

Abstract
Diets high in saturated fat and fructose have been implicated in the development of obesity and nonalcoholic steatohepatitis (NASH) in humans. We hypothesized that mice exposed to a similar diet would develop NASH with fibrosis associated with increased hepatic oxidative stress that would be further reflected by increased plasma levels of the respiratory-chain component, oxidized coenzyme Q9 (oxCoQ9). Adult male C57Bl/6 mice were randomly assigned to chow, High-Fat (HF), or High-Fat High-Carbohydrate (HFHC) diets for 16 weeks. The chow and HF mice had free access to pure water while the HFHC group received water with 55% fructose & 45% sucrose (w/v). The HFHC and HF groups had increased body weight, body fat mass, fasting glucose, and were insulin resistant compared to the chow mice. HF and HFHC consumed similar calories. Hepatic triglyceride content, plasma ALT, and liver weight were significantly increased in HF and HFHC mice compared to chow mice. Plasma cholesterol (p<0.001), histological hepatic fibrosis, liver hydroxyproline content (p=0.006), collagen1 mRNA (p=0.003), CD11b-F4/80+Gr1+ monocytes (p<0.0001), transforming growth factor β1 mRNA (p=0.04) and alpha-smooth muscle actin mRNA (p=0.001) levels were significantly increased in the HFHC mice. Hepatic oxidative stress, as indicated by liver superoxide expression (p=0.002), 4-hydroxynonenal and plasma oxCoQ9 (p<0.001) levels was highest in HFHC mice.
Conclusions
These findings demonstrate that non-genetically modified mice maintained on a HFHC diet in addition to developing obesity, have increased hepatic ROS and a NASH-like phenotype with significant fibrosis. Plasma oxCoQ9 correlated with fibrosis progression. The mechanism of fibrosis may involve fructose inducing increased ROS associated with CD11b+F4/80+Gr1+ hepatic macrophage aggregation resulting in TGFβ1 signaled collagen deposition and histologically visible hepatic fibrosis.
Keywords: Fibrosis, Obesity, Mitochondria, Inflammation, Steatosis, Murine, Ubiquinone, Coenzyme Q, Oxidative Stress
Introduction
Epidemiologic data suggest that there has been a significant rise in calories consumed from saturated fat and fructose rich foods (1). This has been paralleled by an increasing prevalence of obesity and its associated hepatic comorbidity: nonalcoholic fatty liver disease (NAFLD) (2). Natural history studies of NAFLD indicate that the presence of fibrosis within the more severe phenotype, nonalcoholic steatohepatitis (NASH), is an important predictor of adverse long-term outcomes, including diabetes and progression to cirrhosis (3, 4). Fibrosis progression to cirrhosis is thought to be modulated through hepatic reactive oxygen species (ROS) generation, macrophage activation, and transforming growth factor β (TGFβ) mediated collagen deposition (5–7). Though recent data have highlighted potential biomarkers for distinguishing NAFLD from NASH, a liver biopsy continues to be the gold standard to monitor fibrosis progression within NASH (8). The role of saturated fat and fructose in triggering these mechanism(s) of fibrosis progression in NASH still remain to be clearly elucidated (9).
Our understanding of this process has been hampered by the lack of a comprehensive and physiologic small animal model of NASH with fibrosis. To date small animal models of NASH with fibrosis involve either genetic manipulation (10–12), forced overfeeding (13) or contrived diets deficient in methionine and choline (MCD) (14–17). While each of these models have been valuable, they fail to map to key aspects of what occurs in human beings. As an example, few humans experience diets that are deficient in methionine and choline. Moreover, rodents exposed to MCD diets are not obese. Rather they lose weight and actually become more insulin sensitive (17).
Recent attempts especially the ALIOS diet from Tetri et al using ad-lib high fructose and high trans-fat-diets in small animals have had some success in generating steatosis with inflammation but again failed to produce significant fibrosis (18, 19). Lieber-DeCarli have fed a high-fat-liquid diet (71% Kcal from fat) to rats ad libitum but these animals only developed steatosis without any fibrosis or collagen deposition (20). Genetically modified mice (such as liver specific phosphotase and tensin homolog suppressed (10) or carcinoembryonic antigen-related cell adhesion molecule inactivated (21)) do produce fibrosis when metabolically challenged with high-fat diets but non-genetically modified animals either take very long periods of time or require large animal models to generate NASH with fibrosis (22, 23). Thus a key goal of the present research is to develop a model of NASH that produces significant fibrosis in the context of diet-induced obesity in non-genetically modified mice.
Structural mitochondrial damage is a significant pathophysiologic feature of human NASH with fibrosis (24). The generation of ROS by the damaged mitochondrial respiratory chain and concomitant release of lipid peroxidation products produce detrimental effects (25). Plasma levels of antioxidants, such as reduced Coenzyme Q (redCoQ), correlate negatively with increasing fibrosis in NAFLD (26). Further, fructose has been shown in mice to activate macrophages (27) and induce fibrogenesis through ROS-dependent mechanisms (28).
Based on these data we tested the hypothesis that mice provided ad-libitum access to a high calorie diet, with predominantly medium chain hydrogenated saturated trans-fatty acids (contrasting with the ALIOS diet which had long chain saturated trans fats(18)) and fructose would induce increased hepatic ROS and generate significant fibrosis. Our data represent a significant advance to the study of NAFLD in that within 16 weeks, an ad libitum access to this diet yields, obesity, insulin resistance, and NASH with fibrosis in non-genetically modified mice. This phenotype develops in the background of increased hepatic ROS and pro-inflammatory macrophages driving TGFβ and alpha-smooth muscle actin (SMA) driven collagen deposition.
Materials and Methods
Mice
6–8 week old male C57Bl/6 mice (Jackson Laboratory, Bar Harbor, ME) were group-housed in cages in a temperature-controlled vivarium (22±2° C) on a 12-h light/dark schedule at the University of Cincinnati. Animals were randomly assigned to either chow diet (Teklad; Harlan, Madison, WI), high-fat (HF) fed group - Surwit diet, 58 kcal % fat(Research Diets, New Brunswick, NJ) or high-fat high-carbohydrate (HFHC) fed the Surwit diet and drinking water enriched with high fructose corn syrup equivalent. A total of 42 g/L of carbohydrates was mixed in drinking water at a ratio of 55% Fructose (Acros Organics, Morris Plains, NJ) and 45% Sucrose by weight (Sigma-Aldrich, St. Louis, MO). Animals were provided ad-libitum access to these diets for 16 weeks. Body weights were measured weekly while percent body fat was measured at 12 weeks using Echo MRI (Echo Medical Systems, Houston, TX) All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati and Cincinnati Children’s Hospital Medical Center.
Blood Glucose, Plasma Insulin and Insulin Resistance Measurements
Glucose was measured at 12 weeks after a 4 hour fast using Accu-Check glucose meter (Roche Diagnostics, Indianapolis, IN). Plasma insulin was measured using Mouse Insulin ELISA Kit (Crystal Chem, Downers Grove, IL) Insulin resistance was calculated using Homeostasis Model Assessment (HOMA) insulin resistance (IR) (29).
Histology
Liver sections for histology were obtained at sacrifice after 16 weeks, fixed in 10 % Formalin and stained with hematoxylin & eosin (H&E) or trichrome by Cincinnati Digestive Health Center- Histopathology Core. Histology was read by a single independent pathologist, blinded to experimental design and treatment groups. Briefly, steatosis was graded (0–3), lobular inflammation was scored (0–3), and ballooning was rated (0–2). (30) Fibrosis was staged separately on a scale (0–4). TUNEL staining was performed as previously described (31).
Hepatic Triglyceride (TG) and Plasma Alanine Aminotransferase (ALT), TG, and Cholesterol Quantification
Liver TG content was determined at 16 weeks as previously described (32). Briefly, 100 milligrams of wet liver tissue was homogenized and the enzymatic assay was performed using Triglycerides Reagent Set (Pointe Scientific, Inc., Canton, MI). Photometric absorbance was read at 500 nm. Blood collected at 16 weeks and used to measure ALT, TG, and Cholesterol with DiscretPakTM ALT Reagent Kit (Catachem, Bridgeport, CT); Triglycerides Reagent Set (Pointe Scientific, Inc., Canton, MI) and Infinity Cholesterol Liquid Stable Reagent by (Thermo Electron, Waltham, MA).
Hepatic Hydroxyproline Quantification
100 mg of liver was homogenized to which HCl was added and samples were baked at 110 °C for 18 hours. Aliquots were evaporated and pH neutralized. Chloramine-T solution was added and samples were incubated at room temperature. Ehrich’s reagent was then added and samples incubated at 50 °C and absorbance measured at 550 nm.
Fecal Fat Quantification
Total fatty acid-based compounds in the feces were quantified by saponifying a sample of feces to which a known mass of heptadecanoic acid was added. Total fatty acids in a known mass of feces was calculated by gas chromatograpy as previously described (33).
Hepatic Pro-fibrotic qPCR Gene Analysis
RNA was isolated from flash frozen liver tissues. Total RNA was isolated using TRIzol reagent protocol (Molecular Research Center, Cincinnati, OH) Isolated RNA was treated with RNase-Free DNase (Fisher Scientific, Pittsburgh, PA), and purified on a RNeasy Mini Spin Column (Qiagen, Valencia, CA). cDNA was made using TaqMan Reverse Transcription protocol and Eppendorf Mastercycler PCR machine (Eppendorf North America, Westbury, NY) Pre-designed, validated gene-specific TaqMan® probe was used for Collagen 1 and alpha-SMA. Primer sequence for TGFβ1 is as follows reverse CGT AGT AGA CGA TGG GCA GTG G, forward TAT TTG GAG CCT GGA CAC ACA G. Messenger RNA expression was obtained using Stratagene SYBR green real-time kinetic PCR on a Stratagene Mx-3005 Multiplex Quantitative PCR machine (Stratagene, Agilent Technologies, La Jolla, CA). Relative expression was determined by comparison of dT values relative to GAPDH expression using the 2-DDCT method.
Identification of Macrophages by Flow Cytometry
Single liver cell suspensions were prepared by mincing and passing over 40μm cell strainers (Fisher Scientific, Pittsburgh, PA). After centrifugation at 2000 rpm, cell pellet was mixed with 33% percoll (Sigma-Aldrich, St. Louis, MO) in RPMI1640 solution (Invitrogen, Carlsbad, CA). Cell suspension was centrifuged at 2000 rpm for 20 min at room temperature (RT) and cell pellet was removed, washed and red blood cells were lysed with 1X lysis buffer (eBioscience, San Diego, CA). Cells were suspended in 50μl FACS buffer and Fc receptor was blocked with anti-mouse CD16/32 (Clone 93, eBioscience, San Diego, CA). Cells were stained with CD11b-PerCP-Cy5.5 (Clone M1/70), F4/80-PE (Clone BM8) and Gr1-FITC (Clone 1A8) (eBioscience, San Diego, CA). Cells were acquired on FacsCanto FlowCytometer (BD Biosciences, San Jose, CA) and data was analyzed by FlowJo software version 7.5 (TreeStar, Ashland, OR).
ROS Staining of Liver Sections
Frozen liver sections were rehydrated in Phosphate Buffer Solution (PBS). Stock Dihydroethidium (DHE) (Sigma- Aldrich, St. Louis, MO) solution was diluted in DMSO (Sigma- Aldrich, St. Louis, MO). Slides were incubated in DHE solution and washed with 1x PBS and cover-slipped using 80% Glycerol in PBS. Fluorescence was recorded and quantified using Texas red filter on an upright Olympus BX51 microscope, using DPControler software (Olympus; Hamburg, Germany) and IMAGE J software (NIH, Bethesda, MD) (34).
4-Hydroxynonenal (HNE) staining
Liver sections were incubated in 10% normal horse serum after blocking. Sections were incubated with the HNE primary antibody (Alpha Diagnostic International, San Antonio, TX) overnight and then incubated with secondary biotin conjugated antibody (Alpha Diagnostic International, San Antonio, TX). An avidin–biotin peroxidase complex (Vector Laboratories, Burlingame, CA) staining was visualized with diaminobenzidine (Vector Laboratories, Burlingame, CA). The sections were counterstained with Mayer’s hematoxylin.
CoEnzyme Q9 Quantification
Quantification of CoQ9 was performed as previously published (35). Plasma with internal standard CoQ11 was injected into an automated high-performance liquid chromatographic (HPLC) system equipped with coulometer detector. Quantification of oxCoQ9 was obtained by the ChromQuest™ software (Fisher Scientific, Pittsburgh, PA). After injection, the extract was mixed with 1, 4-benzoquinone, incubated and then injected into the HPLC system for measuring total CoQ9. Concentration of reduced coenzyme Q9 was achieved by subtracting oxCoQ9 from total CoQ9.
Statistical Analysis
Statistical comparison between groups and treatments was performed using one way ANOVA and post-hoc Tukey’s test. Student’s T-tests were used when comparing two groups. A p-value of <0.05 was considered statistically significant. Data was presented as mean ± SEM.
Results
Food intake, body weight, body composition, and insulin resistance
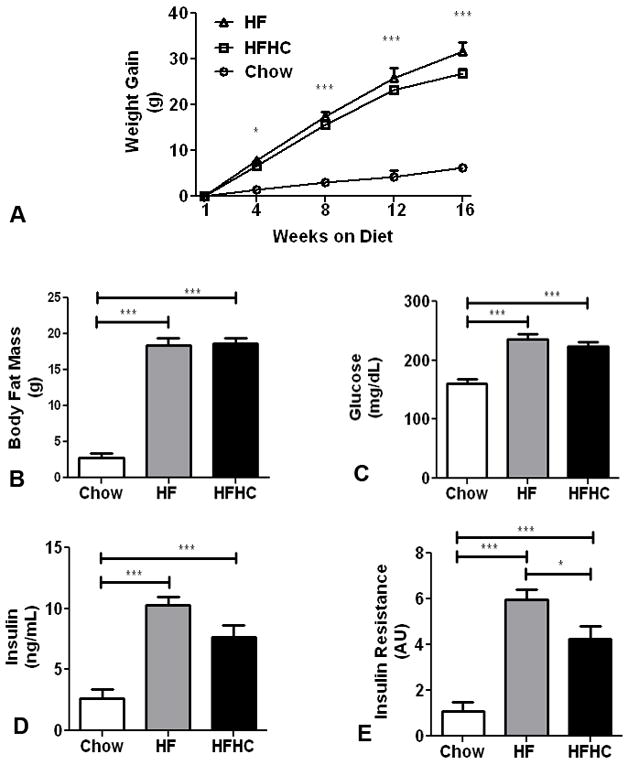
The HFHC and HF diet fed mice consumed more total calories/day (12.54 ± 0.6 and 11.76 ± 1.5 Kcal/d respectively) than their chow fed controls (8.67 ± 1.6). There was no difference between HF and HFHC in terms of total calories consumed or stool output/day. Percent fat content in fecal material was also similar between HF (2.46±0.6%) and HFHC (2.08±0.7%) groups. Mice fed HFHC and HF diets gained more weight than the mice fed the chow diet. HFHC and HF diet fed mice had a mean body weight of 50.5±0.8g and 53.18±1.8g (Figure 1A) compared to chow fed mice that had a mean body weight of 31.94±0.2g at 16 weeks. Total body fat mass estimation by MR at 12 weeks demonstrated that HFHC fed mice (18.66±0.7g) and HF fed mice (18.40±0.9g) have significantly greater body fat compared to chow-fed mice (2.82±0.6g) (p<0.0001; Figure 1B). Fasting plasma glucose levels were higher in HFHC (223.6±7 mg/dL) and HF (235.4±10 mg/dL) diet fed mice than chow diet fed mice (160.4±7.3 mg/dL) (p<0.0001; Figure 1C). Similarly fasting insulin was also higher in HFHC fed mice (7.7±1 ng/mL) and HF diet fed mice (10.3±0.9 ng/ml) compared to chow fed mice (1.9±0.1 ng/mL) (p<0.0001; Figure 1D). The glucose and insulin values were used to estimate insulin resistance as HOMA-IR calculations and both HFHC (4.2±0.6) and HF(5.9±0.5) diet fed mice were significantly insulin resistant compared to chow diet fed mice(1.1±0.4) (p<0.0001; Figure 1E). Thus both HFHC and HF mice were significantly obese and insulin resistant compared to chow mice.
Figure 1.
Hepatic steatosis, inflammation, and apoptosis
Histological examination of livers from HFHC and HF diet fed mice demonstrated substantial steatosis with inflammatory changes. Micro and macrovesicular steatosis were clearly visible on routine histology staining with H&E after 16 weeks (Figure 2A). For sections with a steatosis score of 3, the distribution of steatosis in both the HF and HFHC groups was panlobular. Nearly all hepatocytes have microvesicular steatosis and many had both micro and macrovesicular steatosis with a random distribution. For sections with steatosis scored as grade ≤2 there was a panlobular distribution of steatosis in the HF group, but there was evidence of zone II sparing in the HFHC group. Lobular inflammation was prominent in HFHC sections similar to human NASH descriptions (Figure 2B). Confirming the histological impressions, the weights of the livers of HFHC and HF mice were significantly higher compared to chow fed mice (p<0.0001; Figure 2E). Similarly TG content at 16 weeks was higher in HFHC (1955±430 mg/dL per 100 mg wet liver) and HF mice (1096±115) compared to chow mice (276±34) (One way ANOVA p<0.0001; Figure 2C). Plasma ALT levels were also greater in both HFHC (217.3±40.2 IU/L) and HF mice (187±47 IU/L) at 16 weeks compared to chow fed mice (70.9±5.4 IU/L) (p<0.0001; Figure 2D). TUNEL staining was increased in both HFHC and HF mice compared to chow mice (data not shown). Thus both HFHC and HF mice had significantly more hepatic steatosis, inflammation and apoptosis compared to chow mice.
Figure 2.

Hepatic fibrosis and pro-fibrogenic gene signatures
Trichrome stained liver sections from HFHC fed mice demonstrated significant fibrosis (Figure 3A). Fibrosis was first observed in mice after 14 weeks of HFHC diet. After 16 weeks of HFHC diet fibrosis was clearly visible in half of the mice (Table 1). When seen in a section, fibrosis was seen in most portal areas and present extensively. At 16 weeks 33% of mice had Stage 1a or 1c fibrosis with perisinusoidal or portal/periportal fibrosis, while 16% had Stage 2 fibrosis with perisinusoidal and portal/periportal fibrosis. Perisinusoidal fibrosis was seen in both zones 1 and 2. Periportal fibrosis was seen in all portal triads and there was extension of fibers between portal tracts as well. Thus the distribution of fibrosis seen in HFHC liver sections was akin to human NASH biopsies with the fibrosis being either a predominantly zone 1 (as seen in pediatric patients) or perisinusoidal (seen more in adult patients) (Insert Figure 3A). HF and chow diet fed mice had no evidence of significant fibrosis on histology. RT-PCR for hepatic Collagen 1 mRNA expression was significantly higher in HFHC diet mice (7.36±2.1 fold) compared to both HF (0.92±0.6 fold) when normalized to chow diet fed mice (1.0±0.1) at 16 weeks (p=0.0031; Figure 3B). Similarly mRNA expression of TG F-β1 was significantly higher in HFHC diet fed mice (3.72±1.3 fold) when normalized to chow diet fed mice (1.0±0.2) at 16 weeks (p=0.04; Figure 3C). Hepatic levels of hydroxyproline were higher in the HFHC (0.94±0.05 mg/100mg liver) compared to both HF (0.63±0.04; p<0.01) and Chow (0.61±0.01; p<0.01) (Figure 3D). Thus HFHC mice had significantly more hepatic fibrosis and pro-fibrogenic gene signatures compared to chow and HF mice.
Figure 3.

Table 1.
Histological Characteristics after 16 weeks on Diet
| Parameters | Chow | HF | HFHC |
|---|---|---|---|
| Steatosis Grade (0–3) | 0.00 ± 0.0 | 2.88 ± 0.1* | 2.50 ± 0.3* |
| Lobular Inflammation Score (0–3) | 0.42 ± 0.1 | 0.38 ± 0.3 | 1.33 ± 0.4* # |
| Fibrosis Stage (0–4) | |||
| 0(%) | 100 | 100 | 50 |
| 1(%) | 0 | 0 | 33 |
| 2(%) | 0 | 0 | 16 |
| Fibrosis Present Total (%) | 0 ± 0 | 0 ± 0 | 50 ± 22.4*** ### |
HFHC and HF diet fed mice had higher steatosis grade chow fed mice (1 way ANOVA; p<0.0001 in all three categories; Tukey’s post test ***=p<0.001, **=p<0.01,*=p<0.05).
HFHC liver histology was significantly different from both HF and chow fed mice after 16 weeks of diet for Fibrosis and lobular inflammation score (1 way ANOVA; fibrosis p<0.0001 and inflammation p=0.0195; Tukey’s post test
p<0.001,
p<0.01,
p<0.05).
Legend Key: *=versus Chow, # versus HF. Data are expressed as mean ± SEM. N for groups: (Chow=8; HF=8, HFHC=6).
Hepatic macrophage population and stellate cell activation
The macrophage inflammatory Gr1+ subset is massively recruited into the liver upon toxic injury and may differentiate into fibrocytes (7, 36). We found that HFHC diet fed mice (2.03±0.3%) had an approximately ten fold increase in CD11b+F4/80+ cells compared to HF (0.03±0.0%) and chow mice (0.35±0.1%) (p<0.0001; Figure 4A and 4B). Upon gating on CD11b+F4/80+ cells, the Gr1+ subset of cells were ten fold higher in HFHC diet fed mice (1.12±0.2%) compared to either HF (0.08±0.0%) or chow fed mice (0.1±0.0%)(p<0.0001; Figure 4C). Further mRNA gene expression for alpha-SMA was three fold higher in the HFHC compared to HC and undetectable in Chow livers (Figure 4D). Thus HFHC mice had a significantly more pro-inflammatory monocyte population compared to chow and HF mice which may signal stellate cell activation.
Figure 4.

Hepatic ROS, plasma lipid and CoQ studies
At 16 weeks HFHC diet fed mice livers had more DHE staining (40.3±2.9 FU/HPF) compared to HF (28.3±2.9 FU/HPF) or chow fed mice (17±1.0 FU/HPF) (p=0.002; Figure 5A and 5C). We also observed increased 4HNE staining in the HFHC mice hepatocytes compared to HF and Chow fed mice (Figure 5B). The plasma TG levels were not significantly different between the three groups but serum cholesterol was significantly higher in the HFHC (372.3±21.9 mg/dL) compared to both HF (277.3±50.5 mg/dL; p<0.001) and Chow (127.5±7.1 mg/dL; p <0.001) (Figure 5E). Plasma oxCoQ 9 levels in mice at 16 weeks were significantly higher in HFHC fed mice (0.06±0.004 μg/mL) compared to HF diet fed mice (0.03±0.004 μg/mL) and chow diet fed mice (0.02±0.004 μg/mL) (p<0.0001; Table 2 and Figure 5D). The correlation of liver tissue collagen 1 mRNA relative expression and absolute plasma oxCoQ 9 levels had an R2 value of 0.51. Thus the fructose containing HFHC diet had the most hepatic ROS, hypercholesterolemia, and hepatic fibrosis. This was mirrored by the levels of plasma oxCoQ9 which differed significantly among all three groups and correlated with the presence of fibrosis in this model.
Figure 5.

Table 2.
Plasma CoEnzyme Q9 profile after 16 weeks of Diet
| Parameters | Chow | HF | HFHC |
|---|---|---|---|
| redQ9 (μg/mL) | 0.1204 ± 0.03 | 0.2318 ± 0.04 | 0.2518 ± 0.04 |
| oxQ9 (μg/mL) | 0.019 ± 0.004 | 0.0388 ± 0.004* | 0.064 ± 0.005***## |
| totQ9 (μg/mL) | 0.1394 ± 0.03 | 0.2706 ± 0.05 | 0.3158 ± 0.03* |
| redQ9/oxQ9 Ratio (AU) | 6.825 ± 1.2 | 5.846 ± 0.8 | 4.161 ± 0.8 |
HFHC and HF diet fed mice have higher oxCoQ9 plasma levels (1 way ANOVA; p<0.0001; Tukey’s post test
p<0.001,
p<0.01,
p<0.05).
HFHC diet fed mice also have significantly increased plasma total CoEnzyme Q9 levels compared to both HF and chow fed mice after 16 weeks of diet (1 way ANOVA; p=0.0141; Tukey’s post test *=p<0.05).
Legend Key *=versus Chow, # versus HF. Data are expressed as mean ± SEM. N for groups: (Chow=5; HF=5, HFHC=5).
Discussion
The rising rates of NASH make addressing the underlying causes of this serious condition more pressing. Hepatic steatosis is common in obese patients but only a subset of these patients develop NASH, emphasizing the contribution of genetic and potential environmental risk factors. Human NASH histopathology has been associated with steatosis, lobular and portal inflammation, hepatocyte ballooning, and fibrosis. Specifically, zone 3 predominant macrovesicular steatosis, ballooning, and perisinusoidal fibrosis is deemed consistent with adult or Type 1 NASH. Type 2 or pediatric NASH histopathology has been reported to have panacinar or periportal (zone 1) steatosis, rare ballooning and portal tract expansion by chronic inflammation or fibrosis. (37). Individuals who have NASH with fibrosis have progressive disease and greater morbidity and mortality including the potential for cirrhosis, liver failure, and liver transplantation (3). However, the specific biological determinants that lead to development of NASH with fibrosis are not well-defined.
Fructose consumption accounts for approximately 10.2% of all calories in our average diet in the United States (38). In comparison to other simple sugars such as glucose, use of fructose for hepatic metabolism is not restricted by the rate-limiting step of phosphofructokinase, thus avoiding the regulating action of insulin (39). Fructose intake is 2–3 fold higher in patients with NASH compared to BMI matched controls and recently daily fructose ingestion has been associated with increased hepatic fibrosis (40, 41). These epidemiologic data prompted us to investigate the potential mechanistic role that fructose and other simple sugars may play in the development of NASH.
The present manuscript was focused on the development of a dietary model of NASH. To this end we compared HF-fed mice to mice maintained on the same diet but also given ad libitum access to fructose in their drinking water (HFHC). While weight gain, body fat, insulin resistance and liver steatosis were similar between the two groups (and elevated relative to mice maintained on chow), mice fed the HFHC diet had increased hepatic oxidative stress, CD11b+F4/80+ Gr1+ macrophages in the liver, TGFβ1 driven fibrogenesis and collagen deposition compared to the weight matched controls in the HF fed group. Thus, while HF diets produce a range of the components of the metabolic syndrome, fructose consumption would appear necessary to move the process, from liver fat deposition alone to fibrogenesis.
ROS has been thought to be an important trigger for hepatic stellate cell activation and for promoting expression of fibrogenic molecules such as alpha-SMA, TGFβ1, and collagen 1 (15, 28, 42, 43). Recently, fructose-fed rats have been reported to develop hepatocyte damage with a decrease in the mitochondrial membrane potential similar to that induced by low non-cytotoxic doses of exogenous ROS (44). In-vitro studies have also reported that the cytotoxic mechanism involving fructose driven ROS formation preceded the hepatocytotoxicity, and that this cell injury could be prevented by ROS scavengers (44). We therefore investigated this as a potential process in our model and demonstrated that HFHC fed mice had significantly higher ROS levels compared to both HF diet and chow fed mice (Figure 5).
Previous studies done with fructose diets have reported insulin resistance and severe necro-inflammatory NAFLD but not NASH with fibrosis (18, 19). In contrast to the ALIOS diet from Tetri et al which provided fructose water in gelatin form and long chain saturated trans-fats in their solid diet our high-fat-diet provided 58% calories from medium chain saturated trans-fats and fructose and sucrose in their regular drinking water. This diet resulted in 50% of the mice in our HFHC fed group having fibrosis with a minority having stage 2 fibrosis (Table 2). Karlmark et al highlighted the role of CD11+F4/80+Gr1+ monocytes in perpetuating hepatic stellate cell-driven TGFβ1 dependent fibrosis (7). More recently Niedermeier et al reported that Gr1+ monocytes may be essential in production of murine fibrocytes (36). In our experiment intrahepatic CD11+F4/80+Gr1+ monocyte-derived macrophages were 10 fold higher than either chow or HF diet fed mice, with 50% of the macrophages in HFHC livers being Gr1+ (Figure 4). We propose that the conversion of CD11b+F4/80+Gr1+ monocytes into fibrocytes maybe responsible for the increased collagen 1 deposition through ROS driven TGFβ signaling and stellate cell activation.
In humans, studies have shown extensive mitochondrial damage including paracrystalline inclusion bodies, megamitochondria, damaged respiratory chain and low ATP production with NASH (24). We have previously reported that increased ROS released from damaged mitochondrial respiratory chain is important in NAFLD development (45). CoQ is an important element in the mitochondrial respiratory chain, contributing to the transport of electrons across complex III involving the Qo and Qi sites within the inner mitochondrial membrane to enable generation of ATP. Lower redCoQ plasma levels are present in patients with cirrhosis and redCoQ acts as a lipid soluble anti-oxidant in hepatocytes in culture (46, 47). Supplementation with CoQ has also been reported to inhibit liver fibrosis through suppression of TGFβ1 expression in mice (48). We demonstrate that plasma levels of oxCoQ9 correlate well with collagen 1 mRNA in liver tissue. We also present data that plasma levels of oxCoQ9 can discriminate between NASH with fibrosis and NASH without fibrosis, with our HFHC (NASH with fibrosis) mice having higher levels compared to the HF (NASH without fibrosis) or chow (normal histology) mice (Figure 5).
In summary, we believe therefore, that our ad libitum dietary model results in NASH with fibrosis in non-genetically modified obese mice. Our data suggest that the mechanism of fibrosis in this model may involve fructose producing an increased ROS signature in the liver associated with CD11b+F4/80+Gr1+ macrophage aggregation resulting in TGFβ1 signaled collagen deposition and histologically visible hepatic fibrosis.
Acknowledgments
Financial Support:
This was work was supported by the NIH NICHD K12 HD028827 (RK), NIDDK 1K08DK084310 (RK), Children’s Digestive Health and Nutrition Foundation- George Ferry Young Investigator Award (RK). This work also received support from the Cincinnati Digestive Health Center- PHS Grant P30 DK078392.
List of Abbreviations
- NASH
Nonalcoholic steatohepatitis
- MCT
medium chain triacylglycerols
- HFHC
high fat high carbohydrate
- HF
high fat
- ROS
reactive oxygen species
- oxCoQ9
oxidized Coenzyme Q9
- redCoQ9
reduced Coenzyme Q9
Contributor Information
Rohit Kohli, Email: rohit.kohli@cchmc.org.
Michelle Kirby, Email: michelle.kirby@cchmc.org.
Stavra A. Xanthakos, Email: stavra.xanthakos@cchmc.org.
Samir Softic, Email: samir.softic@cchmc.org.
Ariel E. Feldstein, Email: feldsta@ccf.org.
Vijay Saxena, Email: vijay.saxena@cchmc.org.
Peter H. Tang, Email: peter.tang@cchmc.org.
Lili Miles, Email: lili.miles@cchmc.org.
Michael V. Miles, Email: michael.miles@cchmc.org.
William F. Balistreri, Email: william.balistreri@cchmc.org.
Stephen C. Woods, Email: woodssc@ucmail.uc.edu.
Randy J. Seeley, Email: seelyrj@ucmail.uc.edu.
References
- 1.Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. 2004;79:537–543. doi: 10.1093/ajcn/79.4.537. [DOI] [PubMed] [Google Scholar]
- 2.Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, McClain C. Nonalcoholic fatty liver disease: predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184–195. doi: 10.1016/j.jnutbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 4.McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:521–533. viii. doi: 10.1016/j.cld.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Day CP. From fat to inflammation. Gastroenterology. 2006;130:207–210. doi: 10.1053/j.gastro.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 6.Musso G, Gambino R, De Michieli F, Cassader M, Rizzetto M, Durazzo M, Faga E, et al. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. 2003;37:909–916. doi: 10.1053/jhep.2003.50132. [DOI] [PubMed] [Google Scholar]
- 7.Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, Merad M, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 8.Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN, McCullough AJ. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology. 2009;50:1072–1078. doi: 10.1002/hep.23050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zivkovic AM, German JB, Sanyal AJ. Comparative review of diets for the metabolic syndrome: implications for nonalcoholic fatty liver disease. Am J Clin Nutr. 2007;86:285–300. doi: 10.1093/ajcn/86.2.285. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe S, Horie Y, Suzuki A. Hepatocyte-specific Pten-deficient mice as a novel model for nonalcoholic steatohepatitis and hepatocellular carcinoma. Hepatol Res. 2005;33:161–166. doi: 10.1016/j.hepres.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 11.Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oben JA, Roskams T, Yang S, Lin H, Sinelli N, Li Z, Torbenson M, et al. Norepinephrine induces hepatic fibrogenesis in leptin deficient ob/ob mice. Biochem Biophys Res Commun. 2003;308:284–292. doi: 10.1016/s0006-291x(03)01360-3. [DOI] [PubMed] [Google Scholar]
- 13.Baumgardner JN, Shankar K, Hennings L, Badger TM, Ronis MJ. A new model for nonalcoholic steatohepatitis in the rat utilizing total enteral nutrition to overfeed a high-polyunsaturated fat diet. Am J Physiol Gastrointest Liver Physiol. 2008;294:G27–38. doi: 10.1152/ajpgi.00296.2007. [DOI] [PubMed] [Google Scholar]
- 14.Leclercq IA, Farrell GC, Field J, Bell DR, Gonzalez FJ, Robertson GR. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105:1067–1075. doi: 10.1172/JCI8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.George J, Pera N, Phung N, Leclercq I, Yun Hou J, Farrell G. Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J Hepatol. 2003;39:756–764. doi: 10.1016/s0168-8278(03)00376-3. [DOI] [PubMed] [Google Scholar]
- 16.Sahai A, Malladi P, Melin-Aldana H, Green RM, Whitington PF. Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am J Physiol Gastrointest Liver Physiol. 2004;287:G264–273. doi: 10.1152/ajpgi.00002.2004. [DOI] [PubMed] [Google Scholar]
- 17.Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res. 2008;49:1068–1076. doi: 10.1194/jlr.M800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295:G987–995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawasaki T, Igarashi K, Koeda T, Sugimoto K, Nakagawa K, Hayashi S, Yamaji R, et al. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J Nutr. 2009;139:2067–2071. doi: 10.3945/jn.109.105858. [DOI] [PubMed] [Google Scholar]
- 20.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, et al. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–509. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- 21.Lee SJ, Heinrich G, Fedorova L, Al-Share QY, Ledford KJ, Fernstrom MA, McInerney MF, et al. Development of nonalcoholic steatohepatitis in insulin-resistant liver-specific S503A carcinoembryonic antigen-related cell adhesion molecule 1 mutant mice. Gastroenterology. 2008;135:2084–2095. doi: 10.1053/j.gastro.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koteish A, Mae Diehl A. Animal models of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16:679–690. doi: 10.1053/bega.2002.0332. [DOI] [PubMed] [Google Scholar]
- 23.Lee L, Alloosh M, Saxena R, Van Alstine W, Watkins BA, Klaunig JE, Sturek M, et al. Nutritional model of steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Hepatology. 2009;50:56–67. doi: 10.1002/hep.22904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 25.Fromenty B, Robin MA, Igoudjil A, Mansouri A, Pessayre D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004;30:121–138. doi: 10.1016/s1262-3636(07)70098-8. [DOI] [PubMed] [Google Scholar]
- 26.Yesilova Z, Yaman H, Oktenli C, Ozcan A, Uygun A, Cakir E, Sanisoglu SY, et al. Systemic markers of lipid peroxidation and antioxidants in patients with nonalcoholic Fatty liver disease. Am J Gastroenterol. 2005;100:850–855. doi: 10.1111/j.1572-0241.2005.41500.x. [DOI] [PubMed] [Google Scholar]
- 27.Spruss A, Kanuri G, Wagnerberger S, Haub S, Bischoff SC, Bergheim I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology. 2009;50:1094–1104. doi: 10.1002/hep.23122. [DOI] [PubMed] [Google Scholar]
- 28.Urtasun R, Conde de la Rosa L, Nieto N. Oxidative and nitrosative stress and fibrogenic response. Clin Liver Dis. 2008;12:769–790. viii. doi: 10.1016/j.cld.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akagiri S, Naito Y, Ichikawa H, Mizushima K, Takagi T, Handa O, Kokura S, et al. A Mouse Model of Metabolic Syndrome; Increase in Visceral Adipose Tissue Precedes the Development of Fatty Liver and Insulin Resistance in High-Fat Diet-Fed Male KK/Ta Mice. J Clin Biochem Nutr. 2008;42:150–157. doi: 10.3164/jcbn.2008022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis. 2001;21:3–16. doi: 10.1055/s-2001-12925. [DOI] [PubMed] [Google Scholar]
- 31.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 32.Sahai A, Malladi P, Pan X, Paul R, Melin-Aldana H, Green RM, Whitington PF. Obese and diabetic db/db mice develop marked liver fibrosis in a model of nonalcoholic steatohepatitis: role of short-form leptin receptors and osteopontin. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1035–1043. doi: 10.1152/ajpgi.00199.2004. [DOI] [PubMed] [Google Scholar]
- 33.Metcalfe LDSA, Pelka JR. Rapid preparation of Fatty Acid Esters from Lipids for Gas Chromatographic Analysis. Annal Chem. 1966;38:514–515. [Google Scholar]
- 34.Wainwright MS, Kohli R, Whitington PF, Chace DH. Carnitine treatment inhibits increases in cerebral carnitine esters and glutamate detected by mass spectrometry after hypoxia-ischemia in newborn rats. Stroke. 2006;37:524–530. doi: 10.1161/01.STR.0000198892.15269.f7. [DOI] [PubMed] [Google Scholar]
- 35.Tang PH, Miles MV, Miles L, Quinlan J, Wong B, Wenisch A, Bove K. Measurement of reduced and oxidized coenzyme Q9 and coenzyme Q10 levels in mouse tissues by HPLC with coulometric detection. Clin Chim Acta. 2004;341:173–184. doi: 10.1016/j.cccn.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Niedermeier M, Reich B, Rodriguez Gomez M, Denzel A, Schmidbauer K, Gobel N, Talke Y, et al. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc Natl Acad Sci U S A. 2009;106:17892–17897. doi: 10.1073/pnas.0906070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brunt EM. Histopathology of non-alcoholic fatty liver disease. Clin Liver Dis. 2009;13:533–544. doi: 10.1016/j.cld.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 38.Vos MB, Kimmons JE, Gillespie C, Welsh J, Blanck HM. Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J Med. 2008;10:160. [PMC free article] [PubMed] [Google Scholar]
- 39.Havel PJ. Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr Rev. 2005;63:133–157. doi: 10.1301/nr.2005.may.133-157. [DOI] [PubMed] [Google Scholar]
- 40.Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, Johnson RJ, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. 2008;48:993–999. doi: 10.1016/j.jhep.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, Diehl AM. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. doi: 10.1002/hep.23535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang C, Ding Z, Suhaimi NA, Kng YL, Zhang Y, Zhuo L. A class of imidazolium salts is anti-oxidative and anti-fibrotic in hepatic stellate cells. Free Radic Res. 2009;43:899–912. doi: 10.1080/10715760903137002. [DOI] [PubMed] [Google Scholar]
- 43.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee O, Bruce WR, Dong Q, Bruce J, Mehta R, O’Brien PJ. Fructose and carbonyl metabolites as endogenous toxins. Chem Biol Interact. 2009;178:332–339. doi: 10.1016/j.cbi.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 45.Kohli R, Pan X, Malladi P, Wainwright MS, Whitington PF. Mitochondrial reactive oxygen species signal hepatocyte steatosis by regulating the phosphatidylinositol 3-kinase cell survival pathway. J Biol Chem. 2007;282:21327–21336. doi: 10.1074/jbc.M701759200. [DOI] [PubMed] [Google Scholar]
- 46.Matsura T, Yamada K, Kawasaki T. Antioxidant role of cellular reduced coenzyme Q homologs and alpha-tocopherol in free radical-induced injury of hepatocytes isolated from rats fed diets with different vitamin E contents. Biochim Biophys Acta. 1992;1127:277–283. doi: 10.1016/0005-2760(92)90232-k. [DOI] [PubMed] [Google Scholar]
- 47.Bianchi GP, Fiorella PL, Bargossi AM, Grossi G, Marchesini G. Reduced ubiquinone plasma levels in patients with liver cirrhosis and in chronic alcoholics. Liver. 1994;14:138–140. doi: 10.1111/j.1600-0676.1994.tb00062.x. [DOI] [PubMed] [Google Scholar]
- 48.Choi HK, Pokharel YR, Lim SC, Han HK, Ryu CS, Kim SK, Kwak MK, et al. Inhibition of liver fibrosis by solubilized coenzyme Q10: Role of Nrf2 activation in inhibiting transforming growth factor-beta1 expression. Toxicol Appl Pharmacol. 2009;240:377–384. doi: 10.1016/j.taap.2009.07.030. [DOI] [PubMed] [Google Scholar]