

Abstract
Covalent incorporation (cross-linking) of plasmin inhibitor α2-antiplasmin (α2-AP) into fibrin clots increases their resistance to fibrinolysis. We hypothesized that α2-AP may also interact non-covalently with fibrin prior to its covalent cross-linking. To test this hypothesis, we studied binding of α2-AP to fibrin(ogen) and its fragments by ELISA and Surface Plasmon Resonance. The experiments revealed that α2-AP binds to polymeric fibrin and surface-adsorbed fibrin(ogen) while no binding was observed with fibrinogen in solution. To localize the α2-AP-binding sites, we studied the interaction of α2-AP with the fibrin(ogen)-derived D1, D-D and E3 fragments, and the recombinant αC region and its constituents, αC-connector and αC-domain and its sub-domains, which together encompass practically the whole fibrin(ogen) molecule. In ELISA, α2-AP bound to immobilized D1, D-D, αC region, αC-domain and its C-terminal sub-domain. The binding was Lys-independent and was not inhibited by plasminogen or tPA. Furthermore, the affinity of α2-AP to D-D was significantly increased in the presence of plasminogen while that to the αC-domain remained unaffected. Altogether, these results indicate that the fibrin(ogen) D region and the C-terminal sub-domain of the αC-domain contain high affinity α2-AP-binding sites that are cryptic in fibrinogen and exposed in fibrin or adsorbed fibrinogen, and the presence of plasminogen facilitates interaction of α2-AP with the D regions. The discovered non-covalent interaction of α2-AP with fibrin may contribute to regulation of the initial stage of fibrinolysis and provide proper orientation of the cross-linking sites to facilitate covalent cross-linking of α2-AP to the fibrin clot.
The fibrinolytic system including fibrinolytic proenzyme plasminogen and its activators plays an important role in the dissolution of blood clots and vascular remodeling (1–3). Formation of a blood clot triggers plasminogen activation, which occurs through a number of orchestrated interactions between plasminogen, tissue-type plasminogen activator (tPA1), and fibrin, and results in generation of active fibrinolytic enzyme plasmin (4, 5). Plasmin activity is controlled by a number of inhibitors; the major physiological inhibitor of plasmin is α2-antiplasmin (α2-AP). The importance of such a control is highlighted by the fact that congenital deficiency of α2-AP results in a severe hemorrhagic disorder due to increased susceptibility to fibrinolysis (5–7).
Plasmin inhibitor α2-AP is a single chain glycoprotein consisting of 464 amino acid residues with NH2-terminal Met residue, Met-α2-AP (3, 8, 9). In the circulation, it undergoes proteolytic cleavage between Pro12 and Asn13 by an antiplasmin-cleaving enzyme resulting in a 452-residue version with NH2-terminal Asn residue, Asn-α2-AP, which accounts for approximately 70% of circulating α2-AP (10–13). α2-AP is a member of the serpin (serine protease inhibitor) family whose inhibitory mechanism includes formation of a covalent complex with target proteases and inhibition of the latter. However, in contrast to the other family members, α2-AP has a COOH-terminal extension (approximately 50 residues long) that contains a number of Lys residues (14). This extension, which according to the X-ray structure is located in close proximity to the reactive loop (15), binds to Lys-binding kringles of plasmin increasing the inhibitory efficiency of α2-AP (16, 17). Thus, α2-AP efficiently inhibits free plasmin in the circulation thereby preventing fibrinogenolysis. Upon blood coagulation, α2-AP is covalently cross-linked to forming fibrin by activated factor XIII (factor XIIIa) and becomes an effective inhibitor of fibrinolysis. The cross-linking occurs through Gln14 or Gln2 in Met-α2-AP or Asn-α2-AP, respectively; however, the latter is cross-linked to fibrin much faster than the former (18–20). While the molecular mechanism of plasmin inhibition by α2-AP in solution is well established, that by α2-AP cross-linked to fibrin needs to be further clarified.
Fibrinogen consists of two identical disulfide-linked subunits, each of which is formed by three non-identical polypeptide chains denoted Aα, Bβ, and γ (21). These chains are folded into a number of structural domains that compose several regions (22). The central region E is formed by the disulfide-linked NH2-terminal portions of all six chains converging from both subunits. The COOH-terminal regions of the Bβ and γ chains and a portion of the Aα chain form the terminal D region, one in each subunit, while the remaining COOH-terminal portion of the two Aα chains (residues Aα221–610) form two αC regions. Each αC region is composed of the flexible αC-connector (residues Aα221–391) and compact αC-domain (residues Aα392–610) (23). Thus, the structure of fibrinogen can be presented as consisting of three linearly arranged regions, D-E-D, with the αC-domains attached to the D regions via the αC-connectors (Fig. 1A). The D and E regions correspond to the D and E fragments, respectively, which can be prepared by proteolytic digestion of fibrinogen with plasmin (Fig. 1B) (24). The αC regions are susceptible to proteolysis and degraded into smaller fragments (21); they, as well as their constituents αC-connector and αC-domain, can be prepared by recombinant techniques (25) (Fig. 1C).
Fig. 1.

Schematic representation of fibrinogen, fibrin, and their fragments prepared for this study. Panel A: Ribbon diagram of fibrinogen based on its crystal structure (47); the individual fibrinogen chains, Aα, Bβ, and γ, are colored blue green, and red, respectively, the vertical lines denote approximate boundaries between the D and E regions. The αC regions, whose structure have not been identified, are shown schematically as two blue spheres representing αC-domains, each attached to the bulk of the molecule with the flexible αC-connector. Panel B: Schematic representation of the fibrinogen molecule and its products of plasminolysis, D1 and E3 fragments. Panel C: Recombinant αC region (Aα221–610 fragment), αC-connector (Aα221–391 fragment), and αC-domain (Aα392–610 fragment). Panel D: Schematic representation of fibrin and its products of fibrinolysis, the D-D:E complex, and the D-D and E3 fragments. For the sake of simplicity, only two strands of fibrin molecules without the αC regions are shown; the molecules are linked through the non-covalent DD:E interactions and covalent γ-γ cross-linking between the D regions (shown by small horizontal bars). Small arrows in panels B and D indicate plasmin cleavage resulting in fibrin(ogen) fragments.
Fibrinogen is rather inert in the circulation, however, it becomes highly reactive towards different proteins and cell types after thrombin-mediated conversion into fibrin. Thrombin cleaves two pairs of fibrinopeptides, fibrinopeptide A (FpA) and fibrinopeptide B (FpB), from the NH2-terminal portions of the Aα and Bβ chains, respectively, to expose polymerization sites or “knobs” “A” and “B” in the central E region. Monomeric fibrin molecules interact with each other through these sites and complementary “a” and “b” sites, also called “holes”, which are located in the D regions and are always reactive. Such interaction through D and E regions, also called DD:E interaction, results in formation of fibrin polymers (Fig. 1D). Fibrin polymerization is accompanied by the conformational changes and exposure of multiple binding sites in the polymer (26, 27). Among them are tPA- and plasminogen-binding sites that play a major role in initiation of fibrinolysis (4). Namely, as soon as these sites are exposed, binding of tPA and plasminogen to them brings these two proteins together and provides efficient activation of plasminogen into active plasmin by tPA. Numerous studies localized tPA- and plasminogen-binding sites in the D regions and the αC-domains (reviewed in Ref: 4). Whether fibrin(ogen) contains binding sites for plasminogen inhibitor, α2-AP, remains to be established.
Factor XIIIa can incorporate α2-AP to both fibrinogen and fibrin (28). The incorporation occurs by covalent cross-linking of α2-AP to a single Lys residue (Lys303) located in the COOH-terminal portion of the fibrin(ogen) Aα chain (29, 30), namely in its αC-connector. While covalent cross-linking of α2-AP to fibrin(ogen) is well established, its molecular mechanism is not well understood. Particularly, it is not known whether the efficient cross-linking requires spatial arrangement of the cross-linking sites. In this connection, it was shown that factor XIIIa-mediated γ-γ cross-linking is markedly enhanced in the presence of a fibrin fragment E template that bring together its two D regions through the DD:E interaction (31). Further, our previous studies revealed that fibronectin, another plasma protein that is cross-linked to fibrin by factor XIIIa, binds with high affinity to fibrin to provide proper orientation of the cross-linking sites (32). Thus, we hypothesized that α2-AP may interact non-covalently with fibrin prior to its covalent cross-linking. The major goal of the present study was to test this hypothesis and to further clarify the mechanism of incorporation of α2-AP into a fibrin clot that plays a critical role in controlling fibrinolysis.
EXPERIMENTAL PROCEDURES
Proteins, Enzymes, and Antibodies
Human α2-antiplasmin, namely, its Asn-α2-AP form, was prepared as previously described in detail (33). Plasminogen-depleted human fibrinogen and bovine serum albumin were purchased from Calbiochem. Recombinant single-chain tPA was a Genentech product, human Glu-plasminogen was from an Enzyme Research Laboratories. Bovine α-thrombin, aprotinin, and carboxypeptidase B were from Sigma. The rabbit anti-plasminogen polyclonal antibodies were purchased from Chemicon, the anti-tPA monoclonal antibody mAb GMA-043 was from Calbiochem. The alkaline phosphatase conjugated ExtrAvidin was from Sigma.
Preparation of Fibrin(ogen) Fragments
Fibrinogen D1 and E3 fragments (Fig. 1B) were prepared by plasmin digestion of human fibrinogen by the procedures described in (34). Their NH2-terminal residues determined by direct sequencing for 10 cycles using a Hewlett Packard model G 1000S sequenator were essentially the same as those we reported earlier (35). The fibrin-derived D-D fragment and D-D:E complex (Fig. 1D) were prepared by plasmin digestion of cross-linked fibrin as previously described in (26).
The recombinant Aα221–610, Aα221–391, and Aα392–610 fragments corresponding to the human fibrinogen αC region, αC-connector, and αC-domain, respectively, (Fig. 1C) were expressed in Escherichia coli and subsequently purified and refolded by the procedures described previously (23). Recombinant Aα392–503 and Aα504–610 fragments corresponding to N- and C-terminal sub-domains of the αC-domain, respectively, (36) were expressed in Escherichia coli using the pET-20b expression vector (Novagen Inc.). The cDNAs encoding these fragments were amplified by polymerase chain reaction using a plasmid carrying the full-length human αC region sequence (23, 37). The following oligonucleotides were used as primers: 5'-AGAGACATATGGGCACATTTGAAGAGG-3' (forward) and 5'-AGAGAAAGCTTTTACCAAGTGTCGAAGAAGGCAGC-3' (reverse) for the Aα392–503 fragment, and 5'-AGAGACATATGGCCTCAACTGGAAAAACA-3' (forward) and 5'-AGAGAAAGCTTTTAGACAGGGCGAGATTTAG-3' (reverse) for the Aα504–610 fragment. The forward primers incorporated the NdeI restriction site immediately before the coding region; the final three bases of the NdeI site, ATG, code for the fMet residue that initiates translation. The reverse primers included a TAA stop codon immediately after the coding segment, followed by a HindIII site. The amplified cDNA fragments were purified by electrophoresis in agarose gel, digested with NdeI and HindIII restriction enzymes, and ligated into the pET-20b expression vector. The resulting plasmids were used for transformation of DH5α and then B834(DE3) pLysS E. coli host cells. Both cDNA fragments were sequenced in both directions to confirm the integrity of the coding sequences. The expressed Aα392–503 and Aα504–610 fragments were found in inclusion bodies, from which they were purified by the procedure described earlier (25). The purified fragments were refolded at 4 °C by slow dialysis from urea using the protocol described in (23) and then additionally purified by size-exclusion chromatography on a Superdex S-75 column equilibrated with TBS (20 mM Tris buffer, pH 7.4, containing 150 mM NaCl) and 0.2 mM PMSF. The fragments were concentrated to 1–2 mg/mL with a Centriprep 10 concentrator (Millipore), filtered through 0.2 μm filter unit, and stored at 4 °C.
Protein Concentration Determination
Concentrations of the recombinant Aα392–503 and Aα504–610 fragments were determined spectrophotometrically using extinction coefficients (E280, 1%) calculated from the amino acid composition by the following equation: E280, 1% = (5,690W + 1,280Y + 120S-S)/(0.1 M), where W, Y and S-S represent the number of Trp and Tyr residues and disulfide bonds, respectively, and M represents the molecular mass (38). Molecular masses of these fragments were calculated based on their amino acid composition. The following values of molecular masses and E280, 1% were obtained: 12,394 Da and 5.74 for Aα392–503, and 11,841 Da and 8.42 for Aα504–610. Note that these values take into account the NH2-terminal fMet residue (see above) while the numbering of these fragments does not. The molecular masses and E280, 1% for the recombinant Aα221–610, Aα221–391, and Aα392–610 fragments were determined previously (23).
Solid-Phase Binding Assay
Solid phase binding was performed in plastic microtiter plates using an enzyme-linked immunosorbent assay (ELISA) as described in (39) with some modifications. Microtiter Immulon 2HB plate wells (Thermo) were coated overnight with 100 μL/well of fibrinogen or fibrin(ogen) fragments, all at 10 μg/mL in TBS with 1 mM Ca2+ (TBS-Ca), followed by washing with the same buffer. To convert adsorbed fibrinogen into fibrin, it was incubated with 100 μL of a mixture of thrombin (1 NIH u/mL) and aprotinin (400 u/mL) in TBS-Ca for 30 min at 37 °C, followed by washing with the same buffer. Adsorbed fibrin was treated with 5 μg/mL carboxypeptidase B for 30 min at room temperature to remove possible COOH-terminal Lys residues. The wells were then blocked with 2% bovine serum albumin in TBS-Ca containing 0.01% Tween-20. Followed by washing with TBS-Ca and 0.01% Tween-20, the indicated concentrations of the proteins used as ligands were added to the wells and incubated for 1 h at 37 °C. α2-AP was labeled with biotin using EZ-Link Sulfo-NHS-Biotinylation Kit (Pierce) and bound α2-AP was detected by reaction with the alkaline phosphatase-conjugated avidin. A PPNP Microwell alkaline phosphatase Substrate (Kirkegaard & Perry Laboratories Inc.) was added to the wells and the amount of bound ligand was measured spectrophotometrically at 405 nm. Bound plasminogen or tPA were detected by reactions with specific polyclonal or monoclonal antibodies and the peroxidase-conjugated anti-rabbit or anti-mouse polyclonal antibodies as described earlier (35). A TMB Microwell Peroxide Substrate (Kirkegaard & Perry Laboratories Inc.) was added to the wells and the amount of bound ligand was measured spectrophotometrically at 450 nm. Data were analyzed by nonlinear regression analysis using Equation (1): A = Amax /(1 + Kd/[L]), where A represents absorbance of the oxidized substrate, which is assumed to be proportional to the amount of ligand bound, Amax is the absorption at saturation, [L] is a molar concentration of the ligand, and Kd is the dissociation constant.
Biosensor Assay
The interaction of fibrin(ogen) and its fragments with α2-AP was also studied by surface plasmon resonance (SPR) using the BIAcore 3000 biosensor (Biacore AB, Uppsala, Sweden), which measures association and dissociation of proteins in real time. α2-AP at 5 μg/mL or fibrinogen at 10 μg/mL was immobilized to the CM5 sensor chip at 250 or 1,000 RU, respectively, using the amine coupling kit (BIAcore AB, Uppsala, Sweden) according to the manufacturer instructions. Binding experiments were performed in 20 mM HEPES, pH 7.4, buffer with 150 mM NaCl, 1 mM Ca2+ and 0.01% Tween-20 (HBS-Ca) at 10 μL/min flow rate. The association between the immobilized proteins and added ligands was monitored as the change in the SPR response; the dissociation was measured upon replacement of the ligand solution for the buffer without ligand. To regenerate the chip surface, complete dissociation of the complex was achieved by adding 2 M NaCl in binding buffer for 30 sec followed by re-equilibration with binding buffer. Experimental data were analyzed using BIAevaluation 4.1 software supplied with the instrument. The dissociation equilibrium constant, Kd, was calculated as Kd = kdiss/kass, where kass and kdiss represent kinetic constants that were estimated by global analysis of the association/dissociation data using the 1:1 Langmurian interaction model (kinetic analysis). To confirm the kinetic analysis, Kd was also estimated by analysis of the association data using the steady-state affinity model provided by the same software (equilibrium analysis).
To prepare a fibrin surface for SPR experiments, fibrinogen was additionally purified on Superdex 200 and covalently immobilized to the CM5 sensor chip at 10,000 RU using the amine coupling kit (BIAcore AB, Uppsala, Sweden) according to the manufacturer instructions. Conversion of immobilized fibrinogen to fibrin was done by treatment with a mixture of thrombin (1 NIH u/mL) and aprotinin (400 u/mL) for 30 min. To prepare oligomers/polymers on the surface of the chip, 1 mg/mL fibrinogen was applied to immobilized fibrin for 20 min followed by a mixture of 3 NIH u/mL thrombin and 5 μg/mL factor XIII for 30 min; non-bound proteins were then washed out with HBS-Ca. This application was repeated three times to achieve immobilization of additional fibrin (~600 RU).
RESULTS
Interaction of α2-Antiplasmin with Fibrin(ogen)
Interaction of α2-AP with fibrinogen and fibrin was tested by two methods, ELISA and Surface Plasmon resonance (SPR). In ELISA, when microplate wells were coated with fibrinogen and then increasing concentrations of α2-AP were added, fibrinogen exhibited a dose-dependent binding with Kd = 179 nM (Fig. 2 and Table 1). In contrast, when microplate wells were coated with α2-AP and then increasing concentrations of fibrinogen (up to 2 μM) were added, no binding was observed (not shown). Such a discrepancy may be explained by a well established fact that adsorption of fibrinogen to various surfaces results in conformational changes and exposure of its fibrin-specific binding sites (reviewed in Ref: 27). Alternatively, adsorption of α2-AP on the surface of microplate wells could result in the loss of its binding activity. To select between these alternatives we first performed sandwich ELISA, in which microplate wells were coated with streptavidin that captured biotin-labeled α2-AP. When fibrinogen was added to α2-AP immobilized in such a manner, no binding was observed (not shown). Then we used SPR, in which proteins are immobilized by covalent cross-linking to the spacer on the surface of a sensor chip and, therefore, may preserve their original conformation. When fibrinogen was immobilized in such a manner and then increasing concentrations of α2-AP (up to 2 μM) were added, no binding was observed (not shown). In another SPR experiment, when fibrinogen was added to immobilized α2-AP, again no binding was observed (not shown). Thus, these experiments indicate that fibrinogen interacts with α2-AP only when it is adsorbed onto a surface.
Fig. 2.
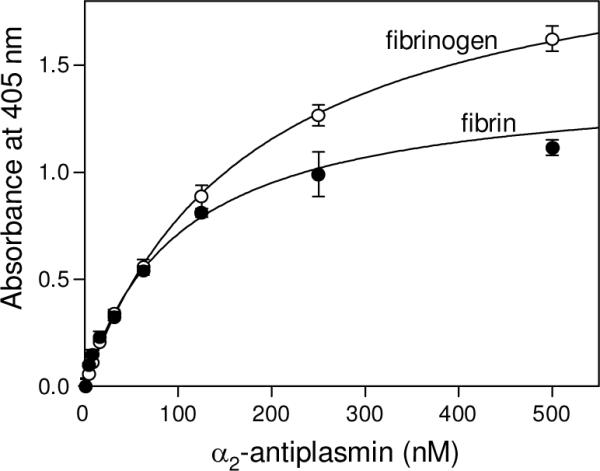
Analysis of the interaction of α2-antiplasmin with fibrinogen and fibrin by ELISA. Increasing concentrations of biotinylated α2-AP were added to surface-adsorbed fibrinogen (empty circles) or fibrin (filled circles). Bound biotinylated α2-AP was detected with avidin conjugated to alkaline phosphatase as described in Experimental Procedures. The curves represent the best fit of the data to eq. 1. All results are means ± the standard deviation of duplicate determinations.
Table 1.
Dissociation constants (Kd) for the interaction of α2-antiplasmin with surface-adsorbed fibrinogen, fibrin, and their fragments obtained by ELISA.
| Proteins/Fragments |
Kd (nM)a |
|||
|---|---|---|---|---|
| + εACA | + tPA | + plasminogen | ||
| fibrinogen | 179 ± 19 | 102 ± 4 | - | - |
| fibrin | 102 ± 4 | 101 ± 12 | 109 ± 9 | 25 ± 2 |
| E3 fragment | n. b.b | - | - | - |
| D1 fragment | 197 ± 33 | 165 ± 11 | - | - |
| D-D fragment | 94 ± 4 | 98 ± 10 | 105 ± 4 | 5.5 ± 0.4 |
| Aα221–610 fragment | 131 ± 25 | - | - | - |
| Aα392–610 fragment | 150 ± 5 | 141 ± 14 | 157 ± 21 | 111 ± 14 |
| Aα221–391 fragment | n. b. | - | - | - |
| Aα392–503 fragment | n. b. | - | - | - |
| Aα504–610 fragment | 123 ± 10 | 130 ± 15 | - | - |
Values are means ± the standard deviation of at least three independent experiments.
No binding was observed.
To test interaction of α2-AP with fibrin, we converted fibrinogen adsorbed on microplate wells or immobilized on the surface of a sensor chip into fibrin by treatment with thrombin, as described in Experimental Procedures. In ELISA, α2-AP exhibited a dose-dependent binding to surface-adsorbed fibrin with Kd = 102 nM (Fig. 2 and Table 1) while in SPR no binding to immobilized fibrin was observed even at 2 μM α2-AP (not shown). Although one cannot exclude that the luck of binding in SPR was connected with the immobilization procedure, another explanation seems to be more reasonable. Namely, because in these experiments fibrinogen was adsorbed/immobilized at comparatively low concentrations, the resulting fibrin was most probably monomeric. Since the conformational changes and exposure of cryptic binding sites occur mainly upon fibrin polymerization (26, 27), this may explain why such monomeric fibrin exhibited fibrinogen-like binding property towards α2-AP, i.e. interacted with the latter only when adsorbed to the surface. In agreement, in control experiments, plasminogen, which is known to interact only with adsorbed fibrin(ogen) or polymeric fibrin, exhibited binding to adsorbed fibrin only in ELISA, as expected, while no binding to immobilized fibrin was observed in SPR (not shown). These results suggest that monomeric fibrin does not interact with α2-AP.
To test whether fibrin polymer may interact with α2-AP, we prepared a fibrin surface on which fibrin was presumably in polymeric form (see Experimental Procedures). Briefly, fibrinogen was immobilized onto the surface of a sensor chip at high density to increase the probability of intermolecular contacts and then treated with thrombin to convert it into fibrin and enable the DD:E interactions. Next, additional fibrinogen was added, which specifically bound to immobilized fibrin through the DD:E interactions, and then such fibrin-fibrinogen complexes were treated with factor XIIIa, which reinforced the complexes by covalent cross-linking; thrombin was added to convert the additionally immobilized fibrinogen into fibrin. Such addition of fibrinogen and subsequent treatment with factor XIIIa and thrombin was repeated three times to immobilize additional molecules that presumably formed oligomers/polymers of fibrin. In contrast to immobilized fibrin monomers described above, such immobilized oligomers/polymers exhibited in control experiments significant interaction with plasminogen (not shown) suggesting that their fibrin-specific binding sites were exposed. When α2-AP at increasing concentrations was added, it also exhibited a dose-dependent binding (Fig. 3). The Kd value determined by the kinetic analysis of association/dissociation data (see Experimental Procedures) was found to be 45 ± 8 nM, that determined by the equilibrium analysis was close, 68 ± 12 nM. These experiments indicate that α2-AP binds to polymeric fibrin with high affinity.
Fig. 3.
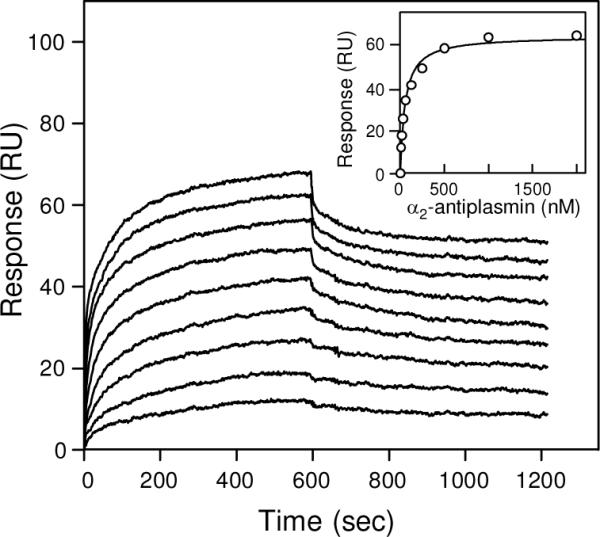
Analysis of the interaction of α2-antiplasmin with polymeric fibrin by surface plasmon resonance. α2-AP at increasing concentrations, 8, 16, 32, 63, 125, 250, 500, 1000 and 2000 nM, was added to immobilized fibrin polymers (see Experimental Procedures) and its association/dissociation was monitored in real time. The inset shows the results of the equilibrium analysis; the amount of bound a2-antiplasmin at equilibrium for each concentration is presented by circles and the best fit is presented by solid curve.
Localization of α2-AP Binding Sites in Fibrin(ogen)
To localize the α2-AP binding site(s) in fibrin(ogen), we studied the interaction of α2-AP with the D1 or D-D fragments and E3 fragment corresponding to the fibrin(ogen) D and E regions, respectively, and the recombinant αC region (Aα221–610), which together encompass practically the whole fibrin(ogen) molecule (Fig. 1B–D). In ELISA, when these fragments were adsorbed on the surface of microplate wells and increasing concentrations of α2-AP were added, a prominent binding was observed with the D-D and Aα221–610 fragments while E3 exhibited no binding (Fig. 4). The binding was dose-dependent and the Kd values determined for D-D and Aα221–610 were 94 and 131 nM, respectively (Table 1). In another experiment, α2-AP also exhibited binding to the surface-adsorbed fibrinogen-derived D1 fragment (Table 1). In reverse ELISA, when the D-D, D1, E3, and Aα221–610 fragments were added to immobilized α2-AP, no binding was detected (not shown). In agreement, in SPR experiments none of these fragments exhibited binding to immobilized α2-AP (not shown). These results suggest that the D and αC regions contain α2-AP binding sites that are cryptic in fibrinogen and the D1, D-D, and Aα221–610 fragments and become exposed upon their adsorption to a surface.
Fig. 4.
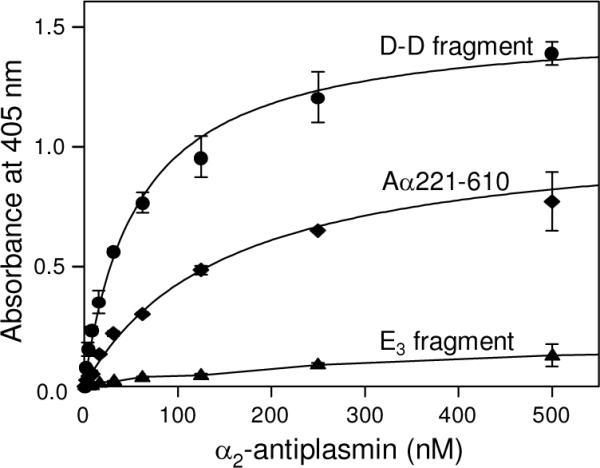
ELISA-detected binding of α2-antiplasmin to immobilized fibrin(ogen) fragments. Increasing concentrations of biotinylated α2-AP were added to the immobilized D-D (circles), Aα221–610 (diamonds), and E3 (triangles) fragments. Bound biotinylated α2-AP was detected with avidin conjugated to alkaline phosphatase. The curves represent the best fit of the data to eq. 1. All results are means ± the standard deviation of duplicate determinations.
Our finding that D1 and D-D fragments did not interact with surface-adsorbed/immobilized α2-AP suggests that in solution these fragments have fibrinogen-specific conformation and their α2-AP binding sites are cryptic. Indeed, X-ray studies of fibrinogen-derived D and fibrin-derived D-D revealed that the overall fold of the D fragment in the monomer and the dimer is very similar (40). At the same time, it was shown that the fibrin-derived D-D:E complex (Fig. 1D) preserves its fibrin-specific conformation, and that its fibrin-specific binding sites, including those for tPA and plasminogen, are accessible (26). To test if the α2-AP-binding sites are also accessible in this complex, we used SPR. When the D-D:E complex at increasing concentrations was added to immobilized α2-AP, it exhibited a dose-dependent binding (Fig. 5). The Kd value determined by the kinetic analysis of the data was found to be 1.4 ± 0.4 μM, that determined by the equilibrium analysis was practically the same, 1.4 ± 0.2 μM. These results clearly indicate that the α2-AP-binding sites in the D-D:E complex are accessible for α2-AP. They further reinforce the above suggestion that the α2-AP-binding sites of the D regions are cryptic in fibrinogen and become exposed in fibrin.
Fig. 5.
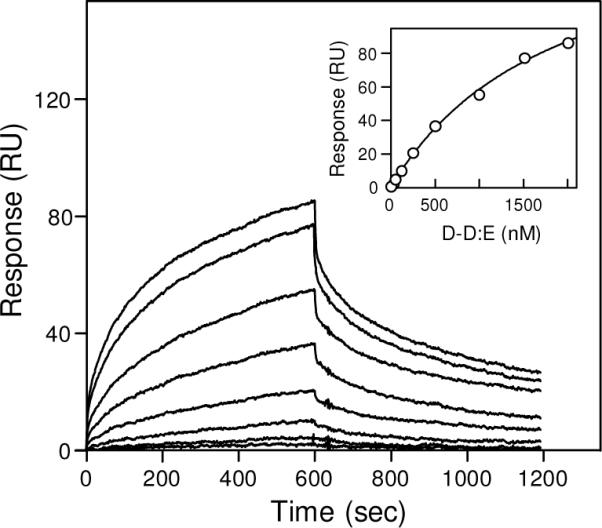
Analysis of the interaction between α2-antiplasmin and the D-D:E complex by surface plasmon resonance. The D-D:E complex at increasing concentrations, 32, 63, 125, 250, 500, 1000, 1500 and 2000 nM, was added to immobilized α2-AP and its association/dissociation was monitored in real time. The inset shows the results of the equilibrium analysis; the amount of bound D-D:E for each concentration is presented by circles and the best fit is presented by solid curve.
Localization of α2-AP-Binding Site in the αC Region
To localize the α2-AP-binding site in the αC region, we studied the interaction of α2-AP with the recombinant Aα221–391 and Aa392–610 fragments, which correspond to the αC-connector and αC-domain, respectively, and compose the entire αC region (Fig. 1C). In ELISA, α2-AP exhibited binding only to the surface-adsorbed Aα392–610 fragment while no binding was observed to Aα221–391 (Fig. 6); the affinity of the binding (Kd = 150 nM) was similar to that determined for the αC region (Table 1). This indicates that only the αC-domain of the αC region is involved in the interaction with α2-AP, i. e. this domain contains α2-AP-binding site.
Fig. 6.
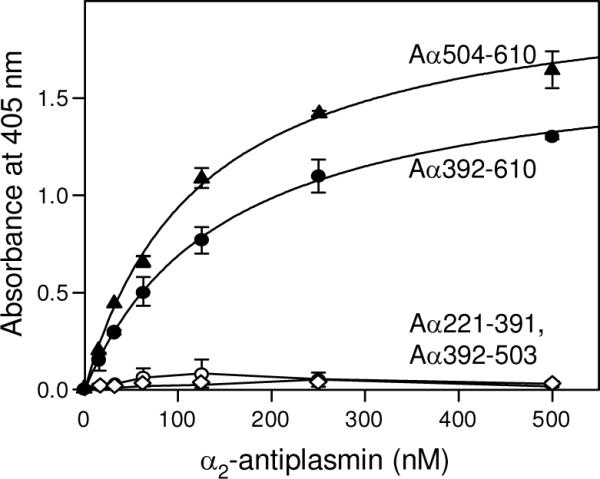
ELISA-detected binding of α2-antiplasmin to immobilized recombinant αC-fragments. Increasing concentrations of biotinylated α2-AP were added to the immobilized Aα221–391 (empty circles), Aα392–610 (filled circles), Aα392–503 (empty diamonds), and Aα504–610 (filled triangles) fragments. Bound biotinylated α2-AP was detected with avidin conjugated to alkaline phosphatase. The curves represent the best fit of the data to eq. 1. All results are means ± the standard deviation of duplicate determinations.
We have shown recently that the αC-domain consists of two independently folded sub-domains, N-terminal and C-terminal (36). To further localize the α2-AP binding site in this domain, we constructed and expressed Aα392–503 and Aα504–610 fragments corresponding to its N- and C-terminal subdomains, respectively (see Experimental Procedures). In ELISA, when increasing concentrations of α2-AP were added to both fragments immobilized on microtiter plate wells, only Aα504–610 exhibited binding (Fig. 6). The Kd value of 123 nM determined for this binding was similar to those determined for the interaction of α2-AP with the αC region or αC-domain (Table 1). Thus, these results indicate that the α2-AP-binding site is located in the C-terminal sub-domain of the αC-domain.
Further Characterization of the Interaction Between α2-AP and Fibrin(ogen)
Since plasminogen, tPA, and α2-AP are the major players in initiation and regulation of fibrinolysis, we next tested whether the former two interfere in the interaction of α2-AP with fibrin(ogen). It is well established that plasminogen and tPA both interact with fibrin(ogen) D regions and αC-domains in a Lys-dependent manner, although a Lys-independent binding of tPA to the D region was also observed (4). To test if α2-AP-fibrin(ogen) interaction involves Lys-binding sites, we studied binding of α2-AP to several fibrin(ogen) fragments that exhibited α2-AP-binding properties in the presence or absence of Lys-binding inhibitor, ε-aminocaproic acid (ε-ACA). In ELISA, when microplate wells were coated with these fragments and then increasing concentrations of α2-AP were added in the presence of 100 mM ε-ACA, all fragments exhibited a dose-dependent binding with Kd values very similar to those determined in the absence of ε-ACA (Table 1). These results indicate that the binding of α2-AP to fibrin(ogen) is Lys-independent. Since the binding of plasminogen and tPA to fibrin(ogen) is mostly Lys-dependent, these results also suggest that α2-AP-binding site are different from those for plasminogen and tPA.
To test the above suggestion, we first studied simultaneous binding of all three species, α2-AP, tPA, and plasminogen, to surface-adsorbed fibrin in the presence or absence of ε-ACA. In ELISA, when a mixture of α2-AP, tPA, and plasminogen, each at saturating concentrations, 0.5 μM, 2.5 μM, and 2.5 μM, respectively, was added to immobilized fibrin, the binding of each species was detected (Fig. 7, filled bars). The addition of 100 mM ε-ACA did not affect the binding of α2-AP while the binding of plasminogen and tPA was abolished, as expected; there was some residual binding of tPA, most probably due to its Lys-independent site (Fig. 7, empty bars). In another ELISA experiments, when fibrin, D-D and Aα392–610 fragments were adsorbed onto the surface of microplate wells and increasing concentrations of α2-AP were added in the presence of saturating concentrations of tPA (0.5 μM for Aα392–610 and 2.5 μM for fibrin and D-D), the binding curves and Kd values for all three species were very similar to those obtained in the absence of tPA (Fig. 8A and Table 1). These results indicate that there was no competition between α2-AP and tPA for the binding sites on fibrin or the D-D and Aα392–610 fragments, i.e. the α2-AP and tPA binding sites do not overlap.
Fig. 7.
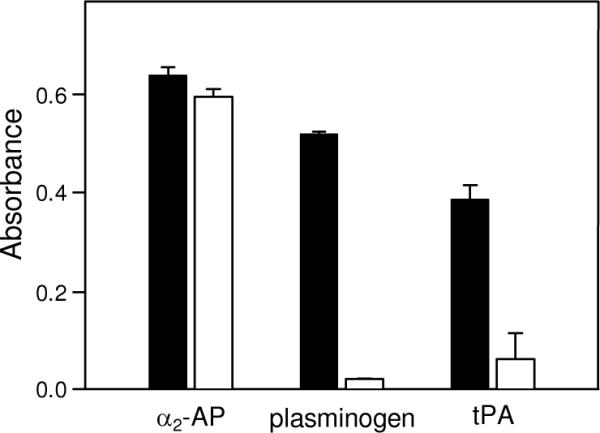
Simultaneous binding of α2-antiplasmin, tPA, and plasminogen to immobilized fibrin detected by ELISA. A mixture of 0.5 μM biotinylated α2-AP, 2.5 μM tPA, and 2.5 μM plasminogen was added to surface-adsorbed fibrin in the absence (filled bars) or presence (empty bars) of 100 mM ε-ACA. Bound biotinylated α2-AP was detected spectrophotometrically at 405 nm using avidin conjugated to alkaline phosphatase, bound tPA and plasminogen were detected spectrophotometrically at 450 nm using the anti-tPA monoclonal antibody and specific polyclonal antibodies, respectively, as described in Experimental Procedures. All results are means ± the standard deviation of two independent experiments, each performed in duplicate.
Fig. 8.

The effect of tPA or plasminogen on the binding of α2-antiplasmin to immobilized fibrin and its fragments detected by ELISA. Panel A: Increasing concentrations of biotinylated α2-AP were added to surface-adsorbed fibrin (triangles), the D-D (circles), or Aα392–610 fragments (diamonds) in the absence (filled symbols, solid lines) or presence (empty symbols, broken lines) of 2.5 μM tPA. Panel B: Increasing concentrations of biotinylated α2-AP were added to surface-adsorbed fibrin (triangles), the D-D (circles), or Aα392–610 fragments (diamonds) in the absence (filled symbols, broken lines) or presence (empty symbols, broken lines) of 2.5 μM plasminogen. Bound biotinylated α2-AP was detected with avidin conjugated to alkaline phosphatase. The curves represent the best fit of the data to eq. 1. All results are means ± the standard deviation of duplicate determinations.
Next, we performed similar binding experiments with α2-AP in the presence of saturating concentrations of plasminogen (0.5 μM for Aα392–610, and 2.5 μM for fibrin and D-D). In the case of the surface-adsorbed Aα392–610 fragment the binding of α2-AP was not affected by the presence of plasminogen and the determined Kd value of 111 nM was very close to that determined in the absence of plasminogen (Fig. 8B and Table 1). In contrast, binding of α2-AP to surface-adsorbed fibrin or the D-D fragment was much stronger in the presence of plasminogen; the Kd values for fibrin and D-D were found to be 25 and 5.5 nM, respectively. These results confirm that plasminogen does not compete with α2-AP for its binding site on fibrin(ogen). They also indicate that in the presence of plasminogen the binding of α2-AP to the surface-adsorbed D-D fragment and fibrin is significantly increased.
DISCUSSION
The major physiological inhibitor of plasmin, α2-AP, plays an important role in the regulation of fibrinolysis. This occurs through direct inhibition of plasmin activity by α2-AP, either in the circulation or on the surface of a fibrin clot. In the circulation, α2-AP rapidly inhibits free plasmin thus preventing fibrinogenolysis (8, 41). α2-AP also inhibits fibrin-bound plasmin, although much less efficiently (12). Meanwhile, incorporation of α2-AP into fibrin endows the fibrin clot with resistance to fibrinolysis (18, 19). According to the current view, this incorporation occurs by covalent cross-linking of α2-AP to fibrin(ogen) with factor XIIIa (6). In the present study, we found that α2-AP can also be incorporated into fibrin or adsorbed fibrin(ogen) through a non-covalent binding to its D regions and αC-domains.
The non-covalent interaction of α2-AP with polymeric fibrin occurs with high affinity; the equilibrium dissociation constant determined by SPR (Kd = 45–68 nM) is far below the physiological concentration of α2-AP (1 μM). This suggests that in vivo α2-AP should efficiently bind to fibrin clots thereby contributing to the regulation of fibrinolysis. Although the exact physiological role of this binding is yet to be established, the accumulated data allows the following speculation. Lys303 residue to which factor XIIIa cross-links α2-AP is located in the fibrin(ogen) αC-connector while one of the α2-AP-binding sites localized in the present study is in the fibrin(ogen) αC-domain, i. e. the binding and cross-linking sites are located in different portions of the αC region. A similar situation was observed with fibronectin, in which binding and cross-linking sites are also located in different portions of the αC region and whose non-covalent binding to the αC-connector promotes its cross-linking by factor XIIIa to the αC-domain (32). Thus, as in the case with fibrin-fibronectin interaction, the non-covalent binding of α2-AP to the αC-domain may provide its proper orientation towards the cross-linking site (Lys303) to enhance the covalent stage of the interaction (cross-linking). This is presented schematically in Fig. 9.
Fig. 9.
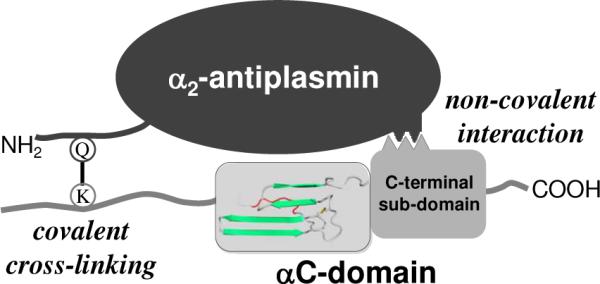
Schematic representation of the non-covalent and covalent interactions between α2-antiplasmin and fibrin(ogen) αC-domain. α2-AP with flexible NH2-termial portion containing reactive Gln14 (Q) is presented on the top. The αC region containing the flexible αC-connector with reactive Lys303 (K) and compact αC-domain consisting of two sub-domains is presented on the bottom; the structure of the αC-domain is adapted from (36). The non-covalent interaction is shown on the right, the covalent cross-linking (Q–K) between reactive Gln14 and Lys303 is shown on the left.
Numerous previous studies localized the tPA- and plasminogen-binding sites in the fibrin(ogen) D and αC regions (αC-domains) (reviewed in Ref: 4). Binding of tPA and plasminogen to these sites brings them together enabling efficient activation of the latter by the former thereby initiating fibrinolysis in places of fibrin deposition (4). In the present study, we localized the α2-AP-binding sites in the same regions. We also found that these binding sites do not overlap and have a different nature; while tPA-and plasminogen-binding sites are mainly Lys-dependent, those for α2-AP are Lys-independent. These findings suggest that fibrin can accommodate all three proteins simultaneously in close proximity to each other. Such spatial arrangement may be necessary for efficient activation of plasminogen by tPA upon initiation of fibrinolysis and subsequent rapid inhibition of newly formed plasmin by bound α2-AP. Thus, non-covalent binding of α2-AP to the D regions and αC-domains may play a role in controlling the initiation of fibrinolysis on fibrin clots.
Our SPR experiments revealed that polymeric fibrin or its soluble model, D-D:E complex, interacted with α2-AP while neither fibrinogen nor fibrin monomer exhibited such interaction. In ELISA, soluble fibrinogen did not interact with immobilized α2-AP while the latter exhibited strong interaction with surface adsorbed fibrin(ogen) and its fragments. These indicate that α2-AP-binding sites are cryptic in fibrinogen and become exposed in fibrin upon polymer formation or in adsorbed fibrinogen. This finding is not unexpected since previous studies revealed that conformational changes upon conversion of fibrinogen into fibrin or upon its adsorption result in the exposure of its numerous cryptic binding sites (27). For example, a similar situation was previously observed with tPA, plasminogen, apolipoprotein(a), fibronectin, and Mac-1 receptor, which exhibited binding only to fibrin or adsorbed fibrin(ogen) and its fragments (25, 26, 32, 35, 42, 43). Thus, the interaction of α2-AP with fibrin(ogen) represents another example of the conformation-dependent exposure of cryptic sites. Such exposure in vivo may serve to localize the activity of α2-AP to the surface of a fibrin clot where the initiation and propagation of fibrinolysis by tPA and plasmin(ogen) take place. One can also expect that binding of α2-AP to fibrinogen deposited on blood contacting foreign surfaces, for example, on implanted biomaterials, may contribute to the regulation of plasminogen activation on such surfaces and may promote cross-linking of α2-AP to adsorbed fibrinogen thereby rendering the latter more resistant to plasmin digestion. It should be noted that although fibrinogen in solution does not interact with α2-AP, the latter may also be cross-linked by factor XIIIa to the former, as revealed in the recent study (28). However, since such cross-linking is not preceded by the non-covalent interaction, it should be less efficient than that to fibrin.
The present study also revealed that the presence of plasminogen significantly facilitates the non-covalent binding of α2-AP to the fibrin(ogen) D regions. When α2-AP was added to the immobilized D-D fragment or fibrin in the presence of plasminogen, the affinity of the binding to both species was dramatically increased while no change was observed in the presence of tPA (Fig. 8 and Table 1). No change in the affinity was also observed when α2-AP was added to the immobilized αC-domain fragment in the presence of plasminogen or tPA. The reason for such increased affinity of α2-AP to the D region is not clear. It is known that plasminogen binds to fibrin or immobilized fibrinogen and its D-containing fragments (4). Thus, one cannot exclude that this binding may elicit conformational changes in the D regions resulting in further exposure of the α2-AP-binding sites and increased affinity to α2-AP. Alternatively, the increased affinity may be connected with the reported ability of α2-AP to interact with plasminogen (6, 8). In this case simultaneous binding of α2-AP to fibrin and fibrin-bound plasminogen could increase the overall affinity. This alternative may seem to be less probable since the interaction of α2-AP with plasminogen was shown to occur through its Lys-binding sites (44) and to competitively inhibit binding of plasminogen to fibrin (6, 45). However, the experiments performed in this study revealed no competition between α2-AP and plasminogen for binding to immobilized fibrinogen or D-D (Fig. 7 and 8). Such a discrepancy may be connected with the presence in plasminogen of five kringle domains at least three of which contain Lys-binding sites with different selectivity towards fibrin and α2-AP. Indeed, it was proposed that α2-AP interacts with plasminogen through Lys-binding site of kringle-1 while the primary interaction of fibrin with plasminogen may occur through kringle-5 (reviewed in Ref: 46). Further experiments are required to test this speculation and to clarify the reasons for plasminogen-induced increased affinity of α2-AP to fibrin(ogen) D regions and its possible role in regulation of fibrinolysis.
In summary, the present study revealed the non-covalent interaction between α2-AP and fibrin or surface adsorbed fibrin(ogen), localized α2-AP-binding sites in the fibrin(ogen) D region and the αC-domains, namely, its C-terminal sub-domain, and further clarified the mechanism of incorporation of α2-AP into fibrin clot. This mechanism includes the exposure of cryptic α2-AP-binding sites upon conversion of fibrinogen into fibrin polymer and non-covalent binding of α2-AP to the same regions of fibrin which bind tPA and plasminogen. Such exposure may serve to localize the activity of α2-AP to places of fibrin deposition. The non-covalent binding prior to the cross-linking may provide proper orientation of the cross-linking site and enhance the covalent cross-linking of α2-AP to the fibrin clot. The non-covalent binding may also serve to bring α2-AP to the clot for efficient inhibition of plasmin formed upon initiation of fibrinolysis while subsequent covalent cross-linking keeps α2-AP bound to the clot thereby increasing its resistance to fibrinolysis upon its propagation. This mechanism can also be applied to fibrinogen deposited on surfaces of implanted biomaterials, which is expected to be resistant to plasmin digestion due to the binding and cross-linking of α2-AP.
Acknowledgments
This work was supported by National Institute of Health Grant HL-56051 to L. Medved, and by National Institute of Health Grant HL-072995 and Warren Medical Research Center to P. McKee.
Abbreviations
- tPA
tissue-type plasminogen activator
- α2-AP
α2-antiplasmin
- ε-ACA
ε-aminocaproic acid
- ELISA
enzyme-link immunosorbent assay
- SPR
surface plasmon resonance
- HBS
HEPES buffer saline (20 mM HEPES, pH 7.4, containing 150 mM NaCl)
- TBS
tris buffer saline (20 mM tris, pH 7.4, containing 150 mM NaCl)
REFERENCES
- 1.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb. Haemost. 2001;86:324–333. [PubMed] [Google Scholar]
- 2.Lijnen HR. Elements of the fibrinolytic system. Ann. N.Y. Acad. Sci. 2001;936:226–236. doi: 10.1111/j.1749-6632.2001.tb03511.x. [DOI] [PubMed] [Google Scholar]
- 3.Rijken DC, Lijnen HR. New insights into the molecular mechanisms of the fibrinolytic system. J. Thromb. Haemost. 2009;7:4–13. doi: 10.1111/j.1538-7836.2008.03220.x. [DOI] [PubMed] [Google Scholar]
- 4.Medved L, Nieuwenhuizen W. Molecular mechanisms of initiation of fibrinolysis by fibrin. Thromb. Haemost. 2003;89:409–419. [PubMed] [Google Scholar]
- 5.Aoki N, Saito H, Kamiya T, Koie K, Sakata Y, Kobakura M. Congenital deficiency of α2-plasmin inhibitor associated with severe hemorrhagic tendency. J. Clin. Invest. 1979;63:877–884. doi: 10.1172/JCI109387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoki N. The past, present and future of plasmin inhibitor. Thromb. Res. 2005;116:455–464. doi: 10.1016/j.thromres.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 7.Carpenter SL, Mathew P. α2-Antiplasmin and its deficiency: fibrinolysis out of balance. Haemophilia. 2008;14:1250–1254. doi: 10.1111/j.1365-2516.2008.01766.x. [DOI] [PubMed] [Google Scholar]
- 8.Moroi M, Aoki N. Isolation and characterization of α2-plasmin inhibitor from human plasma. A novel proteinase inhibitor which inhibits activator-induced clot lysis. J. Biol. Chem. 1976;251:5956–5965. [PubMed] [Google Scholar]
- 9.Coughlin PB. Antiplasmin: the forgotten serpin? FEBS J. 2005;272:4852–4857. doi: 10.1111/j.1742-4658.2005.04881.x. [DOI] [PubMed] [Google Scholar]
- 10.Bangert K, Johnsen AH, Christensen U, Thorsen S. Different N-terminal forms of α2-plasmin inhibitor in human plasma. Biochem J. 1993;291:623–625. doi: 10.1042/bj2910623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koyama T, Koike Y, Toyota S, Miyagi F, Suzuki N, Aoki N. Different NH2-terminal form with 12 additional residues of α2-plasmin inhibitor from human plasma and culture media of Hep G2 cells. Biochem. Biophys. Res. Commun. 1994;200:417–422. doi: 10.1006/bbrc.1994.1465. [DOI] [PubMed] [Google Scholar]
- 12.Lee KN, Jackson KW, Christiansen VJ, Chung KH, McKee PA. α2-antiplasmin: potential therapeutic roles in fibrin survival and removal. Curr. Med. Chem. Cardiovasc. Hematol. Agents. 2004;2:303–310. doi: 10.2174/1568016043356228. [DOI] [PubMed] [Google Scholar]
- 13.Lee KN, Jackson KW, Christiansen VJ, Lee CS, Chun JG, McKee PA. Antiplasmin-cleaving enzyme is a soluble form of fibroblast activation protein. Blood. 2006;107:1397–1404. doi: 10.1182/blood-2005-08-3452. [DOI] [PubMed] [Google Scholar]
- 14.Sumi Y, Nakamura Y, Aoki N, Sakai M, Muramatsu M. Structure of the carboxyl-terminal half of human α2-plasmin inhibitor deduced from that of cDNA. J. Biochem. 1986;100:1399–13402. doi: 10.1093/oxfordjournals.jbchem.a121846. [DOI] [PubMed] [Google Scholar]
- 15.Law RH, Sofian T, Kan WT, Horvath AJ, Hitchen CR, Langendorf CG, Buckle AM, Whisstock JC, Coughlin PB. X-ray crystal structure of the fibrinolysis inhibitor α2-antiplasmin. Blood. 2008;111:2049–2052. doi: 10.1182/blood-2007-09-114215. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki T, Morita T, Iwanaga S. Identification of the plasminogen-binding site of human α2-plasmin inhibitor. J. Biochem. 1986;99:1699–1705. doi: 10.1093/oxfordjournals.jbchem.a135645. [DOI] [PubMed] [Google Scholar]
- 17.Frank PS, Douglas JT, Locher M, Llinás M, Schaller J. Structural/functional characterization of the α2-plasmin inhibitor C-terminal peptide. Biochemistry. 2003;42:1078–1085. doi: 10.1021/bi026917n. [DOI] [PubMed] [Google Scholar]
- 18.Sakata Y, Aoki N. Cross-linking of α2-plasmin inhibitor to fibrin by fibrin-stabilizing factor. J. Clin. Invest. 1980;65:290–297. doi: 10.1172/JCI109671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakata Y, Aoki N. Significance of cross-linking of α2-plasmin inhibitor to fibrin in inhibition of fibrinolysis and in hemostasis. J. Clin. Invest. 1982;69:536–542. doi: 10.1172/JCI110479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee KN, Jackson KW, Christiansen VJ, Lee CS, Chun JG, McKee PA. Why α-antiplasmin must be converted to a derivative form for optimal function. J. Thromb. Haemost. 2007;5:2095–2104. doi: 10.1111/j.1538-7836.2007.02707.x. [DOI] [PubMed] [Google Scholar]
- 21.Henschen A, McDonagh J. Fibrinogen, fibrin and factor XIII. In: Zwaal RFA, Hemker HC, editors. Blood Coagulation. Elsievier Science Publishers; Amsterdam: 1986. pp. 171–241. [Google Scholar]
- 22.Medved L, Weisel JW. Recommendations for nomenclature on fibrinogen and fibrin. Fibrinogen and Factor XIII Subcommittee of Scientific Standardization Committee of International Society on Thrombosis and Haemostasis. J. Thromb. Haemost. 2009;7:355–359. doi: 10.1111/j.1538-7836.2008.03242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsurupa G, Tsonev L, Medved L. Structural organization of the fibrin(ogen) αC-domain. Biochemistry. 2002;41:6449–6459. doi: 10.1021/bi025584r. [DOI] [PubMed] [Google Scholar]
- 24.Pizzo SV, Schwartz ML, Hill RL, McKee PA. The effect of plasmin on the subunit structure of human fibrinogen. J. Biol. Chem. 1972;247:636–645. [PubMed] [Google Scholar]
- 25.Tsurupa G, Medved L. Identification and characterization of novel tPA- and plasminogen- binding sites within fibrin(ogen) αC-domains. Biochemistry. 2001;40:801–808. doi: 10.1021/bi001789t. [DOI] [PubMed] [Google Scholar]
- 26.Yakovlev S, Makogonenko E, Kurochkina N, Nieuwenhuizen W, Ingham K, Medved L. Conversion of fibrinogen to fibrin: mechanism of exposure of tPA- and plasminogen-binding sites. Biochemistry. 2000;39:15730–15741. doi: 10.1021/bi001847a. [DOI] [PubMed] [Google Scholar]
- 27.Medved L, Tsurupa G, Yakovlev S. Conformational changes upon conversion of fibrinogen into fibrin, The mechanisms of exposure of cryptic sites. Ann. NY Acad. Sci. 2001;936:185–204. doi: 10.1111/j.1749-6632.2001.tb03505.x. [DOI] [PubMed] [Google Scholar]
- 28.Mosesson MW, Siebenlist KR, Hernandez I, Lee KN, Christiansen VJ, McKee PA. Evidence that α2-antiplasmin becomes covalently ligated to plasma fibrinogen in the circulation: a new role for plasma factor XIII in fibrinolysis regulation. J. Thromb. Haemost. 2008;6:1565–1570. doi: 10.1111/j.1538-7836.2008.03056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura S, Aoki N. Cross-linking site in fibrinogen for α2-plasmin inhibitor. J. Biol. Chem. 1986;261:15591–15595. [PubMed] [Google Scholar]
- 30.Ritchie H, Lawrie LC, Crombie PW, Mosesson MW, Booth NA. Cross-linking of plasminogen activator inhibitor 2 and α2-antiplasmin to fibrin(ogen) J. Biol. Chem. 2000;275:24915–24920. doi: 10.1074/jbc.M002901200. [DOI] [PubMed] [Google Scholar]
- 31.Mosesson MW, Siebenlist KR, Hernandez I, Wall JS, Hainfeld JF. Fibrinogen assembly and crosslinking on a fibrin fragment E template. Thromb. Haemost. 2002;87:651–658. [PubMed] [Google Scholar]
- 32.Makogonenko E, Tsurupa G, Ingham K, Medved L. Interaction of fibrin(ogen) with fibronectin: further characterization and localization of the fibronectin-binding site. Biochemistry. 2002;41:7907–7913. doi: 10.1021/bi025770x. [DOI] [PubMed] [Google Scholar]
- 33.Lee KN, Jackson KW, Christiansen VJ, Chung KH, McKee PA. A novel plasma proteinase potentiates α2-antiplasmin inhibition of fibrin digestion. Blood. 2004;103:3783–3788. doi: 10.1182/blood-2003-12-4240. [DOI] [PubMed] [Google Scholar]
- 34.Yakovlev S, Gorlatov S, Ingham K, Medved L. Interaction of fibrin(ogen) with heparin: further characterization and localization of the heparin-binding site. Biochemistry. 2003;42:7709–7716. doi: 10.1021/bi0344073. [DOI] [PubMed] [Google Scholar]
- 35.Tsurupa G, Yakovlev S, Lamanuzzi LB, Angles-Cano E, Medved L. Interaction of fibrin(ogen) with apolipoprotein(a): further characterization and identification of a novel lysine-dependent apolipoprotein(a)-binding site within the γ chain 287–411 region. Biochemistry. 2006;45:10624–10632. doi: 10.1021/bi060526h. [DOI] [PubMed] [Google Scholar]
- 36.Tsurupa G, Hantgan RR, Burton RA, Pechik I, Tjandra N, Medved L. Structure, stability, and interaction of the fibrin(ogen) αC-domains. Biochemistry. 2009;48:12191–12201. doi: 10.1021/bi901640e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuka YV, Medved LV, Migliorini MM, Ingham KC. Factor XIIIa-catalyzed cross-linking of recombinant αC fragments of human fibrinogen. Biochemistry. 1996;35:5810–5816. doi: 10.1021/bi952294k. [DOI] [PubMed] [Google Scholar]
- 38.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 39.Tsurupa G, Ho-Tin-Noe B, Angles-Cano E, Medved L. Identification and characterization of novel lysine-independent apolipoprotein(a)-binding sites in fibrin(ogen) αC-domains. J. Biol. Chem. 2003;278:37154–37159. doi: 10.1074/jbc.M305154200. [DOI] [PubMed] [Google Scholar]
- 40.Spraggon G, Everse SJ, Doolittle RF. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature. 1997;389:455–462. doi: 10.1038/38947. [DOI] [PubMed] [Google Scholar]
- 41.Wiman B, Collen D. On the mechanism of the reaction between human α2-antiplasmin and plasmin. J. Biol. Chem. 1979;254:9291–9297. [PubMed] [Google Scholar]
- 42.Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. Regulated unmasking of the cryptic binding site for integrin αMβ2 in the γC-domain of fibrinogen. Biochemistry. 2002;41:12942–12951. doi: 10.1021/bi026324c. [DOI] [PubMed] [Google Scholar]
- 43.Yakovlev S, Zhang L, Ugarova T, Medved L. Interaction of fibrin(ogen) with leukocyte receptor αMβ2 (Mac-1): further characterization and identification of a novel binding region within the central domain of the fibrinogen γ-module. Biochemistry. 2005;44:617–626. doi: 10.1021/bi048266w. [DOI] [PubMed] [Google Scholar]
- 44.Wiman B, Lijnen HR, Collen D. On the specific interaction between the lysine-binding sites in plasmin and complementary binding sites in α2-antiplasmin and in fibrinogen. Biochim Biophys. Acta. 1979;579:142–154. doi: 10.1016/0005-2795(79)90094-1. [DOI] [PubMed] [Google Scholar]
- 45.Moroi M, Aoki N. Inhibition of plasminogen binding to fibrin by α2-plasmin inhibitor. Thromb. Res. 1977;10:851–856. doi: 10.1016/0049-3848(77)90142-6. [DOI] [PubMed] [Google Scholar]
- 46.Pointing CP, Marshall JM, Cederholm-Williams SA. Plasminogen: structural review. Blood Coag. Fibrinolysis. 1992;3:605–614. [PubMed] [Google Scholar]
- 47.Yang Z, Kollman JM, Pandi L, Doolittle RF. Crystal structure of native chicken fibrinogen at 2.7 Å resolution. Biochemistry. 2001;40:12515–12523. doi: 10.1021/bi011394p. [DOI] [PubMed] [Google Scholar]