

Abstract
In this work, we report the preparation of photoactive dextran and demonstrate its utility by photochemically attaching it onto various polymeric substrates. The attachment of homogeneous and patterned dextran films was performed on polyurethane and polystyrene, with detailed analysis of surface morphology, swelling behavior, and the protein resistance of these substrates. The described photoactive dextran and attachment procedure is applicable to a wide variety of substrates while accommodating surfaces with complex surface and geometries. Dextran with azide content between 22 to 0.3 wt% was produced by esterification with p-azidobenzoic acid. Dextran (1.2 wt% azide) was photografted onto plasma oxidized polyurethane and polystyrene and displayed thicknesses of 5 ± 3 nm and 7 ± 3 nm, respectively. The patterned dextran on oxidized polyurethane was patchy with a nominal height difference between dextranized and non-dextranized regions. The azidated dextran on oxidized polystyrene exhibited a distinct step in height. In the presence of PBS buffer, the dextranized regions became smoother and more uniform without affecting the height difference at the oxidized polyurethane boundary. However, the dextranized regions on oxidized polyurethane were observed to swell by a factor of 3 relative to the dried thickness. These dissimilarities were attributed to hydrogen bonding between the dextran and oxidized polyurethane and were confirmed by the photoimmobiliization in the presence of LiCl. The resulting surface was the smoothest of all the azidated dextran samples (RRMS = 1 ± 0.3 nm) and swelled up to 2 times its dried thickness in PBS buffer. The antifouling properties of dextran functionalized surfaces were verified by the selective adsorption of FITC-labeled human albumin only on the non-dextranized regions of the patterned polyurethane and polystyrene substrates.
Keywords: Photoimmobilization, dextran, surface modification, polyurethane, polystyrene, biocoating, protein adsorption, patterning
Introduction
To date, the ideal biocompatible material has remained elusive as exemplified by the need for antiplatelet and anticoagulant therapies upon implanting cardiovascular devices. This is because therapies can result in unwanted complications such as thrombotic, thromboembolic and bleeding risks1. True biocompatibility requires the thorough understanding of the relationship between material surfaces, the biological tissue that contact them (i.e., blood), and the associated biological responses (e.g., inflammatory responses). The endothelial glycocalyx is a polysaccharide rich layer (0.5-1 μm), containing membrane-bound proteoglycans, and glycoproteins. This layer lines the blood vessels and serves as the interface between the flowing blood and the underlying tissue constituting the vessel wall. The endothelial glycocalyx plays a critical role in controlling blood flow, mechanosensing and barrier function for water and ion transport, as well as, cell and molecular adhesion (e.g., plaque formation) onto the vessel wall2. To develop the next generation of biocompatible materials for cardiovascular applications, such as indwelling endovascular stents and catheters, conduit vascular grafts and extracorporeal support of the circulation, we aim to mimick the endothelial glycocalyx by patterning individual molecules on surfaces that are in direct contact with blood. By demonstrating a versatile method to pattern one type of molecule on a variety of surfaces, heterogeneous surfaces approaching the complexity of the glycocalyx can be realized in the future.
Among natural polysaccharides, dextran coatings have demonstrated a remarkable ability to reduce protein adsorption3-11. For instance, antifouling dextran coatings have been grafted onto silicon wafers by silane chemistry3,4, physisorbed via PS-b-DEX block copolymer onto polystyrene7 and adsorbed onto metal oxide surfaces using (L-lysine)-graft-dextran via electrostatic interactions11.
Dextran can also be grafted on surfaces photochemically6,12. Compared to other methods, photochemistry has several advantages. First, compared to the conventional chemistry route, photochemical attachment of biomolecules containing a reactive photochemical group is versatile because the biomolecules can be directly grafted onto a wide variety of surfaces including metals, ceramics and polymeric materials and the grafting occurs without perturbing the backbone structure of the polysaccharide. Secondly, compared to the physical adsorption/electrostatic interactions, photoimmobilization produces a covalent bond between the biomolecule and substrate that can withstand fluid flow, as well as variations in temperatures and pH. Thirdly, by using combinations of polysaccharides with photochemical groups, either homogeneous layers or customized patterns can be created on substrates. This versatility will facilitate the development of coating that better mimic the multi-component endothelial glycocalyx and address specific biological requirements13.
Thus far, the photoimmobilization of dextran onto surfaces has been studied by Barie et al.12 and Bhat et al.6. To design a biosensor, Barie et al.12 photografted a mixture of dextran via a photoactive molecule, trifluoromethyl-aryldiazirine-functionalized bovine serum albumin (T-BSA), onto a polyimide substrate. Because functional groups on either the substrate or dextran are not required, this method is quite simple. However, this approach lacks control over the location of molecules and produces a random distribution of dextran and BSA on the outer layer. Alternatively, Bhat et al.6 modified dextran via an esterification reaction with an aryl azide (p-azidobenzoic acid) but failed to photoimmobilize dextran onto poly(ethylene terephthalate) (PET). These results were attributed to the lack of nucleophilic groups on PET, which were demonstrated by successfully photoimmobilizing dextran onto aminated PET. p-Azidobenzoic acid is a photoactive molecule commonly employed in biochemistry and molecular biology (e.g., photoaffinity reagent for labeling)14 and has also been used to photoimmobilize other polysaccharides15-18. In these prior studies, protein adsorption has received little interest and is limited to the sulfated hyaluronic acid on PET15. Surfaces photografted with dextran and hyaluronic acid are of particular importance because dextran and sulfonated hyaluronic acid attached to surfaces by conventional chemistry, physisorption and via electrostatic interactions have been shown to reduce protein adsorption.
The potential of dextranized surfaces to strongly suppress the adsorption of non-specific proteins is recognized and well-documented3-5,7-9,19,20. Blood contact with biomaterials can elicit protein adsorption, as well as cellular interactions involved in inflammatory and blood clotting responses21. Because of the importance of human albumin, (HA) the most abundant protein in human plasma, in the activation of host responses22, HA has been previously employed to determine the efficiency of dextranized surfaces prepared by other methods to suppress protein adsorption4,8,9. For instance, previous work in our group demonstrated that dextranized surfaces prepared by immobilization of oxidized dextran on amine-functionalized silicon surfaces reduces bovine albumin adsorption by approximately 72% with respect to bare silicon surfaces4. In our studies, HA adsorption was conducted to qualitatively observe the efficiency of PU and PS surfaces with photoimmobilized dextran to prevent nonspecific protein adsorption.
In this work, dextran modified with p-azidobenzoic acid is photoimmobilized either homogeneously or patterned on two chemically different polymeric substrates to prove the versatility of the method towards any polymeric surface. The azide content of dextran is varied from 0.3 to 22 wt% and characterized by UV, 1H-NMR and FTIR. Being soluble in aqueous medium and having sufficient photoactive units per molecule, dextran with 1.2 wt% azide is selected for photoimmobilization on polyurethane (PU) and polystyrene (PS) substrates. PU is a widely utilized biomaterial used for vascular applications (e.g., indwelling catheters). PS, an inert glassy polymer common to biology laboratories (e.g., petri dishes), is chosen to demonstrate the flexibility of the photochemistry approach using azide groups, which can be used to graft polymers lacking reactive functional groups. To facilitate uniform grafting, the PU and PS surfaces are first exposed to an oxygen plasma to overcome the inherent hydrophobicity of PU and PS and, create surfaces that can be wet by the aqueous azide modified dextran solution. In contrast to previous studies6, the polymeric substrates do not require alteration to include nucleophilic groups (i.e., amination) prior to attachment of dextran by photoimmobilization.
Hence, we present the photoattachment of dextran layers onto oxidized PU and PS and discuss, for the first time, the detailed dextran morphology as imaged by AFM in air and phosphate saline buffer (PBS). Over a small area (2×2 μm2), similar dextran surface morphologies and swelling behavior are observed on oxidized PU and PS. In addition to homogeneous surfaces, dextran is photopatterned onto oxidized PU and PS to produce dextran squares, 90 × 90 μm2. The dextran morphology is less uniform on PU and attributed to hydrogen bonding that confounds the formation of a uniform layer. This hypothesis is verified using a novel approach to photoimmobilize dextran onto oxidized PU, in the presence of a salt. The resulting surface is smooth (RMS < 1 ± 0.3 nm) and swells by 2x in PBS. This approach is highly versatile because dextran can be modified with other photoactive molecules and can be attached to many polymer substrates. The antifouling properties of photoimmobilized dextran onto oxidized PU and oxidized PS are demonstrated using fluorescently labeled human albumin protein.
Experimental Section
Materials
Dextran from Leuconostoc ssp (Mw=100,000 gmol−1) was purchased from Fluka. Monodisperse polystyrene (PS) (Mw=200,000 gmol−1) from Polymer Laboratories and polyester based polyurethane (PU) tubing (Thermo Scientific Nalgene 280) from Fisher Scientific was used. Anhydrous dimethylsulfoxide (DMSO) was used. Silicon wafers (CZ Silicon, type: N, dopant: Ph) were acquired from Silicon Quest Int’l. All chemicals, unless otherwise specified, were purchased from Sigma-Aldrich and used as received.
Synthesis of p-azidobenzoic acid
The synthesis of p-azidobenzoic acid from p-aminobenzoic acid is well described in the literature 23. In a typical experiment, 4-aminobenzoic acid (0.0145 mol) was dissolved in 200 mL of 1M HCl solution. The mixture was cooled to 0-10°C and an aqueous solution of sodium nitrite (0.055 mol, 0.4 g/mL) was added. After 45 minutes, an aqueous solution of sodium azide (0.06 mol, 0.4 g/mL) was added dropwise while maintaining temperature. The resulting precipitate was filtered under vacuum, washed with water three times, dried in air and stored in dark.
Synthesis of azidated dextran
The synthesis of azidated dextran was adapted from Bhat et al6. Dextran was dissolved in 100 mL of DMSO in an opaque round flask with 4-dimethylaminopyridine (DMAP) (6.13×10−4 mol) and p-azidobenzoic acid (6.13×10−3 mol). The mixture was cooled to 0-5°C and N,N’-dicyclohexyl carbodiimide (DCC) (6.058×10−3 mol) was added to the mixture. After an hour at 0-5°C, the mixture was slowly brought to room temperature and further reacted for 4h. The precipitate, dicyclohexyl urea (DCU), was filtered and rinsed with DMSO. Azidated dextran was precipitated from the filtrate by adding methanol. The product was washed with methanol, until the total elimination of free p-azidobenzoic acid of the residual solution was confirmed by UV spectroscopy, dried in air and stored in the dark.
Photoimmobilization of azidated dextran onto surfaces
Silicon wafers were first cleaned by immersion in “piranha” solution (70 % H2SO4 and 30% H2O2) for 20 min at 80 °C, washed with copious amounts of water, and then soaked in water till used. Prior to use, the surfaces were blown dry with compressed N2 (g) and exposed to ultraviolet light in a UVO-Cleaner (UVO-Cleaner Model 42, Jelight Co. Inc.) for 10 min to form a uniform oxide layer (16 ± 2 Å). 150 μL solutions of PS (toluene, 1wt%) or PU (tetrahydrofuran:dichloroethane 1:1, 0.75wt% ) were spin coated (PS: 3000 rpm, 15”; PU: 2000 rpm, 10”) onto the freshly cleaned silicon wafers immediately after oxidation. The polymer films were dried over night at 25 °C. PS samples were further annealed under vacuum at 150°C for 24h to remove residual solvent and promote adhesion between the PS film and the substrate. The polymer films were then treated with oxygen plasma (Expanded Plasma Cleaner and PlasmaFlo, Harrick Plasma) for 60 seconds at a power of 18W. Subsequently, an aqueous solution of azidated dextran (3 mg/mL) was deposited onto the oxidized PS/PU films and dried in a desiccator over night. Films were exposed to UV with a handheld UV lamp for 2 minutes (6 W, UVP). Following UV exposure, the films were washed with copious amounts of water, submerged in water over night and blown dry with compressed N2 (g). Alternatively, 150 μL solutions of azidated dextran (3mg/mL, 1 wt% LiCl) were spun coated (2000 rpm, 60”) on oxygen plasma treated polymer films and dried with argon. Subsequent steps were repeated as described above.
Adsorption of human albumin on dextranized surfaces
Human albumin-fluorescein isothiocyanate (HA) was dissolved in phosphate buffer solution (PBS, 1 mg / mL, pH=7.4) and stored in a freezer at −40 °C. The samples were transferred to vials, covered with protein solution and incubated for 1h at 37 °C. Afterward, the samples were rinsed 3 times with 1 mL deionized water to remove the unbound proteins. The adsorbed proteins were qualitatively observed by fluorescence microscopy (BX41, Olympus). Since fluorescence intensity was observed to vary between images likely due to photobleaching and variations on the intensity of the mercury lamp, the brightness and contrast of the images was adjusted as required.
Characterization
Fourier Transform Infrared (FTIR)
FTIR spectra were recorded on a Perkin Elmer infrared spectrophotometer (Spectrum RX I FTIR system) at a resolution of 8 cm−1 averaging 256 scans.
UV spectrophotometer
UV spectra were recorded on a Varian Spectrophotometer (Cary 5000 UV-Vis-NIR). The azide wt% (p-azidobenzoic acid weight %) was quantified by applying Beer’s law to solutions of AZBC with known concentration. The resulting calibration curve was then used to determine the azide wt% of known concentrations of azidated dextran.
Contact angle
Water contact angles were measured using a 1μL sessile drop at ambient conditions. Images were captured by a CCD camera. The contact angle at the three phase boundary was measured by ImageJ (NIST).
Ellipsometry
Thickness was measured using an Auto-El-II Null Ellipsometer (Rudolph Research, Flanders NJ) at a fixed incident angle of 70° with a helium-neon laser source (λ=6328Å). All measurements were collected within 1-2 h of sample preparation to minimize contamination. The thickness of the polysaccharide layer is determined using a fixed refractive index of 1.5624.
AFM
Surface topography and roughness were determined by atomic force microscopy (Digital Instruments, Santa Barbara, CA: Dimension 3000 AFM). In air, tapping mode with a single crystal Si tip with a spring constant of 48 N/m, a radius of curvature of about 10 nm and a resonance frequency of approx. 190 kHz was used. Images in PBS buffer were obtained using TOP MAC tapping mode with Type II or Type V Mac Levers (Agilent Technologies). AFM images were limited to a scan size of 80×80 μm2 and thus in patterned samples, only part of the 90×90 μm2 square pattern was observed at a time. Artifacts were noticed on some images (i.e., non-linear pattern, sinusoidal along x axis) and attributed to working at the upper limit of the scanning area (e.g., 80 × 80 μm2) and intrinsic non-linearity of the scanner. Because the z scale, height, was orders of magnitude smaller than the working limits of the scanner, the effects of non-linearity were less severe and not taken in consideration25. Images were analyzed using Picoview 1.6 software (Agilent Technologies).
Stadistical Analysis
The means and standard deviations (SD) of the substrates thickness, roughness and water contact angle data are presented as averages ± SD. For each substrate, a minimum of 15 readings were used for ellipsometric measurements (3 different spots per sample) and a minimum of 15 separate readings were used for contact angle measurements. The RMS roughness values were determined from 5 separate 2 μm2 images for each substrate type.
Results and Discussion
Synthesis and characterization of azidated dextran
The synthesis of azidated dextran is illustrated in Figure 1a. Photoactive dextran is obtained via esterification reaction of p-azidobenzoic acid (AZBC) with dextran (DEX) in the presence of N,N’-dicyclohexylcarbodiimide (DCC) and a catalytic amount of 4-dimethylaminopyridine (DMAP) in dimethyl sulfoxide (DMSO). Prior to the esterification reaction, AZBC is prepared via azidation of p-aminobenzoic and characterized by FTIR (see Figure 1d). The azido group –N3 can be identified by the strong N=N=N asymmetric stretching band at 2150 cm−1. Furthermore, AZBC is characterized by UV spectroscopy and 1H-NMR, which shows the characteristic azide band at 272 nm as well as proton shifts at 7.20-7.23ppm (H3, H5) and 7.96-7.94 ppm (H2, H6), respectively. These results are in agreement with data from the literature6 (data not shown).
Figure 1.

(a) Synthesis scheme for preparing azidated dextran. The esterification reaction of dextran (DEX) with p-azidobenzoic acid (AZBC) in the presence of of N,N’-dicyclohexylcarbodiimide (DCC) and the catalyst 4-dimethylaminopyridine (DMAP) in dimethyl sulfoxide (DMSO). The product is a precipitate, N,N’-dicyclohexylurea (DCU) and azidated dextran (AZDEX). The percentage of azidation is varied from 0.3 to 22 wt%. FT-IR spectra of (b) dextran (DEX), (c) azidated dextran (AZDEX-22) and (d) p-azidobenzoic acid (AZBC). The asymmetric stretching band for the azide (N3) appears sharp at 2050 cm−1 in the AZBC and AZDEX-22 spectra, whereas in the same region only a broad band is noticeable in the DEX spectrum. The azide band in AZDEX-22 supports the esterification of DEX with AZBC.
The synthesis of azidated dextran (AZDEX) is adapted from Bhat et al.6 and carried out using DMAP-catalyzed DCC, a very effective method for preparing esters including sterically hindered alcohols26. Because dextran is a glucan with mainly secondary hydroxyls (α(1-6) glycosidic linkages) and some (< 5%) primary hydroxyls (short branches of α(1-4) glycosidic linkages and in some cases, α-1,2 and α-1,3 linkages as well). Ester formation at the primary and secondary hydroxyl is expected in the final product. DMSO is used as the reaction solvent due to the common solubility of DCC, AZBC and DEX. The side product, N,N’-dicyclohexylurea (DCU), is removed, and AZDEX is precipitated from solution using methanol.
In contrast to the findings of Bhat et al.6, AZDEX is insoluble in water. The total elimination of free AZBC in the final product is confirmed by UV characterization of the residual solution obtained after extensive rinsing of AZDEX with methanol. The product is confirmed to be AZDEX by FTIR, UV and 1H-NMR. Figure 1c shows the FTIR spectrum of AZDEX. For comparison, the FTIR spectra of AZBC and DEX have been included (Figure 1b and 1d, respectively). As noted, the band for azide near 2150 cm−1 appears sharp and strong in the AZBC spectrum whereas dextran only exhibits a broad shoulder in the same region. The esterification reaction is confirmed by the appearance of the characteristic azide band in the product spectrum (AZDEX). UV analysis of AZDEX shows a shift of the absorbance azide band to higher wavelength compared to AZBC, 275 vs 272 nm further confirming the attachment of AZBC to DEX (data not shown). In addition, 1H-NMR spectra show the shifts characteristic of AZBC and dextran (3-5.3 ppm)6 (data not shown). We note that even though our reaction uses a lower molecular weight dextran than that used by Bhat et al.6 (100k vs 500k gmol−1), the molar concentrations are adjusted so that the same azide wt% should have been observed.
The azide wt% in the AZDEX is quantified by UV spectroscopy. The wt% of azide in AZDEX is determined to be 22% and the number of free hydroxyls per functionalized AZBC molecule is 13 as noted in Table 1. This sample is denoted AZDEX-22. The OH/AZBC ratio is estimated by assuming that dextran is a linear chain consisting of only α(1,6) glycosidic linkages. Thus, on average, AZDEX-22 has a relatively high density of azide groups, namely, 1 in every 14 hydroxyls have been replaced by AZBC. Alternatively, about every 5th repeat unit of dextran contains an azide side group. Given that AZBC is insoluble in water, the limited solubility of AZDEX-22 in water can be attributed to the high AZBC concentration. Correspondingly, AZDEX-22 is soluble in DMSO, a good solvent for AZBC.
Table 1.
Summary of azidated dextrans synthesized in the lab
| AZDEX | DEX:AZBC1(mol) | Azide wt% | OH/AZBC2 |
|---|---|---|---|
| AZDEX-22 | 1:1533 | 22 ± 2 | 13 |
| AZDEX-3.2 | 1:383 | 3.2 ± 2 | 93 |
| AZDEX-1.2 | 1:192 | 1.2 ± 0.1 | 247 |
| AZDEX-0.3 | 1:96 | 0.3 ± 0.1 | 926 |
stochiometric conditions employed
estimated number of free hydroxyls per functionalized (i.e. AZBC attachment) assuming that dextran is a perfectly linear chain that consists of only α (1,6) glycosidic linkages
Azidated dextrans with lower AZBC content are obtained by reducing the concentration of excess AZBC in the esterification reaction. To prepare AZDEX-22, the reaction conditions involve 1 mol of dextran per 1533 mols of AZBC. This excess of AZBC is reduced to 383, 192 and 96 mols of AZBC per mol of dextran. For each concentration of AZBC, AZDEX is prepared with an esterified AZBC molecule every 93, 247 and 926 hydroxyls, respectively and are denoted as AZDEX-3.2, AZDEX-1.2 and AZDEX-0.3, respectively. Table 1 summarizes the stoichiometric conditions, the amount (wt%) of azide and the ratio of free hydroxyls per functionalized AZBC molecule. Whereas AZDEX-3.2 is only partially soluble in water, AZDEX-1.2 and AZDEX-0.3 are completely soluble in water. Water-soluble AZDEXs are highly desirable because the solvent is non-toxic (i.e., green chemistry) and AZDEX brushes can swell in water (i.e., protein resistant). Conversely, water soluble AZDEXs do not readily wet hydrophobic polymers, such as PS. As noted below, by briefly exposing PU and PS to oxygen plasma, surfaces can be rendered wettable by aqueous solution of AZDEX.
Photoimmobilization of azidated dextran onto polyurethane (PU) and polystyrene (PS) surfaces
Because it contains the highest azide concentration while maintaining water solubility, AZDEX-1.2 is chosen as the ideal AZDEX for photoimmobilization onto polymer substrates. The photoimmobilization process involves spin coating a solution of PU/PS onto silicon wafers to create a uniform PU or PS film, exposure of the PU or PS surfaces to an oxygen plasma (OX-PU/OX-PS), casting of the AZDEX-1.2 aqueous solution onto OX-PU/OX-PS, and finally, photoimmobilization of the dried dextran layer by exposure to UV (DEX-PU/DEX-PS). We note that, from now on, photoimmobilized AZDEX-1.2 onto polymer substrates will denoted simply as DEX. The film and surface properties are characterized by ellipsometry and contact angle measurements under atmospheric conditions and results summarized in Figure 2. The initial thickness (h) values of the PU and PS films are 40 ± 1 and 47 ± 1 nm, respectively. Thin films are chosen in order to enhance sensitivity to changes in thickness after surface treatments. The contact angle of water on PU and PS are 78 ± 2° and 81 ± 3°, respectively, consistent with hydrophobic surfaces.
Figure 2.

Thickness, contact angle and RMS roughness (RRMS) with their corresponding standard deviation of (a) PU and (b) PS based films including bare films (PU/PS), oxidized films (OX-PU/OX-PS), dextranized films in air (DEX-PU/DEX-PS) and dextranized PU films in PBS buffer (DEX-PU-WET/DEX-PS-WET). The film thickness decreases upon oxygen plasma treatment but increases with photochemical attachment of AZDEX. On the other hand, the contact angle decreases drastically with oxygen plasma treatment and remains low with AZDEX attachment. The RRMS remains unaltered after oxygen plasma treatment. For dried DEX-PU, the RRMS increases to 2 ± 1 nm but reduces to 1.4 ± 0.5 nm in buffer whereas for dried DEX-PS, the RRMS increases to 1.4 ± 0.5 nm but returns to 0.6 ± 0.1nm in buffer.
Due to their hydrophobic nature, the PU and PS films are lightly oxidized to introduce a hydrophilic surface for improved wetting of DEX without degradation of the film intrinsic properties. Hydrophobic surfaces can be made hydrophilic by exposing them to oxygen plasma, which produces oxygen containing surface groups (i.e., CHO, COOH) that increase the surface energy and wettability. Using IR-VIS sum frequency generation, Zhang et al27. demonstrated that an oxygen plasma attacked the aromatic ring of PS to produce aldehydes and carboxylate species. As shown in Figure 2, the contact angles of PU and PS decrease from 80° to less than 10° upon plasma treatment. After long exposure times (>10 min) oxygen plasma can erode the surfaces because O2 reacts with C-C bonds on the surface to form volatile reaction products. Etching is minimized by using short exposure times (1 min) that only reduces film thickness by ~ 3 nm as noted in Figure 2. Because oxygen plasma treated surfaces can reconstruct under humid conditions28, OX-PU and OX-PS films are treated with DEX within 1-2 h after exposure.
After plasma treatment of the hydrophobic polymers, DEX is then cast and dried, and covalently bound to OX-PU and OX-PS surfaces by photoimmobilization (namely DEX-PU and DEX-PS, respectively). Free DEX is removed from the surfaces by soaking the samples in water at room temperature for 24h, followed by rigorous rinsing. The remaining dextran molecules are covalently bound to the surface and stable under testing conditions. For the OX-PU and OX-PS films, the thickness increases to 5 ± 3 nm and 7 ± 3 nm, respectively. As shown in Figure 2, the contact angles remain <10°, consistent with the attachment of hydrophilic DEX.
The surfaces morphology and roughness of PU/PS, OX-PU/OX-PS and DEX-PU/DEX-PS are studied using tapping mode AFM. Representative topography images over 2×2 μm2 scan areas are shown in Figure 3. For PU and PS, and their modified surfaces the root main square roughness (Rrms) values are shown in Figure 2. The topography image for PU exhibits a network-like structure, which can be attributed to the assembly of nanodomains of hard segments during solvent evaporation. This network-like structure is readily observed in the phase image (data not shown) suggesting harder and softer regions on the PU surface. As cast PS surfaces are very smooth and featureless and remain relatively unaltered after exposure to an oxygen plasma for 1 min. as seen in the topography image. These morphological differences are reflected in their Rrms values. The Rrms of as-cast PU is 3x larger than PS, namely 0.3 ± 0.1 nm vs 0.9 ± 0.1 nm although for both, PS and PU, Rrms values are similar before and after plasma treatment, consistent with previous plasma treatment studies of PS29.
Figure 3.

AFM topography images of (a, b, c, d) PU and (e, f, g, h) PS based films: (a, e) bare film (PU/PS), (b, f) oxidized film (OX-PU/OX-PS), (c, g) dextranized film in air (DEX-PU/DEX-PS) and (d, h) dextranized film in PBS buffer (DEX-PU-WET/DEX-PS-WET) for 2×2 μm2 scan size. Initially, PU surfaces show a network-like morphology that remains unaltered upon oxygen plasma treatment (OX-PU) but fades upon photochemical attachment of dextran (DEX-PU). The dextranized layer (DEX-PU) shows uneven features that turn into a smooth layer when immersed in PBS buffer (DEX-PU-WET). In contrast, PS surfaces are featureless and remain so upon oxygen plasma treatment (OX-PS). Features appear upon photochemical attachment of dextran (DEX-PS) and turn into smooth layer when immersed in PBS buffer (DEX-PS-WET).
Topographic images of DEX-PS and DEX-PU reveal features absent in the as-cast or oxidized substrates. Although, DEX-PU surfaces show a network-like structure similar to PU and OX-PU, the increase in roughness reflected by the z scale change (8 nm to 35 nm), suggests that DEX covers the nanodomain features. In addition, photoattachment of a continuous layer is supported by the constant phase contrast observed in phase images of DEX-PU (data not shown). Previously, oxidized dextran was immobilized on APTES-modified silicon30-32. Using oxidation times from 0.5 to 4h, the surface displayed flat (~nm) uniform dextran patches with a diameter of ~ 50 nm31,32. Tasker et al30. observed slightly larger features, 75 ± 12 nm, for chloroformate-activated dextran. Using the dextran radius of gyration as a guide ( RG = 0.66Mw0.43)33, they concluded that the features contained 10-12 dextran molecules. In our studies, round features ranging from 60 to 300 nm are observed on the surface of DEX-PU and DEX-PS. In the present study, DEX is esterified with an average of one azide per 82 repeat units, which is equivalent to a minimum distance between attachments of 3.9 nm (assuming equal distribution of the azide groups along the chain and a Mw = 162 gmol−1 for a dextran monomer). Therefore, our features consist of 15-75 bound azide groups. After the photoattachment of DEX to OX-PU and OX-PS, the RRMS increases from 0.8 ± 0.4 nm to 2.6 ± 1.2 nm and 0.3 ± 0.1 nm to 2 ± 1 nm, respectively. Interestingly, although the roughness values for OX-PU and OX-PS differ by ~ 3x, the roughness of dextran on the PU and PS surfaces turns out to be similar because of the attachment of dextran to the valleys on the rough PU surface.
DEX-PU and DEX-PS are also imaged in PBS buffer to observe changes in their morphology upon swelling. By comparing the DEX-PU/DEX-PS and DEX-PU-WET/DEX-PS-WET images in Figure 3, features observed in the dried state are no longer apparent in the wet state and are attributed to swelling of the dextran layer, which reduces the height difference of the dried collapsed features. A comparison of the maximum values of the z-scales, 35 (dry) and 16 nm (wet) for PU (Figures 3c and 3d) and 10 nm (dry) and 4 nm (wet) for PS (Figures 3g and 3h) showed that surfaces become smoother by a factor of ~ 2. Furthermore, the RRMS decreases to 1.4 ± 0.5 nm and to 0.6 ± 0.1 nm for DEX-PU-WET and DEX-PS-WET, respectively. For both DEX-PU-WET and DEX-PS-WET, the phase images are homogeneous indicative of uniform swelling of the grafted dextran (data not shown). At high coverage of poly(DEX-b-PS) physisorbed on PS, Bosker et al7 also observed a homogeneous dextranized layer upon exposure to water.
Photopatterning of azidated dextran onto polyurethane (PU) and polystyrene (PS) surfaces
Using a photomask, as-cast DEX is photopatterned onto previously oxidized PU/PS substrates. In areas exposed to UV, DEX covalently attaches to OX-PU or OX-PS, regions, whereas on protected regions DEX is rinsed away to expose OX-PU and OX-PS surfaces. The topography of photopatterned DEX on OX-PU and OX-PS is shown in Figure 4, respectively. The dextranized regions (i.e., patterned areas) are readily observed in both the topography (Figure 4) and phase (not shown) images with an interface between the patterned and OX-PU or OX-PS regions. To better visualize this interface, 3D images and line-scans (white lines from the 2D images) are also presented in Figure 4. On PU, the dextranized regions display irregularly shaped features that are up to 20 nm height and these features gradually disappear at the interface; in contrast, the dextran features on PS have a more uniform size and are smaller , ~ 5 nm, than the PU case with a distinct 1.3 ± 0.4 nm height change at the interface. The difference between the dextran features on the PU and PS substrates may be attributed to the different surface chemistries on the oxidized films, which in turn determines how tightly dextran is bound to the substrate. The treatment of PU by an oxygen plasma mainly produces C=O, OH, C-O surface groups34; however, polyester-urethane treated with oxygen plasma causes scission of chains and the generation of free amines35. The free amines on OX-PU surfaces can facilitate the formation of larger dextran features by acting as electron acceptor or donor species that form hydrogen bonds with dextran and/or behaving as a nucleophilic group that enhances the reactivity of the azides6. Either or both factors would result in larger dextran features. An additional consequence of the hydrogen bonding of OX-PU is a reduction in the swelling of grafted DEX in PBS as noted later.
Figure 4.

AFM images of photopatterned dextran on (a, b, c) PU and (d, e, f) PS: (a, d) topography, (b, e) line scan across while line in (a, d) and (c, e) 3D topographic image of 80×80 μm2 scan size (z scale: 20 nm for PU, z scale: 15 nm for PS). A photomask is used to pattern dextran on the substrate. The unmasked regions on PU (photochemically bound dextran, DEX-PU) display dextran clusters whereas smoother regions correspond to the masked regions (“protected”, OX-PU). The unmasked regions on PS (photochemically bound dextran, DEX-PS) display a layer of dextran (1.3 ± 0.4 nm) with uniform dextran features whereas smoother regions correspond to the masked regions (“protected”, OX-PS).
Surface characterization of the dextranized and unmodified regions is reported in Figure 5. The RMS roughness values, RRMS, determined from the patterned PU/PS (namely, the dextranized and unmodified regions) agree with the values measured on the corresponding homogeneous samples. Specifically, RRMS values of 2.2 ± 1.4 nm and 1.4 ± 0.8 nm are observed for patterned DEX-PU and DEX-PS, respectively and 0.5 ± 0.2 nm for the unmodified regions of both OX-PU and OX-PS. For the patterned samples, the dextran feature size from AFM can be compared with thickness measurements from ellipsometry. For DEX-PU and DEX-PS the average feature heights (~50 measurements) are 7 ± 4 nm and 4 ± 2 nm which is comparable to the thickness values, namely, 5 ± 3 nm and 7 ± 3nm for DEX-PU and DEX-PS.
Figure 5.
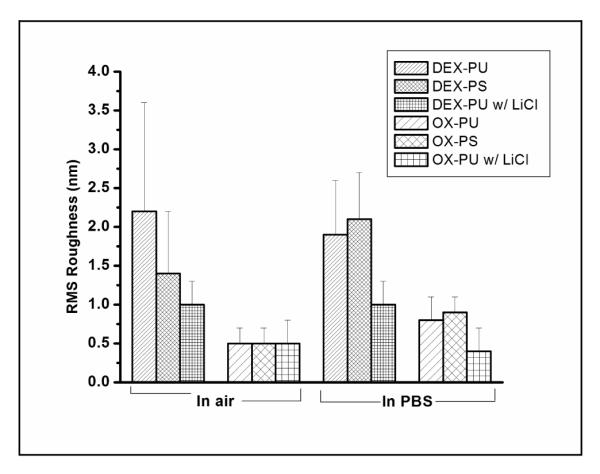
RMS roughness (RRMS) of the patterned dextranized regions (DEX-PU, DEX-PS and DEX-PU with LiCl) and oxidized regions (OX-PU, OX-PS and OX-PU with LiCl) in air and PBS measured by AFM (80×80 μm2). For the dextranized regions, the RRMS in air is lowest for DEX-PU with LiCl substrates, slightly larger for DEX-PS substrates and highest for DEX-PU substrates, whereas in PBS, RRMS remains unaltered for DEX-PU with LiCl, decreases for DEX-PU but increases for DEX-PS substrates. This behavior can be explained in terms of hydrogen bonding, which is inhibited by adding LiCl. On the other hand, the oxidized regions show very similar RRMS in air, independent of the substrate. Here the RRMS increases in PBS for the OX-PU and OX-PS substrates due to the swelling of residual dextran molecules, but remains unaltered for OX-PU with LiCl substrates.
To investigate dextran swelling, photopatterned samples are imaged while immersed in PBS. Figure 6 show 3D topography, images and line scans (taken from while line in 2D images) for photopatterned on OX-PU and OX-PS, respectively. On the PU, the dextran features appear swollen and smooth. In PBS the swollen features are more similar in height (~5 nm) compared to the dried features. In contrast, on PS, the dextranized regions swell up to 3x the dried thickness, from 1.3 ± 0.4 nm to 3.8 ± 1 nm. The strong swelling of dextran grafted to PS is attributed to the different functional groups on the surface of the oxidized PU and PS.
Figure 6.

AFM images in PBS buffer of photopatterned dextran on (a, b, c) polyurethane (PU) and (e, f, g) polystyrene (PS): (a, d) topography, (b, e) line scan across white line in (a) and (c, f) 3D topographic image of 70×70 μm2 scan size (z scale: 20 nm) and 80×80 μm2 scan size (z scale: 20 nm) for PU and PS, respectively. On PU in PBS, the irregular dextran features observed in air (h ≤ 20 nm) swell and turn into flatter and more uniform features (h ≤ 5 nm). Although the PU regions (OX-PU) remain predominantly flat in PBS, the PU regions also display some features due to swelling of arbitrary dextran chains. In constrast, on PS in PBS, the dextran region (DEX-PS) swell by 3x its dried thickness from 1.3 ± 0.4 nm to 3.8 ± 1 nm. Similar to the dried sample, the surface is smooth as previouly noted in Figure 4. The PS regions (OX-PS) also remain predominantly flat in PBS but display some features due to swelling of residual dextran molecules.
The RRMS values of the patterned PU and PS surfaces in PBS are shown in Figure 5. In the swollen dextranized regions, the RRMS values decrease and increase for dextran on DEX-PU and DEX-PS surfaces, respectively, compared to their value in the dry state. In agreement with the leveling of the irregular dextran features (Figure 5) and flattening of homogeneous DEX-PU-WET (Figures 2 and 3), the RRMS of patterned DEX-PU in PBS decreases to 1.9 ± 0.7 nm from 2.2 ± 1.4 nm (in air). In contrast, the RRMS of patterned DEX-PS increases in PBS to 2.1± 0.6 nm from 1.4 ± 0.8 nm (in air). This result suggests that although the dextran layer strongly swells (Figure 5), some regions may swell more than others as indicated by the uniform swelling observed at the smaller scan size (Figures 2 and 3). For the unmodified regions (OX-PU and OX-PS), RRMS in PBS increases due to the swelling of residual dextran molecules, from 0.5 ± 0.2 nm (dry) to 0.8 ± 0.3 nm (wet) and to 0.9 ± 0.2 nm (wet) for OX-PU and OX-PS, respectively.
We hypothesize that photoimmobilized dextran is more strongly bound to PU than PS surfaces because of free amines that allow for hydrogen bonding and higher dextran grafting density. To test this hypothesis, LiCl is added to the DEX solution to disrupt hydrogen bonding between DEX and OX-PU. The DEX solution is deposited onto OX-PU by spin-coating to favor fast drying and prevent possible DEX reorganization. Representative topography AFM images of these dextran patterns, imaged in air and PBS, together with the line scan in the 2D images (white lines) are presented in Figure 7. In air, a uniform, smooth dextran layer is observed. The interface with the unexposed region is sharp with a height difference of 2.5 ± 0.6 nm. In PBS, the dextranized regions swell smoothly in a similar fashion as patterned DEX-PS resulting in a sharp increase in height, up to 2x the dried thickness, at the boundary between patterned DEX-PU with LiCl and OX-PU. The RRMS values in air and PBS for dextranized and unmodified regions are given in Figure 5. In agreement with topography images (Figure 7), the dextranized regions in air are the smoothest with an RRMS of 1 ± 0.3 nm. This roughness is very low in comparison to the other dextranized regions either on PU without LiCl or PS: 2.6 ± 1.2 nm and 2.2 ± 1.4 nm for homogeneous and patterned DEX-PU, respectively and 1.4 ± 0.8 nm and 2.1 ± 0.6 nm for homogeneous and patterned DEX-PS. On the unmodified regions, the RRMS in air is 0.5 ± 0.3 nm, similar to homogeneous OX-PU (0.8 ± 0.4 nm) and patterned DEX-PU without LiCl (0.5 ± 0.2 nm). In PBS, even though swelling is observed for patterned DEX-PU with LiCl, as noted in Figure 7, the RRMS remains unaltered. The RRMS in air and in PBS are also similar in the unmodified region with values of 0.4 ± 0.3 nm in PBS and 0.5 ± 0.3 nm in air, indicating that the unmodified regions contained fewer residual dextran molecules. These results not only verify hydrogen bonding hypothesis of DEX on PU but also suggest a novel approach to obtain smooth and uniform films that swell uniformly in PBS. This novel approach is applicable to other photoactive molecules as well as polymeric surfaces that have a tendency to form hydrogen bonds in the presence of nucleophilic groups.
Figure 7.
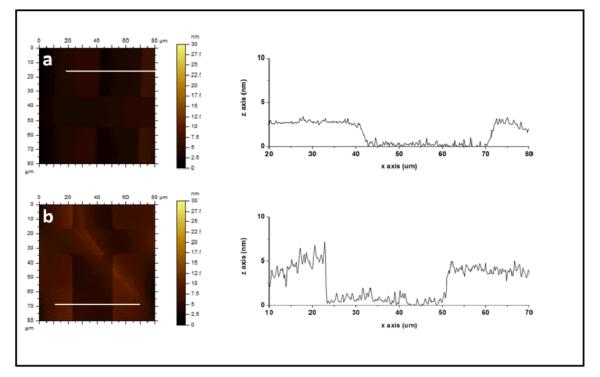
Topographic AFM images of photopatterned dextran (LiCl, 1 wt%) on PU in (a) air and (b) PBS buffer, along with their corresponding line scans. In PBS, surfaces are smoother overall with thick dextranized regions (DEX-PU) that swell by up to 2x its dried thickness from 2.3 ± 0.6 nm to 4.3 ± 1.2 nm. The PU regions (OX-PU) remain unaltered in PBS.
Previous swelling studies of dextran films have been reported3,24 using spectroscopic ellipsometry and reflection interference contrast microscopy. From these studies, the swelling of dextran was observed to depend on the coupling chemistry, molecular weight, measuring technique and solvent. For instance, Elender et al24. observed that dextran (500kDa) films swell up to a factor of 100 in water relative to the dried film thickness, 8 Å, as measured by reflection interference contrast microscopy and ellipsometry, whereas Piehler et al3. observed that dextran swelling in PBS increased by a factor of 7.5 (10kDa) and 5.8 (300kDa) relative to the dried film thickness, 2.5 and 6.2 nm, respectively as measured by spectroscopic ellipsometry. In our studies the dextran (100kDa) layer are observed to swell up to a factor of 3 in PBS relative to the dried film thickness, 5-7nm. The reduction in swelling compared to prior studies is attributed to the different coupling chemistry and higher attachment of dextran to the surface. In our studies the dextran layer is directly attached to the surface via a single molecule (p-azidobenzoic acid) whereas larger silane molecules are employed in the other studies 3,24. In addition, our dextran film is attached to the surface with a maximum of 13 azide groups per chain, which may further limit the final extent of swelling in PBS.
Adsorption of human albumin (HA) on dextran patterned polyurethane (PU) and polystyrene (PS) substrates
To ensure exact HA adsorption conditions, in situ comparison of antifouling properties of dextran (DEX-PU and DEX-PS) relative to the unmodified regions (OX-PU and OX-PS) is carried out on patterned samples. Figure 8 shows representative fluorescence images of dextran patterned on OX-PU and OX-PS, prior to HA adsorption (Figure 8a) and after 1h HA adsorption (Figure 8b, c) at 37 °C. Prior to HA adsorption, fluorescence (brighter regions) appears within the dextranized regions, DEX-PU and DEX-PS (i.e. square patterns) due to the aromatic group, i.e. AZBC, employed to photochemically activate dextran. However, after HA protein adsorption, fluorescence appears on the unmodified regions, OX-PU and OX-PS (i.e., in the edges between square patterns) due to adsorption of the fluorescently labeled HA. These results confirm the suppression of HA adsorption onto dextranized regions, which is limited to the unmodified polymer.
Figure 8.

Fluorescence microscopy images of a photo-patterned dextranized surface on polyurethane (PU): (a) before and (b) and (c) after adsorption of human albumin (HA) in PBS for 1h and on PS film: (d) before and (e) and (f) after adsorption of human albumin (HA) in PBS for 1h. Prior to adsorption of HA, dextranized regions (squares) show fluorescence (white regions) due to the aryl group (AZBC) used to photoactivate the dextran. Upon HA adsorption, bright fluorescence appears on the non-dextranized regions, due to localized adsorption of fluorescently labeled HA on PU and PS regions. The pattern dimensions are 90×90 μm2 separated by 35 μm. The image magnifications are 20x for (a), (b) and (e) and 40x for (c), (d) and (f).
Because the unmodified regions are plasma treated and oxidized surfaces tend to reorganize and/or decay with time28, the exact nature of the surface at the time the HA adsorption measurements are taken (2-3 days after preparation) is not clear. To confirm the extent of oxidation of PU and PS substrates, blank samples of OX-PU and OX-PS are prepared and their contact angles measured prior to HA adsorption. The contact angles for OX-PU and OX-PS after 3 days increase to 45° ± 10 from < 10° after oxidation treatment, indicating that some oxygen functionalities are still present and only partially reorganization/decay occurs at the surface. Regardless of the extent of reorganization/decay, work by Browne et al.36 has shown that human albumin adsorb onto PS via dispersion interactions and also but at less extent onto oxidized PS surfaces by polar interactions. Their findings further support our conclusion that photochemically attached dextranized surfaces present in this work successfully suppress human albumin adsorption relative to the dextran-free surfaces.
Conclusions
Photochemistry is a versatile method to graft dextran onto polymeric surfaces. Photochemically active dextran containing azide groups every 13, 93, 247 and 946 hydroxyls are prepared and characterized. The dextran with an azide group every 247 hydroxyls is photoimmobilized onto plasma oxidized PU and PS (OX-PU and OX-PS, respectively). Dextranized surfaces show features that become smooth when immersed in PBS buffer. Furthermore, dextran is photopatterned on OX-PU and OX-PS. Patterned dextran on the OX-PU substrate shows irregularly shaped dextran features that swell and turn into smooth, uniform features in PBS, The interface with the unmodified region is gradual. In contrast, patterned dextran on OX-PS is smoother and displays a distinct step height at the interface. By adding PBS buffer, the dextranized regions on OX-PS are observed to swell by 3x its dry thickness from 1.3 nm to 3.8 nm. These morphological differences are attributed to hydrogen bond formation at the surface between dextran and OX-PU. To verify this hypothesis, LiCl is added to dextran to disrupt hydrogen bonds. The result is smooth patterned dextran on OX-PU that swells by 2x from 2.3 nm to 4.3 nm relative to the dried film. This novel approach greatly improves upon the original method because smoother and more uniform films are produced that swell evenly on OX-PU. Additionally, photoimmobilized dextran on PU and PS are shown to disrupt nonspecific protein adsorption, specifically human albumin (HA). In patterned dextranized surfaces, HA adsorption is only observed on unmodified regions. Being able to control protein placement provides an interesting platform for patterning cells on biomaterials surfaces, many of which are polymers.
ACKNOWLEDGMENTS
This research was supported by the NIH under Ruth L. Kirschstein National Research Service Award 2T32HL007954 from NIH-NHLBI (MCCF) and R01 HL060230 (MCCF, SY, DME and RJC) RJC thanks NSF/DMR (DMR09-07493) and NSF/NBIC (DMR05-32802) for facilities.
References
- (1).Hanson SR. In: Handbook of biomaterial properties. Black J, Hastings G, editors. Chapman & Hall London: 1998. pp. 545–555. [Google Scholar]
- (2).Reitsma S, Slaaf DW, Vink H, van Zandvoort M, Egbrink M. Pflugers Archiv-European Journal of Physiology. 2007;454:345–359. doi: 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Piehler J, Brecht A, Hehl K, Gauglitz G. Colloid Surf. B-Biointerfaces. 1999;13:325–336. [Google Scholar]
- (4).Ombelli M, Composto RJ, Meng QC, Eckmann DM. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2005;826:198–205. doi: 10.1016/j.jchromb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- (5).Dubois J, Gaudreault C, Vermette P. Colloid Surf. B-Biointerfaces. 2009;71:293–299. doi: 10.1016/j.colsurfb.2009.03.002. [DOI] [PubMed] [Google Scholar]
- (6).Bhat VT, James NR, Jayakrishnan A. Polym. Int. 2008;57:124–132. [Google Scholar]
- (7).Bosker WTE, Patzsch K, Stuart MAC, Norde W. Soft Matter. 2007;3:754–762. doi: 10.1039/b618259c. [DOI] [PubMed] [Google Scholar]
- (8).Martwiset S, Koh AE, Chen W. Langmuir. 2006;22:8192–8196. doi: 10.1021/la061064b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Frazier RA, Matthijs G, Davies MC, Roberts CJ, Schacht E, Tendler SJB. Biomaterials. 2000;21:957–966. doi: 10.1016/s0142-9612(99)00270-7. [DOI] [PubMed] [Google Scholar]
- (10).Massia SP, Stark J, Letbetter DS. Biomaterials. 2000;21:2253–2261. doi: 10.1016/s0142-9612(00)00151-4. [DOI] [PubMed] [Google Scholar]
- (11).Perrino C, Lee S, Choi SW, Maruyama A, Spencer ND. Langmuir. 2008;24:8850–8856. doi: 10.1021/la800947z. [DOI] [PubMed] [Google Scholar]
- (12).Barie N, Sigrist H, Rapp M. Analusis. 1999;27:622–629. [Google Scholar]
- (13).Sigrist H, Collioud A, Clemence JF, Gao H, Luginbuhl R, Sanger M, Sundarababu G. Optical Engineering. 1995;34:2339–2348. [Google Scholar]
- (14).Brunner J. Annual Review of Biochemistry. 1993;62:483–514. doi: 10.1146/annurev.bi.62.070193.002411. [DOI] [PubMed] [Google Scholar]
- (15).Chen GP, Ito Y, Imanishi Y, Magnani A, Lamponi S, Barbucci R. Bioconjugate Chem. 1997;8:730–734. doi: 10.1021/bc9700493. [DOI] [PubMed] [Google Scholar]
- (16).Pasqui D, Rossi A, Barbucci R, Lamponi S, Gerli R, Weber E. Lymphology. 2005;38:50–65. [PubMed] [Google Scholar]
- (17).Zhu A, Zhang M, Shen R. Journal of Biomaterials Science-Polymer Edition. 2003;14:411–421. doi: 10.1163/156856203766652039. [DOI] [PubMed] [Google Scholar]
- (18).Zhu A, Zhang M, Wu J, Shen J. Biomaterials. 2002;23:4657–4665. doi: 10.1016/s0142-9612(02)00215-6. [DOI] [PubMed] [Google Scholar]
- (19).Osterberg E, Bergstrom K, Holmberg K, Schuman TP, Riggs JA, Burns NL, Vanalstine JM, Harris JM. J. Biomed. Mater. Res. 1995;29:741–747. doi: 10.1002/jbm.820290610. [DOI] [PubMed] [Google Scholar]
- (20).Osterberg E, Bergstrom K, Holmberg K, Riggs JA, Vanalstine JM, Schuman TP, Burns NL, Harris JM. Colloids and Surfaces a-Physicochemical and Engineering Aspects. 1993;77:159–169. [Google Scholar]
- (21).Hanson SR, Harker LA. In: Biomaterials Science: An Introduction to Materials in Medicine. Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, editors. Academic Press, Inc.; San Diego, CA: 1996. [Google Scholar]
- (22).Didisheim P, Watson JT. In: Biomaterials Science: An Introduction to Materials in Medicine. Ratner B, Hoffman B, Schoen F, Lemons J, editors. Academic Press Inc.; San Diego, CA: 2004. [Google Scholar]
- (23).Scriven EFV, Turnbull K. Chemical Reviews. 1988;88:297–368. [Google Scholar]
- (24).Elender G, Kuhner M, Sackmann E. Biosens. Bioelectron. 1996;11:565–577. doi: 10.1016/0956-5663(96)83292-1. [DOI] [PubMed] [Google Scholar]
- (25).Ricci D, Braga P. Atomic Force Microscopy. Vol. 242. Humana Press; Totowa, NJ: 2004. pp. 25–37. [Google Scholar]
- (26).Neises B, Steglich W. Angewandte Chemie-International Edition in English. 1978;17:522–524. [Google Scholar]
- (27).Zhang D, Dougal SM, Yeganeh MS. Langmuir. 2000;16:4528–4532. [Google Scholar]
- (28).Dupont-Gillain CC, Adriaensen Y, Derclaye S, Rouxhet PG. Langmuir. 2000;16:8194–8200. [Google Scholar]
- (29).Dupont-Gillain CC, Rouxhet PG. Langmuir. 2001;17:7261–7266. [Google Scholar]
- (30).Tasker S, Matthijs G, Davies MC, Roberts CJ, Schacht EH, Tendler SJB. Langmuir. 1996;12:6436–6442. [Google Scholar]
- (31).Ombelli M, Eckmann DM, Composto RJ. In: Polymer/Metal Interfaces and Defect Mediated Phenomena in Ordered Polymers. E. M, G.G. M, editors. Vol. 734. Boston: 2002. pp. 205–210. [Google Scholar]
- (32).Miksa D, Irish ER, Chen D, Composto RJ, Eckmann DM. Biomacromolecules. 2006;7:557–564. doi: 10.1021/bm050601o. [DOI] [PubMed] [Google Scholar]
- (33).Senti FR, Hellman NN, Ludwig NH, Babcock GE, Tobin R, Glass CA, Lamberts BL. Journal of Polymer Science. 1955;17:527–546. [Google Scholar]
- (34).Sanchis MR, Calvo O, Fenollar O, Garcia D, Balart R. Journal of Applied Polymer Science. 2007;105:1077–1085. [Google Scholar]
- (35).Skaja A, Fernando D, Croll S. Jct Research. 2006;3:41–51. [Google Scholar]
- (36).Browne MM, Lubarsky GV, Davidson MR, Bradley RH. Surface Science. 2004;553:155–167. [Google Scholar]