGC-1α has previously been shown to function by binding to, and promoting alternative splicing of RNA [22]. To evaluate whether the regulation of alternatively splice transcripts could be implicated in this phenotype we isolated RNA from the cardiac tissue of the doxycycline induced PGC-1 overexpressing and control heart tissue. Interestingly, at least in the heart, no evidence of alternate splice variants were identified using the Affymetrix exon array chips (data not shown). We then explored gene array profiles by Affymetrix expression array analysis. Multiple nuclear encoded mitochondrial transcripts were induced by PGC-1α and these are shown in the supplemental data. The two most interesting transcripts with respect to putative affectors of the phenotype found were the significant downregulation of UCP3 and the induction of ANT1 following PGC-1α overexpression. We focused on ANT1 as the overexpression of this translocator isoform has previously been shown to promote cell death in numerous transformed cell lines [23]. More recently the overexpression of ANT1 in cardiomyocytes has been shown to induce a gene-dose dependent increase in cell death, independent of mitochondrial permeability transition [24]. In parallel, in ANT1 knockout mice, brain mitochondria show increased resistance to calcium-induced mitochondrial permeability transition [17]. Furthermore, data suggests that an increase in the ratio of ANT1 to ANT2 may have detrimental effects [25]. This change in ratio has been shown to diminish the efficiency of nucleotide transfer [26] and a change in this ratio of ANT1 to ANT2 has recently been implicated in skeletal muscle mitochondrial dysfunction in pacing-induced heart failure in dogs [27]. Finally, the activation of ANT has been shown to be instrumental in mitochondrial uncoupling associated with ischemia-reperfusion injury in the heart [28]. Although the majority of data suggest an increase in ANT is detrimental, one study with transgenic overexpression of ANT1 in the mouse heart has been shown to have a beneficial effect in preventing contractile dysfunction in response to severe hyperglycemia in streptozotocin induced diabetic mice [29]. In this study, the induction of ANT1 following short-term PGC-1α activation is modest, and is not associated with any change in contractile function at baseline in these mice. In parallel, the upregulation of ANT1 in H9c2 cells in response to infection with PGC-1α is not associated with reduced basal cell viability. However, cardiac contractile recovery and cell viability and the maintenance of the mitochondrial membrane potential in H9c2 cells following ischemia-reperfusion, anoxia-reoxygenation and H2O2 stress respectively is attenuated in association with this induction of ANT1. The direct implication of ANT1 in this phenotype is shown in the H9c2 cells, where genetic knockdown of ANT1 in parallel with PGC-1α infection rescues this detrimental phenotype.
The adverse effects of PGC-1α induction appear to be operational in the model systems employed in this study. However, several limitations of this study need to be recognized. Firstly, and most importantly, the adverse effects of transient isolated PGC-1α induction in the heart is primarily evident as diminished contractile recovery and was not replicated by an increase in the degree of myocardial infarction. Possible explanations for this discrepancy include that as PGC-1α primarily regulates metabolic programs it is feasible that its effect predominantly modulates the degree of stunning [30] following ischemia/reperfusion rather than infarct size. Additionally, as the FVB strain of mouse has been shown to exhibit greater resistance to infarction following myocardial ischemia compared to the more commonly used C57Bl/6 strain [31], and as the inducible PGC-1α transgene is incorporated into the FVB background in this study, it may be possible that the capacity to demonstrate a difference in infarct size in this strain is less robust. The second limitation to this study is the use of the H9c2 myoblast cell line. Although these cells are derived from the heart and include numerous cardiomyocyte characteristics [32], it is a transformed dividing cell line which is quite distinct from the predominantly post-mitotic heart. Moreover, the H9c2 line is known not to be a faithful reporter of the heart phenotype in that it expresses both cardiac and skeletal muscle calcium channels [33]. Hence, a caveat exists with respect to extending the findings from this cell line to the intact heart and our findings need to be interpreted in this context.
In conclusion, studies to date suggest that the modest and transient induction of PGC-1α does have salutary effects in multiple tissue types and in response to various biological stressors. These data have even stimulated the search for small molecular activators of PGC-1α [34]. However, our data cautions whether even lows levels or transient isolated induction of PGC-1α is appropriate as a putative adaptive mediator in cardiac ischemia-reperfusion and/or oxidative stress. Furthermore, this study demonstrates that the deleterious effect of PGC-1α induction is mediated, at least in part, by the upregulation of ANT1 in cardiac derived H9c2 cells.
Supplementary Material
Acknowledgments
We would like to thank the members of the NHLBI Genotyping Core, including Nalini Raghavachari and Xiuli Xu for their assistance with the gene array expression analysis. We would also like to thank Ann Williams from the NHLBI Flow Cytometry core for assistance.. This work is funded by NHLBI Division of Intramural Research Funds. We thank Daniel P. Kelly and Maria L. Valencik for the gift of the cardiac inducible PGC-1α transgenic mouse.
Footnotes
Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. CardiovascRes. 2006;72(2):210–9. doi: 10.1016/j.cardiores.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88(2):581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McLeod CJ, Pagel I, Sack MN. The mitochondrial biogenesis regulatory program in cardiac adaptation to ischemia--a putative target for therapeutic intervention. Trends CardiovascMed. 2005;15(3):118–23. doi: 10.1016/j.tcm.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz DR, Sack MN. Targeting the mitochondria to augment myocardial protection. CurrOpinPharmacol. 2008;8(2):160–5. doi: 10.1016/j.coph.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1(4):259–71. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoSBiol. 2005;3(4):e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. JClinInvest. 2000;106(7):847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 9.St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. JBiolChem. 2003;278(29):26597–603. doi: 10.1074/jbc.M301850200. [DOI] [PubMed] [Google Scholar]
- 10.McLeod CJ, Jeyabalan AP, Minners JO, Clevenger R, Hoyt RF, Jr, Sack MN. Delayed ischemic preconditioning activates nuclear-encoded electron-transfer-chain gene expression in parallel with enhanced postanoxic mitochondrial respiratory recovery. Circulation. 2004;110(5):534–9. doi: 10.1161/01.CIR.0000136997.53612.6C. [DOI] [PubMed] [Google Scholar]
- 11.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, et al. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. CircRes. 2004;94(4):525–33. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 12.Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation. 2009;120(3):245–54. doi: 10.1161/CIRCULATIONAHA.109.868729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pagel-Langenickel I, Bao J, Joseph JJ, Schwartz DR, Mantell BS, Xu X, et al. PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. JBiolChem. 2008;283(33):22464–72. doi: 10.1074/jbc.M800842200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynn EG, McLeod CJ, Gordon JP, Bao J, Sack MN. SIRT2 is a negative regulator of anoxia-reoxygenation tolerance via regulation of 14–3–3 zeta and BAD in H9c2 cells. FEBS Lett. 2008;582(19):2857–62. doi: 10.1016/j.febslet.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Z, Sack MN. ATF-1 is a hypoxia-responsive transcriptional activator of skeletal muscle mitochondrial-uncoupling protein 3. JBiolChem. 2008;283(34):23410–8. doi: 10.1074/jbc.M801236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLeod CJ, Aziz A, Hoyt RF, Jr, McCoy JP, Jr, Sack MN. Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. JBiolChem. 2005;280(39):33470–6. doi: 10.1074/jbc.M505258200. [DOI] [PubMed] [Google Scholar]
- 17.Lee J, Schriner SE, Wallace DC. Adenine nucleotide translocator 1 deficiency increases resistance of mouse brain and neurons to excitotoxic insults. BiochimBiophysActa. 2009;1787(5):364–70. doi: 10.1016/j.bbabio.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98(1):115–24. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 19.Benton CR, Nickerson JG, Lally J, Han XX, Holloway GP, Glatz JF, et al. Modest PGC-1alpha overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in subsarcolemmal, not intermyofibrillar, mitochondria. JBiolChem. 2008;283(7):4228–40. doi: 10.1074/jbc.M704332200. [DOI] [PubMed] [Google Scholar]
- 20.Bolli R. The late phase of preconditioning. CircRes. 2000;87(11):972–83. doi: 10.1161/01.res.87.11.972. [DOI] [PubMed] [Google Scholar]
- 21.Yellon DM, Downey JM. Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev. 2003;83(4):1113–51. doi: 10.1152/physrev.00009.2003. [DOI] [PubMed] [Google Scholar]
- 22.Monsalve M, Wu Z, Adelmant G, Puigserver P, Fan M, Spiegelman BM. Direct coupling of transcription and mRNA processing through the thermogenic coactivator PGC-1. MolCell. 2000;6(2):307–16. doi: 10.1016/s1097-2765(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 23.Bauer MK, Schubert A, Rocks O, Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. JCell Biol. 1999;147(7):1493–502. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baines CP, Molkentin JD. Adenine nucleotide translocase-1 induces cardiomyocyte death through upregulation of the pro-apoptotic protein Bax. 2009 doi: 10.1016/j.yjmcc.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dorner A, Giessen S, Gaub R, Grosse SH, Schwimmbeck PL, Hetzer R, et al. An isoform shift in the cardiac adenine nucleotide translocase expression alters the kinetic properties of the carrier in dilated cardiomyopathy. EurJHeart Fail. 2006;8(1):81–9. doi: 10.1016/j.ejheart.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 26.De Marcos LC, Trezeguet V, Dianoux AC, Brandolin G, Lauquin GJ. The human mitochondrial ADP/ATP carriers: kinetic properties and biogenesis of wild-type and mutant proteins in the yeast S. cerevisiae. Biochemistry. 2002;41(48):14412–20. doi: 10.1021/bi0261490. [DOI] [PubMed] [Google Scholar]
- 27.Rosca MG, Okere IA, Sharma N, Stanley WC, Recchia FA, Hoppel CL. Altered expression of the adenine nucleotide translocase isoforms and decreased ATP synthase activity in skeletal muscle mitochondria in heart failure. JMolCell Cardiol. 2009 doi: 10.1016/j.yjmcc.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Nadtochiy SM, Tompkins AJ, Brookes PS. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. BiochemJ. 2006;395(3):611–8. doi: 10.1042/BJ20051927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Ebermann L, Sterner-Kock A, Wika S, Schultheiss HP, Dorner A, et al. Myocardial overexpression of adenine nucleotide translocase 1 ameliorates diabetic cardiomyopathy in mice. ExpPhysiol. 2009;94(2):220–7. doi: 10.1113/expphysiol.2008.044800. [DOI] [PubMed] [Google Scholar]
- 30.Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev. 1999;79(2):609–34. doi: 10.1152/physrev.1999.79.2.609. [DOI] [PubMed] [Google Scholar]
- 31.Gao XM, Xu Q, Kiriazis H, Dart AM, Du XJ. Mouse model of post-infarct ventricular rupture: time course, strain- and gender-dependency, tensile strength, and histopathology. CardiovascRes. 2005;65(2):469–77. doi: 10.1016/j.cardiores.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 32.Stuck BJ, Lenski M, Bohm M, Laufs U. Metabolic switch and hypertrophy of cardiomyocytes following treatment with angiotensin II are prevented by AMP-activated protein kinase. JBiolChem. 2008;283(47):32562–9. doi: 10.1074/jbc.M801904200. [DOI] [PubMed] [Google Scholar]
- 33.Mejia-Alvarez R, Tomaselli GF, Marban E. Simultaneous expression of cardiac and skeletal muscle isoforms of the L-type Ca2+ channel in a rat heart muscle cell line. J Physiol. 1994 Jul 15;478(Pt 2):315–29. doi: 10.1113/jphysiol.1994.sp020252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arany Z, Wagner BK, Ma Y, Chinsomboon J, Laznik D, Spiegelman BM. Gene expression-based screening identifies microtubule inhibitors as inducers of PGC-1alpha and oxidative phosphorylation. ProcNatlAcadSciUSA. 2008;105(12):4721–6. doi: 10.1073/pnas.0800979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.