

Abstract
The aged canine is a higher animal model that naturally accumulates β-amyloid (Aβ) and shows age-related cognitive decline. However, profiles of Aβ accumulation in different species (40 vs. 42), its assembly states, and Aβ precursor protein (APP) processing as a function of age remain unexplored. In this study, we show that Aβ increases progressively with age as detected in extracellular plaques and biochemically extractable Aβ40 and Aβ42 species. Soluble oligomeric forms of the peptide, with specific increases in an Aβ oligomer migrating at 56kDa, also increase with age. Changes in APP processing could potentially explain why Aβ accumulates, and we show age-related shifts towards decreased total APP protein and non-amyloidogenic (α-secretase) processing coupled with increased amyloidogenic (β-secretase) cleavage of APP. Importantly, we describe Aβ pathology in the cingulate and temporal cortex and provide a description of oligomeric Aβ across the canine lifespan. Our findings are in line with observations in the human brain, suggesting that canines are a valuable higher animal model for the study of Aβ pathogenesis.
Keywords: beta amyloid, canine, dog, oligomer, abeta star, 56 kda, cingulate, temporal, secretase, app, ide, nep, ctf, adam
1. Introduction
The gradual accumulation of beta-amyloid (Aβ) appears to trigger a pathological cascade of molecular and cellular alterations that produce the phenotype of Alzheimer disease (AD), often referred to as the amyloid cascade hypothesis (Hardy and Selkoe, 2002). We have studied aging beagles as a natural model of Aβ accumulation, as canines and humans share a 100% homology of the Aβ protein (Johnstone et al., 1991; Cotman and Head, 2008) and the canine APP sequence shares a 98% homology with that of the human (http://www.ensembl.org/Canis_familiaris/). Our focus has been on age and region-dependent increases in Aβ that closely correlate with cognitive decline in several domains of learning and memory (Cummings et al., 1996a; Head et al., 1998; Head et al., 2000). In particular, the prefrontal, parietal, entorhinal, and occipital regions have been important areas for establishing spatial and temporal Aβ deposition patterns (Head et al., 1998; Head et al., 2000). To date, analysis of canine Aβ has primarily relied on immunohistochemical study of extracellular plaque development. In contrast, very little is known about the temporal accumulation of soluble and insoluble Aβ40 and Aβ42 species, the accumulation of oligomeric Aβ, or the production and clearance enzymes involved in the amyloid precursor processing (APP) pathway. Elucidating these profiles across the canine’s lifespan provides essential information for intervention studies targetting Aβ and cognitive decline, which in turn will be significant for translating research using the canine model to humans.
Of particular interest are soluble Aβ oligomers, which have emerged as important players in AD pathogenesis for their toxicity and impact on cognition. Aβ oligomers may block long-term potentiation and are more toxic to cells than insoluble fibrils. Thus, a potential critical role of oligomeric Aβ species in neurotoxicity and cognitive decline has been identified in humans and transgenic AD mice (Selkoe, 2008). However, the properties and temporal profiles of oligomeric Aβ assembly states have not been established in the canine, and it will be important to characterize these protein states in this natural model of Aβ pathogenesis.
The sequential cleavage of Aβ via the APP processing pathway has been well characterized in humans and several AD models (Thinakaran and Koo, 2008). Amyloidogenic APP processing occurs via beta secretase (βSEC) cleavage at an extracellular site and subsequent cleavage by the gamma secretase complex in the APP transmembrane region releases Aβ isoforms of 38 to 43 amino acids in length. Once formed, Aβ monomers may assemble into larger protein structures such as Aβ oligomers and fibrils, and are susceptible to enzymatic clearance primarily by neprilysin (NEP) and insulin degrading enzyme (IDE). Interestingly, the majority (~90%) of APP processing undergoes non-amyloidogenic processing via alpha secretase (αSEC) cleavage within the Aβ domain. Thus, the constitutive pathway suggests that a change in the balance between amyloidogenic and non-amyloidogenic APP processing has the potential to dramatically impact Aβ generation and accumulation. It is possible that an age-related shift in the normal equilibrium of APP processing pathways is responsible for Aβ accumulation in the canine brain.
In this study, we characterized the temporal accumulation of Aβ as soluble and insoluble Aβ40 and Aβ42 species as well as oligomeric assembly states, and assessed how APP processing and Aβ degradation capacity changes across the lifespan. We present findings in established regions of interest, namely the prefrontal, parietal, entorhinal, and occipital cortices, and provide additional analyses of Aβ in the cingulate and temporal cortex of beagles. We also investigate age-related changes in enzymes responsible for Aβ production and clearance in canines, and determine whether the production of non-amyloidogenic fragments at younger ages shifts to favor amyloidogenic processing and production of Aβ in later years. Our findings provide essential data about the molecular Aβ cascade in a natural canine model of Aβ pathogenesis, which will guide future intervention studies in canines, and serve as a valuable reference for understanding the role of Aβ in the human brain.
2. Materials and Methods
2.1 Animals
Frozen and free-floating brain sections were selected from archived samples, accumulated from studies conducted in beagles from the Lovelace Respiratory Research Institute (Albuquerque, NM) for a total of five dogs for the young group (mean=4.12 +/− 0.19 SEM yrs) and six for the old group (mean=13.95 +/− 0.30 SEM yrs). An additional set of archived parietal cortex tissue samples were used for measures of BACE1 protein and included five dogs for the young group (mean=1.26 +/− 0.26 SEM yrs) and five for the old group (mean=12.10 +/− 0.42 SEM yrs), None of the animals had participated in any other study (i.e. drug or inhalation study common for the institute), although the group of six old dogs (mean=13.95 +/− 0.30 SEM yrs) served as the control arm of another study intervention using dietary antioxidants and behavioral enrichment. Besides routine behavioral testing in these aged animals, all dogs were maintained in the same environment, on the same diet, and in the same facility. Selected brain regions include the prefrontal, parietal, cingulate, entorhinal, temporal, and occipital cortices. The 34 dogs used to study Aβ assembly states ranged in age from 1 to 16 years old (including young, middle aged, and old), and frozen sections from the lateral temporal cortex at the level of the hippocampus were available for all animals for this study. The parietal cortex at the level of the hippocampus of 12 animals was available for the size exclusion chromatography, specifically four animals in each of the following groups: young (mean=2.62 +/− 0.61 SEM yrs), middle age (mean=6.98 +/− 0.49 SEM yrs), and old (mean=14.39 +/− 0.58 yrs).
2.2 Aβ immunohistochemistry and Aβ load quantification
These protocols have been previously published (Head et al., 2008). Briefly, tissue from the left hemisphere was sectioned at 40μm by Neuroscience Associates (Knoxville, TN). We selected free floating sections containing the prefrontal, parietal, cingulate, entorhinal, and occipital cortices. After pretreatment with 90% formic acid, Aβ plaques were detected with anti-Aβ1-17 (mouse monoclonal 6E10 antibody, 1:5000, Signet Labs. Inc., Dedham, MA), followed by anti-mouse secondary antibody, detection with an ABC peroxidase kit, and visualization with a DAB substrate kit (Vector Labs., Burlingame, CA). Control experiments where primary or secondary antibody was omitted resulted in negative staining. The procedure for quantifying Aβ loads has been reported previously (Head et al., 2000). Briefly, ten images were captured using a 20X objective in each brain region and the area occupied by Aβ quantified using gray scale thresholding (NIH Image), to obtain “Aβ loads”. Results were confirmed with an additional set of sections at least 200μm away.
2.3 Aβ sandwich ELISA
To detect differences in the young and old prefrontal, parietal, temporal, and occipital cortices, we used previously published methods (Head et al., 2008). Briefly, frozen cortical samples were sequentially extracted in radioimmunoprecipitation assay (RIPA) buffer (pH=8, 50mM Tris-HCl, 150mM NaCl, 0.5% deoxycholate, 0.1% SDS, 1% Triton X-100, protease inhibitor cocktail (MP Biomedicals, Costa Mesa, CA)) to obtain a soluble RIPA fraction and the resulting RIPA pellets were resuspended in a 70% formic acid buffer (FA) to measure insoluble Aβ. In a larger study of 34 brains that included a broader range of ages, a three-step extraction procedure was conducted to optimize our detection of soluble proteins, due to the dynamic nature of soluble Aβ and its ability to aggregate. Pulverized tissue was sequentially extracted starting with a detergent-free PBS buffer (pH 7.4, protease inhibitor cocktail, Amresco, OH), then an SDS extraction buffer (2% SDS, protease inhibitor cocktail, Amresco, OH), and finally in 70% FA buffer. All FA samples were neutralized in neutralization buffer and all samples run in triplicate on ELISA plates coated with a monoclonal anti-Aβ1-16 antibody (20.1, kindly provided by Dr. William Van Nostrand, Stony Brook University, Stony Brook, NY) and detection was by monoclonal HRP-conjugated antibodies anti-Aβx-40 (MM32-13.1.1) or anti-Aβx-42 (MM40-21.3.1) (both antibodies kindly provided by Dr. Christopher Eckman, Mayo Clinic Jacksonville, Jacksonville, CA) (Kukar et al., 2005; McGowan et al., 2005). For standards, dilutions of Aβ1-40 and Aβ1-42 peptides (Bachem California, Inc., Torrance, CA) were used after a pretreatment with HFIP to prevent fibril formation. The inclusion of a series of controls to test the absorbance of buffers, samples, and antibodies yielded negative results.
2.4 Size fractionation columns and Aβ ELISA
Of the 12 animals with parietal cortex available, pulverized tissue was homogenized at 1mL buffer/200mg pulverized tissue weight in PBS (pH 7.4, protease inhibitors 1mM PMSF, 20μM leupeptin, 10uM Pepstatin-A). Following centrifugation at 40,000g for 1 hour, the PBS soluble fractions were recovered and pooled for the four dogs in each of the following age groups: young, middle age, and old. Pooled samples were loaded onto a G-75 column and run in PBS + 2mg/mL BSA and 400 μl fractions collected over approximately 30 timepoints. Larger, heavier proteins were eluted first and most likely contained aggregated or oligomeric Aβ. Smaller proteins were eluted in subsequent fractions and most likely contained the pool of monomeric Aβ. The experiment was replicated with fresh samples and eluted fractions were tested for concentration of Aβ by a sandwich ELISA. For these studies, the capture antibody was the same anti-Aβ1-16 used previously, and the detection antibody was HRP-conjugated 4G8 against Aβ17-24 (Covance, CA), with the OD at 450nm recording the total Aβ concentration in each eluent. Using a control experiment in which 4G8 served as both the capture and detection antibody, we optimized this Aβ ELISA as a single-site sandwich assay better suited to detect oligomeric protein forms in initial column eluents and monomeric proteins in later column fractions. Thus, earlier column fractions contained larger Aβ aggregates and produced a signal because the Aβ17-24 site was available at least twice, from some Aβ peptides during the initial capture with 4G8 and from adjacent Aβ peptides later at the HRP-4G8 incubation step for detection. In contrast, later eluted fractions produced no detectable signal because all Aβ17-24 sites were occupied on the Aβ monomers during the initial capture with 4G8 and no additional sites were available at the HRP-4G8 incubation step for detection.
2.5 Western blotting
For the oligomer studies, pulverized tissue samples were extracted in PBS buffer (powder packet from Sigma-Aldrich, St. Louis, MO, pH 7.4, 0.2% NaN3 with Complete Mini protease inhibitor (Roche Diagnostics, Indianapolis, IN)). For protein analyses on APP processing proteins, frozen tissue was homogenized in either SDS extraction buffer (pH=6.8, 100 mM Tris, 1% SDS, protease inhibitor cocktail kit (MP Biomedicals, Costa Mesa, CA)) at 1 mL buffer/100mg wet weight tissue or in RIPA buffer (pH=8, 50mM Tris-HCl, 150mM NaCl, 0.5% deoxycholate, 0.1% SDS, 1% Triton X-100, protease inhibitor cocktail (MP Biomedicals, Costa Mesa, CA)) at 1 mL buffer/150mg pulverized tissue weight. Samples were centrifuged at 100,000g for 1 hour at 4C, and soluble fractions recovered with all samples brought to an equal protein concentration by BCA (Pierce Biotechnology Inc., Rockford, IL). Samples were prepared for the gel by the addition of loading buffer (2.5mM Tris pH 6.8, 2% SDS, 0.007% bromophenol blue, 4% beta-mercaptoethanol, 10% glycerol) and boiled at 100C for 5 minutes. Equal protein amounts were loaded together on 4-20% Tris-HCl Criterion gels (BioRad Laboratories, Hercules, CA) and transferred to sequi-blot PVDF membranes (Biorad Laboratories, Hercules, CA) for all proteins, with the exception of nitrocellulose membranes used for BACE1 (Biorad Laboratories, Hercules, CA). For the oligomer studies, incubations were in Tris Buffered Saline with 0.01% Tween-20 (TTBS), specifically using 3% BSA/TTBS for all blocking and antibody incubations with β-actin (mouse, 1:5000, Abcam Inc., Cambridge, MA) and A11 antibody (rabbit, 1:1000, Chemicon, Temecula, CA) (Kayed et al., 2003). For other protein analyses on APP processing, incubations were in 5% milk/0.1%Tween20-TBS for β-actin (rabbit, 1:5000, Abcam Inc., Cambridge, MA), GAPDH antibody (rabbit 1:3000, Abcam Inc., Cambridge, MA), 6E10 antibody against anti-Aβ1-16 to detect full-length APP (mouse, 1:500, Signet Laboratories, Dedham, MA), ADAM10 antibody (rabbit, 1:500, Chemicon, Temecula, CA), BACE1 antibody (mouse, 1:4000, MAB931, R&D Systems, Minneapolis, MN), CT20 antibody for CTFs (rabbit, 1:2000, raised against the C-terminal 20 amino acids of APP). Secondary antibodies were HRP-conjugated IgG anti-mouse (goat anti-mouse, 1:5000, BioRad Laboratories, Hercules, CA) or anti-rabbit (goat anti-rabbit, 1:10,000 from BioRad Laboratories, Hercules, CA or Rockland Immunochemicals, Gilbertsville, PA) as needed. Supersignal Chemiluminscent Substrate (Pierce Biotechnology Inc., Rockford, IL) was used to visualize HRP activity on Hyperfilm ECL (Amersham Bioscience, Piscataway, NJ). The omission of primary antibody resulted in negative staining. For densitometry of BACE1, a subset of samples was pooled and each membrane included 4 lanes with increasing protein concentrations (2.5-20 μg or 10-60 μg) to ensure that optical densities were within the linear range of detection. Immunoblots were quantified using NIH Image J software, with optical density (OD) measures adjusted for individual β-actin OD levels.
2.6 Secretase activity assays
These protocols have been previously published (Nistor et al., 2007). Briefly, frozen tissue was prepared according to protocols provided by the commercial supplier of the Secretase Activity Kits (R&D Systems, Minneapolis, MN). For αSEC we used 125 μg total protein (2.5μg/μl in 50μl total per well, OD read at 2 hours) and for βSEC we used 7.5μg total protein (0.15μg/μl in 50μl total per well, OD read at 30 min.). All samples were run in triplicate and the assays were replicated to confirm results.
2.7 Statistical analyses
All statistical analyses were performed using SPSS for Windows and graphs were produced using SPSS and Sigma Plot. Normality and homogeneity of variance were assessed by the Shapiro-Wilk statistic and the Levene’s statistic, respectively. The student’s t-test for two independent samples was used to quantify differences between groups. In cases where normality and variance assumptions were violated and not rectifiable by log transformation of the data, group differences were assessed using nonparametric Mann-Whitney U tests. Correlations were assessed using the Pearson coefficient, Partial correlation with age as a covariate, or nonparametric statistics as needed.
3. Results
3.1 Changes in Aβ with age
For initial analyses dogs were divided into young (mean=4.12 +/− 0.19 SEM yrs) and old (mean=13.95 +/− 0.30 SEM yrs) groups, and Aβ extracellular plaques were quantified as a measure of Aβ load in the prefrontal, parietal, cingulate, entorhinal, and occipital cortices. Representative sections of Aβ immunostaining from the old and young beagles (Figure 1A) demonstrate overall increases in Aβ plaques across brain regions and cortical layers with age. Aβ plaque load was significantly increased across all cortical layers (Figure 1B), specifically in the prefrontal (p=0.006), parietal (p=0.006), cingulate (p=0.006), entorhinal (p=0.028), and occipital cortices (p=0.045).
Figure 1. Aβ load.
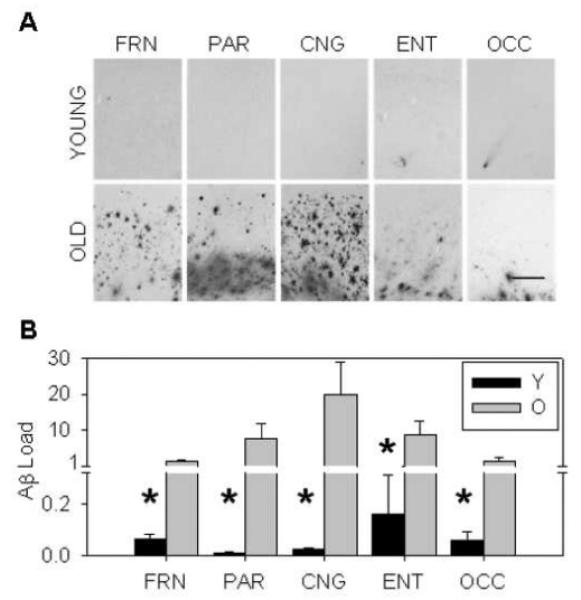
(A and B) Old dogs exhibit an increased Aβ burden throughout multiple brain regions and across cortical layers compared to young. (FRN=prefrontal, PAR=parietal, CNG=cingulate, ENT=entorhinal, OCC=occipital, Y=young, O=old, scale bar=500μm, bars in B=group mean+/−SEM, *=p<0.05)
To detect changes in Aβ species, we measured soluble and insoluble levels of Aβ40 and Aβ42 in the young and old dogs in the prefrontal, parietal, temporal, and occipital cortices. Soluble levels were obtained using RIPA buffer and insoluble Aβ levels were from the FA buffer fraction. There were general increases in Aβ with age across most brain regions and tissue fractions (Figure 2A-D). The frontal cortex showed the most consistent increases with age, having significantly more soluble Aβ40 (p=0.011), soluble Aβ42 (p=0.044), insoluble Aβ40 (p=0.006), and insoluble Aβ42 (p=0.006) compared to the young animals. The parietal cortex also showed significant increases in soluble Aβ40 and Aβ42 (p=0.028 and p=0.045, respectively) and insoluble Aβ42 (p=0.006), but no significant changes for insoluble Aβ40 (p>0.1). Aβ levels in the temporal and occipital cortices were very low overall in comparison to other regions. Insoluble Aβ42 levels in the temporal cortex were significantly increased with age (p=0.028), with no changes in soluble levels of Aβ40 or Aβ42, or insoluble Aβ40 (p>0.1 for all) as measured by ELISA. Similarly, occipital levels of insoluble Aβ42 were increased with age (p=0.018), but no changes were found in soluble levels of Aβ40 or Aβ42 (p>0.1 for both). Marginal increases for insoluble Aβ40 were found in the occipital cortex (p=0.068). The results from the ELISA suggest that deposition of Aβ42 occurs prior to Aβ40, and the emergence of Aβ first in the RIPA soluble fractions and then in the FA insoluble pool.
Figure 2. Soluble and insoluble Aβ40 and Aβ42 in young and old canines.
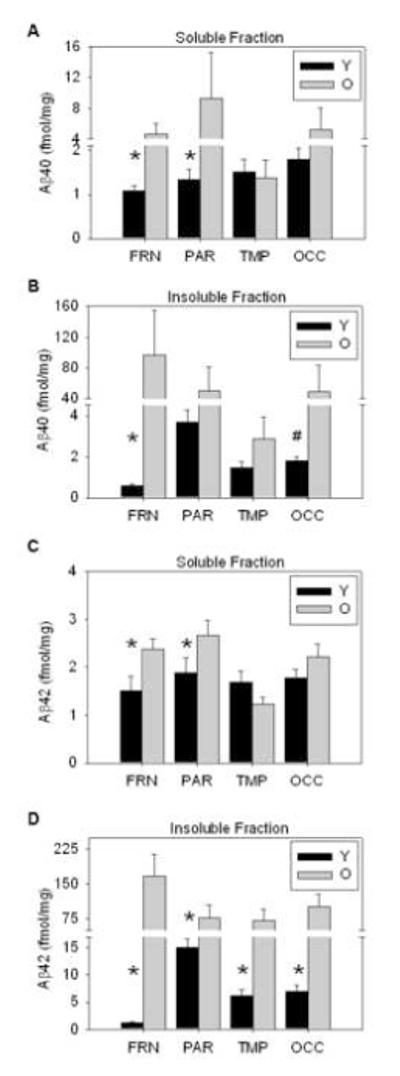
Aβ increases with age across brain regions and tissue fractions, with insoluble Aβ being higher than soluble levels. Specifically, (A) soluble Aβ40 is more abundant in the frontal cortex and significantly increased in both the frontal and parietal cortices of aged dogs. (B) Insoluble Aβ40 significantly increases with age in the frontal cortex, with moderate increases in the occipital area. (C) Soluble Aβ42 is only significantly increased in the frontal and parietal regions, while (D) all regions exhibit age-associated increases in insoluble Aβ42, which is the earliest deposited Aβ species. (FRN=prefrontal, PAR=parietal, TMP=temporal, OCC=occipital, bars=group mean+/−SEM, *=p<0.05, #=p=0.068)
To obtain a more precise measure of changes in Aβ profiles in the temporal cortex, we quantified Aβ in a larger group of canines with ages ranging from 1 to 16 years old. We obtained a chronological profile of soluble and insoluble Aβ accumulation in the lateral temporal cortex by ELISA (Figure 3). Aβ40 and Aβ42 were detected in cellular fractions obtained by serial extraction with PBS (Figure 3A-B), SDS (Figure 3C-D), and FA buffers (Figure 3E-F). There was a significant linear relationship with age in soluble levels of Aβ40 (r=0.469, p=0.007, Figure 3A) and in Aβ42 (r=0.563, p=0.001, Figure 3B) extracted in PBS buffer. In SDS buffer, Aβ40 was associated with increased age (r=0.339, p=0.058, Figure 3C) and Aβ42 was significantly increased with age (r=0.651, p<0.001, Figure 3D). In the insoluble FA fraction, Aβ levels remained stable until 12 years of age, at which point dogs exhibited a spike in Aβ accumulation with a high amount of individual variability with an increase in Aβ40 (r=0.348, p=0.051, Figure 3E) and a significant increase in Aβ42 (r=0.502, p=0.003, Figure 3F).
Figure 3. Soluble and insoluble Aβ40 and Aβ42 in canines across the lifespan.
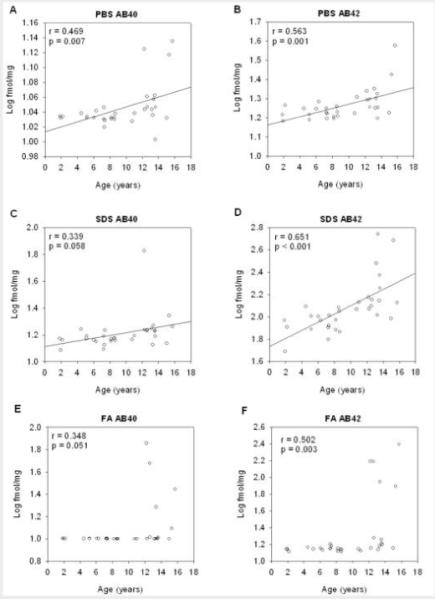
Both Aβ40 and Aβ42 protein significantly increase with age in all cellular fractions obtained by serial extraction with three buffers (A and B) PBS, (C and D) SDS, and (E and F) FA in canine temporal cortex. (fmol/mg=femto moles of Aβ per milligram raw tissue weight, PBS=phosphate buffered saline buffer, SDS=sodium dodecyl sulfate buffer, FA=70% formic acid buffer, open circles=individual data points, solid lines=regression line, r=correlation coefficient, p=correlation p-value)
3.2 Detection and quantification of oligomeric proteins
We next hypothesized that increases in Aβ in the soluble fraction reflected increases in soluble oligomeric proteins, we proceeded to find Aβ-specific oligomers in the canine brain. An initial study determined that oligomeric proteins were most abundant in the soluble PBS cellular fraction as detected by an anti-oligomer antibody A11 (Supplemental Figure 1). A follow-up experiment using immunoprecipitation with anti-Aβ antibodies prior to immunoblotting with A11 selectively revealed Aβ oligomers with a prominent band at 56kDa (Supplemental Figure 2). We also confirmed increases in oligomeric proteins with age using dot blots of the soluble PBS tissue fraction of young and old dogs, and detected significant increases with the anti-oligomer antibodies M204 (p=0.051) and I11 (p=0.004) (Supplemental Figure 3).
We proceeded to look at age-related changes in early Aβ assembly states in the larger cohort of animals. We first analyzed the Aβ oligomer at 56kDa using the PBS fraction of the lateral temporal cortex from 34 dogs ranging in age from 1 to 16 years old. Quantification revealed significant increases with age in the 56kDa Aβ oligomer (r=0.41, p=0.02, Figure 4).along with a large amount of individual variability. In addition, parietal cortex available from a subset of the same animals was grouped into young (mean=2.61 +/− 0.62 SEM yrs), middle age (mean=6.98 +/− 0.49 SEM yrs), and old groups (mean age=14.39 +/− 0.58 SEM yrs). Following size fractionation, aliquots were analyzed for total Aβ by ELISA and showed a differential age profile for Aβ monomers and Aβ aggregates (Figure 5). Young dogs showed high levels of monomeric Aβ and almost no aggregated protein, while middle-aged dogs showed the opposite trend with low levels of monomeric Aβ and increased Aβ aggregates. Old animals exhibited both Aβ pools, with prominent peaks for monomeric and aggregated Aβ assembly states. Thus, we detected Aβ oligomeric proteins in canines and show that they gradually accumulate beginning in middle age and throughout the rest of the lifespan.
Figure 4. Aβ oligomer at 56kDa.
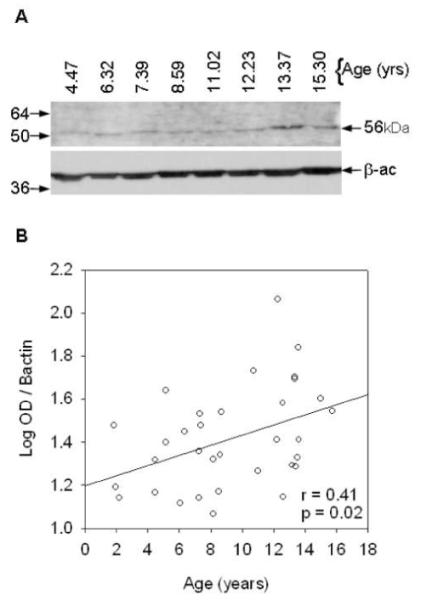
(A) A sample Western blot and (B) quantification of all cases shows the anti-oligomer A11 antibody detects a significant increase across the lifespan in an Aβ oligomer migrating at 56kDa in the PBS soluble fraction of canine temporal cortex. (numbers on left in A=kilodalton marker, OD=optical density, kDa=kilodaltons, solid line=regression line, r=correlation coefficient, p=correlation p-value)
Figure 5. Age-related changes in Aβ assembly states.
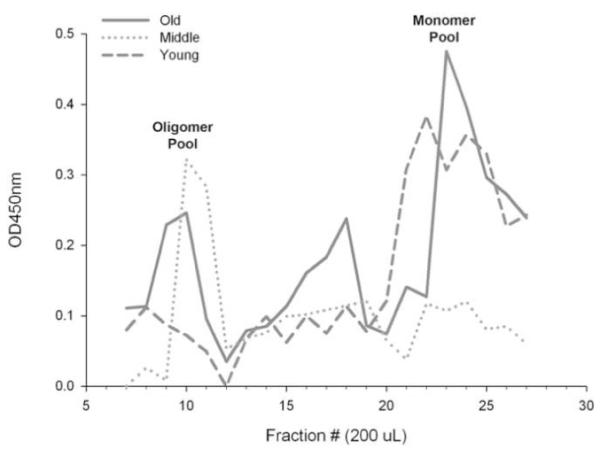
An analysis of Aβ in the PBS fraction of the parietal cortex was performed in a subset of dogs classified into young (mean=2.6 years), middle age (mean=6.98 years), and old (mean=14.41 years) groups. Size exclusion chromatography followed by an Aβ ELISA protocol shows a differential age profile for oligomeric and monomeric Aβ. Young dogs have high levels of monomeric Aβ and almost no oligomers, while middle aged dogs show the opposite trend. Old animals have both monomeric and oligomeric pools of Aβ. (OD=optical density, nm=nanometer, μl=microliter)
3.3 Changes in the APP processing pathway with age
Several mechanisms may underlie the accumulation of Aβ with age, including increased availability of APP, decreased clearance by Aβ degrading enzymes, or altered processing favoring the amyloidogenic pathway. To determine whether age-related increases in Aβ, regardless of its assembly state, are due to changes in the APP processing pathway, we used cortical tissue from young and old canines.
To determine whether increases in APP might account for increases in Aβ, we measured total APP protein levels by Western blot (Figure 6A) in tissue available from the prefrontal, parietal, hippocampal, and occipital cortex of young and old canines. Unexpectedly, total APP was significantly decreased with age in all examined regions. Significant decreases were found in the prefrontal cortex (p=0.008, Figure 6B and F), parietal cortex (p=0.005, Figure 6C and F), hippocampus (p=0.013, Figure 6D and F), and occipital cortex (p=0.0001, Figure 6E and F). These results indicate that that increased availability of APP does not account for increased Aβ accumulation with age.
Figure 6. Total APP protein levels.
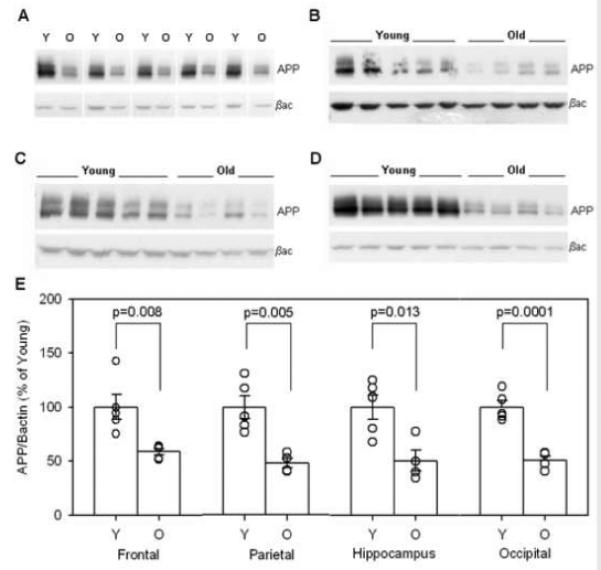
Western blots of total APP protein with beta-actin as a loading control are shown for the (B) frontal cortex, (C) parietal cortex, (D) hippocampus, and (E) occipital cortex. (F) Quantification of total APP levels indicates significant decreases with age in all examined brain regions. (Y=young, O=old, APP=beta-amyloid precursor protein, βac=beta-actin, bars in F=group mean+/−SEM, circles in F=individual data points)
We next investigated whether decreased Aβ clearance may be responsible for increased age-related accumulation of Aβ. We measured protein levels of NEP and IDE by Western blot in tissue available from the parietal cortex of young and old canines (figure not shown). No significant changes were detected with age in NEP (p>0.1) or IDE (p>0.1), suggesting that decreased enzymatic clearance by lower protein levels of NEP or IDE does not account for increased Aβ accumulation with age.
Because Aβ accumulation likely reflects a balance between production and clearance mechanisms, we next asked if differential APP cleavage might explain our previous findings. Using commercially available αSEC and βSEC enzyme activity kits, we found that old animals displayed a significant 20% decrease in αSEC activity (p=0.004, Figure 7A) indicative of decreased non-amyloidogenic APP processing with age. In addition, aged dogs showed a 24% increase in (SEC activity with age (p=0.024, Figure 7B) indicative of increased amyloidogenic APP processing. With changes in APP enzymatic cleavage indicating a significant shift towards amyloidogenic processing in aged dogs, we expected that the APP CTF proteins would show a similar trend (Figure 8). In agreement with increased (SEC activity, aged dogs displayed significantly higher levels of CTF(protein compared to young animals in the parietal cortex (p=0.02, Figure 8A and C) and significantly higher levels of the (-secretase protein BACE1 in the parietal cortex (p=0.05, Figure 10A and B). However, we found no significant changes in CTF(protein (p>0.1, Figure 8A and B) and thus proceeded to look at the protein levels of one of the candidate (SEC enzymes, ADAM10. The antibody detected both the precursor and mature forms of ADAM10 in the parietal cortex of young and old canines (Figure 9A). Quantification of both proteins revealed a significant decrease in the ADAM10 precursor (p=0.045, Figure 9B) with very low levels in aged animals. Likewise, the mature form of the protein was significantly decreased in aged animals compared to young (p=0.0001, Figure 9C). Overall, these findings indicate a shift in APP processing that favors amyloidogenesis with age.
Figure 7. Enzyme activity levels of alpha and beta secretase.
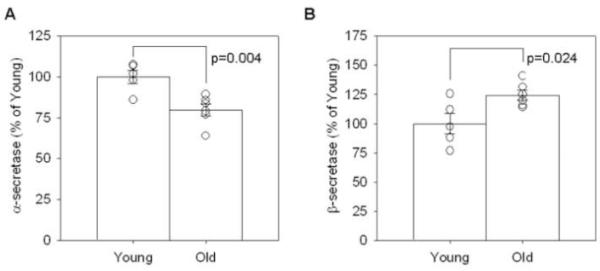
In the parietal cortex of a group of young and old canines, (A) alpha secretase activity decreases significantly with age while (B) beta secretase activity increases significantly with age. (bars=group mean+/−SEM, circles=individual data points)
Figure 8. Protein levels of C-terminal fragments alpha and beta.
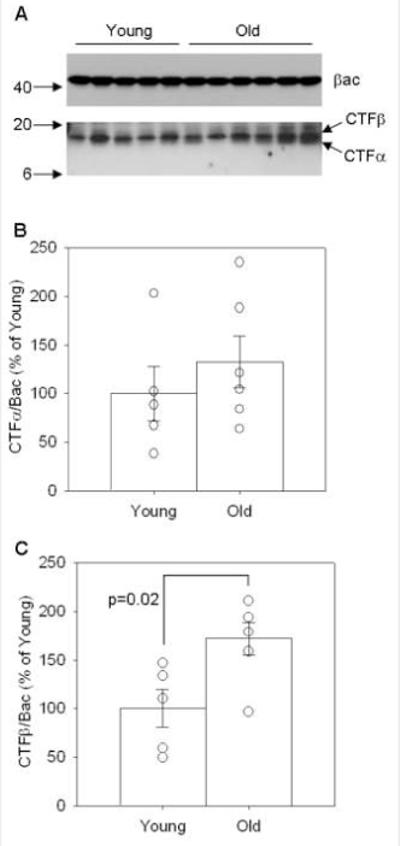
(A) CTF alpha and beta proteins were detected and quantified using beta-actin as a loading control in the parietal cortex of a group of young and old canines. (B) No significant changes were detected in CTFalpha, (C) but there are significant increases with age in CTF beta. (CTFα=carboxyl terminal fragment alpha of APP, CTFβ=carboxyl terminal fragment beta of APP, βac=beta-actin, numbers on left in A=kDa values, bars in B and C=group mean+/−SEM, circles in B and C=individual data points)
Figure 10. Levels of BACE1 protein as a function of age.
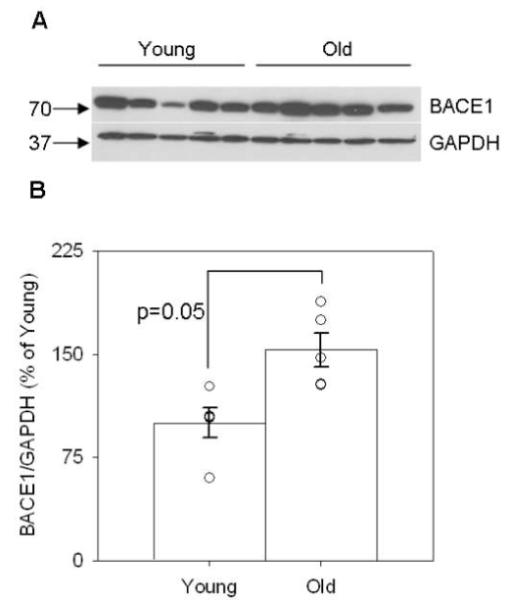
(A) BACE1 protein is detected in the parietal cortex of young and old canines, using GAPDH as a loading control. Quantification of (B) BACE1 protein reveals a significant increase with age. (BACE1=beta-site APP-cleaving enzyme 1, GAPDH=glyceraldehyde-3-phosphate dehydrogenase, numbers on left in A=kDa values, bars in B=group mean+/−SEM, circles in B=individual data points)
Figure 9. Levels of ADAM10 precursor and mature protein.
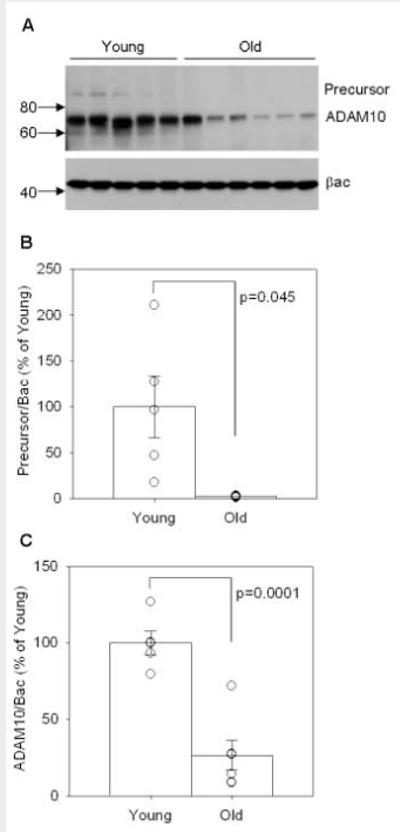
(A) Both the precursor and mature forms of ADAM10 are detected in the parietal cortex of young and old canines, using beta-actin as a loading control. Quantification of (B) ADAM10 precursor protein and (C) mature ADAM10 protein levels reveals a significant decrease in both proteins with age. (ADAM10=a disintegrin and metalloprotease 10, βac=beta-actin, numbers on left in A=kDa values, bars in B and C=group mean+/−SEM, circles in B and C=individual data points)
4. Discussion
Aged canines are valuable as a model of brain aging and dementia because they display progressive cognitive impairment alongside a spontaneous process of Aβ formation and accumulation with age (Cummings et al., 1996a; Head et al., 1998; Head et al., 2000). Here, we demonstrate that Aβ pathogenesis occurs in many of the same brain regions as observed in human AD. Our findings also suggest a common profile between dogs and humans of Aβ accumulation in different species and assembly states over time, with a shared molecular pathway through differential APP processing that favors amyloidogenesis.
4.1 Aβ deposition increases in a spatial and temporal pattern with age
Using both immunohistochemical and biochemical assays, we show that Aβ levels increase with age across several brain regions, including the prefrontal, parietal, cingulate, entorhinal, temporal, and occipital cortices and are consistent with previous reports in beagles (Russell et al., 1996; Head et al., 1998; Head et al., 2000). In the canine cingulate cortex, we show increased extracellular plaque pathology which is consistent with previous findings in dogs of various breeds (Shimada et al., 1991). The temporal cortex is noteworthy in human studies, as memory scores prior to death are related to increased pathology in this region (Fleischman et al., 2005) and the rate of atrophy of mesial temporal lobe structures can differentiate healthy from pathological aging (Stoub et al., 2008). In the canine temporal cortex, Aβ40 and Aβ42 solubilized in PBS and RIPA buffers increased in a linear fashion across the lifespan, while insoluble Aβ (in FA buffer) showed a spike in deposition after age 12 years old, graphically representing a log relationship similar to plaque pathology data previously characterized in other cortical areas of beagles (Head et al., 2000).
Our biochemical analyses revealed selective differences in Aβ40 and Aβ42 species depending on brain region and Aβ pool (soluble, insoluble). Aβ42 in the insoluble pool from the FA fraction was higher than Aβ40, consistent with immunohistochemical studies of plaques in the DS brain (Lemere et al., 1996) and data in canines showing that Aβ42 is deposited before Aβ40 (Cummings et al., 1996b; Tekirian et al., 1996). MCI patients show increased levels of soluble Aβ42 compared to controls (Murphy et al., 2007) and the Aβ42:Aβ40 ratio is selectively increased in some APP mutations that cause early-onset AD (Younkin, 1998). In addition, our data imply that Aβ emerging in the soluble pool (RIPA fraction) serves as an initial source of Aβ accumulation that moves to the insoluble pool (FA fraction) with age. Current evidence suggests that early Aβ assembly states are soluble and intracellular, while larger Aβ aggregates that are insoluble and more resistant to degradation are located extracellularly (Selkoe, 2008). Overall, our ELISA data suggest that Aβ42 deposits are the oldest species, emerging first in the soluble fractions before the insoluble, and are more abundant than Aβ40 in aged canines.
As in the aged human brain, the vast majority of Aβ in the aged canine brain is highly insoluble and consists of greater amounts of Aβ42 than Aβ40 (Roher et al., 1993; Steinerman et al., 2008; Roher et al., 2009). In addition, as occurs in the normal human brain and in AD, the proportion of Aβ42 increases with age in the canine brain, and shows greater heterogeneity with age, possibly linked to one or more aspects of brain dysfunction in a subpopulation of animals similar to that in humans and the development of AD. Thus, the canine brain shows striking similarities to the human brain with respect to Aβ (Murphy and LeVine, 2010).
4.2 Detection of specific Aβ oligomeric proteins in canines
To our knowledge, we provide the first characterization of oligomeric protein profiles across the lifespan in the canine temporal cortex. We showed increased accumulation of oligomers using the dot blot method, which has been successful in previous reports of oligomers in vitro and in human brain tissue (Kayed et al., 2003; Lambert et al., 2007), as well as in the triple-transgenic AD model (Oddo et al., 2006). Most notably, our data revealed a significant increase with age in a higher molecular weight Aβ aggregate migrating at 56kDa. Importantly, similar findings in Tg2576 transgenic AD mice suggest that the oligomer migrating at 56kDa (Aβ*) is a candidate toxic aggregate associated with cognitive deficits, even in the presence of Aβ plaques (Lesne et al., 2006; Lesne et al., 2008). In addition to the accumulation of Aβ* with age, we observed a shift in size of Aβ aggregates. The size exclusion column fractionation data indicate that younger animals produce Aβ monomers, which likely accumulate over time to form a stable population of oligomeric aggregates during middle-age, with both assembly states present in old canines.
The presence of oligomeric Aβ in the temporal cortex of dogs may have an impact on cognition that parallels reports in humans. As previously mentioned, studies indicate the accumulation of Aβ pathology or structural change in the temporal area may be important indicators of cognitive status or predictors for disease progression (Fleischman et al., 2005; Stoub et al., 2008). In structural MRI scans, patients that developed MCI had decreased gray matter in the temporal lobe while still cognitively normal (Smith et al., 2007) and increases in medial temporal atrophy that correlated with cognitive assessments was predictive for conversion from MCI to AD (DeCarli et al., 2007). Our data on toxic oligomeric Aβ species in the temporal cortex identifies this as an important region for consideration in future studies.
4.3 Aβ accumulation depends on amyloidogenic APP processing
In our investigation of possible mechanisms for increased Aβ with age, we evaluated the possibility that Aβ levels were regulated by the availability of the precursor protein APP, Aβ degrading enzymes NEP and IDE, or altered APP processing favoring the amyloidogenic pathway.
Our data indicate that increased levels of APP protein do not underlie increases in Aβ, since levels of APP were dramatically reduced in brain regions known to accumulate Aβ with age. In canines of various breeds and studies with relatively small sample sizes, there are inconsistent reports on APP protein, with some reporting no change (Rofina et al., 2004) while others detect increases with age (Calderon-Garciduenas et al., 2003). Human studies are also few and inconsistent depending on APP protein or expression levels, with one study reporting no age-associated changes in APP protein in control or DS cases (Nistor et al., 2007). As for APP expression, one study finds increases in nondemented individuals and decreases in the vicinity of plaques of AD patients (Barger et al., 2008), while microarray data from our lab reveals decreases with age in nondemented individuals (unpublished findings). However, the present findings in canines focus on total APP protein levels and are derived using multiple brain regions from the same group of young and old beagles. Furthermore, our observed decreases in total APP protein are internally consistent across four brain regions. A recent microarray analysis of the canine cerebral cortex showed a 2.2. fold increase in APP mRNA in aged beagles compared to young animals (Swanson et al., 2009), indicating that a compensatory effect may occur with age. These results suggest that APP protein decreases more likely stem from posttranslational events.
Since the accumulation of Aβ likely reflects a balance of production and clearance mechanisms, we investigated whether age-dependent changes in clearance enzymes could account for Aβ accumulation. We were unable to detect significant changes with age in NEP or IDE protein, thus lower protein levels of these enzymes are unlikely to account for increases in Aβ. Current literature reveals many discrepancies based on age, brain region, and whether mRNA, protein, or activity levels of IDE and NEP are the most critical, although there is evidence suggesting that enzymatic activity and oxidation state may be more important than total protein levels (Wang et al., 2003).
In our investigation of possible mechanisms for increased Aβ with age, we determined that APP processing shifts in favor of amyloidogenesis. We found concomitant decreases in the constitutive non-amyloidogenic pathway and increased APP processing via the amyloidogenic pathway. Our findings revealed decreased αSEC activity alongside lower ADAM10 precursor and mature protein contributing to reduced non-amyloidogenic processing, with increased βSEC activity, increased βACE1 protein, and increased CTFβ protein production reflecting an increase in amyloidogenic processing and Aβ production. Importantly, these findings are consistent with data from humans showing age-related increases in components of the amyloidogenic pathway in normal aging (Fukumoto et al., 2004), DS (Russo et al., 2001; Nistor et al., 2007) and AD brain (Russo et al., 2001; Fukumoto et al., 2002; Tyler et al., 2002; Yang et al.) and with reports of decreased nonamyloidogenic processing in AD (Tyler et al., 2002).
4.4 Multiple effects on neuronal health and function
The concomitant age-related decreases in APP protein availability and lower non-amyloidogenic processing, together with increases in Aβ production and accumulation may have multiple negative effects on cell function and survival in the aging brain. The secreted form of APP produced via non-amyloidogenic processing is important for development, plasticity, and learning and memory systems (Thinakaran and Koo, 2008). APP also appears to be neuroprotective against excitotoxic and ischemic events (Mattson et al., 1993; Smith-Swintosky et al., 1994; Masliah et al., 1997). The aversive properties of Aβ produced via the amyloidogenic pathway are well established, with evidence for the detrimental effects of Aβ plaques (Spires et al., 2005; Kuchibhotla et al., 2008) as well as the negative impact of Aβ oligomers (Selkoe, 2008). The importance of the α-secretase pathway is also highlighted by recent reports of ADAM10 mutations in AD patients, which functionally impair non-amyloidogenic cleavage of APP and promote elevated Aβ levels (Kim et al., 2009). Thus, a reduction in neuroprotective forms of processed APP coupled with increased Aβ may converge and together compromise neuronal function, indicating that both APP and Aβ may be important factors underlying cognitive decline in aged dogs.
Supplementary Material
Supplemental Figure 1. Detection of soluble oligomers by Western blot. The A11 antibody was used to detect oligomers of various sizes in the canine, human, and mouse brain extracts as well as an Aβ peptide control. Three samples of canine temporal cortex were serially extracted in PBS, RIPA and FA. The A11 antibody detects the majority of canine oligomeric bands in the soluble PBS fraction, a few oligomers in the soluble RIPA fraction, and no staining is seen in the FA fraction. For comparison, soluble RIPA protein samples are included from two Down Syndrome patients, a transgenic mouse, and an Aβ peptide aggregate preparation. Notably, a band at 56kDa appears in canine, human, mouse, and Aβ peptide preparations. A band for monomeric Aβ can be seen at 4kDa in the Aβ peptide sample, with multiple bands indicative of aggregated Aβ at higher kDa ranges. (numbers on left=kiloDalton marker, PBS=phosphate buffered saline, RIPA=radioimmunoprecipitation assay buffer, FA=70% formic acid buffer, DS=Down Syndrome RIPA brain extract, Tg=mouse Tg2576 RIPA brain extract, Aβ=aggregated beta-amyloid peptide)
Supplemental Figure 2. Detection of specific Aβ oligomers in the canine brain. (A) A11 detects only the 56kDa band in temporal cortex of young and old canines, specifically in the soluble PBS samples after IP with 3μl of anti-Aβ 6E10 antibody (+), but multiple bands are detected in untreated samples (−). (B) In another experiment, samples were further concentrated by an IP with 5μl of the anti-Aβ antibodies 4G8 or 6E10 (+). A11 staining again reveals the 56kDa band. (numbers on left in A and B=kiloDalton marker, PBS=phosphate buffered saline, IP=immunoprecipitation, Y=young, O=old)
Supplemental Figure 3. Soluble proteins and quantification by dot blot. (A) Representative dot blot of proteins in the soluble PBS fraction of the parietal cortex as detected with the secondary-only antibody, and two anti-oligomer antibodies M204 and I11 (kindly provided by Dr. C. Glabe, UCI). (B) Following subtraction of background reactivity, quantification reveals a large amount of individual variability, with significant increases in soluble oligomers with the M204 and I11 antibodies. (2° only=anti-rabbit secondary, M204=monoclonal rabbit anti-oligomer antibodies, I11=polyclonal rabbit anti-oligomer antibody, bars in B=group mean+/−SEM, circles in B=individual data points)
5. Acknowledgements
The authors would like to thank the following people for technical assistance and advice: Jennifer Thompson for brain oligomer extractions, Anna Pensalfini for dot blots, Paul Adlard and Wayne Poon for the enzyme activity assays and Xiaohong Wang for BACE1 Western blots. Funding was provided by NIH grants NS058383 and AG005119 (to M.P.M.), AG12694 and AG17066 (to C.W.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement: The authors do not have any actual or potential conflicts of interest.
6. References
- Barger SW, DeWall KM, Liu L, Mrak RE, Griffin WS. Relationships between expression of apolipoprotein E and beta-amyloid precursor protein are altered in proximity to Alzheimer beta-amyloid plaques: potential explanations from cell culture studies. J Neuropathol Exp Neurol. 2008;67:773–783. doi: 10.1097/NEN.0b013e318180ec47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon-Garciduenas L, Maronpot RR, Torres-Jardon R, Henriquez-Roldan C, Schoonhoven R, Acuna-Ayala H, Villarreal-Calderon A, Nakamura J, Fernando R, Reed W, Azzarelli B, Swenberg JA. DNA damage in nasal and brain tissues of canines exposed to air pollutants is associated with evidence of chronic brain inflammation and neurodegeneration. Toxicol Pathol. 2003;31:524–538. doi: 10.1080/01926230390226645. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Head E. The canine (dog) model of human aging and disease: dietary, environmental and immunotherapy approaches. J Alzheimers Dis. 2008;15:685–707. doi: 10.3233/jad-2008-15413. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Head E, Afagh AJ, Milgram NW, Cotman CW. Beta-amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiol Learn Mem. 1996a;66:11–23. doi: 10.1006/nlme.1996.0039. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Satou T, Head E, Milgram NW, Cole GM, Savage MJ, Podlisny MB, Selkoe DJ, Siman R, Greenberg BD, Cotman CW. Diffuse plaques contain C-terminal A beta 42 and not A beta 40: evidence from cats and dogs. Neurobiol Aging. 1996b;17:653–659. doi: 10.1016/0197-4580(96)00062-0. [DOI] [PubMed] [Google Scholar]
- DeCarli C, Frisoni GB, Clark CM, Harvey D, Grundman M, Petersen RC, Thal LJ, Jin S, Jack CR, Jr., Scheltens P. Qualitative estimates of medial temporal atrophy as a predictor of progression from mild cognitive impairment to dementia. Arch Neurol. 2007;64:108–115. doi: 10.1001/archneur.64.1.108. [DOI] [PubMed] [Google Scholar]
- Fleischman DA, Wilson RS, Gabrieli JD, Schneider JA, Bienias JL, Bennett DA. Implicit memory and Alzheimer’s disease neuropathology. Brain. 2005;128:2006–2015. doi: 10.1093/brain/awh559. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. Beta-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol. 2004;164:719–725. doi: 10.1016/s0002-9440(10)63159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Head E, Callahan H, Muggenburg BA, Cotman CW, Milgram NW. Visual-discrimination learning ability and beta-amyloid accumulation in the dog. Neurobiol Aging. 1998;19:415–425. doi: 10.1016/s0197-4580(98)00084-0. [DOI] [PubMed] [Google Scholar]
- Head E, McCleary R, Hahn FF, Milgram NW, Cotman CW. Region-specific age at onset of beta-amyloid in dogs. Neurobiol Aging. 2000;21:89–96. doi: 10.1016/s0197-4580(00)00093-2. [DOI] [PubMed] [Google Scholar]
- Head E, Pop V, Vasilevko V, Hill M, Saing T, Sarsoza F, Nistor M, Christie LA, Milton S, Glabe C, Barrett E, Cribbs D. A two-year study with fibrillar beta-amyloid (Abeta) immunization in aged canines: effects on cognitive function and brain Abeta. J Neurosci. 2008;28:3555–3566. doi: 10.1523/JNEUROSCI.0208-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res Mol Brain Res. 1991;10:299–305. doi: 10.1016/0169-328x(91)90088-f. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, Norton D, Tesco G, Elliott K, Wagner SL, Moir RD, Becker KD, Tanzi RE. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate {alpha}-secretase activity. Hum Mol Genet. 2009;18:3987–3996. doi: 10.1093/hmg/ddp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, Sagi SA, Weggen S, Smith TE, Ladd T, Khan MA, Kache R, Beard J, Dodson M, Merit S, Ozols VV, Anastasiadis PZ, Das P, Fauq A, Koo EH, Golde TE. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid beta-peptides and APOE in Down Syndrome: Implications for initial events in amyloid plaque formation. Neurobiology of Disease. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- Lesne S, Kotilinek L, Ashe KH. Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience. 2008;151:745–749. doi: 10.1016/j.neuroscience.2007.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L. Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience. 1997;78:135–146. doi: 10.1016/s0306-4522(96)00553-2. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, St Clair DK, LeVine H, 3rd, Keller JN. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Murphy MP, LeVine H., 3rd Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis. 2010;19(1):311–23. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistor M, Don M, Parekh M, Sarsoza F, Goodus M, Lopez GE, Kawas C, Leverenz J, Doran E, Lott IT, Hill M, Head E. Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain. Neurobiol Aging. 2007;28:1493–1506. doi: 10.1016/j.neurobiolaging.2006.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Rofina JE, Singh K, Skoumalova-Vesela A, van Ederen AM, van Asten AJ, Wilhelm J, Gruys E. Histochemical accumulation of oxidative damage products is associated with Alzheimer-like pathology in the canine. Amyloid. 2004;11:90–100. doi: 10.1080/13506120412331285779. [DOI] [PubMed] [Google Scholar]
- Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci. 1993;90(22):10836–40. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher AE, Esh CL, Kokjohn TA, Castaño EM, Van Vickle GD, Kalback WM, Patton RL, Luehrs DC, Daugs ID, Kuo YM, Emmerling MR, Soares H, Quinn JF, Kaye J, Connor DJ, Silverberg NB, Adler CH, Seward JD, Beach TG, Sabbagh MN. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement. 2009;5(1):18–29. doi: 10.1016/j.jalz.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MJ, Bobik M, White RG, Hou Y, Benjamin SA, Geddes JW. Age-specific onset of beta-amyloid in beagle brains. Neurobiol Aging. 1996;17:269–273. doi: 10.1016/0197-4580(95)02072-1. [DOI] [PubMed] [Google Scholar]
- Russo C, Salis S, Dolcini V, Venezia V, Song XH, Teller JK, Schettini G. Amino-terminal modification and tyrosine phosphorylation of [corrected] carboxy-terminal fragments of the amyloid precursor protein in Alzheimer’s disease and Down’s syndrome brain. Neurobiol Dis. 2001;8:173–180. doi: 10.1006/nbdi.2000.0357. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A, Kuwamura M, Umemura T, Takada K, Ohama E, Itakura C. Modified Bielschowsky and immunohistochemical studies on senile plaques in aged dogs. Neurosci Lett. 1991;129:25–28. doi: 10.1016/0304-3940(91)90712-3. [DOI] [PubMed] [Google Scholar]
- Smith-Swintosky VL, Pettigrew LC, Craddock SD, Culwell AR, Rydel RE, Mattson MP. Secreted forms of beta-amyloid precursor protein protect against ischemic brain injury. J Neurochem. 1994;63:781–784. doi: 10.1046/j.1471-4159.1994.63020781.x. [DOI] [PubMed] [Google Scholar]
- Smith CD, Chebrolu H, Wekstein DR, Schmitt FA, Jicha GA, Cooper G, Markesbery WR. Brain structural alterations before mild cognitive impairment. Neurology. 2007;68:1268–1273. doi: 10.1212/01.wnl.0000259542.54830.34. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinerman JR, Irizarry M, Scarmeas N, Raju S, Brandt J, Albert M, Blacker D, Hyman B, Stern Y. Distinct pools of beta-amyloid in Alzheimer disease-affected brain: a clinicopathologic study. Arch Neurol. 2008;65(7):906–12. doi: 10.1001/archneur.65.7.906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoub TR, Rogalski EJ, Leurgans S, Bennett DA, Detoledo-Morrell L. Rate of entorhinal and hippocampal atrophy in incipient and mild AD: Relation to memory function. Neurobiol Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KS, Vester BM, Apanavicius CJ, Kirby NA, Schook LB. Implications of age and diet on canine cerebral cortex transcription. Neurobiol Aging. 2009;30:1314–1326. doi: 10.1016/j.neurobiolaging.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Tekirian TL, Cole GM, Russell MJ, Yang F, Wekstein DR, Patel E, Snowdon DA, Markesbery WR, Geddes JW. Carboxy terminal of beta-amyloid deposits in aged human, canine, and polar bear brains. Neurobiol Aging. 1996;17:249–257. doi: 10.1016/0197-4580(95)02062-4. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler SJ, Dawbarn D, Wilcock GK, Allen SJ. Alzheimer’s disease. Biochem Biophys Res Commun. 2002;299:373–376. doi: 10.1016/s0006-291x(02)02635-9. [DOI] [PubMed] [Google Scholar]
- Wang DS, Iwata N, Hama E, Saido TC, Dickson DW. Oxidized neprilysin in aging and Alzheimer’s disease brains. Biochem Biophys Res Commun. 2003;310:236–241. doi: 10.1016/j.bbrc.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The role of A beta 42 in Alzheimer’s disease. J Physiol Paris. 1998;92:289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Detection of soluble oligomers by Western blot. The A11 antibody was used to detect oligomers of various sizes in the canine, human, and mouse brain extracts as well as an Aβ peptide control. Three samples of canine temporal cortex were serially extracted in PBS, RIPA and FA. The A11 antibody detects the majority of canine oligomeric bands in the soluble PBS fraction, a few oligomers in the soluble RIPA fraction, and no staining is seen in the FA fraction. For comparison, soluble RIPA protein samples are included from two Down Syndrome patients, a transgenic mouse, and an Aβ peptide aggregate preparation. Notably, a band at 56kDa appears in canine, human, mouse, and Aβ peptide preparations. A band for monomeric Aβ can be seen at 4kDa in the Aβ peptide sample, with multiple bands indicative of aggregated Aβ at higher kDa ranges. (numbers on left=kiloDalton marker, PBS=phosphate buffered saline, RIPA=radioimmunoprecipitation assay buffer, FA=70% formic acid buffer, DS=Down Syndrome RIPA brain extract, Tg=mouse Tg2576 RIPA brain extract, Aβ=aggregated beta-amyloid peptide)
Supplemental Figure 2. Detection of specific Aβ oligomers in the canine brain. (A) A11 detects only the 56kDa band in temporal cortex of young and old canines, specifically in the soluble PBS samples after IP with 3μl of anti-Aβ 6E10 antibody (+), but multiple bands are detected in untreated samples (−). (B) In another experiment, samples were further concentrated by an IP with 5μl of the anti-Aβ antibodies 4G8 or 6E10 (+). A11 staining again reveals the 56kDa band. (numbers on left in A and B=kiloDalton marker, PBS=phosphate buffered saline, IP=immunoprecipitation, Y=young, O=old)
Supplemental Figure 3. Soluble proteins and quantification by dot blot. (A) Representative dot blot of proteins in the soluble PBS fraction of the parietal cortex as detected with the secondary-only antibody, and two anti-oligomer antibodies M204 and I11 (kindly provided by Dr. C. Glabe, UCI). (B) Following subtraction of background reactivity, quantification reveals a large amount of individual variability, with significant increases in soluble oligomers with the M204 and I11 antibodies. (2° only=anti-rabbit secondary, M204=monoclonal rabbit anti-oligomer antibodies, I11=polyclonal rabbit anti-oligomer antibody, bars in B=group mean+/−SEM, circles in B=individual data points)